
The Role of Microbiota in Primary Sclerosing Cholangitis and Related Biliary Malignancies


Department of Hepatology and Gastroenterology, Campus Virchow Klinikum (CVK) and Campus Charité Mitte (CCM), Charité Universitätsmedizin Berlin, Augustenburger Platz 1, 13353 Berlin, Germany
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2021, 22(13), 6975; https://doi.org/10.3390/ijms22136975
Submission received: 28 May 2021
/
Revised: 18 June 2021
/
Accepted: 21 June 2021
/
Published: 28 June 2021
(This article belongs to the Special Issue Microbiota and Cancer 2.0)
Abstract
:Primary sclerosing cholangitis (PSC) is an immune-related cholangiopathy characterized by biliary inflammation, cholestasis, and multifocal bile duct strictures. It is associated with high rates of progression to end-stage liver disease as well as a significant risk of cholangiocarcinoma (CCA), gallbladder cancer, and colorectal carcinoma. Currently, no effective medical treatment with an impact on the overall survival is available, and liver transplantation is the only curative treatment option. Emerging evidence indicates that gut microbiota is associated with disease pathogenesis. Several studies analyzing fecal and mucosal samples demonstrate a distinct gut microbiome in individuals with PSC compared to healthy controls and individuals with inflammatory bowel disease (IBD) without PSC. Experimental mouse and observational human data suggest that a diverse set of microbial functions may be relevant, including microbial metabolites and bacterial processing of pharmacological agents, bile acids, or dietary compounds, altogether driving the intrahepatic inflammation. Despite critical progress in this field over the past years, further functional characterization of the role of the microbiota in PSC and related malignancies is needed. In this review, we discuss the available data on the role of the gut microbiome and elucidate important insights into underlying pathogenic mechanisms and possible microbe-altering interventions.
1. Primary Sclerosing Cholangitis (PSC)—An Overview
Primary sclerosing cholangitis is an immune-related cholangiopathy that is characterized by biliary inflammation, cholestasis, and multifocal bile duct strictures of the intra and/or extra-hepatic biliary tree. It is associated with high rates of progression to liver cirrhosis and end-stage liver disease as well as a significant risk of cholangiocarcinoma (CCA) and gallbladder carcinoma [1,2,3]. PSC is a rare disease with an incidence that ranges from 0 to 1.3 cases per 100,000 persons per year [1,4,5]. However, awareness of this disease has grown in the past years. Due to improved access to noninvasive means of imaging the biliary tree (magnetic resonance cholangiopancreatography (MRCP)), it has become clear that PSC accounts for approximately 10% of all liver transplants performed each year [6,7]. Co-occurrence of inflammatory bowel disease (IBD), most commonly ulcerative colitis (UC), has been reported in up to 80% of patients with PSC, and 2–7.5% of IBD-patients develop PSC [3,6,8,9,10,11,12]. It has even been postulated that PSC-associated colitis represents a distinct IBD entity alongside Crohn’s disease (CD) and UC [13,14]. Emerging data suggest that PSC-IBD has distinct clinical features such as a higher incidence of pancolitis, backwash ileitis, rectal sparing and subclinical inflammation with an increased potential for malignant transformation, exceeding the risk of colorectal cancer of IBD patients without PSC [8,13,15,16].
Currently, no effective medical treatment with impact on overall survival is available. Liver transplantation seems to be the only curative treatment option, but there is also a risk of recurrent PSC after transplantation [1,17,18]. However, the development of better therapeutic agents requires a greater understanding of disease pathogenesis. In this context, the gut microbiota, representing a metabolically highly active human ‘organ’, has recently emerged as an important new player in the pathophysiology of PSC [19,20,21,22,23,24,25,26,27]. Thus, in this review, we discuss the available data on the role of the microbiota in PSC, PSC-IBD and biliary cancer types such as CCA and gall bladder cancer. Moreover, we elucidate important insights into underlying pathophysiologic mechanisms and potential microbe-altering interventions.
2. Pathogenesis of PSC and the Role of the Gut-Liver Axis
Many aspects of the pathogenesis of PSC remain unclear. However, it is generally accepted that the interplay between genetic predisposition and environmental factors contributes to the development of the disease and its progression [6,28]. Several genetic risk factors are associated with PSC, but they only contribute to a small fraction of disease susceptibility [2,6,28]. Environmental factors play a pivotal role in the pathogenesis of PSC. Of these, the microbiota has emerged as one of the most important potential environmental players in chronic inflammatory diseases [12,29,30,31].
The close association between PSC and IBD indicates the involvement of the gut-liver axis (Figure 1). Since the gastrointestinal tract harbors a rich and dense microbiota, which is altered in IBD, a potential role of gut microbiota for PSC has been prosed and investigated. Bidirectional communication between the gut and liver is maintained through the biliary tract, the portal vein, and systemic circulation. The liver communicates with the intestine by transportation of bile salts and antimicrobial molecules, such as immunoglobulin A, to the intestinal lumen through the biliary tract, maintaining gut eubiosis by restricting and regulating bacterial growth. Additionally, endogenous and exogenous metabolites from the liver such as free fatty acids (FFA), ethanol or choline metabolites are transported to the intestine via the capillary system [32,33] (Figure 1).
The ‘leaky gut’ hypothesis suggests that leakage of bacterial products across an impaired intestinal epithelial barrier (e.g., damaged by inflammation or infection as well as chemical injury) can lead to translocation of gut microbes or their products (also called pathogen-associated molecular patterns (PAMPs)), which can induce activation of immune cells in the liver [33,34,35]. The activation of Kupffer and hepatic stellate cells and consequent overproduction of proinflammatory cytokines and chemokines, such as tumor necrosis factor, plays a key role in this process and may lead to chronic biliary tract infection and inflammation, resulting in portal fibrosis, and ultimately PSC [32,36,37,38,39]. As a proof-of-principle, Lichtman and colleagues revealed that small intestinal bacterial overgrowth in a rat model, achieved by using a blind jejunal loop, increased translocation of bacterial products and led to cholangiographic changes resembling PSC [40]. Furthermore, analysis of classical markers of bacterial translocation in the serum of 166 PSC patients and 100 healthy controls revealed an elevation in sCD14 and lipopolysaccharide-binding protein (LBP) levels. Strikingly, high LBP levels were associated with poor transplantation-free survival, which indicates that this ongoing gut leakage could have a clinical impact in PSC [41]. In contrast, Björnsson et al. observed no increase in intestinal permeability in PSC patients compared to healthy controls after assessing their differential urinary excretion of lactulose/L-rhamnose [42]. Nevertheless, their study was limited by a small cohort (n = 22 PSC patients).
Further observations in respect to PSC pathogenesis include the homing of gut-derived lymphocytes to the portal areas and the possible presence of an antigenic trigger derived from the colonic content [43,44,45]. Adams and colleagues proposed that the hepatic complications of IBD are mediated by mucosal T cells that are recruited to the portal areas in response to aberrantly expressed mucosal addressin cell-adhesion molecule 1 (MAdCAM1), which is normally restricted to the gut [44]. They revealed that lymphocytes infiltrating to the liver in PSC include T cells recruited to the liver by CCL25 expression, which is a chemokine that activates α4β7 binding to MAdCAM1 on the hepatic endothelium [43]. Adams and colleagues were the first ones demonstrating in humans that activation of T cells in the gut can be recruited to an extraintestinal site [44]. However, vedolizumab, a α4β7 integrin antibody that blocks lymphocyte homing towards MAdCAM-1, unfortunately, appears to be ineffective in PSC [46,47].
3. Gut Microbiota and PSC Pathogenesis—Mechanistic Insights
The microbiota can be regarded as an environmental factor in itself, being influenced by other environmental factors such as medications, diet and physical activity [48,49,50,51]. Genome-wide association studies have identified 16 risk loci for PSC [52]. Some of these risk loci are associated with microbiome composition e.g., fucosyltransferase 2 (FUT2), which is an important genetic risk factor for disease progression in PSC and host-microbial diversity [53]. Patients with PSC and FUT-2 variants have a different microbial pattern in the bile and colon. Moreover, they are associated with an increased frequency of biliary infections and incidence of dominant stenoses [54]. Another study identified a protective role of the microbiota in pathogenesis of PSC using germ-free mouse models. They demonstrated that germ-free multidrug resistance 2 (mdr2) knockout mice exhibit biochemical and histological features of PSC. Further, they revealed an elevation in cholangiocyte senescence, which is a characteristic of progressive biliary disease [55]. However, the mechanisms remain incompletely explored. One possibility could be the defective bile acid metabolism in germ-free mice, which requires the gut microbiota [55]. Consequently, absence of the anti-inflammatory properties of secondary bile acids might play an important role in increased inflammation and subsequent responses of biliary cells [55]. Further, Nakamoto and colleagues generated gnotobiotic mice by using fecal samples from a patient with PSC and UC harboring Klebsiella pneumoniae, which they found to be abundant in patients with PSC [56]. A bacterial-organoid co-culture system demonstrated a damaging effect of Klebsiella pneumoniae on epithelial cells. Furthermore, colonization with these bacteria in vivo is associated with bacterial translocation and susceptibility to T helper cell 17-mediated hepatobiliary injuries in mice. Of note, antibiotic treatment targeting Klebsiella protected from severe injury. These results highlight a potential role of pathobionts in promoting gut epithelial injury and liver inflammation [56,57].
Furthermore, since PSC is associated with an altered bile metabolism and bile acids can directly affect bacterial survival, such changes could not only impact the colonic microbiota but also influence the upper gastrointestinal microbiota, which then in turn may affect biliary or colonic microbial communities.
