
The Role of Leaky Gut in Nonalcoholic Fatty Liver Disease: A Novel Therapeutic Target

 , , , , ,
, , , , , {kind=link}
{kind=link}
Abstract
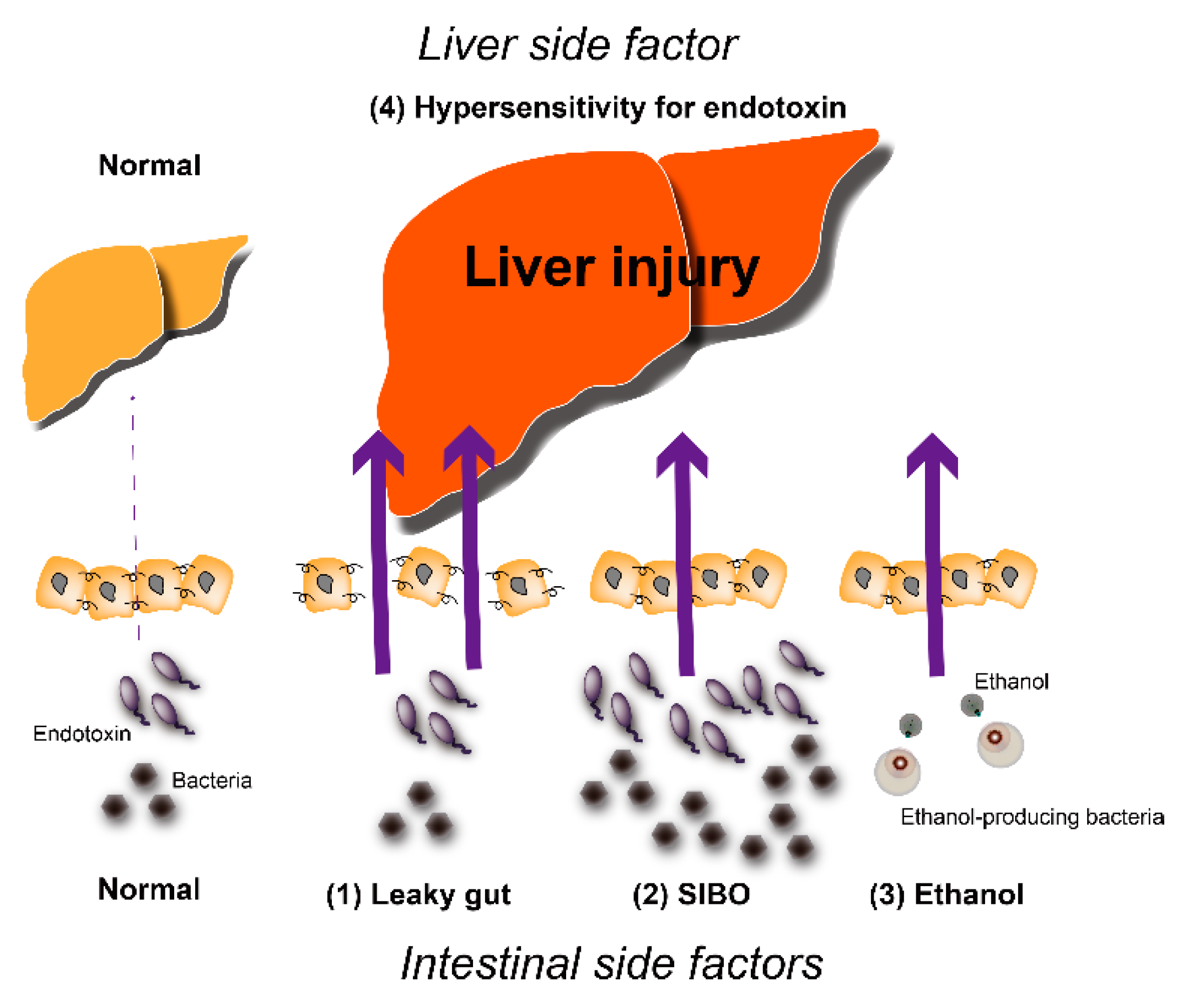
:1. Introduction
2. Hepatic Inflammation and NAFLD
3. Endotoxins and NAFLD
4. Intestinal Factors Associated with Endotoxins
4.1. Intestinal Permeability
4.2. SIBO
4.3. Ethanol-Producing Bacteria
5. Hepatic Factors Associated with Endotoxins
5.1. Enhanced Response to Endotoxin
5.2. Responses to Gut-Derived Ethanol
6. NASH/NAFLD Treatment Focused on the Gut
6.1. Probiotics and Prebiotics
6.2. Fecal Microbiota Transplantation
6.3. Anti-LPS Immunoglobulins
6.4. Vitamin B6
6.5. Vitamin D
6.6. Constipation Drug Lubiprostone (LUB)
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Loomba, R.; Sanyal, A.J. The global NAFLD epidemic. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 686–690. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clemente, M.G.; Mandato, C.; Poeta, M.; Vajro, P. Pediatric non-alcoholic fatty liver disease: Recent solutions, unresolved issues, and future research directions. World J. Gastroenterol. 2016, 22, 8078–8093. [Google Scholar] [CrossRef] [PubMed]
- Rotman, Y.; Sanyal, A.J. Current and upcoming pharmacotherapy for non-alcoholic fatty liver disease. Gut 2017, 66, 180–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, R.; Azevedo, I. Chronic inflammation in obesity and the metabolic syndrome. Mediat. Inflamm. 2010, 2010, 289645. [Google Scholar] [CrossRef]
- Wellen, K.E.; Hotamisligil, G.S. Inflammation, stress, and diabetes. J. Clin. Investig. 2005, 115, 1111–1119. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef]
- Racanelli, V.; Rehermann, B. The liver as an immunological organ. Hepatology 2006, 43 (Suppl. 1), S54–S62. [Google Scholar] [CrossRef]
- Akira, S.; Takeda, K.; Kaisho, T. Toll-like receptors: Critical proteins linking innate and acquired immunity. Nat. Immunol. 2001, 2, 675–680. [Google Scholar] [CrossRef]
- Mencin, A.; Kluwe, J.; Schwabe, R.F. Toll-like receptors as targets in chronic liver diseases. Gut 2009, 58, 704–720. [Google Scholar] [CrossRef]
- Szabo, G.; Bala, S. Alcoholic liver disease and the gut-liver axis. World J. Gastroenterol. 2010, 16, 1321–1329. [Google Scholar] [CrossRef]
- O’Neill, L.A.; Bowie, A.G. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 2007, 7, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Kagan, J.C.; Medzhitov, R. Phosphoinositide-mediated adaptor recruitment controls Toll-like receptor signaling. Cell 2006, 125, 943–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzgerald, K.A.; Chen, Z.J. Sorting out Toll signals. Cell 2006, 125, 834–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seki, E.; Brenner, D.A. Toll-like receptors and adaptor molecules in liver disease: Update. Hepatology 2008, 48, 322–335. [Google Scholar] [CrossRef] [PubMed]
- Szabo, G.; Dolganiuc, A.; Mandrekar, P. Pattern recognition receptors: A contemporary view on liver diseases. Hepatology 2006, 44, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Su, G.L.; Klein, R.D.; Aminlari, A.; Zhang, H.Y.; Steinstraesser, L.; Alarcon, W.H.; Remick, D.G.; Wang, S.C. Kupffer cell activation by lipopolysaccharide in rats: Role for lipopolysaccharide binding protein and toll-like receptor 4. Hepatology 2000, 31, 932–936. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Wheeler, M.D.; Kono, H.; Bradford, B.U.; Gallucci, R.M.; Luster, M.I.; Thurman, R.G. essential role of tumor necrosis factor alpha in alcohol-induced liver injury in mice. Gastroenterology 1999, 117, 942–952. [Google Scholar] [CrossRef]
