
TGF-β Signaling: From Tissue Fibrosis to Tumor Microenvironment

 , and
, and
Abstract
:1. Introduction
2. TGF-β Signaling
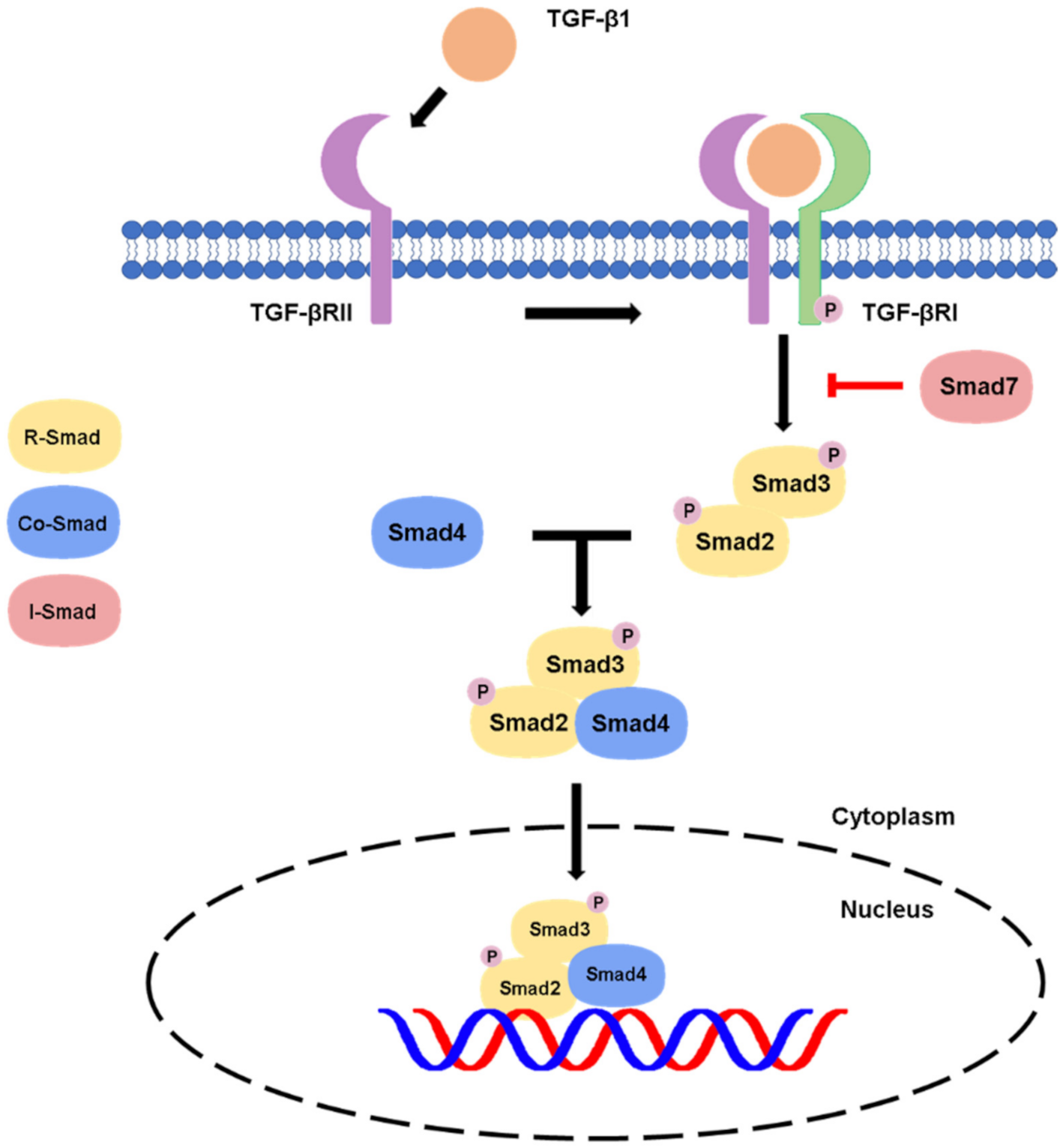
2.1. Canonical Pathway
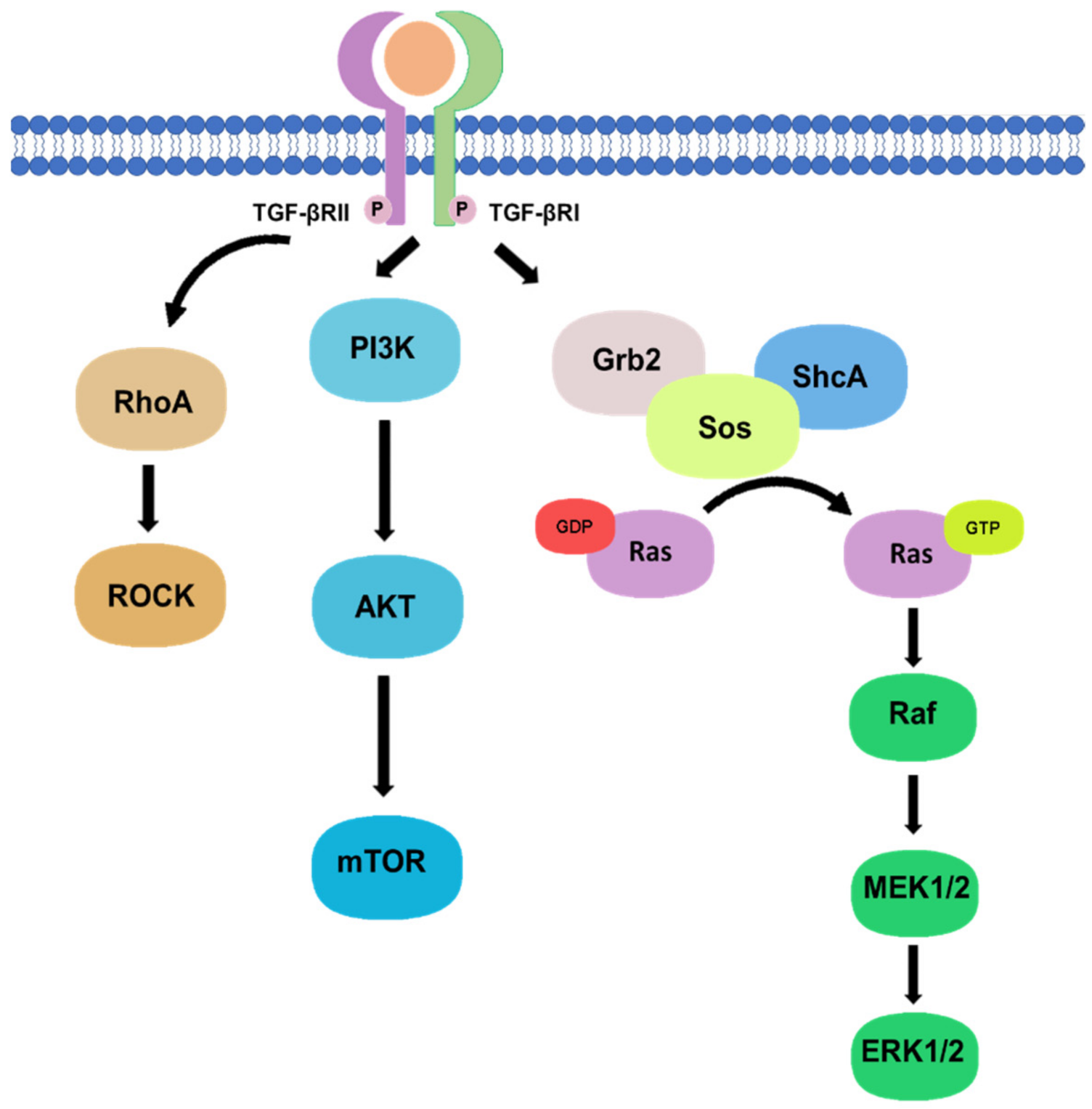
2.2. Non-Canonical Pathway
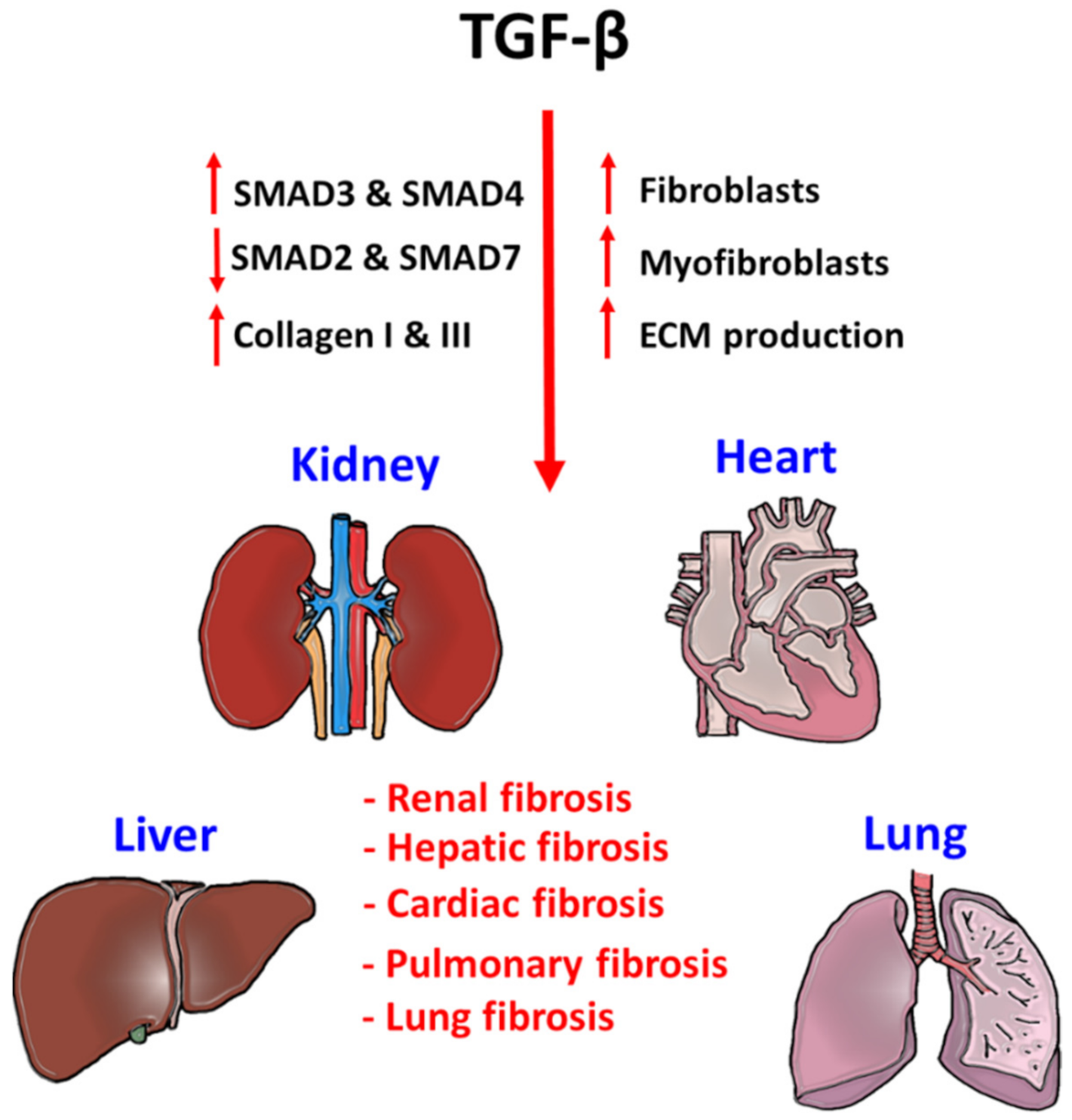
3. TGF-β Signaling in Tissue Fibrosis
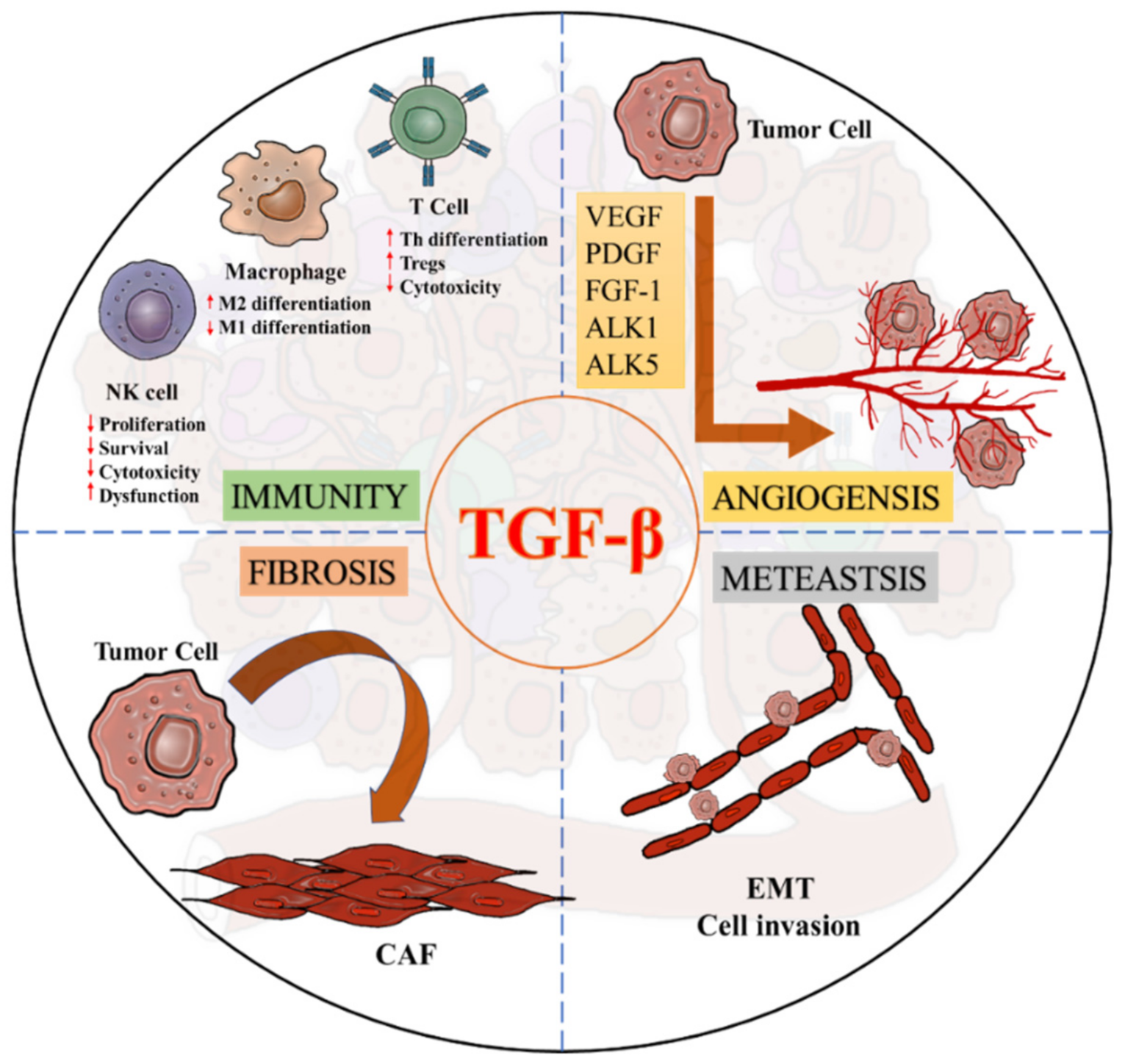
4. TGF-β Signaling in the Tumor Microenvironment
5. Importance of TGF-β Signaling in the Fibrotic TME
5.1 Invasion and Metastasis
5.2. Angiogenesis
5.3. Immunosuppression
5.4. Drug Resistance
6. Future Prospect
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tang, P.M.; Zhang, Y.Y.; Mak, T.S.; Tang, P.C.; Huang, X.R.; Lan, H.Y. Transforming growth factor-beta signalling in renal fibrosis: From Smads to non-coding RNAs. J. Physiol. 2018, 596, 3493–3503. [Google Scholar] [CrossRef] [PubMed]
- Voisin, A.; Damon-Soubeyrand, C.; Bravard, S.; Saez, F.; Drevet, J.R.; Guiton, R. Differential expression and localisation of TGF-beta isoforms and receptors in the murine epididymis. Sci. Rep. 2020, 10, 995. [Google Scholar] [CrossRef] [PubMed]
- Mourskaia, A.A.; Dong, Z.; Ng, S.; Banville, M.; Zwaagstra, J.C.; O’Connor-McCourt, M.D.; Siegel, P.M. Transforming growth factor-beta1 is the predominant isoform required for breast cancer cell outgrowth in bone. Oncogene 2009, 28, 1005–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, X.M.; Tang, P.M.; Li, J.; Lan, H.Y. TGF-beta/Smad signaling in renal fibrosis. Front. Physiol. 2015, 6, 82. [Google Scholar] [CrossRef] [Green Version]
- Prud’homme, G.J. Pathobiology of transforming growth factor beta in cancer, fibrosis and immunologic disease, and therapeutic considerations. Lab. Investig. 2007, 87, 1077–1091. [Google Scholar] [CrossRef] [Green Version]
- Vander Ark, A.; Cao, J.; Li, X. TGF-beta receptors: In and beyond TGF-beta signaling. Cell Signal. 2018, 52, 112–120. [Google Scholar] [CrossRef]
- Zhao, H.; Wei, J.; Sun, J. Roles of TGF-beta signaling pathway in tumor microenvirionment and cancer therapy. Int. Immunopharmacol. 2020, 89 Pt B, 107101. [Google Scholar] [CrossRef]
- David, C.J.; Massague, J. Contextual determinants of TGFbeta action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol. 2018, 19, 419–435. [Google Scholar] [CrossRef]
- Tang, P.M.; Zhou, S.; Meng, X.M.; Wang, Q.M.; Li, C.J.; Lian, G.Y.; Huang, X.R.; Tang, Y.J.; Guan, X.Y.; Yan, B.P.; et al. Smad3 promotes cancer progression by inhibiting E4BP4-mediated NK cell development. Nat. Commun. 2017, 8, 14677. [Google Scholar] [CrossRef] [Green Version]
- Welch-Reardon, K.M.; Ehsan, S.M.; Wang, K.; Wu, N.; Newman, A.C.; Romero-Lopez, M.; Fong, A.H.; George, S.C.; Edwards, R.A.; Hughes, C.C. Angiogenic sprouting is regulated by endothelial cell expression of Slug. J. Cell Sci. 2014, 127 Pt 9, 2017–2028. [Google Scholar]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-beta: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef]
- Tang, P.M.-K.; Tang, P.C.-T.; Chung, J.Y.-F.; Huang, X.-R.; To, K.-F.; Lan, H.-Y. Abstract 1081: Smad3 silences neutrophil anticancer activity in the tumor microenvironment. Cancer Res. 2019, 79 (Suppl. 13), 1081. [Google Scholar]
- Boulter, L.; Bullock, E.; Mabruk, Z.; Brunton, V.G. The fibrotic and immune microenvironments as targetable drivers of metastasis. Br. J. Cancer 2021, 124, 27–36. [Google Scholar] [CrossRef]
- Bierie, B.; Moses, H.L. Tumour microenvironment: TGFbeta: The molecular Jekyll and Hyde of cancer. Nat. Rev. Cancer 2006, 6, 506–520. [Google Scholar] [CrossRef]
- Wakefield, L.M.; Hill, C.S. Beyond TGFbeta: Roles of other TGFbeta superfamily members in cancer. Nat. Rev. Cancer 2013, 13, 328–341. [Google Scholar] [CrossRef]
- Neuzillet, C.; Tijeras-Raballand, A.; Cohen, R.; Cros, J.; Faivre, S.; Raymond, E.; de Gramont, A. Targeting the TGFbeta pathway for cancer therapy. Pharmacol. Ther. 2015, 147, 22–31. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.E. Non-Smad Signaling Pathways of the TGF-beta Family. Cold Spring Harb Perspect. Biol. 2017, 9, a022129. [Google Scholar] [CrossRef]
- Finnson, K.W.; Almadani, Y.; Philip, A. Non-canonical (non-SMAD2/3) TGF-beta signaling in fibrosis: Mechanisms and targets. Semin Cell Dev. Biol. 2020, 101, 115–122. [Google Scholar] [CrossRef]
- Reich, N.; Maurer, B.; Akhmetshina, A.; Venalis, P.; Dees, C.; Zerr, P.; Palumbo, K.; Zwerina, J.; Nevskaya, T.; Gay, S.; et al. The transcription factor Fra-2 regulates the production of extracellular matrix in systemic sclerosis. Arthritis Rheum. 2010, 62, 280–290. [Google Scholar] [CrossRef]
- Xia, H.; Ooi, L.L.; Hui, K.M. MicroRNA-216a/217-induced epithelial-mesenchymal transition targets PTEN and SMAD7 to promote drug resistance and recurrence of liver cancer. Hepatology 2013, 58, 629–641. [Google Scholar] [CrossRef]
- Yang, W.L.; Wang, J.; Chan, C.H.; Lee, S.W.; Campos, A.D.; Lamothe, B.; Hur, L.; Grabiner, B.C.; Lin, X.; Darnay, B.G.; et al. The E3 ligase TRAF6 regulates Akt ubiquitination and activation. Science 2009, 325, 1134–1138. [Google Scholar] [CrossRef] [Green Version]