These exciting findings give more mechanistic insights into the relationship between microbes and PSC, representing important progress in this field. They do not only reveal a harmful but also a protective effect of gut microbes in respect to pathogenesis. However, while they can serve as a proof-of-principle for potential microbiota-derived mechanisms, findings from animal experiments cannot be directly translated to human PSC populations [51,58]. Thus, in addition, it is important to investigate the relationship between PSC and gut microbiome in human populations.
4. Gut Microbiota and PSC—Cross-Sectional Studies
In the past years, numerous publications aimed to characterize the gut microbiota in PSC/PSC-IBD compared to healthy controls and IBD populations (Table 1). Most of them used 16S rRNA methods to explore the composition of the microbiota. Results of published studies analyzing fecal and mucosal microbiota are summarized in Table 1 and Figure 2.
4.1. Fecal Analysis of Gut Microbiota in Patients with PSC
Previous studies of the fecal microbiome in PSC demonstrate that the overall bacterial community is distinct compared to healthy controls without showing any consistency with respect to the specific microbes altered [12,25,27,51,59,60,61,67,68] (Table 1). Sabino et al. analyzed fecal microbiota of 175 individuals (4 cohorts consisting of PSC only, PSC and IBD, IBD, healthy controls), showing that the gut microbiota in PSC (regardless of the presence of IBD) is distinct compared with healthy controls and patients with UC without liver disease. The microbiota of patients with PSC was characterized by decreased bacterial diversity, and a significant overrepresentation of Enterococcus, Fusobacterium and Lactobacillus genera. Strikingly, a link between one operational taxonomic unit of the Enterococcus genus and elevated levels of serum alkaline phosphatase (ALP), a marker of disease severity, could be demonstrated [59].
Up to now, the abundance of Veillonella genus in PSC has been supported by a majority of published reports (Table 1) [25,27,59,60,61]. Nevertheless, this fact is controversially discussed, since the analysis from Sabino et al. revealed that there was no longer a significant association between Veillonella genus and PSC when patients with liver cirrhosis were excluded from the analysis [25,59]. Veillonella are anaerobic, gram-negative cocci. They contain genes that encode for amine oxidases and are producers of primary amines that could act as vascular adhesion protein-1 (VAP-1) substrates, which is an amine oxidase and adhesion molecule, critical for effector cell recruitment to the liver [25,69,70,71]. Moreover, Veillonella species are associated with other inflammatory and progressive fibrotic conditions like pulmonary cystic fibrosis and idiopathic pulmonary fibrosis and the recurrence of disease in patients with CD undergoing ileocaecal resection [72,73,74]. Of note, in previous studies on CD, Veillonella species alongside Enterococcus species were associated with an increased risk of recurrent disease after surgical resection and predisposition to penetrating complications in paediatric patients [74,75]. However, further studies with larger patient cohorts with and without liver cirrhosis, as well as mechanistic studies, are necessary to further understand the role of Veillonella species in PSC.
To investigate geographical differences and to overcome single-center bias, Rühlemann and colleagues analyzed the fecal microbiota of a German and Norwegian cohort (n = 137). They demonstrated an enrichment of eight taxa in PSC patients compared to healthy controls, independent from the presence of IBD and the use of ursodeoxycholic acid (UDCA) [12]. Compared to healthy controls, many new alterations were identified in PSC such as an increase of Proteobacteria and Parabacteroides, which is a bile-tolerant taxon, associated with changes in cholesterol and bile acid metabolism [76,77]. Furthermore, compared to healthy controls Bacteroidetes species were increased and play an important role in protein metabolism as well as in bile acid deconjugation [78,79]. The authors also confirmed previously reported associations for the genera Veillonella and Streptococcus [27,59].
Bajer and colleagues revealed a marked overrepresentation of Rothia genera in PSC, regardless of the presence of IBD [25]. According to the authors and in line with previous reports, the increased abundance of the genus Rothia, especially R. mucilaginosa, may indicate that oral microbiota is overrepresented in the lower gastrointestinal tract of patients with progressive liver disease [25,31]. Due to the fact that Rothia is sensitive to gastric fluid, the authors assume that a contamination of the intestinal flora by previous endoscopic retrograde cholangiopancreatography (ERCP), particularly with repeated stenting, might be possible [25,80]. In this context, Liwinski et al. investigated oral cavity fluid, duodenal fluid, duodenal mucosa and bile fluid in parallel, revealing that the upper alimentary tract and the bile ducts of PSC patients are likewise affected by microbial dysbiosis. Their study revealed that the bile harbors a unique and diverse microbiome, clearly different from all other communities. They did not reveal any association between previous ERCPs and bile duct composition. However, Staphylococcus and Streptococcus sanguinis were over-represented in bile samples of patients with PSC who formerly received ERCP [66]. To minimize the risk of contamination of bile fluids during ERCPs the authors additionally performed sequencing of the microbiome from the proximal upper digestive system.
Lemoinne et al. were the first to address fungal changes in the fecal microbiota of patients with PSC and IBD, revealing an increase in biodiversity and an altered composition [61]. They observed an increased proportion of Exophiala, a fungi genus of the Herpotrichiellaceae family that is mainly involved in infections in immunocompromised hosts [61]. Strikingly, previous case reports already indicated an association between PSC pathology and Exophiala. Oztas et al. reported a case of a systemic infection by Exophiala dermatitidis mimicking PSC in a young woman without immunodeficiency [81]. Moreover, a case of end-stage liver disease caused by Exophilia dermatitidis and characterized by cholestasis and dilatation of intrahepatic bile ducts has been published previously [82]. Furthermore, Lemoinne et al. report a decreased proportion of Saccharomyces cerevisiae, which has anti-inflammatory characteristics and was reduced in the microbiota of patients with IBD [61,83].
4.2. Mucosal Analysis of Gut Microbiota in Patients with PSC
Compared to fecal microbiota analysis, studies of the mucosal microbiome in PSC are less consistent, smaller sample-sized, and fewer in number [51]. Two studies found little to no PSC-specific microbiome alterations, while others showed similar results as fecal analysis [24,26,51,63,65]. In addition, the variability in mucosal microbiota studies may be related to the different sample localizations as well as the different sampling techniques [51].
Quraishi’s recently published study is the first to apply an integrative and comparative approach to colonic gene expression, mucosal gut microbiota and immune cell signatures in PSC patients with IBD compared to patients with UC and healthy controls. According to the authors, the microbial alterations and the different gene expression between PSC patients with IBD compared to patients with UC imply a dysregulation of the bile acid metabolism in PSC patients with IBD [65]. Previous studies of biopsies from the colon revealed a significant increase in Escherichia, Megasphera and Lachnospiraceae, which are able to perform 7α-dehydroxylation, an important step in converting primary to secondary bile acids in the intestine [24,84]. Similar to the Veillonella species, these bacteria contain genes that encode for amine oxidases and are all potent producers of primary amines that could act as vascular adhesion protein-1 (VAP-1) [65,69,70,85]. Quraishi et al. demonstrated a reduced abundance of Prevotella and Roseburia, a butyrate producer. Anaerobic intestinal microbes produce short-chain fatty acids (SCFA) such as acetate, butyrate and propionate by fermenting nondigestible carbohydrates [86,87,88]. Butyrate is a short chain fatty acid, which promotes intestinal barrier function by intracellular tight junction assembly. Therefore, lower butyrate may contribute to weakening of the barrier integrity and dysregulation of the mucosal immunity [65,89]. SCFAs can be metabolized by enterocytes via beta-oxidation, which is thought to maintain an anaerobic environment in the gut—since it is important for survival of anaerobes. Disturbance of this homeostatic process could account for the loss of anaerobes and subsequent loss of SCFAs. SCFAs affect the innate and adaptive immune responses as well as the differentiation of regulatory T cells and disruption of these processes, which may be relevant events for PSC disease processes [90]. In their recently published study, Quraishi and colleagues also found an overrepresentation of Sphingomonas genus. These bacteria express amine oxidases and are associated with aberrant homing of gut lymphocytes to the liver, a mechanism that underlies the gut-liver axis [65,91].
Another study included biopsies from the terminal ileum and colon of patients with PSC. The authors demonstrated an increase of the Barnesiellaceae family, the Blautia genus, and operational taxonomic units (OTU), representing Clostridiales [64]. Bacteria from the Clostridiaceae family might play a crucial role in colonic homeostasis since they can affect the number, function and differentiation of colonic regulatory T cells and the production of proinflammatory cytokines [92].
In contrast, Rossen et al. conducted a study, in which they revealed that the colonic mucosa-associated microbiota in PSC is characterized by low diversity and a decreased abundance of Uncultured Clostridiales II by using phylogenetic microarrays of ileocecal biopsies [63,93]. Strikingly, in the gut flora of patients with advanced cirrhosis, an increase of Enterobacteriaceae and a reduction of Clostridiales and Bacteroides with a concomitant reduced level of fecal secondary bile acids has been described [94].
In summary, the above-mentioned studies reveal exciting new data on the mucosal and fecal gut microbiota in patients with PSC and represent an important first step in analyzing the association between the gut microbiome and PSC. The reported microbiota profiling studies demonstrate that the gut microbiota in PSC (regardless of the presence of IBD) is distinct compared with healthy controls and patients with IBD without liver disease. Unfortunately, the key question whether PSC is the cause or a result of the microbiota differences remains unanswered. On one hand, the fact that reduced microbial diversity is observed in various inflammatory and metabolic diseases might suggest that reduced microbial diversity is a secondary result of the inflammatory disease [58,95,96,97]. On the other hand, the specific relations between specific bacteria and their impact on the metabolism (e.g., bile acid, protein metabolism (Figure 2)) point towards a leading role of single microorganisms in disease pathogenesis. For instance, Veillonella, one of the most reported species enriched in PSC is not only associated with fibrotic conditions but also present in several chronic liver conditions such as primary biliary cholangitis [58,98,99]. However, the importance of the impact of single microorganisms remains unelucidated and will require further investigation.