- Santucci, L.; Fiorucci, S.; Chiorean, M.; Brunori, P.M.; Di Matteo, F.M.; Sidoni, A.; Migliorati, G.; Morelli, A. Interleukin 10 reduces lethality and hepatic injury induced by lipopolysaccharide in galactosamine-sensitized mice. Gastroenterology 1996, 111, 736–744. [Google Scholar] [CrossRef] [PubMed]
- Dela Peña, A.; Leclercq, I.; Field, J.; George, J.; Jones, B.; Farrell, G. NF-KappaB activation, rather than TNF, mediates hepatic inflammation in a murine dietary model of steatohepatitis. Gastroenterology 2005, 129, 1663–1674. [Google Scholar] [CrossRef]
- Leclercq, I.A.; Farrell, G.C.; Sempoux, C.; dela Peña, A.; Horsmans, Y. Curcumin inhibits NF-KappaB activation and reduces the severity of experimental steatohepatitis in mice. J. Hepatol. 2004, 41, 926–934. [Google Scholar] [CrossRef] [PubMed]
- Koca, S.S.; Bahcecioglu, I.H.; Poyrazoglu, O.K.; Ozercan, I.H.; Sahin, K.; Ustundag, B. The treatment with antibody of TNF-alpha reduces the inflammation, necrosis and fibrosis in the non-alcoholic steatohepatitis induced by methionine- and choline-deficient diet. Inflammation 2008, 31, 91–98. [Google Scholar] [CrossRef]
- Tomita, K.; Tamiya, G.; Ando, S.; Ohsumi, K.; Chiyo, T.; Mizutani, A.; Kitamura, N.; Toda, K.; Kaneko, T.; Horie, Y.; et al. Tumour necrosis factor alpha signalling through activation of Kupffer cells plays an essential role in liver fibrosis of non-alcoholic steatohepatitis in mice. Gut 2006, 55, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003, 3, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Soloski, M.J.; Diehl, A.M. Dietary factors alter hepatic innate immune system in mice with nonalcoholic fatty liver disease. Hepatology 2005, 42, 880–885. [Google Scholar] [CrossRef]
- Guebre-Xabier, M.; Yang, S.; Lin, H.Z.; Schwenk, R.; Krzych, U.; Diehl, A.M. Altered hepatic lymphocyte subpopulations in obesity-related murine fatty livers: Potential mechanism for sensitization to liver damage. Hepatology 2000, 31, 633–640. [Google Scholar] [CrossRef]
- Kremer, M.; Hines, I.N.; Milton, R.J.; Wheeler, M.D. Favored T helper 1 response in a mouse model of hepatosteatosis is associated with enhanced T cell-mediated hepatitis. Hepatology 2006, 44, 216–227. [Google Scholar] [CrossRef]
- Xu, J.; Gordon, J.I. Honor thy symbionts. Proc. Natl. Acad. Sci. USA 2003, 100, 10452–10459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harte, A.L.; da Silva, N.F.; Creely, S.J.; McGee, K.C.; Billyard, T.; Youssef-Elabd, E.M.; Tripathi, G.; Ashour, E.; Abdalla, M.S.; Sharada, H.M.; et al. Elevated endotoxin levels in non-alcoholic fatty liver disease. J. Inflamm. 2010, 7, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pendyala, S.; Walker, J.M.; Holt, P.R. A high-fat diet is associated with endotoxemia that originates from the gut. Gastroenterology 2012, 142, 1100–1101.e2. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Bjarnason, I.; Takeuchi, K.; Bjarnason, A.; Adler, S.N.; Teahon, K. The G.U.T. of gut. Scand. J. Gastroenterol. 2004, 39, 807–815. [Google Scholar] [CrossRef]
- Zeuzem, S. Gut-liver axis. Int. J. Colorectal Dis. 2000, 15, 59–82. [Google Scholar] [CrossRef] [PubMed]
- Dumas, M.E.; Barton, R.H.; Toye, A.; Cloarec, O.; Blancher, C.; Rothwell, A.; Fearnside, J.; Tatoud, R.; Blanc, V.; Lindon, J.C.; et al. Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc. Natl. Acad. Sci. USA 2006, 103, 12511–12516. [Google Scholar] [CrossRef] [Green Version]
- Solga, S.F.; Diehl, A.M. Non-alcoholic fatty liver disease: Lumen-liver interactions and possible role for probiotics. J. Hepatol. 2003, 38, 681–687. [Google Scholar] [CrossRef]