- Thuault, S.; Valcourt, U.; Petersen, M.; Manfioletti, G.; Heldin, C.H.; Moustakas, A. Transforming growth factor-beta employs HMGA2 to elicit epithelial-mesenchymal transition. J. Cell Biol. 2006, 174, 175–183. [Google Scholar] [CrossRef] [Green Version]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valcourt, U.; Kowanetz, M.; Niimi, H.; Heldin, C.H.; Moustakas, A. TGF-beta and the Smad signaling pathway support transcriptomic reprogramming during epithelial-mesenchymal cell transition. Mol. Biol. Cell 2005, 16, 1987–2002. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Law, B.K.; Chytil, A.M.; Brown, K.A.; Aakre, M.E.; Moses, H.L. Activation of the Erk pathway is required for TGF-beta1-induced EMT in vitro. Neoplasia 2004, 6, 603–610. [Google Scholar] [CrossRef] [Green Version]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. Inflammatory processes in renal fibrosis. Nat. Rev. Nephrol. 2014, 10, 493–503. [Google Scholar] [CrossRef]
- Eddy, A.A.; Neilson, E.G. Chronic kidney disease progression. J. Am. Soc. Nephrol. 2006, 17, 2964–2966. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y. Cellular and molecular mechanisms of renal fibrosis. Nat. Rev. Nephrol. 2011, 7, 684–696. [Google Scholar] [CrossRef]
- Tang, P.C.; Zhang, Y.Y.; Chan, M.K.; Lam, W.W.; Chung, J.Y.; Kang, W.; To, K.F.; Lan, H.Y.; Tang, P.M. The Emerging Role of Innate Immunity in Chronic Kidney Diseases. Int. J. Mol. Sci. 2020, 21, 4018. [Google Scholar] [CrossRef]
- Tang, P.M.; Zhang, Y.Y.; Hung, J.S.; Chung, J.Y.; Huang, X.R.; To, K.F.; Lan, H.Y. DPP4/CD32b/NF-kappaB Circuit: A Novel Druggable Target for Inhibiting CRP-Driven Diabetic Nephropathy. Mol. Ther. 2021, 29, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Tang, P.C.; Chan, A.S.; Zhang, C.B.; Garcia Cordoba, C.A.; Zhang, Y.Y.; To, K.F.; Leung, K.T.; Lan, H.Y.; Tang, P.M. TGF-beta1 Signaling: Immune Dynamics of Chronic Kidney Diseases. Front. Med. 2021, 8, 628519. [Google Scholar] [CrossRef] [PubMed]
- Lan, H.Y. Tubular epithelial-myofibroblast transdifferentiation mechanisms in proximal tubule cells. Curr. Opin. Nephrol. Hypertens. 2003, 12, 25–29. [Google Scholar] [CrossRef]
- Meng, X.M.; Chung, A.C.; Lan, H.Y. Role of the TGF-beta/BMP-7/Smad pathways in renal diseases. Clin. Sci. 2013, 124, 243–254. [Google Scholar] [CrossRef] [Green Version]
- Vindevoghel, L.; Lechleider, R.J.; Kon, A.; de Caestecker, M.P.; Uitto, J.; Roberts, A.B.; Mauviel, A. SMAD3/4-dependent transcriptional activation of the human type VII collagen gene (COL7A1) promoter by transforming growth factor beta. Proc. Natl. Acad. Sci. USA 1998, 95, 14769–14774. [Google Scholar] [CrossRef]
- Chen, S.J.; Yuan, W.; Mori, Y.; Levenson, A.; Trojanowska, M.; Varga, J. Stimulation of type I collagen transcription in human skin fibroblasts by TGF-beta: Involvement of Smad 3. J. Investig. Dermatol. 1999, 112, 49–57. [Google Scholar] [CrossRef] [Green Version]
- Yuan, W.; Varga, J. Transforming growth factor-beta repression of matrix metalloproteinase-1 in dermal fibroblasts involves Smad3. J. Biol. Chem 2001, 276, 38502–38510. [Google Scholar] [CrossRef] [Green Version]
- Hall, M.C.; Young, D.A.; Waters, J.G.; Rowan, A.D.; Chantry, A.; Edwards, D.R.; Clark, I.M. The comparative role of activator protein 1 and Smad factors in the regulation of Timp-1 and MMP-1 gene expression by transforming growth factor-beta 1. J. Biol. Chem. 2003, 278, 10304–10313. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.M.; Huang, X.R.; Chung, A.C.; Qin, W.; Shao, X.; Igarashi, P.; Ju, W.; Bottinger, E.P.; Lan, H.Y. Smad2 protects against TGF-beta/Smad3-mediated renal fibrosis. J. Am. Soc. Nephrol. 2010, 21, 1477–1487. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Puerto, M.C.; Iyengar, P.V.; Garcia de Vinuesa, A.; Ten Dijke, P.; Sanchez-Duffhues, G. Bone morphogenetic protein receptor signal transduction in human disease. J. Pathol. 2019, 247, 9–20. [Google Scholar] [CrossRef]
- Meng, X.M.; Huang, X.R.; Xiao, J.; Chung, A.C.; Qin, W.; Chen, H.Y.; Lan, H.Y. Disruption of Smad4 impairs TGF-beta/Smad3 and Smad7 transcriptional regulation during renal inflammation and fibrosis in vivo and in vitro. Kidney Int. 2012, 81, 266–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebisawa, T.; Fukuchi, M.; Murakami, G.; Chiba, T.; Tanaka, K.; Imamura, T.; Miyazono, K. Smurf1 interacts with transforming growth factor-beta type I receptor through Smad7 and induces receptor degradation. J. Biol. Chem. 2001, 276, 12477–12480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, P.A.; Lin, H.; Wrana, J.L.; Forman-Kay, J.D. An expanded WW domain recognition motif revealed by the interaction between Smad7 and the E3 ubiquitin ligase Smurf2. J. Biol. Chem. 2006, 281, 17069–17075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.Y.; Li, X.Z.; Peng, Y.M.; Liu, H.; Liu, Y.H. Arkadia regulates TGF-beta signaling during renal tubular epithelial to mesenchymal cell transition. Kidney Int. 2008, 73, 588–594. [Google Scholar] [CrossRef] [Green Version]
- Park, N.H.; Song, I.H.; Chung, Y.H. Molecular Pathogenesis of Hepatitis-B-virus-associated Hepatocellular Carcinoma. Gut Liver 2007, 1, 101–117. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; Liu, C.; Zhou, D.; Zhang, L. TGF-beta/SMAD Pathway and Its Regulation in Hepatic Fibrosis. J. Histochem. Cytochem. 2016, 64, 157–167. [Google Scholar] [CrossRef]
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E.; Cushman, M.; Das, S.R.; Deo, R.; de Ferranti, S.D.; Floyd, J.; Fornage, M.; Gillespie, C.; et al. Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association. Circulation 2017, 135, e146–e603. [Google Scholar] [CrossRef]
- Khan, R.; Sheppard, R. Fibrosis in heart disease: Understanding the role of transforming growth factor-beta in cardiomyopathy, valvular disease and arrhythmia. Immunology 2006, 118, 10–24. [Google Scholar] [CrossRef]