While most studies confirm that changes in the microbiota do occur, the qualitative and quantitative changes are highly variable in different studies. The reported broad spectrum of bacteria might be associated with multiple environmental factors such as geographical region, diet, medications, physical activity and hygiene, which can induce substantial shifts in the microbiome composition [48,49,50,51]. One of the main limitations of most of the reported microbiota studies is that the influence of a wide range of environmental factors has not been considered. Thus, it remains unclear, if the significant microbiota alterations in PSC patients are stable even under different environmental conditions (e.g., diet, geographical regions). To further elucidate this question in the context of PSC, Rühlemann et al. performed a multicenter study (Germany and Norway) that included dietary patterns (but only from the German cohort) [12]. Analysis of dietary patterns did not reveal general differences between individuals based on disease status. According to the authors, only minor influences on beta diversity were observed, without association to the clear disease-associated microbial shift [12]. Similarly, studies that analyzed the impact of UDCA treatment revealed no effect on the gut microbiota, since even after exclusion of patients taking UDCA the most common bacterial genera associated with PSC dysbiosis remained [12,25,27,59]. The influence of antibiotic use on the gut microbiota has previously been demonstrated in multiple studies, however, there is no general consensus on how to adjust for antibiotic use in microbiome analysis. While Sabino et al. included all patients who had not taken any antibiotics within the previous month, Bajer et al. used a cutoff of three months and Kummen a cutoff of one year. However, none of the studies observed substantial differences related to antibiotic use. Due to the lack of multicenter studies and studies systematically analyzing the impact of environmental factors (differing diets, medication, physical activity), these questions remain unelucidated, necessitating further investigations to assess the robustness of the observed alterations.
Another important aspect is that studies focusing on fecal analysis reveal consistent results, while results on the mucosal microbiome are highly divergent. Despite studies that report a more accurate microbiome representation by analysis of mucosal samples, review of the studies mentioned above fails to reveal a clear superiority of mucosal analysis [30,51] (Table 1). Thus, more studies comparing the different analysis techniques (fecal vs. mucosal microbiome) in patients with PSC should be performed in the near future.
Although these results represent critical progress in this field, they are also associated with a number of limitations such as small sample size (n < 30 on average, n < 137 overall), the lack of cross-regional analysis and analysis of additional environmental factors influencing the gut microbiome. Most importantly, functional analyses to further understand the mechanisms underlying the association between gut microbiome and PSC are urgently needed [51]. More studies using metagenomics, proteomics, metatranscriptomics and metabolomics similar to Quraishi’s integrative pilot analysis would enrich this still underexplored field [51,58].
4.3. Gut Virome
Although the main focus of gut microbiome research and this review has so far been the bacterial compartment, the gut virome is also present but largely uncharacterized. At present, there are no published studies analyzing the gut virome in PSC patients. Sequencing of the DNA of virus-like particle preparations from fecal samples of IBD patients revealed a significant expansion of Caudovirales bacteriophages and a reduction of Microviridae compared with controls [58,100]. The viromes of patients with CD and UC were disease- and cohort-specific and most importantly, a positive correlation between bacteriophage enrichment and disease activity was shown [58,100]. The authors of the study speculate that the gut virome may contribute to intestinal inflammation by lysis of bacteria during the normal life cycle of a bacteriophage, releasing PAMPs that trigger inflammatory cascades or by induction of a humoral immune response in the host [58,100]. In the context of alcoholic hepatitis, Jiang et al. not only demonstrated that viral taxa are altered in fecal samples from patients, but also revealed an association with disease severity and mortality [101]. However, since there are no studies on the gut virome in PSC, IBD or chronic liver diseases, its potential role in these conditions remains unclear.
5. PSC and Occult Viral Infections
The role of occult viral infection in PSC patients is still unelucidated since only a small number of studies and cases with concomitant or previous occult viral infections have been reported.Moreover, current evidence does not reveal consistent or reproducible data to support a role of viruses in ongoing pathogenesis of PSC. Ponsioen et al. analyzed potential agents that increase the risk for PSC. These include bacteria such as Chlamydia spp. as well as multiple virus types such as Epstein-Barr virus, Cytomegalovirus, Mumps, Measles, Coxsackie 1-6 and hepatitis A, B and C. Their results did not reveal evidence of higher titers of the tested viruses amongst PSC patients [102]. Gerorgiadou et al. investigated the presence of occult hepatitis B infection in patients with PSC and failed to detect a significant association [103]. The prevalence of PSC in association with hepatitis C infection is very rare; so far, there are only three reported cases of patients with an initially diagnosed hepatitis C infection who developed PSC during the course of the disease [104,105]. Since larger clinical studies evaluating the role of viral infections in PSC are still lacking, future clinical and experimental studies in this field are urgently required.
6. Gut Microbiota as a Therapeutic Target and Future Approach
More insights into the impact of the gut microbiota in PSC and PSC-IBD may lead to the development of useful microbe-altering interventions. In this context, previous studies have already reported evidence that manipulation of the gut microbiota could potentially affect the disease process in PSC. A short overview of therapeutic agents targeting the gut microbiota is provided in Table 2.
6.1. Antibiotic Treatment
Emerging data on oral antibiotic treatment (vancomycin, metronidazole, minocycline) revealed a significant improvement in PSC disease activity and progression [107,108,110,112]. In addition to improvement of clinical symptoms, a significant decrease in Mayo-Risk-Score and in serum alkaline phosphatase (ALP) levels could be demonstrated [107,108,112]. The Mayo-Risk-Score is used to predict survival up to 4 years and normalization of ALP is linked to a better long-term outcome and transplant-free survival [113,114]. In particular, vancomycin has evolved as a potential therapeutic agent [107,115]. Its exact mechanisms of action remain to be elucidated but are assumed to be mediated through targeting of the microbiome as well as through anti-inflammatory and possibly immunomodulatory activities. Vancomycin has a relatively narrow antibiotic spectrum and specifically targets gram-positive bacteria such as Clostridiales. Since Clostridiales biotransforms primary bile acids into secondary bile acids in the distal small intestine and colon, it is possible that the effects of vancomycin on gut microbes influence bile acid metabolism, which in turn might reduce disease activity [112,116,117].
Supporting this notion, a recent RCT including 20 patients with metabolic syndrome revealed not only a reduced fecal microbial diversity with a drop in Firmicutes and an increase in Proteobacteria, but also a reduction of hydrophobic secondary bile acids after oral vancomycin treatment [112,115]. Furthermore, in a recent mouse study, vancomycin reversed mycophenolate mofetil (MMF)-induced gastrointestinal toxicity by restoring the MMF-induced gut dysbiosis [118]. The mechanisms contributing to MMF-related gastrointestinal toxicity have yet to be fully characterized. However, one leading hypothesis is that MMF changes the gut microbiota composition by selection for β-glucuronidase-expressing bacteria. An up-regulation of β-glucuronidase activity in the gut induces chronic inflammation and weight loss [118]. In addition, a study in 14 PSC UC children demonstrated an increase in T regulatory cells and transforming growth factor-beta levels in response to treatment with vancomycin, indicating an immunomodulatory effect [119].
Although, according to a recent meta-analysis of previous RCTs, vancomycin might be the most effective antibacterial agent for PSC, a recent matched analysis in a pediatric population (n = 264) did not show any efficacy of oral vancomycin treatment (n = 88) compared to UDCA treatment (n = 88) or no treatment (n = 88) [115,120]. Moreover, despite the absence of major adverse events, antibiotic drug resistance is still an important concern. However, strikingly, a recent case series describing the use of oral vancomycin to treat colitis in 17 children suffering from PSC, did not reveal the presence of any vancomycin-resistant enterococcus in this small cohort [115,121]. At present, most available studies are limited by small sample size. Hence, large sample-sized RCTs with longer follow-up period and treatment duration are needed. Furthermore, the exact mechanisms of these microbiome-based therapies remain to be explored [112].
6.2. Fecal Microbiota Transplantation (FMT)
Fecal microbiota transplantation (FMT is the transfer of the fecal microbiota from a healthy individual to a patient in order to reverse gut dysbiosis and restore a normal balance in the lower gastrointestinal tract [122]. Several RCTs and case reports implicate that FMT has evolved as a promising treatment alternative for recurrent Clostridioides difficile infection and UC [123,124,125,126,127,128,129]. Allegretti et al. performed a pilot study with 10 patients with PSC and IBD in remission who received FMT from one donor. Transplanted OTU included genera capable of producing SCFA, which are known to be depleted in IBD [111]. Strikingly, 30% experienced a ≥50% decrease in ALP levels. Up to now, only one pilot study has been published, thus, larger sample-sized studies are needed to define efficacy and FMT mechanisms in PSC [111].
At present, most of the trials on the use of FMT have been stopped or at least revised, because two clinical studies in the USA reported bacteremia with Extended spectrum beta-lactamase-producing E. coli after transmission as part of FMT, resulting in the death of one patient [130].
7. Bile Microbiota in PSC
Bile is a biological fluid, consisting mainly of bile acids, cholesterol, phospholipids, and proteins. Another key component of human bile is bicarbonate, and impaired bicarbonate secretion is associated with liver damage. The concept of a biliary “bicarbonate umbrella” specifies that bile duct integrity depends on a protective bicarbonate layer that facilitates deprotonation of hydrophobic bile acids, thereby preventing cholangiocellular cytotoxicity [131,132]. Therefore, several treatment options in cholestatic liver diseases directly or indirectly stabilize the biliary HCO3-umbrella [132]. For instance, UDCA enhances the secretory capacity of hepatocytes and cholangiocytes, improving biliary HCO3-secretion [132].