- Farhadi, A.; Gundlapalli, S.; Shaikh, M.; Frantzides, C.; Harrell, L.; Kwasny, M.M.; Keshavarzian, A. Susceptibility to gut leakiness: A possible mechanism for endotoxaemia in non-alcoholic steatohepatitis. Liver Int. 2008, 28, 1026–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brun, P.; Castagliuolo, I.; Di Leo, V.; Buda, A.; Pinzani, M.; Palù, G.; Martines, D. Increased intestinal permeability in obese mice: New evidence in the pathogenesis of nonalcoholic steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G518–G525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miele, L.; Valenza, V.; La Torre, G.; Montalto, M.; Cammarota, G.; Ricci, R.; Mascianà, R.; Forgione, A.; Gabrieli, M.L.; Perotti, G.; et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 2009, 49, 1877–1887. [Google Scholar] [CrossRef]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef] [Green Version]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, R.; Young, C.; Neu, J. Molecular modulation of intestinal epithelial barrier: Contribution of microbiota. J. Biomed. Biotechnol. 2010, 2010, 305879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghoshal, S.; Witta, J.; Zhong, J.; de Villiers, W.; Eckhardt, E. Chylomicrons promote intestinal absorption of lipopolysaccharides. J. Lipid Res. 2009, 50, 90–97. [Google Scholar] [CrossRef] [Green Version]
- Luther, J.; Garber, J.J.; Khalili, H.; Dave, M.; Bale, S.S.; Jindal, R.; Motola, D.L.; Luther, S.; Bohr, S.; Jeoung, S.W.; et al. Hepatic injury in nonalcoholic steatohepatitis contributes to altered intestinal permeability. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 222–232. [Google Scholar] [CrossRef] [Green Version]
- Nazim, M.; Stamp, G.; Hodgson, H.J. Non-alcoholic steatohepatitis associated with small intestinal diverticulosis and bacterial overgrowth. Hepato-gastroenterology 1989, 36, 349–351. [Google Scholar]
- Lichtman, S.N.; Sartor, R.B.; Keku, J.; Schwab, J.H. Hepatic inflammation in rats with experimental small intestinal bacterial overgrowth. Gastroenterology 1990, 98, 414–423. [Google Scholar] [CrossRef]
- Lichtman, S.N.; Keku, J.; Schwab, J.H.; Sartor, R.B. Hepatic injury associated with small bowel bacterial overgrowth in rats is prevented by metronidazole and tetracycline. Gastroenterology 1991, 100, 513–519. [Google Scholar] [CrossRef]
- Diehl, A.M.; Li, Z.P.; Lin, H.Z.; Yang, S.Q. Cytokines and the pathogenesis of non-alcoholic steatohepatitis. Gut 2005, 54, 303–306. [Google Scholar] [CrossRef] [Green Version]
- Farrell, G.C.; Larter, C.Z. Nonalcoholic fatty liver disease: From steatosis to cirrhosis. Hepatology 2006, 43 (Suppl. 1), S99–S112. [Google Scholar] [CrossRef] [PubMed]
- Shanab, A.A.; Scully, P.; Crosbie, O.; Buckley, M.; O’Mahony, L.; Shanahan, F.; Gazareen, S.; Murphy, E.; Quigley, E.M. Small intestinal bacterial overgrowth in nonalcoholic steatohepatitis: Association with toll-like receptor 4 expression and plasma levels of interleukin 8. Dig. Dis. Sci. 2011, 56, 1524–1534. [Google Scholar] [CrossRef]
- Grover, M.; Kanazawa, M.; Palsson, O.S.; Chitkara, D.K.; Gangarosa, L.M.; Drossman, D.A.; Whitehead, W.E. Small intestinal bacterial overgrowth in irritable bowel syndrome: Association with colon motility, bowel symptoms, and psychological distress. Neurogastroenterol. Motil. 2008, 20, 998–1008. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.C. Small intestinal bacterial overgrowth: A framework for understanding irritable bowel syndrome. JAMA 2004, 292, 852–858. [Google Scholar] [CrossRef]
- Lupascu, A.; Gabrielli, M.; Lauritano, E.C.; Scarpellini, E.; Santoliquido, A.; Cammarota, G.; Flore, R.; Tondi, P.; Pola, P.; Gasbarrini, G.; et al. Hydrogen glucose breath test to detect small intestinal bacterial overgrowth: A prevalence case–control study in irritable bowel syndrome. Aliment. Pharmacol. Ther. 2005, 22, 1157–1160. [Google Scholar] [CrossRef] [PubMed]