- Yue, Y.; Meng, K.; Pu, Y.; Zhang, X. Transforming growth factor beta (TGF-beta) mediates cardiac fibrosis and induces diabetic cardiomyopathy. Diabetes Res. Clin. Pract. 2017, 133, 124–130. [Google Scholar] [CrossRef]
- Khalil, H.; Kanisicak, O.; Prasad, V.; Correll, R.N.; Fu, X.; Schips, T.; Vagnozzi, R.J.; Liu, R.; Huynh, T.; Lee, S.J.; et al. Fibroblast-specific TGF-beta-Smad2/3 signaling underlies cardiac fibrosis. J. Clin. Investig. 2017, 127, 3770–3783. [Google Scholar] [CrossRef]
- Kalchiem-Dekel, O.; Galvin, J.R.; Burke, A.P.; Atamas, S.P.; Todd, N.W. Interstitial Lung Disease and Pulmonary Fibrosis: A Practical Approach for General Medicine Physicians with Focus on the Medical History. J. Clin. Med. 2018, 7, 476. [Google Scholar] [CrossRef] [Green Version]
- Flanders, K.C. Smad3 as a mediator of the fibrotic response. Int. J. Exp. Pathol. 2004, 85, 47–64. [Google Scholar] [CrossRef]
- Chanda, D.; Otoupalova, E.; Smith, S.R.; Volckaert, T.; De Langhe, S.P.; Thannickal, V.J. Developmental pathways in the pathogenesis of lung fibrosis. Mol. Asp. Med. 2019, 65, 56–69. [Google Scholar] [CrossRef]
- Francisco, J.; Zhang, Y.; Jeong, J.I.; Mizushima, W.; Ikeda, S.; Ivessa, A.; Oka, S.; Zhai, P.; Tallquist, M.D.; Del Re, D.P. Blockade of Fibroblast YAP Attenuates Cardiac Fibrosis and Dysfunction Through MRTF-A Inhibition. JACC Basic Transl. Sci. 2020, 5, 931–945. [Google Scholar] [CrossRef]
- Batlle, E.; Massague, J. Transforming Growth Factor-beta Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Eshima, K.; Misawa, K.; Ohashi, C.; Iwabuchi, K. Role of T-bet, the master regulator of Th1 cells, in the cytotoxicity of murine CD4(+) T cells. Microbiol. Immunol. 2018, 62, 348–356. [Google Scholar] [CrossRef] [Green Version]
- Gorelik, L.; Flavell, R.A. Transforming growth factor-beta in T-cell biology. Nat. Rev. Immunol. 2002, 2, 46–53. [Google Scholar] [CrossRef]
- Tzachanis, D.; Freeman, G.J.; Hirano, N.; van Puijenbroek, A.A.; Delfs, M.W.; Berezovskaya, A.; Nadler, L.M.; Boussiotis, V.A. Tob is a negative regulator of activation that is expressed in anergic and quiescent T cells. Nat. Immunol. 2001, 2, 1174–1182. [Google Scholar] [CrossRef]
- Iacobuzio-Donahue, C.A.; Fu, B.; Yachida, S.; Luo, M.; Abe, H.; Henderson, C.M.; Vilardell, F.; Wang, Z.; Keller, J.W.; Banerjee, P.; et al. DPC4 gene status of the primary carcinoma correlates with patterns of failure in patients with pancreatic cancer. J. Clin. Oncol. 2009, 27, 1806–1813. [Google Scholar] [CrossRef] [Green Version]
- Rodell, C.B.; Arlauckas, S.P.; Cuccarese, M.F.; Garris, C.S.; Li, R.; Ahmed, M.S.; Kohler, R.H.; Pittet, M.J.; Weissleder, R. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat. Biomed. Eng. 2018, 2, 578–588. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, H.; Wang, X.; Jiang, G.; Liu, H.; Zhang, G.; Wang, H.; Fang, R.; Bu, X.; Cai, S.; et al. TGF-beta induces M2-like macrophage polarization via SNAIL-mediated suppression of a pro-inflammatory phenotype. Oncotarget 2016, 7, 52294–52306. [Google Scholar] [CrossRef] [Green Version]
- Regis, S.; Dondero, A.; Caliendo, F.; Bottino, C.; Castriconi, R. NK Cell Function Regulation by TGF-beta-Induced Epigenetic Mechanisms. Front. Immunol. 2020, 11, 311. [Google Scholar] [CrossRef] [Green Version]
- Rybinski, B.; Franco-Barraza, J.; Cukierman, E. The wound healing, chronic fibrosis, and cancer progression triad. Physiol. Genom. 2014, 46, 223–244. [Google Scholar] [CrossRef]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [PubMed]
- Hawinkels, L.J.; Paauwe, M.; Verspaget, H.W.; Wiercinska, E.; van der Zon, J.M.; van der Ploeg, K.; Koelink, P.J.; Lindeman, J.H.; Mesker, W.; ten Dijke, P.; et al. Interaction with colon cancer cells hyperactivates TGF-beta signaling in cancer-associated fibroblasts. Oncogene 2014, 33, 97–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Untergasser, G.; Gander, R.; Lilg, C.; Lepperdinger, G.; Plas, E.; Berger, P. Profiling molecular targets of TGF-beta1 in prostate fibroblast-to-myofibroblast transdifferentiation. Mech. Ageing Dev. 2005, 126, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, Y.; Li, D.; Jing, S. Notch and TGF-beta/Smad3 pathways are involved in the interaction between cancer cells and cancer-associated fibroblasts in papillary thyroid carcinoma. Tumour Biol. 2014, 35, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Young, C.D.; Zhou, H.; Wang, X. Transforming Growth Factor-beta Signaling in Fibrotic Diseases and Cancer-Associated Fibroblasts. Biomolecules 2020, 10, 1666. [Google Scholar] [CrossRef]
- Evans, R.A.; Tian, Y.C.; Steadman, R.; Phillips, A.O. TGF-beta1-mediated fibroblast-myofibroblast terminal differentiation-the role of Smad proteins. Exp. Cell Res. 2003, 282, 90–100. [Google Scholar] [CrossRef]
- Ronnov-Jessen, L.; Petersen, O.W. Induction of alpha-smooth muscle actin by transforming growth factor-beta 1 in quiescent human breast gland fibroblasts. Implications for myofibroblast generation in breast neoplasia. Lab. Investig. 1993, 68, 696–707. [Google Scholar]
- Lamprecht, S.; Sigal-Batikoff, I.; Shany, S.; Abu-Freha, N.; Ling, E.; Delinasios, G.J.; Moyal-Atias, K.; Delinasios, J.G.; Fich, A. Teaming Up for Trouble: Cancer Cells, Transforming Growth Factor-beta1 Signaling and the Epigenetic Corruption of Stromal Naive Fibroblasts. Cancers 2018, 10, 61. [Google Scholar] [CrossRef] [Green Version]