Bile acids, which are the major organic solutes of human bile, are believed to play a crucial role in the pathogenesis of cholestatic liver diseases such as PSC. As conversion of primary bile acids into secondary, potentially noxious bile acids is thought to be primarily driven by the bacterial gut microbiome, microbial dysbiosis is expected to play a crucial role in PSC [23]. Moreover, even if bile acids do not represent the primary cause of cholestatic liver injury, it is believed that they may perpetuate disease progression by generating or maintaining a chemokine and cytokine response [133].
Emerging data using next-generation sequencing indicate that bile might not always be sterile, as assumed before, but can harbor a diverse bile microbiome [93,134,135,136]. Patients with PSC are characterized by ecological alterations of the ductal bile, including reduced biodiversity and expansion of pathogenic bacteria such as Enterococcus faecalis, which is associated with impaired intestinal permeability and mucosal inflammation due to its production of matrix metalloproteinases such as gelatinase [66,137]. Of note, microbial dysbiosis such as Enterococcus abundance is associated with an increase of the proinflammatory and potentially cancerogenic bile acid taurolithocholic acid [66]. Moreover, earlier culture-based studies suggested that biliary isolates of bacteria, such as Enterococcus faecalis, the most frequently identified genus in bile culture studies, and Enterococcus gallinarum, induce a T helper cell type 17 immune response in patients with PSC [56,138,139].
Currently, only a limited number of studies have investigated bile microbiota in PSC patients [66,93]. Peirara et al. detected only slight microbial alterations in patients with either biliary dysplasia or cholangiocarcinoma, while patients without disease complications showed virtually no microbial differences compared with controls. Nevertheless, their study results are limited by a small patient cohort. In contrast, after analysis of 46 PSC patients, Liwinski et al. suggested that biliary dysbiosis might be linked with elevated concentrations of the proinflammatory and potentially cancerogenic agent taurolithocholic acid [66]. They observed a significant increase of the facultative anaerobic phylum Proteobacteria in the bile fluid of patients with PSC. The increase of Proteobacterica, comprising many human pathogens, such as members of the Enterobacteriaceae family, is associated with increased epithelial oxygen availability, a hallmark of inflammation, epithelial dysfunction and disease [140]. In addition, bile fluid analysis of PSC patients revealed an increased frequency of known cholangitis pathogens such as Enterococcus, Prevotella, Staphylococcus and Cutibacterium [66]. Moreover, an increase in Streptococcus abundance was significantly associated with the number of ERC examinations and disease severity [93]. Furthermore, Liwinski et al. report an increased microbial burden in the bile with increased PSC duration.
Since there is only a limited number of studies investigating the association between bile microbiota and PSC, more research in this field is urgently needed. In particular, multi-center studies that include the impact of different environmental factors have to be performed in near future [66].
7.1. Bile Acid Pathways and Their Therapeutic Targets
Pharmacotherapies targeting bile acid pathways represent an active area of research for the treatment of PSC. A short overview of a selection of therapeutic agents is provided in Table 3.
UDCA is widely used. Several studies demonstrated a beneficial effect on liver biochemistries, but no long-term benefits such as liver transplant or death coule be shown up to now [141,142]. However, a study by Lindor et al. revealed an association between UDCA and an increased risk of serious adverse events when used at higher doses (28-30 mg/kg per day) [143]. Moreover, previous studies showed that the altered gut microbiota in PSC patients is independent of treatment with UDCA [27,59]. Further, Fickert et al. revealed that treatment with norursodeoxycholic acid (norUDCA), a derivative of UCDA, resulted in significant reductions of ALP levels and a similar safety profile (low incidence of pruritus) compared to the placebo group after 12 weeks of treatment duration [51,149]. Kowdley and colleagues conducted a RCT including 76 PSC patients to investigate obeticholic acid (OCA), a semi-synthetic FXR ligand. They demonstrated a significant decrease in ALP levels after 24 weeks treatment duration, however, a higher proportion of patients suffering from pruritus have been reported [145]. Trauner et al. investigated Cilofexor, a nonsteroidal FXR agonist, which led to lower ALP levels at week 12 regardless of UCDA treatment [51,146]. Bezafibrate, a ligand of the intranuclear receptor peroxisome-proliferator-activated receptor alpha (PPARα), is another therapeutic agent that showed evidence of an improvement in serum liver enzymes in RCT [147,148]. It leads to an increase in P-glycoprotein levels in the bile duct canaliculi, which in turn increases phospholipid levels in the bile, leading to protection of the bile from toxic bile acids (anti-inflammatory effect) [150]. Moreover, an association with superoxide dismutase expression in the liver has been reported (anti-oxidative effect) [151]. Furthermore, Lemoinne et al. performed a retrospective analysis on 20 patients with incomplete response to UDCA, revealing an improvement in ALP levels as well as in pruritus after addition of bezafibrate or fenofibrate, another PPARα agonist [152]. Another therapeutic agent is seladelpar, a selective peroxisome-proliferator-activated receptor delta (PPARδ) agonist that is the subject of an ongoing RCT (NCT04024813). Whether and how these therapeutic interventions interfere with the microbiota should be analyzed in follow-up studies.
8. PSC and CCA
8.1. PSC and CCA-Overview
CCA is the most common malignancy in patients with PSC and the sixth most common cause of cancer in the gastrointestinal tract in the western world world [153,154,155]. The prevalence of CCA in patients with PSC is between 7 to 13% [155]. Although the pathogenesis of CCA in PSC is largely unelucidated, inflammation-driven carcinogenesis concomitant with genetic and epigenetic alterations are thought to be the underlying factors [156,157,158]. However, the knowledge of predisposing risk factors is scarce and PSC-CCA development does not seem to be linked to duration and severity of PSC [159,160,161,162]. The leading hypothesis is that PSC causes multifocal strictures of the biliary tree, biliary infections, and accumulation of toxic bile acids, which results in an increased expression of adhesion and antigen-presenting molecules and inflammatory mediators. Macrophages trigger the apoptosis and senescence of cholangiocytes in activated T cells and the production of proinflammatory chemokines and cytokines acting on hepatic stellate cells leads to liver fibrosis. In response, cholangiocytes lining the biliary tree activate a wide range of cellular processes such as hepatocellular proliferation, apoptosis, angiogenesis and fibrosis as well as immunoregulatory functions, such as upregulation of human leukocyte antigen molecules and recruitment of immune cells. However, it remains unclear if activated cholangiocytes directly activate B cell antibody production and T cell expansion [162]. Moreover, the biliary epithelial injury-induced regenerative response by peribiliary glands is also believed to be a key process involved in inflammation-induced CCA carcinogenesis [163,164]. The cholangiocytes and associated progenitor cells lining the biliary tree can undergo DNA damage, metaplasia, and low-grade dysplasia. In case of persistent stimuli, the disease may progress to high-grade dysplasia and ultimately CCA [156,157,165,166,167]. Emerging evidence already demonstrated that cholangiocytes in explant livers from PSC-CCA patients show metaplasia and dysplasia more often than in patients with PSC without CCA [162,168,169]. Strikingly, the majority of CCA cases develop from a dominant stricture, which is defined as a stricture with a diameter < 1.5 mm in the common bile duct or <1.0 mm in the hepatic duct [155].
CCA is the most common malignancy in patients with PSC and the sixth most common cause of cancer in the gastrointestinal tract in the western world world [153,154,155]. The prevalence of CCA in patients with PSC is between 7 to 13% [155]. Although the pathogenesis of CCA in PSC is largely unelucidated, inflammation-driven carcinogenesis concomitant with genetic and epigenetic alterations are thought to be the underlying factors [156,157,158]. However, the knowledge of predisposing risk factors is scarce and PSC-CCA development does not seem to be linked to duration and severity of PSC [159,160,161,162]. The leading hypothesis is that PSC causes multifocal strictures of the biliary tree, biliary infections, and accumulation of toxic bile acids, which results in an increased expression of adhesion and antigen-presenting molecules and inflammatory mediators. Macrophages trigger the apoptosis and senescence of cholangiocytes in activated T cells and the production of proinflammatory chemokines and cytokines acting on hepatic stellate cells leads to liver fibrosis. In response, cholangiocytes lining the biliary tree activate a wide range of cellular processes such as hepatocellular proliferation, apoptosis, angiogenesis and fibrosis as well as immunoregulatory functions, such as upregulation of human leukocyte antigen molecules and recruitment of immune cells. However, it remains unclear if activated cholangiocytes directly activate B cell antibody production and T cell expansion [162]. Moreover, the biliary epithelial injury-induced regenerative response by peribiliary glands is also believed to be a key process involved in inflammation-induced CCA carcinogenesis [163,164]. The cholangiocytes and associated progenitor cells lining the biliary tree can undergo DNA damage, metaplasia, and low-grade dysplasia. In case of persistent stimuli, the disease may progress to high-grade dysplasia and ultimately CCA [156,157,165,166,167]. Emerging evidence already demonstrated that cholangiocytes in explant livers from PSC-CCA patients show metaplasia and dysplasia more often than in patients with PSC without CCA [162,168,169]. Strikingly, the majority of CCA cases develop from a dominant stricture, which is defined as a stricture with a diameter < 1.5 mm in the common bile duct or <1.0 mm in the hepatic duct [155].