- Pimentel, M.; Chow, E.J.; Lin, H.C. Eradication of small intestinal bacterial overgrowth reduces symptoms of irritable bowel syndrome. Am. J. Gastroenterol. 2000, 95, 3503–3506. [Google Scholar] [CrossRef]
- Pimentel, M.; Chow, E.J.; Lin, H.C. Normalization of lactulose breath testing correlates with symptom improvement in irritable bowel syndrome. a double-blind, randomized, placebo controlled study. Am. J. Gastroenterol. 2003, 98, 412–419. [Google Scholar] [PubMed]
- Pimentel, M.; Mayer, A.G.; Park, S.; Chow, E.J.; Hasan, A.; Kong, Y. Methane production during lactulose breath test is associated with gastrointestinal disease presentation. Dig. Dis. Sci. 2003, 48, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Pimentel, M.; Wallace, D.; Hallegua, D.; Chow, E.; Kong, Y.; Park, S.; Lin, H.C. A link between irritable bowel syndrome and fibromyalgia may be related to findings on lactulose breath testing. Ann. Rheum. Dis. 2004, 63, 450–452. [Google Scholar] [CrossRef] [Green Version]
- Sabaté, J.M.; Jouët, P.; Harnois, F.; Mechler, C.; Msika, S.; Grossin, M.; Coffin, B. High prevalence of small intestinal bacterial overgrowth in patients with morbid obesity: A contributor to severe hepatic steatosis. Obes. Surg. 2008, 18, 371–377. [Google Scholar] [CrossRef]
- Scarpellini, E.; Giorgio, V.; Gabrielli, M.; Lauritano, E.C.; Pantanella, A.; Fundarò, C.; Gasbarrini, A. Prevalence of small intestinal bacterial overgrowth in children with irritable bowel syndrome: A case–control study. J. Pediatr. 2009, 155, 416–420. [Google Scholar] [CrossRef] [PubMed]
- Wigg, A.J.; Roberts-Thomson, I.C.; Dymock, R.B.; McCarthy, P.J.; Grose, R.H.; Cummins, A.G. The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor alpha in the pathogenesis of non-alcoholic steatohepatitis. Gut 2001, 48, 206–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.C.; Zhao, W.; Li, S. Small intestinal bacteria overgrowth decreases small intestinal motility in the NASH rats. World J. Gastroenterol. 2008, 14, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Drenick, E.J.; Fisler, J.; Johnson, D. Hepatic steatosis after intestinal bypass. Prevention and reversal by metronidazole, irrespective of protein calorie malnutrition. Gastroenterology 1982, 82, 535–548. [Google Scholar] [CrossRef]
- Vanderhoof, J.A.; Tuma, D.J.; Antonson, D.L.; Sorrell, M.F. Effect of antibiotics in the prevention of jejunoileal bypass-induced liver dysfunction. Digestion 1982, 23, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Bergheim, I.; Weber, S.; Vos, M.; Krämer, S.; Volynets, V.; Kaserouni, S.; McClain, C.J.; Bischoff, S.C. Antibiotics protect against fructose-induced hepatic lipid accumulation in mice: Role of endotoxin. J. Hepatol. 2008, 48, 983–992. [Google Scholar] [CrossRef]
- Leung, C.; Rivera, L.; Furness, J.B.; Angus, P.W. The role of the gut microbiota in NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 412–425. [Google Scholar] [CrossRef]
- Dawes, E.A.; Foster, S.M. The formation of ethanol in Escherichia coli. Biochim. Biophys. Acta 1956, 22, 253–265. [Google Scholar] [CrossRef]
- Baker, S.S.; Baker, R.D.; Liu, W.; Nowak, N.J.; Zhu, L. Role of alcohol metabolism in non-alcoholic steatohepatitis. PLoS ONE 2010, 5, e9570. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Baker, R.D.; Zhu, R.; Baker, S.S. Gut microbiota produce alcohol and contribute to NAFLD. Gut 2016, 65, 1232. [Google Scholar] [CrossRef]
- Guercio Nuzio, S.; Di Stasi, M.; Pierri, L.; Troisi, J.; Poeta, M.; Bisogno, A.; Belmonte, F.; Tripodi, M.; Di Salvio, D.; Massa, G.; et al. Multiple gut-liver axis abnormalities in children with obesity with and without hepatic involvement. Pediatr. Obes. 2017, 12, 446–452. [Google Scholar] [CrossRef]