- Costanza, B.; Umelo, I.A.; Bellier, J.; Castronovo, V.; Turtoi, A. Stromal Modulators of TGF-beta in Cancer. J. Clin. Med. 2017, 6, 7. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.; Xie, J.; Gong, T.; Shi, S.; Zhang, T.; Fu, N.; Lin, Y. Smad signal pathway regulates angiogenesis via endothelial cell in an adipose-derived stromal cell/endothelial cell co-culture, 3D gel model. Mol. Cell Biochem. 2016, 412, 281–288. [Google Scholar] [CrossRef]
- Cunha, S.I.; Pietras, K. ALK1 as an emerging target for antiangiogenic therapy of cancer. Blood 2011, 117, 6999–7006. [Google Scholar] [CrossRef] [Green Version]
- Chandler, C.; Liu, T.; Buckanovich, R.; Coffman, L.G. The double edge sword of fibrosis in cancer. Transl. Res. 2019, 209, 55–67. [Google Scholar] [CrossRef] [Green Version]
- Underwood, T.J.; Hayden, A.L.; Derouet, M.; Garcia, E.; Noble, F.; White, M.J.; Thirdborough, S.; Mead, A.; Clemons, N.; Mellone, M.; et al. Cancer-associated fibroblasts predict poor outcome and promote periostin-dependent invasion in oesophageal adenocarcinoma. J. Pathol. 2015, 235, 466–477. [Google Scholar] [CrossRef]
- Lai, D.; Ma, L.; Wang, F. Fibroblast activation protein regulates tumor-associated fibroblasts and epithelial ovarian cancer cells. Int. J. Oncol. 2012, 41, 541–550. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, M.; Ogawa, T.; Zhang, X.; Hanamura, N.; Kashikura, Y.; Takamura, M.; Yoneda, M.; Shiraishi, T. Role of stromal myofibroblasts in invasive breast cancer: Stromal expression of alpha-smooth muscle actin correlates with worse clinical outcome. Breast Cancer 2012, 19, 170–176. [Google Scholar] [CrossRef]
- Tsujino, T.; Seshimo, I.; Yamamoto, H.; Ngan, C.Y.; Ezumi, K.; Takemasa, I.; Ikeda, M.; Sekimoto, M.; Matsuura, N.; Monden, M. Stromal myofibroblasts predict disease recurrence for colorectal cancer. Clin. Cancer Res. 2007, 13, 2082–2090. [Google Scholar] [CrossRef] [Green Version]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFbeta to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Chen, S.; Wang, W.; Ning, B.F.; Chen, F.; Shen, W.; Ding, J.; Chen, W.; Xie, W.F.; Zhang, X. Cancer-associated fibroblasts promote hepatocellular carcinoma metastasis through chemokine-activated hedgehog and TGF-beta pathways. Cancer Lett. 2016, 379, 49–59. [Google Scholar] [CrossRef]
- De Wever, O.; Nguyen, Q.D.; Van Hoorde, L.; Bracke, M.; Bruyneel, E.; Gespach, C.; Mareel, M. Tenascin-C and SF/HGF produced by myofibroblasts in vitro provide convergent pro-invasive signals to human colon cancer cells through RhoA and Rac. FASEB J. 2004, 18, 1016–1018. [Google Scholar] [CrossRef]
- Henriksson, M.L.; Edin, S.; Dahlin, A.M.; Oldenborg, P.A.; Oberg, A.; Van Guelpen, B.; Rutegard, J.; Stenling, R.; Palmqvist, R. Colorectal cancer cells activate adjacent fibroblasts resulting in FGF1/FGFR3 signaling and increased invasion. Am. J. Pathol. 2011, 178, 1387–1394. [Google Scholar] [CrossRef]
- Gaggioli, C.; Hooper, S.; Hidalgo-Carcedo, C.; Grosse, R.; Marshall, J.F.; Harrington, K.; Sahai, E. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat. Cell Biol. 2007, 9, 1392–1400. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.O.; Mullins, S.R.; Franco-Barraza, J.; Valianou, M.; Cukierman, E.; Cheng, J.D. FAP-overexpressing fibroblasts produce an extracellular matrix that enhances invasive velocity and directionality of pancreatic cancer cells. BMC Cancer 2011, 11, 245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetz, J.G.; Minguet, S.; Navarro-Lerida, I.; Lazcano, J.J.; Samaniego, R.; Calvo, E.; Tello, M.; Osteso-Ibanez, T.; Pellinen, T.; Echarri, A.; et al. Biomechanical remodeling of the microenvironment by stromal caveolin-1 favors tumor invasion and metastasis. Cell 2011, 146, 148–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanisavljevic, J.; Loubat-Casanovas, J.; Herrera, M.; Luque, T.; Pena, R.; Lluch, A.; Albanell, J.; Bonilla, F.; Rovira, A.; Pena, C.; et al. Snail1-expressing fibroblasts in the tumor microenvironment display mechanical properties that support metastasis. Cancer Res. 2015, 75, 284–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Palmero, I.; Torres, S.; Bartolome, R.A.; Pelaez-Garcia, A.; Larriba, M.J.; Lopez-Lucendo, M.; Pena, C.; Escudero-Paniagua, B.; Munoz, A.; Casal, J.I. Twist1-induced activation of human fibroblasts promotes matrix stiffness by upregulating palladin and collagen alpha1(VI). Oncogene 2016, 35, 5224–5236. [Google Scholar] [CrossRef]
- Erdogan, B.; Ao, M.; White, L.M.; Means, A.L.; Brewer, B.M.; Yang, L.; Washington, M.K.; Shi, C.; Franco, O.E.; Weaver, A.M.; et al. Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin. J. Cell Biol. 2017, 216, 3799–3816. [Google Scholar] [CrossRef] [Green Version]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef]
- Casey, T.M.; Eneman, J.; Crocker, A.; White, J.; Tessitore, J.; Stanley, M.; Harlow, S.; Bunn, J.Y.; Weaver, D.; Muss, H.; et al. Cancer associated fibroblasts stimulated by transforming growth factor beta1 (TGF-beta 1) increase invasion rate of tumor cells: A population study. Breast Cancer Res. Treat. 2008, 110, 39–49. [Google Scholar] [CrossRef]
- Huang, S.; Chakrabarty, S. Regulation of fibronectin and laminin receptor expression, fibronectin and laminin secretion in human colon cancer cells by transforming growth factor-beta 1. Int. J. Cancer 1994, 57, 742–746. [Google Scholar] [CrossRef]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.A.; Lee, Y.H.; Schiemann, W.P. Role of TGF-beta and the tumor microenvironment during mammary tumorigenesis. Gene Expr. 2011, 15, 117–132. [Google Scholar] [CrossRef]