8.2. Gut Microbiota in CCA
The role of biliary microbiota has scarcely been investigated in CCA. However, previous studies performing 16S RNA sequencing demonstrated a significant difference of biliary microbiota composition between CCA patients and controls [162]. Initially, some authors suggested that bile fluid dysbiosis could be linked to various diseases, including biliary lithiasis. They hypothesized that the reduction of the biliary flow by (partial) obstruction could cause a change in biliary microbiota composition [135,170,171,172]. Although bacteria may find a more favorable growth environment in obstructed bile, this likely does not represent the only reason for differential colonization. It has been indicated that altered bile metabolism may provide metabolic advantages to certain bacteria, which might explain tumor growth and outcomes through host immune response to CCA [112,162]. Jia et al. were the first to characterize intestinal microbiota, bile acids, and cytokines in patients with CCA by analysis of fecal microbiota in a series of intrahepatic CCA (iCCA). Their findings revealed that the α-diversities and β-diversities of iCCA were highest and that the abundance of four genera (Lactobacillus, Actinomyces, Peptostreptococcaceae and Alloscardovia) was increased in patients with iCCA compared to non-iCCA patients (HCC, healthy, liver cirrhosis) [173]. Based on previous findings that suggest a different pattern of bile acid concentration in biliary cancer (compared to patients with benign biliary diseases), Jia et al. quantified bile acids in serum and stool [173,174]. They found that total serum bile acids were higher in patients with CCA compared to healthy controls. Moreover, they demonstrated increased plasma-stool ratios of glycoursodeoxycholic acid and tauroursodeoxycholic acid in patients with iCCA as well as a positive correlation of tauroursodeoxycholic acid with Lactobacillus and Alloscardovia, proposing them as diagnostic markers [173].
Avilés-Jiménez et al. compared mucosal microbiota (tissue samples) of extrahepatic CCA (eCCA) to benign biliary tumors and showed an abundance of Methylophilaceae, Fusobacterium, Prevotella, Actinomyces, Novosphingobium and Helicobacter pylori (HP) in eCCA [175]. However, despite the leading role of HP in various cancers, the presence of HP and Actinomyces are assumed to be gastric/intestinal contamination since these findings could not be confirmed in similar follow-up studies [162,173].
According to a recent study by Saab et al. (n = 28 CCA patients, n = 47 controls), the most abundant genera were Enterococcus, Streptococcus, Bacteroides, Klebsiella, and Pyramidobacter in CCA’s biliary microbiota. Strikingly, Bacteroides Geobacillus, Meiothermus, and Anoxybacillus genera levels were significantly higher in CCA patients’ biliary microbiota compared to the control group. Up to now, no link between carcinogenesis, especially CCA, and Geobacillus, Meiothermus, and Anoxybacillus levels could be demonstrated. In contrast, several studies have already demonstrated associations between Bacteroides and colon cancer, cholethiasis through metabolomic changes and various autoimmune diseases, such as arthritis in transgenic rats HLA-B27 [176,177,178,179,180].
Current literature on microbiota and PSC-CCA is scarce due to the fact that the pathogenesis of transformation from PSC to PSC-CCA remains unelucidated. Therefore, future studies should focus on the transformation of inflammatory changes of the biliary tree to cancer. Based on the results of these studies, further analysis on gut microbiota in PSC-CCA should follow [162,181].
9. Gallbladder Carcinoma—Overview and Role of Genotoxins in Carcinogenesis
Gallbladder cancer is the most common cancer of the biliary system and associated with a very poor prognosis. The interplay of genetic predisposition, lifestyle factors and infections in gallbladder carcinogenesis remains obscure. Its incidence is relatively high in the western parts of South America and in the northern part of the Indian subcontinent, while being rather uncommon in Western countries [182]. Strikingly, in these regions, an epidemiological association between gall bladder cancer and chronic infection with the bacteria Salmonella enterica Typhi/Paratyphi A exists. The leading hypothesis is that in these patients, Salmonella resides in the gallbladder by forming biofilms on gallstones, which facilitates enhanced colonization, persistence in this organ and carriage into the duodenum [183,184,185,186]. Boccellato and colleagues developed a three-dimensional organoid model that recapitulates the infection dynamics in the healthy gallbladder epithelium. Infection with Salmonella, in particular the typhoid toxin CdtB subunit, induces DNA double-strand breaks. With their advanced organoid model, the authors were able to see the toxin and DNA damage spread to neighboring cells that were not infected with Salmonella. While infected cells underwent a toxin-independent cell cycle arrest, uninfected intoxicated cells were able to continue proliferating despite the DNA damage, providing a potential opportunity for malignant transformation [187].
There is emerging evidence that bacterial infections and bacterial products from HP, E. coli, Chlamydia trachomatis and others can cause DNA damage in host cells by secreting genotoxic proteins or through mechanisms involving host response to the infection [188,189,190,191,192,193]. In addition to producing genotoxins or causing DNA damage through increased production of reactive oxygen species, bacterial infections can also modify the DNA damage response, thereby interfering with mechanisms of repair [188]. Recent studies provided evidence that colibactin, a bacterial toxin that is expressed in E. coli and Klebsiella pneumoniae, might have an important impact on cancerogenesis by directly causing DNA damage [194,195]. Recently, a specific colibactin-driven genomic signature has been found using organoids [195]. Meyer and colleagues also found DNA breaks at a distinct sequence motif upon infection and confirmed their occurrence in human cancer [194]. Furthermore, short-term exposure to colibactin-producing E. coli was sufficient to promote colonic organoid transformation [196]. Thus, future studies involving the genotoxic effect of specific bacterial products such as colibactin and their association to biliary cancer are urgently needed. It will be important to investigate whether and to what extent direct genotoxic effects also contribute to PSC-associated cancers.
10. Conclusions
Several clinical studies, which analyzed microbial bacterial composition in fecal, mucosal and bile samples have demonstrated that the gut microbiome in patients with PSC and PSC and IBD is different from that in healthy controls and IBD patients only. Moreover, the frequent concomitance of PSC and IBD indicates a critical role of the gut-liver axis in disease pathogenesis. Experimental and clinical data suggest that a diverse set of microbial functions may be relevant, including endogenous molecules produced by the microbiota, bacterial processing of pharmacological agents or dietary compounds and specific bacterial molecules or metabolites driving the immune process. With regard to carcinogenesis, there is emerging evidence that bacterial products can directly promote epithelial injury and act as genotoxins.
Despite crucial progress in this field over the past years, a better understanding of the functional characterization of PSC, the gut microbiome, and related malignancies is needed. Therefore, future research should perform cross-sectional and longitudinal studies with larger patient cohorts to identify the role of the microbiota in disease progression.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors would like to thank Rike Zietlow for editing the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| Alkaline phosphatase | ALP |
| Cholangiocarcinoma | CCA |
| Crohn’s disease | CD |
| Endoscopic retrograde cholangiopancreatography | ERCP |
| Extrahepatic CCA | eCCA |
| Fecal microbiota transplantation | FMT |
| Free fatty acids | FFA |
| Helicobacter pylori | HP |
| Inflammatory bowel disease | IBD |
| Intrahepatic CCA | iCCA |
| Lipopolysaccharide-binding protein | LBP |
| Pathogen-associated molecular patterns | PAMPs |
| Peroxisome-proliferator-activated receptor alpha | PPARα |
| Peroxisome-proliferator-activated receptor delta | PPARδ |
| Multidrug resistance 2 | mdr2 |
| Mucosal addressin cell-adhesion molecule 1 | MAdCAM1 |
| Mycophenolate mofetil | MMF |
| Nonalcoholic fatty liver disease | NAFLD |