- Zhao, L.F.; Jia, J.M.; Han, D.W. The role of enterogenous endotoxemia in the pathogenesis of non-alcoholic steatohepatitis. Zhonghua Gan Zang Bing Za Zhi Zhonghua Ganzangbing Zazhi Chin. J. Hepatol. 2004, 12, 632. [Google Scholar]
- Chitturi, S.; Farrell, G.C. Etiopathogenesis of nonalcoholic steatohepatitis. Semin. Liver Dis. 2001, 21, 27–41. [Google Scholar] [CrossRef]
- Kudo, H.; Takahara, T.; Yata, Y.; Kawai, K.; Zhang, W.; Sugiyama, T. Lipopolysaccharide triggered TNF-alpha-induced hepatocyte apoptosis in a murine non-alcoholic steatohepatitis model. J. Hepatol. 2009, 51, 168–175. [Google Scholar] [CrossRef]
- Verdam, F.J.; Rensen, S.S.; Driessen, A.; Greve, J.W.; Buurman, W.A. Novel evidence for chronic exposure to endotoxin in human nonalcoholic steatohepatitis. J. Clin. Gastroenterol. 2011, 45, 149–152. [Google Scholar] [CrossRef]
- Loguercio, C.; De Simone, T.; D’Auria, M.V.; de Sio, I.; Federico, A.; Tuccillo, C.; Abbatecola, A.M.; Del Vecchio, B.C.; Italian AISF Clinical Group. Non-alcoholic fatty liver disease: A multicentre clinical study by the Italian Association for the Study of the Liver. Dig. Liver Dis. 2004, 36, 398–405. [Google Scholar] [CrossRef]
- Ansell, J.; Widrich, W.; Johnson, W.; Fine, J. Endotoxin and bacteria in portal blood. Gastroenterology 1977, 73, 1190. [Google Scholar] [CrossRef]
- Imajo, K.; Fujita, K.; Yoneda, M.; Nozaki, Y.; Ogawa, Y.; Shinohara, Y.; Kato, S.; Mawatari, H.; Shibata, W.; Kitani, H.; et al. Hyperresponsivity to low-dose endotoxin during progression to nonalcoholic steatohepatitis is regulated by leptin-mediated signaling. Cell Metab. 2012, 16, 44–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Liu, T.; Rose, J.L.; Stevens, R.L.; Hoyt, D.G. Sensitivity of mice to lipopolysaccharide is increased by a high saturated fat and cholesterol diet. J. Inflamm. 2007, 4, 22. [Google Scholar] [CrossRef] [Green Version]
- Roncon-Albuquerque, R., Jr.; Moreira-Rodrigues, M.; Faria, B.; Ferreira, A.P.; Cerqueira, C.; Lourenço, A.P.; Pestana, M.; von Hafe, P.; Leite-Moreira, A.F. Attenuation of the cardiovascular and metabolic complications of obesity in CD14 knockout mice. Life Sci. 2008, 83, 502–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delude, R.L.; Savedra, R., Jr.; Zhao, H.; Thieringer, R.; Yamamoto, S.; Fenton, M.J.; Golenbock, D.T. CD14 enhances cellular responses to endotoxin without imparting ligand-specific recognition. Proc. Natl. Acad. Sci. USA 1995, 92, 9288–9292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrero, E.; Jiao, D.; Tsuberi, B.Z.; Tesio, L.; Rong, G.W.; Haziot, A.; Goyert, S.M. Transgenic mice expressing human CD14 are hypersensitive to lipopolysaccharide. Proc. Natl. Acad. Sci. USA 1993, 90, 2380–2384. [Google Scholar] [CrossRef] [Green Version]
- Haziot, A.; Ferrero, E.; Lin, X.Y.; Stewart, C.L.; Goyert, S.M. CD14-deficient mice are exquisitely insensitive to the effects of LPS. Prog. Clin. Biol. Res. 1995, 392, 349–351. [Google Scholar]
- Su, G.L. Lipopolysaccharides in liver injury: Molecular mechanisms of Kupffer cell activation. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 283, G256–G265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, S.D.; Ramos, R.A.; Tobias, P.S.; Ulevitch, R.J.; Mathison, J.C. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science 1990, 249, 1431–1433. [Google Scholar] [CrossRef] [PubMed]
- Brun, P.; Castagliuolo, I.; Floreani, A.R.; Buda, A.; Blasone, L.; Palù, G.; Martines, D. Increased risk of NASH in patients carrying the C(−159)T polymorphism in the CD14 gene promoter region. Gut 2006, 55, 1212. [Google Scholar] [CrossRef] [Green Version]