- Jia, H.; Janjanam, J.; Wu, S.C.; Wang, R.; Pano, G.; Celestine, M.; Martinot, O.; Breeze-Jones, H.; Clayton, G.; Garcin, C.; et al. The tumor cell-secreted matricellular protein WISP1 drives pro-metastatic collagen linearization. EMBO J. 2019, 38, e101302. [Google Scholar] [CrossRef]
- Plou, J.; Juste-Lanas, Y.; Olivares, V.; Del Amo, C.; Borau, C.; Garcia-Aznar, J.M. From individual to collective 3D cancer dissemination: Roles of collagen concentration and TGF-beta. Sci. Rep. 2018, 8, 12723. [Google Scholar] [CrossRef]
- De Palma, M.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef]
- Russell, J.S.; Brown, J.M. Circulating mouse Flk1+/c-Kit+/CD45- cells function as endothelial progenitors cells (EPCs) and stimulate the growth of human tumor xenografts. Mol. Cancer 2014, 13, 177. [Google Scholar] [CrossRef] [Green Version]
- Moore-Smith, L.D.; Isayeva, T.; Lee, J.H.; Frost, A.; Ponnazhagan, S. Silencing of TGF-beta1 in tumor cells impacts MMP-9 in tumor microenvironment. Sci. Rep. 2017, 7, 8678. [Google Scholar] [CrossRef] [Green Version]
- Zonneville, J.; Safina, A.; Truskinovsky, A.M.; Arteaga, C.L.; Bakin, A.V. TGF-beta signaling promotes tumor vasculature by enhancing the pericyte-endothelium association. BMC Cancer 2018, 18, 670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enholm, B.; Paavonen, K.; Ristimaki, A.; Kumar, V.; Gunji, Y.; Klefstrom, J.; Kivinen, L.; Laiho, M.; Olofsson, B.; Joukov, V.; et al. Comparison of VEGF, VEGF-B, VEGF-C and Ang-1 mRNA regulation by serum, growth factors, oncoproteins and hypoxia. Oncogene 1997, 14, 2475–2483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Ma, J.; Han, J.D.; Wang, N.; Chen, Y.G. Distinct regulation of gene expression in human endothelial cells by TGF-beta and its receptors. Microvasc. Res. 2006, 71, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.J.; Valdimarsdottir, G.; Itoh, S.; Rosendahl, A.; Sideras, P.; ten Dijke, P. Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. EMBO J. 2002, 21, 1743–1753. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Tang, L.; Letterio, J.J.; Benveniste, E.N. The Smad3 protein is involved in TGF-beta inhibition of class II transactivator and class II MHC expression. J. Immunol. 2001, 167, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Valderrama-Carvajal, H.; Cocolakis, E.; Lacerte, A.; Lee, E.H.; Krystal, G.; Ali, S.; Lebrun, J.J. Activin/TGF-beta induce apoptosis through Smad-dependent expression of the lipid phosphatase SHIP. Nat. Cell Biol. 2002, 4, 963–969. [Google Scholar] [CrossRef]
- Stephen, T.L.; Rutkowski, M.R.; Allegrezza, M.J.; Perales-Puchalt, A.; Tesone, A.J.; Svoronos, N.; Nguyen, J.M.; Sarmin, F.; Borowsky, M.E.; Tchou, J.; et al. Transforming growth factor beta-mediated suppression of antitumor T cells requires FoxP1 transcription factor expression. Immunity 2014, 41, 427–439. [Google Scholar] [CrossRef] [Green Version]
- Gunderson, A.J.; Yamazaki, T.; McCarty, K.; Fox, N.; Phillips, M.; Alice, A.; Blair, T.; Whiteford, M.; O’Brien, D.; Ahmad, R.; et al. TGFbeta suppresses CD8(+) T cell expression of CXCR3 and tumor trafficking. Nat. Commun. 2020, 11, 1749. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.L.; Pittet, M.J.; Gorelik, L.; Flavell, R.A.; Weissleder, R.; von Boehmer, H.; Khazaie, K. Regulatory T cells suppress tumor-specific CD8 T cell cytotoxicity through TGF-beta signals in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 419–424. [Google Scholar] [CrossRef] [Green Version]
- Das, J.; Ren, G.; Zhang, L.; Roberts, A.I.; Zhao, X.; Bothwell, A.L.; Van Kaer, L.; Shi, Y.; Das, G. Transforming growth factor beta is dispensable for the molecular orchestration of Th17 cell differentiation. J. Exp. Med. 2009, 206, 2407–2416. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Davidson, T.S.; Wei, G.; Jankovic, D.; Cui, K.; Schones, D.E.; Guo, L.; Zhao, K.; Shevach, E.M.; Paul, W.E. Down-regulation of Gfi-1 expression by TGF-beta is important for differentiation of Th17 and CD103+ inducible regulatory T cells. J. Exp. Med. 2009, 206, 329–341. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Gao, X.; Shen, G.; Wang, W.; Li, J.; Zhao, J.; Wei, Y.Q.; Edwards, C.K. Interleukin-10 deficiency impairs regulatory T cell-derived neuropilin-1 functions and promotes Th1 and Th17 immunity. Sci. Rep. 2016, 6, 24249. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Hegde, S.; DeNardo, D.G. Tumor-associated fibrosis as a regulator of tumor immunity and response to immunotherapy. Cancer Immunol. Immunother. 2017, 66, 1037–1048. [Google Scholar] [CrossRef]
- Cheng, J.T.; Deng, Y.N.; Yi, H.M.; Wang, G.Y.; Fu, B.S.; Chen, W.J.; Liu, W.; Tai, Y.; Peng, Y.W.; Zhang, Q. Hepatic carcinoma-associated fibroblasts induce IDO-producing regulatory dendritic cells through IL-6-mediated STAT3 activation. Oncogenesis 2016, 5, e198. [Google Scholar] [CrossRef] [Green Version]
- Ruffell, B.; Chang-Strachan, D.; Chan, V.; Rosenbusch, A.; Ho, C.M.; Pryer, N.; Daniel, D.; Hwang, E.S.; Rugo, H.S.; Coussens, L.M. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell 2014, 26, 623–637. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Bu, W.; Meng, L.; Liu, X.; Wang, S.; Jiang, L.; Ren, M.; Fan, Y.; Sun, H. CXCL12/CXCR4 pathway orchestrates CSC-like properties by CAF recruited tumor associated macrophage in OSCC. Exp. Cell Res. 2019, 378, 131–138. [Google Scholar] [CrossRef]