| Norursodeoxycholic acid | norUDCA |
| Operational Taxonomic Units | OTU |
| Primary sclerosing cholangitis | PSC |
| Short-chain fatty acids | SCFA |
| Ulcerative colitis | UC |
| Ursodeoxycholic acid | UDCA |
| Vascular adhesion protein-1 | VAP-1 |
References
- Lazaridis, K.N.; LaRusso, N.F. Primary Sclerosing Cholangitis. N. Engl. J. Med. 2016, 375, 1161–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eaton, J.E.; Talwalkar, J.A.; Lazaridis, K.N.; Gores, G.J.; Lindor, K.D. Pathogenesis of Primary Sclerosing Cholangitis and Advances in Diagnosis and Management. Gastroenterology 2013, 145, 521–536. [Google Scholar] [CrossRef] [Green Version]
- Hirschfield, G.M.; Karlsen, T.H.; Lindor, K.D.; Adams, D. Primary sclerosing cholangitis. Lancet 2013, 382, 1587–1599. [Google Scholar] [CrossRef]
- Molodecky, N.A.; Kareemi, H.; Parab, R.; Barkema, H.; Quan, H.; Myers, R.P.; Kaplan, G.G. Incidence of primary sclerosing cholangitis: A systematic review and meta-analysis. Hepatology 2011, 53, 1590–1599. [Google Scholar] [CrossRef]
- Boonstra, K.; Beuers, U.; Ponsioen, C.Y. Epidemiology of primary sclerosing cholangitis and primary biliary cirrhosis: A systematic review. J. Hepatol. 2012, 56, 1181–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlsen, T.H.; Folseraas, T.; Thorburn, D.; Vesterhus, M. Primary sclerosing cholangitis—A comprehensive review. J. Hepatol. 2017, 67, 1298–1323. [Google Scholar] [CrossRef] [Green Version]
- Staufer, K.; Kivaranovic, D.; Rasoul-Rockenschaub, S.; Soliman, T.; Trauner, M.; Berlakovich, G. Waitlist mortality and post-transplant survival in patients with cholestatic liver disease—Impact of changes in allocation policy. HPB 2018, 20, 916–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmela, C.; Peerani, F.; Castaneda, D.; Torres, J.; Itzkowitz, S.H. Inflammatory Bowel Disease and Primary Sclerosing Cholangitis: A Review of the Phenotype and Associated Specific Features. Gut Liver 2018, 12, 17–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mertz, A.; Nguyen, N.A.; Katsanos, K.H.; Kwok, R.M. Primary sclerosing cholangitis and inflammatory bowel disease comorbidity: An update of the evidence. Ann. Gastroenterol. 2019, 32, 124–133. [Google Scholar] [CrossRef]
- Chapman, R.; Fevery, J.; Kalloo, A.; Nagorney, D.M.; Boberg, K.M.; Shneider, B.; Gores, G.J. Diagnosis and management of primary sclerosing cholangitis. Hepatology 2010, 51, 660–678. [Google Scholar] [CrossRef]
- Lunder, A.K.; Hov, J.R.; Borthne, A.; Gleditsch, J.; Johannesen, G.; Tveit, K.; Viktil, E.; Henriksen, M.; Hovde, Ø.; Huppertz-Hauss, G.; et al. Prevalence of Sclerosing Cholangitis Detected by Magnetic Resonance Cholangiography in Patients With Long-term Inflammatory Bowel Disease. Gastroenterology 2016, 151, 660–669.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rühlemann, M.; Liwinski, T.; Heinsen, F.-A.; Bang, C.; Zenouzi, R.; Kummen, M.; Thingholm, L.; Tempel, M.; Lieb, W.; Karlsen, T.; et al. Consistent alterations in faecal microbiomes of patients with primary sclerosing cholangitis independent of associated colitis. Aliment. Pharmacol. Ther. 2019, 50, 580–589. [Google Scholar] [CrossRef] [Green Version]
- Loftus, E.V.; Harewood, G.C.; Loftus, C.G.; Tremaine, W.J.; Harmsen, W.S.; Zinsmeister, A.R.; Jewell, D.A.; Sandborn, W.J. PSC-IBD: A unique form of inflammatory bowel disease associated with primary sclerosing cholangitis. Gut 2005, 54, 91–96. [Google Scholar] [CrossRef] [PubMed]
- De Vries, A.B.; Janse, M.; Blokzijl, H.; Weersma, R.K. Distinctive inflammatory bowel disease phenotype in primary sclerosing cholangitis. World J. Gastroenterol. 2015, 21, 1956–1971. [Google Scholar] [CrossRef] [Green Version]
- Ricciuto, A.; Kamath, B.M.; Griffiths, A.M. The IBD and PSC Phenotypes of PSC-IBD. Curr. Gastroenterol. Rep. 2018, 20, 16. [Google Scholar] [CrossRef]
- Cleveland, N.K.; Rubin, D.T.; Hart, J.; Weber, C.R.; Meckel, K.; Tran, A.L.; Aelvoet, A.S.; Pan, I.; Gonsalves, A.; Gaetano, J.N.; et al. Patients With Ulcerative Colitis and Primary Sclerosing Cholangitis Frequently Have Subclinical Inflammation in the Proximal Colon. Clin. Gastroenterol. Hepatol. 2018, 16, 68–74. [Google Scholar] [CrossRef]
- Vera, A.; Moledina, S.; Gunson, B.; Hubscher, S.; Mirza, D.; Olliff, S.; Neuberger, J. Risk factors for recurrence of primary sclerosing cholangitis of liver allograft. Lancet 2002, 360, 1943–1944. [Google Scholar] [CrossRef]
- Steenstraten, I.C.; Korkmaz, K.S.; Trivedi, P.J.; Inderson, A.; Van Hoek, B.; Girondo, M.D.M.R.; Maljaars, P.W.J. Systematic review with meta-analysis: Risk factors for recurrent primary sclerosing cholangitis after liver transplantation. Aliment. Pharmacol. Ther. 2019, 49, 636–643. [Google Scholar] [CrossRef] [PubMed]
- Kostic, A.; Xavier, R.J.; Gevers, D. The Microbiome in Inflammatory Bowel Disease: Current Status and the Future Ahead. Gastroenterology 2014, 146, 1489–1499. [Google Scholar] [CrossRef] [Green Version]
- Sokol, H.; Pigneur, B.; Watterlot, L.; Lakhdari, O.; Bermúdez-Humarán, L.G.; Gratadoux, J.-J.; Blugeon, S.; Bridonneau, C.; Furet, J.-P.; Corthier, G.; et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. USA 2008, 105, 16731–16736. [Google Scholar] [CrossRef] [Green Version]
- Joossens, M.; Huys, G.; Cnockaert, M.; De Preter, V.; Verbeke, K.; Rutgeerts, P.; Vandamme, P.; Vermeire, S. Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut 2011, 60, 631–637. [Google Scholar] [CrossRef] [Green Version]
- Dignass, A.; Eliakim, R.; Magro, F.; Maaser, C.; Chowers, Y.; Geboes, K.; Mantzaris, G.; Reinisch, W.; Colombel, J.-F.; Vermeire, S.; et al. Second European evidence-based consensus on the diagnosis and management of ulcerative colitis Part 1: Definitions and diagnosis. J. Crohn’s Colitis 2012, 6, 965–990. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Tang, R.; Leung, P.S.; Gershwin, M.E.; Ma, X. Bile acids and intestinal microbiota in autoimmune cholestatic liver diseases. Autoimmun. Rev. 2017, 16, 885–896. [Google Scholar] [CrossRef] [PubMed]
- Quraishi, M.N.; Sergeant, M.; Kay, G.; Iqbal, T.; Chan, J.; Constantinidou, C.; Trivedi, P.; Ferguson, J.; Adams, D.; Pallen, M.; et al. The gut-adherent microbiota of PSC–IBD is distinct to that of IBD. Gut 2017, 66, 386–388. [Google Scholar] [CrossRef]
- Bajer, L.; Kverka, M.; Kostovcik, M.; Macinga, P.; Dvorak, J.; Stehlikova, Z.; Brezina, J.; Wohl, P.; Spicak, J.; Drastich, P. Distinct gut microbiota profiles in patients with primary sclerosing cholangitis and ulcerative colitis. World J. Gastroenterol. 2017, 23, 4548–4558. [Google Scholar] [CrossRef] [PubMed]
- Kevans, D.; Tyler, A.D.; Holm, K.; Jorgensen, K.K.; Vatn, M.H.; Karlsen, T.H.; Kaplan, G.; Eksteen, B.; Gevers, D.; Hov, J.; et al. Characterization of Intestinal Microbiota in Ulcerative Colitis Patients with and without Primary Sclerosing Cholangitis. J. Crohn’s Colitis 2016, 10, 330–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kummen, M.; Holm, K.; Anmarkrud, J.A.; Nygård, S.; Vesterhus, M.; Høivik, M.L.; Trøseid, M.; Marschall, H.-U.; Schrumpf, E.; Moum, B.; et al. The gut microbial profile in patients with primary sclerosing cholangitis is distinct from patients with ulcerative colitis without biliary disease and healthy controls. Gut 2017, 66, 611–619. [Google Scholar] [CrossRef] [Green Version]
- Lazaridis, K.N.; LaRusso, N.F. The Cholangiopathies. Mayo Clin. Proc. 2015, 90, 791–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gevers, D.; Kugathasan, S.; Denson, L.A.; Vázquez-Baeza, Y.; Van Treuren, W.; Ren, B.; Schwager, E.; Knights, D.; Song, S.J.; Yassour, M.; et al. The Treatment-Naive Microbiome in New-Onset Crohn’s Disease. Cell Host Microbe 2014, 15, 382–392. [Google Scholar] [CrossRef] [Green Version]
- Qin, N.; Yang, F.; Li, A.; Prifti, E.; Chen, Y.; Shao, L.; Guo, J.; Le Chatelier, E.; Yao, J.; Wu, L.; et al. Alterations of the human gut microbiome in liver cirrhosis. Nature 2014, 513, 59–64. [Google Scholar] [CrossRef]
- Seo, Y.S.; Shah, V.H. The role of gut-liver axis in the pathogenesis of liver cirrhosis and portal hypertension. Clin. Mol. Hepatol. 2012, 18, 337–346. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, A.; Debelius, J.; Brenner, D.A.; Karin, M.; Loomba, R.; Schnabl, B.; Knight, R. The gut–liver axis and the intersection with the microbiome. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 397–411. [Google Scholar] [CrossRef]
- Seki, E.; Schnabl, B. Role of innate immunity and the microbiota in liver fibrosis: Crosstalk between the liver and gut. J. Physiol. 2012, 590, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Anand, G.; Zarrinpar, A.; Loomba, R. Targeting Dysbiosis for the Treatment of Liver Disease. Semin. Liver Dis. 2016, 36, 037–047. [Google Scholar] [CrossRef] [PubMed]