- Considine, R.V.; Sinha, M.K.; Heiman, M.L.; Kriauciunas, A.; Stephens, T.W.; Nyce, M.R.; Ohannesian, J.P.; Marco, C.C.; McKee, L.J.; Bauer, T.L.; et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N. Engl. J. Med. 1996, 334, 292–295. [Google Scholar] [CrossRef]
- Halaas, J.L.; Gajiwala, K.S.; Maffei, M.; Cohen, S.L.; Chait, B.T.; Rabinowitz, D.; Lallone, R.L.; Burley, S.K.; Friedman, J.M. Weight-reducing effects of the plasma protein encoded by the obese gene. Science 1995, 269, 543–546. [Google Scholar] [CrossRef] [PubMed]
- Kessoku, T.; Imajo, K.; Honda, Y.; Kato, T.; Ogawa, Y.; Tomeno, W.; Kato, S.; Mawatari, H.; Fujita, K.; Yoneda, M.; et al. Resveratrol ameliorates fibrosis and inflammation in a mouse model of nonalcoholic steatohepatitis. Sci. Rep. 2016, 6, 22251. [Google Scholar] [CrossRef] [Green Version]
- Sarkola, T.; Eriksson, C.J. Effect of 4-methylpyrazole on endogenous plasma ethanol and methanol levels in humans. Alcohol. Clin. Exp. Res. 2001, 25, 513–516. [Google Scholar] [CrossRef] [PubMed]
- Blomstrand, R. Observations of the formation of ethanol in the intestinal tract in man. Life Sci. II 1971, 10, 575–582. [Google Scholar] [CrossRef]
- Watanabe-Suzuki, K.; Seno, H.; Ishii, A.; Kumazawa, T.; Suzuki, O. Ultrasensitive method for determination of ethanol in whole blood by headspace capillary gas chromatography with cryogenic oven trapping. J. Chromatogr. B. Biomed. Sci. Appl. 1999, 727, 89–94. [Google Scholar] [CrossRef]
- Engeland, K.; Maret, W. Extrahepatic, differential expression of four classes of human alcohol dehydrogenase. Biol. Chem. Biophys. Res. Commun. 1993, 193, 47–53. [Google Scholar] [CrossRef]
- Cope, K.; Risby, T.; Diehl, A.M. Increased gastrointestinal ethanol production in obese mice: Implications for fatty liver disease pathogenesis. Gastroenterology 2000, 119, 1340–1347. [Google Scholar] [CrossRef]
- Ruiz, A.G.; Casafont, F.; Crespo, J.; Cayon, A.; Mayorga, M.; Estebanez, A.; Fernadez-Escalante, J.C.; Pons-Romero, F. Lipopolysaccharide-binding protein plasma levels and liver TNFalpha gene expression in obese patients: Evidence for the potential role of endotoxin in the pathogenesis of non-alcoholic steatohepatitis. Obes. Surg. 2007, 17, 1374–1380. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.K.; Seth, A.; Sheth, P. Recent advances in alcoholic liver disease I. Role of intestinal permeability and endotoxemia in alcoholic liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, G881–G884. [Google Scholar] [CrossRef] [PubMed]
- Volynets, V.; Kuper, M.A.; Strahl, S.; Maier, I.B.; Spruss, A.; Wagnerberger, S.; Königsrainer, A.; Bischoff, S.C.; Bergheim, I. Nutrition, intestinal permeability, and blood ethanol levels are altered in patients with nonalcoholic fatty liver disease (NAFLD). Dig. Dis. Sci. 2012, 57, 1932–1941. [Google Scholar] [CrossRef] [PubMed]
- Rafter, J.; Bennett, M.; Caderni, G.; Clune, Y.; Hughes, R.; Karlsson, P.C.; Klinder, A.; O’Riordan, M.; O’Sullivan, G.C.; Pool-Zobel, B.; et al. Dietary synbiotics reduce cancer risk factors in polypectomized and colon cancer patients. Am. J. Clin. Nutr. 2007, 85, 488–496. [Google Scholar] [CrossRef]
- Esposito, E.; Iacono, A.; Bianco, G.; Autore, G.; Cuzzocrea, S.; Vajro, P.; Canani, R.B.; Calignano, A.; Raso, G.M.; Meli, R. Probiotics reduce the inflammatory response induced by a high-fat diet in the liver of young rats. J. Nutr. 2009, 139, 905–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loguercio, C.; De Simone, T.; Federico, A.; Terracciano, F.; Tuccillo, C.; Di Chicco, M.; Cartenì, M. Gut-liver axis: A new point of attack to treat chronic liver damage? Am. J. Gastroenterol. 2002, 97, 2144–2146. [Google Scholar] [CrossRef]