- Kelly, J.; Ali Khan, A.; Yin, J.; Ferguson, T.A.; Apte, R.S. Senescence regulates macrophage activation and angiogenic fate at sites of tissue injury in mice. J. Clin. Investig. 2007, 117, 3421–3426. [Google Scholar] [CrossRef]
- Zhang, S.; Che, D.; Yang, F.; Chi, C.; Meng, H.; Shen, J.; Qi, L.; Liu, F.; Lv, L.; Li, Y.; et al. Tumor-associated macrophages promote tumor metastasis via the TGF-beta/SOX9 axis in non-small cell lung cancer. Oncotarget 2017, 8, 99801–99815. [Google Scholar] [CrossRef] [Green Version]
- Standiford, T.J.; Kuick, R.; Bhan, U.; Chen, J.; Newstead, M.; Keshamouni, V.G. TGF-beta-induced IRAK-M expression in tumor-associated macrophages regulates lung tumor growth. Oncogene 2011, 30, 2475–2484. [Google Scholar] [CrossRef] [Green Version]
- Balsamo, M.; Scordamaglia, F.; Pietra, G.; Manzini, C.; Cantoni, C.; Boitano, M.; Queirolo, P.; Vermi, W.; Facchetti, F.; Moretta, A.; et al. Melanoma-associated fibroblasts modulate NK cell phenotype and antitumor cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 20847–20852. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Yang, Y.; Hua, X.; Wang, G.; Liu, W.; Jia, C.; Tai, Y.; Zhang, Q.; Chen, G. Hepatocellular carcinoma-associated fibroblasts trigger NK cell dysfunction via PGE2 and IDO. Cancer Lett. 2012, 318, 154–161. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, J.; Zhang, X.; Cheng, Y.; Hu, J.; Li, Y.; Zhao, X.; Shang, Q.; Sun, Y.; Tu, B.; et al. Activated hepatic stellate cells impair NK cell anti-fibrosis capacity through a TGF-beta-dependent emperipolesis in HBV cirrhotic patients. Sci. Rep. 2017, 7, 44544. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Lin, Y.; Shi, Y.; Li, B.; Liu, W.; Yin, W.; Dang, Y.; Chu, Y.; Fan, J.; He, R. FAP Promotes Immunosuppression by Cancer-Associated Fibroblasts in the Tumor Microenvironment via STAT3-CCL2 Signaling. Cancer Res. 2016, 76, 4124–4135. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Ertl, H.C. Depletion of FAP+ cells reduces immunosuppressive cells and improves metabolism and functions CD8+T cells within tumors. Oncotarget 2016, 7, 23282–23299. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.H.; Hong, H.C.; Hong, T.M.; Chiang, W.F.; Jin, Y.T.; Chen, Y.L. Targeting galectin-1 in carcinoma-associated fibroblasts inhibits oral squamous cell carcinoma metastasis by downregulating MCP-1/CCL2 expression. Clin. Cancer Res. 2011, 17, 1306–1316. [Google Scholar] [CrossRef] [Green Version]
- Torres, S.; Bartolome, R.A.; Mendes, M.; Barderas, R.; Fernandez-Acenero, M.J.; Pelaez-Garcia, A.; Pena, C.; Lopez-Lucendo, M.; Villar-Vazquez, R.; de Herreros, A.G.; et al. Proteome profiling of cancer-associated fibroblasts identifies novel proinflammatory signatures and prognostic markers for colorectal cancer. Clin. Cancer Res. 2013, 19, 6006–6019. [Google Scholar] [CrossRef] [Green Version]
- Mathew, E.; Brannon, A.L.; Del Vecchio, A.; Garcia, P.E.; Penny, M.K.; Kane, K.T.; Vinta, A.; Buckanovich, R.J.; di Magliano, M.P. Mesenchymal Stem Cells Promote Pancreatic Tumor Growth by Inducing Alternative Polarization of Macrophages. Neoplasia 2016, 18, 142–151. [Google Scholar] [CrossRef] [Green Version]
- Mao, L.; Li, Y.; Zhao, J.; Li, Q.; Yang, B.; Wang, Y.; Zhu, Z.; Sun, H.; Zhai, Z. Transforming growth factor-beta1 contributes to oxaliplatin resistance in colorectal cancer via epithelial to mesenchymal transition. Oncol. Lett. 2017, 14, 647–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Zhang, B.; Xiang, L.; Xia, S.; Kucuk, O.; Deng, X.; Boise, L.H.; Dong, J.T. TGF-beta causes Docetaxel resistance in Prostate Cancer via the induction of Bcl-2 by acetylated KLF5 and Protein Stabilization. Theranostics 2020, 10, 7656–7670. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Morel, A.P.; Hinkal, G.W.; Thomas, C.; Fauvet, F.; Courtois-Cox, S.; Wierinckx, A.; Devouassoux-Shisheboran, M.; Treilleux, I.; Tissier, A.; Gras, B.; et al. EMT inducers catalyze malignant transformation of mammary epithelial cells and drive tumorigenesis towards claudin-low tumors in transgenic mice. PLoS Genet. 2012, 8, e1002723. [Google Scholar] [CrossRef] [Green Version]
- Salem, A.F.; Whitaker-Menezes, D.; Lin, Z.; Martinez-Outschoorn, U.E.; Tanowitz, H.B.; Al-Zoubi, M.S.; Howell, A.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Two-compartment tumor metabolism: Autophagy in the tumor microenvironment and oxidative mitochondrial metabolism (OXPHOS) in cancer cells. Cell Cycle 2012, 11, 2545–2556. [Google Scholar] [CrossRef] [Green Version]
- Shintani, Y.; Fujiwara, A.; Kimura, T.; Kawamura, T.; Funaki, S.; Minami, M.; Okumura, M. IL-6 Secreted from Cancer-Associated Fibroblasts Mediates Chemoresistance in NSCLC by Increasing Epithelial-Mesenchymal Transition Signaling. J. Thorac. Oncol. 2016, 11, 1482–1492. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.; Yang, G.; Hou, Y.; Tang, X.; Wu, C.; Wu, X.A.; Guo, L.; Zhu, Q.; Luo, H.; Du, Y.E.; et al. Cytoplasmic GPER translocation in cancer-associated fibroblasts mediates cAMP/PKA/CREB/glycolytic axis to confer tumor cells with multidrug resistance. Oncogene 2017, 36, 2131–2145. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Qiu, X.; Wang, X.; He, J. FAP positive fibroblasts induce immune checkpoint blockade resistance in colorectal cancer via promoting immunosuppression. Biochem. Biophys. Res. Commun. 2017, 487, 8–14. [Google Scholar] [CrossRef]