- Lichtman, S.N.; Keku, J.; Clark, R.L.; Schwab, J.H.; Sartor, R.B. Biliary tract disease in rats with experimental small bowel bacterial overgrowth. Hepatology 1991, 13, 766–772. [Google Scholar] [CrossRef]
- Lichtman, S.N.; Okoruwa, E.E.; Keku, J.; Schwab, J.H.; Sartor, R.B. Degradation of endogenous bacterial cell wall polymers by the muralytic enzyme mutanolysin prevents hepatobiliary injury in genetically susceptible rats with experimental intestinal bacterial overgrowth. J. Clin. Investig. 1992, 90, 1313–1322. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.-M.; Kaplan, M.M. Primary Sclerosing Cholangitis. N. Engl. J. Med. 1995, 332, 924–933. [Google Scholar] [CrossRef] [PubMed]
- Guicciardi, M.E.; Trussoni, C.E.; Krishnan, A.; Bronk, S.F.; Pisarello, M.J.L.; O’Hara, S.P.; Splinter, P.L.; Gao, Y.; Vig, P.; Revzin, A.; et al. Macrophages contribute to the pathogenesis of sclerosing cholangitis in mice. J. Hepatol. 2018, 69, 676–686. [Google Scholar] [CrossRef]
- Lichtman, S.N.; Keku, J.; Schwab, J.H.; Sartor, R. Hepatic injury associated with small bowel bacterial overgrowth in rats is prevented by metronidazole and tetracycline. Gastroenterology 1991, 100, 513–519. [Google Scholar] [CrossRef]
- Dhillon, A.K.; Kummen, M.; Trøseid, M.; Åkra, S.; Liaskou, E.; Moum, B.; Vesterhus, M.; Karlsen, T.H.; Seljeflot, I.; Hov, J.R. Circulating markers of gut barrier function associated with disease severity in primary sclerosing cholangitis. Liver Int. 2018, 39, 371–381. [Google Scholar] [CrossRef] [Green Version]
- Björnsson, E.; Cederborg, A.; Åkvist, A.; Simren, M.; Stotzer, P.-O.; Bjarnason, I. Intestinal permeability and bacterial growth of the small bowel in patients with primary sclerosing cholangitis. Scand. J. Gastroenterol. 2005, 40, 1090–1094. [Google Scholar] [CrossRef]
- Eksteen, B.; Grant, A.J.; Miles, A.; Curbishley, S.M.; Lalor, P.; Hübscher, S.G.; Briskin, M.; Salmon, M.; Adams, D.H. Hepatic Endothelial CCL25 Mediates the Recruitment of CCR9+ Gut-homing Lymphocytes to the Liver in Primary Sclerosing Cholangitis. J. Exp. Med. 2004, 200, 1511–1517. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.H.; Eksteen, B. Aberrant homing of mucosal T cells and extra-intestinal manifestations of inflammatory bowel disease. Nat. Rev. Immunol. 2006, 6, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Terjung, B.; Söhne, J.; Lechtenberg, B.; Gottwein, J.; Muennich, M.; Herzog, V.; Mähler, M.; Sauerbruch, T.; Spengler, U. p-ANCAs in autoimmune liver disorders recognise human -tubulin isotype 5 and cross-react with microbial protein FtsZ. Gut 2009, 59, 808–816. [Google Scholar] [CrossRef] [Green Version]
- Lynch, K.D.; Chapman, R.W.; Keshav, S.; Montano-Loza, A.J.; Mason, A.L.; Kremer, A.E.; Vetter, M.; de Krijger, M.; Ponsioen, C.Y.; Trivedi, P.; et al. Effects of Vedolizumab in Patients With Primary Sclerosing Cholangitis and Inflammatory Bowel Diseases. Clin. Gastroenterol. Hepatol. 2020, 18, 179–187.e6. [Google Scholar] [CrossRef] [Green Version]
- Laborda, T.J.; Ricciuto, A.; Aumar, M.; Carman, N.; DiGuglielmo, M.; Draijer, L.G.; Furuya, K.N.; Gupta, N.; Koot, B.G.; Loomes, K.M.; et al. Vedolizumab Therapy in Children With Primary Sclerosing Cholangitis: Data From the Pediatric Primary Sclerosing Cholangitis Consortium. J. Pediatr. Gastroenterol. Nutr. 2020, 71, 459–464. [Google Scholar] [CrossRef]
- Zuo, T.; Kamm, M.A.; Colombel, J.-F.; Ng, S.C. Urbanization and the gut microbiota in health and inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 440–452. [Google Scholar] [CrossRef]
- Schroeder, B.O.; Bäckhed, F. Signals from the gut microbiota to distant organs in physiology and disease. Nat. Med. 2016, 22, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef]
- Little, R.; Wine, E.; Kamath, B.M.; Griffiths, A.M.; Ricciuto, A. Gut microbiome in primary sclerosing cholangitis: A review. World J. Gastroenterol. 2020, 26, 2768–2780. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Z.; Hov, J.R.; Folseraas, T.; Ellinghaus, E.; Rushbrook, S.M.; Doncheva, N.T.; Andreassen, O.A.; Weersma, R.K.; Weismüller, T.J.; Eksteen, B.; et al. Dense genotyping of immune-related disease regions identifies nine new risk loci for primary sclerosing cholangitis. Nat. Genet. 2013, 45, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Rupp, C.; Friedrich, K.; Folseraas, T.; Wannhoff, A.; Bode, K.A.; Weiss, K.-H.; Schirmacher, P.; Sauer, P.; Stremmel, W.; Gotthardt, D.N. Fut2 genotype is a risk factor for dominant stenosis and biliary candida infections in primary sclerosing cholangitis. Aliment. Pharmacol. Ther. 2014, 39, 873–882. [Google Scholar] [CrossRef]
- Wannhoff, A.; Rupp, C.; Friedrich, K.; Brune, M.; Knierim, J.; Flechtenmacher, C.; Sauer, P.; Stremmel, W.; Hov, J.; Schirmacher, P.; et al. Inflammation But Not Biliary Obstruction Is Associated with Carbohydrate Antigen 19-9 Levels in Patients with Primary Sclerosing Cholangitis. Clin. Gastroenterol. Hepatol. 2015, 13, 2372–2379. [Google Scholar] [CrossRef] [PubMed]
- Tabibian, J.H.; O’Hara, S.P.; Trussoni, C.E.; Tietz, P.S.; Splinter, P.L.; Mounajjed, T.; Hagey, L.R.; LaRusso, N.F. Absence of the intestinal microbiota exacerbates hepatobiliary disease in a murine model of primary sclerosing cholangitis. Hepatology 2016, 63, 185–196. [Google Scholar] [CrossRef] [Green Version]
- Nakamoto, N.; Sasaki, N.; Aoki, R.; Miyamoto, K.; Suda, W.; Teratani, T.; Suzuki, T.; Koda, Y.; Chu, P.-S.; Taniki, N.; et al. Gut pathobionts underlie intestinal barrier dysfunction and liver T helper 17 cell immune response in primary sclerosing cholangitis. Nat. Microbiol. 2019, 4, 492–503. [Google Scholar] [CrossRef]
- Patel, M.; Watson, A.J.; Rushbrook, S. A Mechanistic Insight Into the Role of Gut Microbiota in the Pathogenesis of Primary Sclerosing Cholangitis. Gastroenterology 2019, 157, 1686–1688. [Google Scholar] [CrossRef] [Green Version]
- Hov, J.R.; Karlsen, T.H. The Microbiome in Primary Sclerosing Cholangitis: Current Evidence and Potential Concepts. Semin. Liver Dis. 2017, 37, 314–331. [Google Scholar] [CrossRef] [Green Version]
- Sabino, J.; Vieira-Silva, S.; Machiels, K.; Joossens, M.; Falony, G.; Ballet, V.; Ferrante, M.; Van Assche, G.; Van Der Merwe, S.; Vermeire, S.; et al. Primary sclerosing cholangitis is characterised by intestinal dysbiosis independent from IBD. Gut 2016, 65, 1681–1689. [Google Scholar] [CrossRef] [Green Version]
- Iwasawa, K.; Suda, W.; Tsunoda, T.; Oikawa-Kawamoto, M.; Umetsu, S.; Inui, A.; Fujisawa, T.; Morita, H.; Sogo, T.; Hattori, M. Characterisation of the faecal microbiota in Japanese patients with paediatric-onset primary sclerosing cholangitis. Gut 2016, 66, 1344–1346. [Google Scholar] [CrossRef] [Green Version]
- Lemoinne, S.; Kemgang, A.; Ben Belkacem, K.; Straube, M.; Jegou, S.; Corpechot, C.; Chazouillères, O.; Housset, C.; Sokol, H.; Network, S.-A.I. Fungi participate in the dysbiosis of gut microbiota in patients with primary sclerosing cholangitis. Gut 2020, 69, 92–102. [Google Scholar] [CrossRef] [Green Version]
- Kummen, M.; Thingholm, L.B.; Rühlemann, M.C.; Holm, K.; Hansen, S.H.; Moitinho-Silva, L.; Liwinski, T.; Zenouzi, R.; Storm-Larsen, C.; Midttun, Ø.; et al. Altered Gut Microbial Metabolism of Essential Nutrients in Primary Sclerosing Cholangitis. Gastroenterology 2021, 160, 1784–1798. [Google Scholar] [CrossRef] [PubMed]
- Rossen, N.G.; Fuentes, S.; Boonstra, K.; D’Haens, G.R.; Heilig, H.G.; Zoetendal, E.G.; De Vos, W.M.; Ponsioen, C.Y. The Mucosa-associated Microbiota of PSC Patients is Characterized by Low Diversity and Low Abundance of Uncultured Clostridiales II. J. Crohn’s Colitis 2015, 9, 342–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, J.; Bao, X.; Goel, A.; Colombel, J.-F.; Pekow, J.; Jabri, B.; Williams, K.; Castillo, A.; Odin, J.; Meckel, K.; et al. The features of mucosa-associated microbiota in primary sclerosing cholangitis. Aliment. Pharmacol. Ther. 2016, 43, 790–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quraishi, M.N.; Acharjee, A.; Beggs, A.D.; Horniblow, R.; Tselepis, C.; Gkoutos, G.; Ghosh, S.; Rossiter, A.E.; Loman, N.; Van Schaik, W.; et al. A Pilot Integrative Analysis of Colonic Gene Expression, Gut Microbiota, and Immune Infiltration in Primary Sclerosing Cholangitis-Inflammatory Bowel Disease: Association of Disease With Bile Acid Pathways. J. Crohn’s Colitis 2020, 14, 935–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liwinski, T.; Zenouzi, R.; John, C.; Ehlken, H.; Rühlemann, M.C.; Bang, C.; Groth, S.; Lieb, W.; Kantowski, M.; Andersen, N.; et al. Alterations of the bile microbiome in primary sclerosing cholangitis. Gut 2020, 69, 665–672. [Google Scholar] [CrossRef] [Green Version]