- Loguercio, C.; Federico, A.; Tuccillo, C.; Terracciano, F.; D’Auria, M.V.; De Simone, C.; Del Vecchio Blanco, C. Beneficial effects of a probiotic VSL #3 on parameters of liver dysfunction in chronic liver diseases. J. Clin. Gastroenterol. 2005, 39, 540–543. [Google Scholar]
- Socha, P.; Horvath, A.; Vajro, P.; Dziechciarz, P.; Dhawan, A.; Szajewska, H. Pharmacological interventions for nonalcoholic fatty liver disease in adults and in children: A systematic review. J. Pediatr. Gastroenterol. Nutr. 2009, 48, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Aller, R.; De Luis, D.A.; Izaola, O.; Conde, R.; Gonzalez Sagrado, M.; Primo, D.; De La Fuente, B.; Gonzalez, J. Effect of a probiotic on liver aminotransferases in nonalcoholic fatty liver disease patients: A double blind randomized clinical trial. Eur. Rev. Med. Pharmacol. Sci. 2011, 15, 1090–1095. [Google Scholar] [PubMed]
- Vajro, P.; Mandato, C.; Licenziati, M.R.; Franzese, A.; Vitale, D.F.; Lenta, S.; Caropreso, M.; Vallone, G.; Meli, R. Effects of Lactobacillus rhamnosus strain GG in pediatric obesity-related liver disease. J. Pediatr. Gastroenterol. Nutr. 2011, 52, 740–743. [Google Scholar] [CrossRef] [Green Version]
- Roberfroid, M.; Gibson, G.R.; Hoyles, L.; McCartney, A.L.; Rastall, R.; Rowland, I.; Wolvers, D.; Watzl, B.; Szajewska, H.; Stahl, B.; et al. Prebiotic effects: Metabolic and health benefits. Br. J. Nutr. 2010, 104 (Suppl. 2), S1–S63. [Google Scholar] [CrossRef] [Green Version]
- Parnell, J.A.; Reimer, R.A. Prebiotic fiber modulation of the gut microbiota improves risk factors for obesity and the metabolic syndrome. Gut Microbes 2012, 3, 29–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adar, T.; Ben Ya’acov, A.; Lalazar, G.; Lichtenstein, Y.; Nahman, D.; Mizrahi, M.; Wong, V.; Muller, B.; Rawlin, G.; Ilan, Y. Oral administration of immunoglobulin G-enhanced colostrum alleviates insulin resistance and liver injury and is associated with alterations in natural killer T cells. Clin. Exp. Immunol. 2012, 167, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Ben Ya’acov, A.; Lichtenstein, Y.; Zolotarov, L.; Ilan, Y. The gut microbiome as a target for regulatory T cell-based immunotherapy: Induction of regulatory lymphocytes by oral administration of anti-LPS enriched colostrum alleviates immune mediated colitis. BMC Gastroenterol. 2015, 15, 154. [Google Scholar]
- Mizrahi, M.; Shabat, Y.; Ben Ya’acov, A.; Lalazar, G.; Adar, T.; Wong, V.; Muller, B.; Rawlin, G.; Ilan, Y. Alleviation of insulin resistance and liver damage by oral administration of Imm124-E is mediated by increased Tregs and associated with increased serum GLP-1 and adiponectin: Results of a phase I/II clinical trial in NASH. J. Inflamm. Res. 2012, 5, 141–150. [Google Scholar]
- Kobayashi, T.; Kessoku, T.; Ozaki, A.; Iwaki, M.; Honda, Y.; Ogawa, Y.; Imajo, K.; Yoneda, M.; Saito, S.; Nakajima, A. Vitamin B6 efficacy in the treatment of nonalcoholic fatty liver disease: An open-label, single-arm, single-center trial. J. Clin. Biochem. Nutr. 2021, 68, 181–186. [Google Scholar] [CrossRef]
- Nijhout, H.F.; Gregory, J.F.; Fitzpatrick, C.; Cho, E.; Lamers, K.Y.; Ulrich, C.M.; Reed, M.C. A mathematical model gives insights into the effects of vitamin B-6 deficiency on 1-carbon and glutathione metabolism. J. Nutr. 2009, 139, 784–791. [Google Scholar] [CrossRef] [Green Version]
- Ai, Y.; Sun, Z.; Peng, C.; Liu, L.; Xiao, X.; Li, J. Homocysteine induces hepatic steatosis involving ER stress response in high methionine diet-fed mice. Nutrients 2017, 9, 346. [Google Scholar] [CrossRef] [Green Version]
- Honda, Y.; Kessoku, T.; Sumida, Y.; Kobayashi, T.; Kato, T.; Ogawa, Y.; Tomeno, W.; Imajo, K.; Fujita, K.; Yoneda, M.; et al. Efficacy of glutathione for the treatment of nonalcoholic fatty liver disease: An open-label, single-arm, multicenter, pilot study. BMC Gastroenterol. 2017, 17, 96. [Google Scholar] [CrossRef]