- Tang, Y.A.; Chen, Y.F.; Bao, Y.; Mahara, S.; Yatim, S.; Oguz, G.; Lee, P.L.; Feng, M.; Cai, Y.; Tan, E.Y.; et al. Hypoxic tumor microenvironment activates GLI2 via HIF-1alpha and TGF-beta2 to promote chemoresistance in colorectal cancer. Proc. Natl. Acad. Sci. USA 2018, 115, E5990–E5999. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Letterio, J.J.; Lechleider, R.J.; Chen, L.; Hayman, R.; Gu, H.; Roberts, A.B.; Deng, C. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-beta. EMBO J. 1999, 18, 1280–1291. [Google Scholar] [CrossRef] [Green Version]
- Thomas, D.A.; Massague, J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef] [Green Version]
- Mitra, M.S.; Lancaster, K.; Adedeji, A.O.; Palanisamy, G.S.; Dave, R.A.; Zhong, F.; Holdren, M.S.; Turley, S.J.; Liang, W.C.; Wu, Y.; et al. A Potent Pan-TGFbeta Neutralizing Monoclonal Antibody Elicits Cardiovascular Toxicity in Mice and Cynomolgus Monkeys. Toxicol. Sci. 2020, 175, 24–34. [Google Scholar] [CrossRef]
- Tang, P.M.; Zhang, Y.Y.; Xiao, J.; Tang, P.C.; Chung, J.Y.; Li, J.; Xue, V.W.; Huang, X.R.; Chong, C.C.; Ng, C.F.; et al. Neural transcription factor Pou4f1 promotes renal fibrosis via macrophage-myofibroblast transition. Proc. Natl. Acad. Sci. USA 2020, 117, 20741–20752. [Google Scholar] [CrossRef]
- Tang, P.M.; Zhou, S.; Li, C.J.; Liao, J.; Xiao, J.; Wang, Q.M.; Lian, G.Y.; Li, J.; Huang, X.R.; To, K.F.; et al. The proto-oncogene tyrosine protein kinase Src is essential for macrophage-myofibroblast transition during renal scarring. Kidney Int. 2018, 93, 173–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burk, U.; Schubert, J.; Wellner, U.; Schmalhofer, O.; Vincan, E.; Spaderna, S.; Brabletz, T. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008, 9, 582–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, L.; Wang, R.; Wang, B.; Hua, P.; Li, J. LncRNA Erbb4-IR promotes esophageal squamous cell carcinoma (ESCC) by downregulating miR-145. J. Cell Biochem. 2019, 120, 17566–17572. [Google Scholar] [CrossRef]
- Gu, Y.Y.; Lu, F.H.; Huang, X.R.; Zhang, L.; Mao, W.; Yu, X.Q.; Liu, X.S.; Lan, H.Y. Non-Coding RNAs as Biomarkers and Therapeutic Targets for Diabetic Kidney Disease. Front. Pharmacol. 2020, 11, 583528. [Google Scholar] [CrossRef]
- Sun, S.F.; Tang, P.M.K.; Feng, M.; Xiao, J.; Huang, X.R.; Li, P.; Ma, R.C.W.; Lan, H.Y. Novel lncRNA Erbb4-IR Promotes Diabetic Kidney Injury in db/db Mice by Targeting miR-29b. Diabetes 2018, 67, 731–744. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Tang, P.M.; Niu, Y.; Garcia Cordoba, C.A.; Huang, X.R.; Yu, C.; Lan, H.Y. Long Non-coding RNA LRNA9884 Promotes Acute Kidney Injury via Regulating NF-kB-Mediated Transcriptional Activation of MIF. Front. Physiol. 2020, 11, 590027. [Google Scholar] [CrossRef]
- Li, S.; Zhang, Y.; Sun, Y.; Zhang, G.; Bai, J.; Guo, J.; Su, X.; Du, H.; Cao, X.; Yang, J.; et al. Naringenin improves insulin sensitivity in gestational diabetes mellitus mice through AMPK. Nutr. Diabetes 2019, 9, 28. [Google Scholar] [CrossRef] [Green Version]
- Tang, P.M.; Nikolic-Paterson, D.J.; Lan, H.Y. Macrophages: Versatile players in renal inflammation and fibrosis. Nat. Rev. Nephrol. 2019, 15, 144–158. [Google Scholar] [CrossRef]
- Torres, A.; Munoz, K.; Nahuelpan, Y.; AP, R.S.; Mendoza, P.; Jara, C.; Cappelli, C.; Suarez, R.; Oyarzun, C.; Quezada, C.; et al. Intraglomerular Monocyte/Macrophage Infiltration and Macrophage-Myofibroblast Transition during Diabetic Nephropathy Is Regulated by the A2B Adenosine Receptor. Cells 2020, 9, 1051. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Hofbauer, T.M.; Ondracek, A.S.; Chausheva, S.; Alimohammadi, A.; Artner, T.; Panzenboeck, A.; Rinderer, J.; Shafran, I.; Mangold, A.; et al. Neutrophil extracellular traps promote fibrous vascular occlusions in chronic thrombosis. Blood 2021, 137, 1104–1116. [Google Scholar] [CrossRef] [PubMed]
- Xue, V.W.; Chung, J.Y.; Cordoba, C.A.G.; Cheung, A.H.; Kang, W.; Lam, E.W.; Leung, K.T.; To, K.F.; Lan, H.Y.; Tang, P.M. Transforming Growth Factor-beta: A Multifunctional Regulator of Cancer Immunity. Cancers 2020, 12, 3099. [Google Scholar] [CrossRef] [PubMed]
- Tang, P.C.-T.; Tang, P.M.-K.; Chung, J.Y.-F.; Huang, X.-R.; TO, K.-F.; LAN, H.-Y. Abstract 1095: Macrophage is a novel and rich source of cancer-associated fibroblasts in the tumor microenvironment. Cancer Res. 2019, 79 (Suppl. 13), 1095. [Google Scholar]
- Liu, Y.; Li, Y.; Li, N.; Teng, W.; Wang, M.; Zhang, Y.; Xiao, Z. TGF-beta1 promotes scar fibroblasts proliferation and transdifferentiation via up-regulating MicroRNA-21. Sci. Rep. 2016, 6, 32231. [Google Scholar] [CrossRef]
- Xiong, M.; Jiang, L.; Zhou, Y.; Qiu, W.; Fang, L.; Tan, R.; Wen, P.; Yang, J. The miR-200 family regulates TGF-beta1-induced renal tubular epithelial to mesenchymal transition through Smad pathway by targeting ZEB1 and ZEB2 expression. Am. J. Physiol. Renal Physiol. 2012, 302, F369–F379. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Li, H.; Wang, X.; Wang, L.; Zeng, Q. Lnc-LFAR1 affects intrahepatic cholangiocarcinoma proliferation, invasion, and EMT by regulating the TGFbeta/Smad signaling pathway. Int. J. Clin. Exp. Pathol. 2019, 12, 2455–2461. [Google Scholar]
- Zhang, K.; Han, X.; Zhang, Z.; Zheng, L.; Hu, Z.; Yao, Q.; Cui, H.; Shu, G.; Si, M.; Li, C.; et al. The liver-enriched lnc-LFAR1 promotes liver fibrosis by activating TGFbeta and Notch pathways. Nat. Commun. 2017, 8, 144. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Luo, M.L.; Song, E.; Zhou, Z.; Ma, T.; Wang, J.; Jia, N.; Wang, G.; Nie, S.; Liu, Y.; et al. Long noncoding RNA lnc-TSI inhibits renal fibrogenesis by negatively regulating the TGF-beta/Smad3 pathway. Sci. Transl Med. 2018, 10, eaat2039. [Google Scholar] [CrossRef] [Green Version]
- Pachera, E.; Assassi, S.; Salazar, G.A.; Stellato, M.; Renoux, F.; Wunderlin, A.; Blyszczuk, P.; Lafyatis, R.; Kurreeman, F.; de Vries-Bouwstra, J.; et al. Long noncoding RNA H19X is a key mediator of TGF-beta-driven fibrosis. J. Clin. Investig. 2020, 130, 4888–4905. [Google Scholar] [CrossRef]
- Li, R.; Wu, C.; Liang, H.; Zhao, Y.; Lin, C.; Zhang, X.; Ye, C. Knockdown of TWIST enhances the cytotoxicity of chemotherapeutic drugs in doxorubicin-resistant HepG2 cells by suppressing MDR1 and EMT. Int. J. Oncol. 2018, 53, 1763–1773. [Google Scholar] [CrossRef] [Green Version]
- Sakata, J.; Utsumi, F.; Suzuki, S.; Niimi, K.; Yamamoto, E.; Shibata, K.; Senga, T.; Kikkawa, F.; Kajiyama, H. Inhibition of ZEB1 leads to inversion of metastatic characteristics and restoration of paclitaxel sensitivity of chronic chemoresistant ovarian carcinoma cells. Oncotarget 2017, 8, 99482–99494. [Google Scholar] [CrossRef]
- Siebzehnrubl, F.A.; Silver, D.J.; Tugertimur, B.; Deleyrolle, L.P.; Siebzehnrubl, D.; Sarkisian, M.R.; Devers, K.G.; Yachnis, A.T.; Kupper, M.D.; Neal, D.; et al. The ZEB1 pathway links glioblastoma initiation, invasion and chemoresistance. EMBO Mol. Med. 2013, 5, 1196–1212. [Google Scholar] [CrossRef]
- Meidhof, S.; Brabletz, S.; Lehmann, W.; Preca, B.T.; Mock, K.; Ruh, M.; Schuler, J.; Berthold, M.; Weber, A.; Burk, U.; et al. ZEB1-associated drug resistance in cancer cells is reversed by the class I HDAC inhibitor mocetinostat. EMBO Mol. Med. 2015, 7, 831–847. [Google Scholar] [CrossRef]
- Hohenauer, T.; Berking, C.; Schmidt, A.; Haferkamp, S.; Senft, D.; Kammerbauer, C.; Fraschka, S.; Graf, S.A.; Irmler, M.; Beckers, J.; et al. The neural crest transcription factor Brn3a is expressed in melanoma and required for cell cycle progression and survival. EMBO Mol. Med. 2013, 5, 919–934. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | Effects of CAF on Immune Cell | Ref. |
|---|---|---|
| Dendritic cell | Inhibiting DC maturation | [114] |
| Macrophage | Promotes macrophage differentiation into immunosuppressive M2-like phenotype and inhibiting differentiation into pro-inflammatory M1-like phenotype. | [61,116] |
| Natural killer cell | Impairs cytotoxic activation and the expression of natural cytotoxicity receptors, as well as promoting NK cells apoptosis | [120,121] |
| T cell | Reduces T-cell activation, differentiation by influencing DC and MDSC | [115,124] |
| MDSC -granulocytic -monocytic | Reprograms myeloid cells into MDSC Increases its recruitment and function to supress T cell proliferation Increases its recruitment to inhibit T cell activity and accumulation in TME | [123,124] |
| Factor | Mediators | Effect After Inhibition | Ref. |
|---|---|---|---|
| MicroRNAs (miRNAs) | miR-21 | Decreases ECM synthesis and fibrosis via upregulation of MMP-9 expression | [154] |
| miR-200 | Protects TGF-β-mediated EMT by inhibiting the expression of ZEB 1 and 2 | [155] | |
| Long non-coding RNAs (LncRNAs) | Lnc-LFAR1 | Downregulates TGF-β/Smad signaling pathway cancer cell proliferation | [156] |
| Blocks tissue fibrosis by reducing Smad2/3 phosphorylation and binding to TGFβR1 | [157] | ||
| Lnc-TSI | Upregulates the interaction between Smad3 and TGFβR1 lead to cancer metastasis | [158] | |
| LncRNA H19X | Reduces ECM synthesis induced by TGF-β and controls the differentiation and survival of ECM-producing myofibroblasts | [159] | |
| Erbb4-IR | Supresses miR-29b transcription and consequent antifibrotic function | [68] | |
| Downregulates miRNA-145 to reduce cancer cell proliferation | [144] | ||
| Transcription factors (TFs) | Twist | Increases sensitivity to chemotherapy by downregulating MDR1 and reducing drug efflux | [160] |
| ZEB1 | Reduces antifibrotic miR200c and miR141 expression | [142] | |
| Reverses metastasis and restores chemosensitivity in chronic chemoresistant | [161] | ||
| Reduces chemoresistance mediated by MGMT | [162] | ||
| Restores miR-203 expression, supresses stemness and promotes chemo-sensitivity | [163] | ||
| Pou4f1 | Prevents macrophage-myofibroblast transition, thereby supressing MMT-mediated fibrosis | [140,164] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chung, J.Y.-F.; Chan, M.K.-K.; Li, J.S.-F.; Chan, A.S.-W.; Tang, P.C.-T.; Leung, K.-T.; To, K.-F.; Lan, H.-Y.; Tang, P.M.-K. TGF-β Signaling: From Tissue Fibrosis to Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 7575. https://doi.org/10.3390/ijms22147575
Chung JY-F, Chan MK-K, Li JS-F, Chan AS-W, Tang PC-T, Leung K-T, To K-F, Lan H-Y, Tang PM-K. TGF-β Signaling: From Tissue Fibrosis to Tumor Microenvironment. International Journal of Molecular Sciences. 2021; 22(14):7575. https://doi.org/10.3390/ijms22147575
Chicago/Turabian StyleChung, Jeff Yat-Fai, Max Kam-Kwan Chan, Jane Siu-Fan Li, Alex Siu-Wing Chan, Philip Chiu-Tsun Tang, Kam-Tong Leung, Ka-Fai To, Hui-Yao Lan, and Patrick Ming-Kuen Tang. 2021. "TGF-β Signaling: From Tissue Fibrosis to Tumor Microenvironment" International Journal of Molecular Sciences 22, no. 14: 7575. https://doi.org/10.3390/ijms22147575