- Su, D.; Nie, Y.; Zhu, A.; Chen, Z.; Wu, P.; Zhang, L.; Luo, M.; Sun, Q.; Cai, L.; Lai, Y.; et al. Vitamin D signaling through induction of paneth cell defensins maintains gut microbiota and improves metabolic disorders and hepatic steatosis in animal models. Front. Physiol. 2016, 7, 498. [Google Scholar] [CrossRef]
- Luger, M.; Kruschitz, R.; Kienbacher, C.; Traussnigg, S.; Langer, F.B.; Schindler, K.; Würger, T.; Wrba, F.; Trauner, M.; Prager, G.; et al. Prevalence of liver fibrosis and its association with non-invasive fibrosis and metabolic markers in morbidly obese patients with vitamin D deficiency. Obes. Surg. 2016, 26, 2425–2432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fei, G.; Raehal, K.; Liu, S.; Qu, M.H.; Sun, X.; Wang, G.D.; Wang, X.Y.; Xia, Y.; Schmid, C.L.; Bohn, L.M.; et al. Lubiprostone reverses the inhibitory action of morphine on intestinal secretion in guinea pig and mouse. J. Pharmacol. Exp. Ther. 2010, 334, 333–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, S.; Kurata, N.; Yamaguchi, A.; Amagase, K.; Takeuchi, K. Lubiprostone prevents nonsteroidal anti-inflammatory drug-induced small intestinal damage by suppressing the expression of inflammatory mediators via EP4 receptors. J. Pharmacol. Exp. Ther. 2014, 349, 470–479. [Google Scholar] [CrossRef] [Green Version]
- Arakawa, K.; Ishigami, T.; Nakai-Sugiyama, M.; Chen, L.; Doi, H.; Kino, T.; Minegishi, S.; Saigoh-Teranaka, S.; Sasaki-Nakashima, R.; Hibi, K.; et al. Lubiprostone as a potential therapeutic agent to improve intestinal permeability and prevent the development of atherosclerosis in apolipoprotein E-deficient mice. PLoS ONE 2019, 14, e0218096. [Google Scholar] [CrossRef] [PubMed]
- Nighot, P.K.; Leung, L.; Ma, T.Y. Chloride channel ClC-2 enhances intestinal epithelial tight junction barrier function via regulation of caveolin-1 and caveolar trafficking of occludin. Exp. Cell Res. 2017, 352, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Kato, T.; Honda, Y.; Kurita, Y.; Iwasaki, A.; Sato, T.; Kessoku, T.; Uchiyama, S.; Ogawa, Y.; Ohkubo, H.; Higurashi, T.; et al. Lubiprostone improves intestinal permeability in humans, a novel therapy for the leaky gut: A prospective randomized pilot study in healthy volunteers. PLoS ONE 2017, 12, e0175626. [Google Scholar] [CrossRef] [Green Version]
- Kessoku, T.; Imajo, K.; Kobayashi, T.; Ozaki, A.; Iwaki, M.; Honda, Y.; Kato, T.; Ogawa, Y.; Tomeno, W.; Kato, S.; et al. Lubiprostone in patients with non-alcoholic fatty liver disease: A randomised, double-blind, placebo-controlled, phase 2a trial. Lancet Gastroenterol. Hepatol. 2020, 5, 996–1007. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kessoku, T.; Kobayashi, T.; Tanaka, K.; Yamamoto, A.; Takahashi, K.; Iwaki, M.; Ozaki, A.; Kasai, Y.; Nogami, A.; Honda, Y.; et al. The Role of Leaky Gut in Nonalcoholic Fatty Liver Disease: A Novel Therapeutic Target. Int. J. Mol. Sci. 2021, 22, 8161. https://doi.org/10.3390/ijms22158161
Kessoku T, Kobayashi T, Tanaka K, Yamamoto A, Takahashi K, Iwaki M, Ozaki A, Kasai Y, Nogami A, Honda Y, et al. The Role of Leaky Gut in Nonalcoholic Fatty Liver Disease: A Novel Therapeutic Target. International Journal of Molecular Sciences. 2021; 22(15):8161. https://doi.org/10.3390/ijms22158161
Chicago/Turabian StyleKessoku, Takaomi, Takashi Kobayashi, Kosuke Tanaka, Atsushi Yamamoto, Kota Takahashi, Michihiro Iwaki, Anna Ozaki, Yuki Kasai, Asako Nogami, Yasushi Honda, and et al. 2021. "The Role of Leaky Gut in Nonalcoholic Fatty Liver Disease: A Novel Therapeutic Target" International Journal of Molecular Sciences 22, no. 15: 8161. https://doi.org/10.3390/ijms22158161