- Rühlemann, M.; Heinsen, F.-A.; Zenouzi, R.; Lieb, W.; Franke, A.; Schramm, C. Faecal microbiota profiles as diagnostic biomarkers in primary sclerosing cholangitis. Gut 2017, 66, 753–754. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Liu, E.J.; Kheradman, R.; Fagan, A.; Heuman, D.M.; White, M.; Gavis, E.A.; Hylemon, P.; Sikaroodi, M.; Gillevet, P.M. Fungal dysbiosis in cirrhosis. Gut 2017, 67, 1146–1154. [Google Scholar] [CrossRef]
- Trivedi, P.J.; Adams, D.H. Gut–liver immunity. J. Hepatol. 2016, 64, 1187–1189. [Google Scholar] [CrossRef] [Green Version]
- The UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Weston, C.J.; Shepherd, E.L.; Claridge, L.C.; Rantakari, P.; Curbishley, S.M.; Tomlinson, J.W.; Hubscher, S.G.; Reynolds, G.M.; Aalto, K.; Anstee, Q.M.; et al. Vascular adehesion protein 1 promotes liver inflammation and drives hepatic fibrosis. J. Clin. Investig. 2015, 125, 501–520. [Google Scholar] [CrossRef] [Green Version]
- Fodor, A.A.; Klem, E.R.; Gilpin, D.; Elborn, J.; Boucher, R.C.; Tunney, M.; Wolfgang, M.C. The Adult Cystic Fibrosis Airway Microbiota Is Stable over Time and Infection Type, and Highly Resilient to Antibiotic Treatment of Exacerbations. PLoS ONE 2012, 7, e45001. [Google Scholar] [CrossRef]
- Molyneaux, P.L.; Cox, M.J.; Willis-Owen, S.A.G.; Mallia, P.; Russell, K.E.; Russell, A.-M.; Murphy, E.; Johnston, S.L.; Schwartz, D.A.; Wells, A.U.; et al. The role of bacteria in the pathogenesis and progression of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 906–913. [Google Scholar] [CrossRef] [Green Version]
- De Cruz, P.; Kang, S.; Wagner, J.; Buckley, M.; Sim, W.H.; Prideaux, L.; Lockett, T.; McSweeney, C.; Morrison, M.; Kirkwood, C.D.; et al. Association between specific mucosa-associated microbiota in Crohn’s disease at the time of resection and subsequent disease recurrence: A pilot study. J. Gastroenterol. Hepatol. 2014, 30, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Kugathasan, S.; Denson, L.A.; Walters, T.D.; Kim, M.-O.; Marigorta, U.M.; Schirmer, M.; Mondal, K.; Liu, C.; Griffiths, A.; Noe, J.D.; et al. Prediction of complicated disease course for children newly diagnosed with Crohn’s disease: A multicentre inception cohort study. Lancet 2017, 389, 1710–1718. [Google Scholar] [CrossRef] [Green Version]
- Worthmann, A.; John, C.; Rühlemann, M.C.; Baguhl, M.; Heinsen, F.-A.; Schaltenberg, N.; Heine, M.; Schlein, C.; Evangelakos, I.; Mineo, C.; et al. Cold-induced conversion of cholesterol to bile acids in mice shapes the gut microbiome and promotes adaptive thermogenesis. Nat. Med. 2017, 23, 839–849. [Google Scholar] [CrossRef]
- Sakamoto, M.; Tanaka, Y.; Benno, Y.; Ohkuma, M. Parabacteroides faecis sp. nov., isolated from human faeces. Int. J. Syst. Evol. Microbiol. 2015, 65, 1342–1346. [Google Scholar] [CrossRef] [PubMed]
- Rajilić-Stojanović, M.; De Vos, W.M. The first 1000 cultured species of the human gastrointestinal microbiota. FEMS Microbiol. Rev. 2014, 38, 996–1047. [Google Scholar] [CrossRef]
- Narushima, S.; Itoh, K.; Miyamoto, Y.; Park, S.-H.; Nagata, K.; Kuruma, K.; Uchida, K. Deoxycholic acid formation in gnotobiotic mice associated with human intestinal bacteria. Lipids 2006, 41, 835–843. [Google Scholar] [CrossRef] [PubMed]
- Rosen, R.; Amirault, J.; Liu, H.; Mitchell, P.; Hu, L.; Khatwa, U.; Onderdonk, A. Changes in gastric and lung microflora with acid suppression: Acid suppression and bacterial growth. JAMA Pediatr. 2014, 168, 932–937. [Google Scholar] [CrossRef] [Green Version]
- Oztas, E.; Odemis, B.; Kekilli, M.; Kurt, M.; Dinc, B.M.; Parlak, E.; Kalkanci, A.; Sasmaz, N. Systemic phaeohyphomycosis resembling primary sclerosing cholangitis caused by Exophiala dermatitidis. J. Med. Microbiol. 2009, 58, 1243–1246. [Google Scholar] [CrossRef] [Green Version]
- Hong, K.H.; Kim, J.W.; Jang, S.J.; Yu, E.; Kim, E.-C. Liver cirrhosis caused by Exophiala dermatitidis. J. Med. Microbiol. 2009, 58, 674–677. [Google Scholar] [CrossRef] [Green Version]
- Sokol, H.; Leducq, V.; Aschard, H.; Pham, H.-P.; Jegou, S.; Landman, C.; Cohen, D.; Liguori, G.; Bourrier, A.; Nion-Larmurier, I.; et al. Fungal microbiota dysbiosis in IBD. Gut 2016, 66, 1039–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridlon, J.M.; Kang, D.-J.; Hylemon, P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2006, 47, 241–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hacisalihoglu, A.; Jongejan, J.A.; Duine, J.A. Distribution of amine oxidases and amine dehydrogenases in bacteria grown on primary amines and characterization of the amine oxidase from Klebsiella oxytoca. Microbiology 1997, 143, 505–512. [Google Scholar] [CrossRef] [Green Version]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Duncan, S.H.; Belenguer, A.; Holtrop, G.; Johnstone, A.M.; Flint, H.J.; Lobley, G.E. Reduced Dietary Intake of Carbohydrates by Obese Subjects Results in Decreased Concentrations of Butyrate and Butyrate-Producing Bacteria in Feces. Appl. Environ. Microbiol. 2007, 73, 1073–1078. [Google Scholar] [CrossRef] [Green Version]
- Pouteau, E.; Nguyen, P.; Ballevre, O.; Krempf, M. Production rates and metabolism of short-chain fatty acids in the colon and whole body using stable isotopes. Proc. Nutr. Soc. 2003, 62, 87–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venegas, D.P.; De La Fuente, M.K.; Landskron, G.; González, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Short Chain Fatty Acids (SCFAs)-Mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front. Immunol. 2019, 10, 277. [Google Scholar] [CrossRef] [Green Version]
- Quraishi, M.N.; Shaheen, W.; Oo, Y.H.; Iqbal, T.H. Immunological mechanisms underpinning faecal microbiota transplantation for the treatment of inflammatory bowel disease. Clin. Exp. Immunol. 2020, 199, 24–38. [Google Scholar] [CrossRef] [Green Version]
- Trivedi, P.J.; Tickle, J.; Vesterhus, M.N.; Eddowes, P.; Bruns, T.; Vainio, J.; Parker, R.; Smith, D.; Liaskou, E.; Thorbjørnsen, L.W.; et al. Vascular adhesion protein-1 is elevated in primary sclerosing cholangitis, is predictive of clinical outcome and facilitates recruitment of gut-tropic lymphocytes to liver in a substrate-dependent manner. Gut 2018, 67, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, K.; Tanoue, T.; Oshima, K.; Suda, W.; Nagano, Y.; Nishikawa, H.; Fukuda, S.; Saito, T.; Narushima, S.; Hase, K.; et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 2013, 500, 232–236. [Google Scholar] [CrossRef]
- Pereira, P.; Aho, V.; Arola, J.; Boyd, S.; Jokelainen, K.; Paulin, L.; Auvinen, P.; Färkkilä, M. Bile microbiota in primary sclerosing cholangitis: Impact on disease progression and development of biliary dysplasia. PLoS ONE 2017, 12, e0182924. [Google Scholar] [CrossRef] [PubMed]
- Kakiyama, G.; Pandak, W.M.; Gillevet, P.M.; Hylemon, P.B.; Heuman, D.M.; Daita, K.; Takei, H.; Muto, A.; Nittono, H.; Ridlon, J.M.; et al. Modulation of the fecal bile acid profile by gut microbiota in cirrhosis. J. Hepatol. 2013, 58, 949–955. [Google Scholar] [CrossRef] [Green Version]
- Gurung, M.; Li, Z.; You, H.; Rodrigues, R.; Jump, D.B.; Morgun, A.; Shulzhenko, N. Role of gut microbiota in type 2 diabetes pathophysiology. EBioMedicine 2020, 51, 102590. [Google Scholar] [CrossRef] [Green Version]
- Bouter, K.E.; van Raalte, D.H.; Groen, A.K.; Nieuwdorp, M. Role of the Gut Microbiome in the Pathogenesis of Obesity and Obesity-Related Metabolic Dysfunction. Gastroenterology 2017, 152, 1671–1678. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.; Rivera, L.; Furness, J.B.; Angus, C.L.P.W. The role of the gut microbiota in NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.; Wei, Y.; Li, Y.; Chen, W.; Chen, H.; Wang, Q.; Yang, F.; Miao, Q.; Xiao, X.; Zhang, H.; et al. Gut microbial profile is altered in primary biliary cholangitis and partially restored after UDCA therapy. Gut 2018, 67, 534–541. [Google Scholar] [CrossRef]
- Lv, L.-X.; Fang, D.-Q.; Shi, D.; Chen, D.-Y.; Yan, R.; Zhu, Y.-X.; Chen, Y.-F.; Shao, L.; Guo, F.-F.; Wu, W.-R.; et al. Alterations and correlations of the gut microbiome, metabolism and immunity in patients with primary biliary cirrhosis. Environ. Microbiol. 2016, 18, 2272–2286. [Google Scholar] [CrossRef]
- Norman, J.; Handley, S.A.; Baldridge, M.; Droit, L.; Liu, C.Y.; Keller, B.C.; Kambal, A.; Monaco, C.; Zhao, G.; Fleshner, P.; et al. Disease-Specific Alterations in the Enteric Virome in Inflammatory Bowel Disease. Cell 2015, 160, 447–460. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Lang, S.; Duan, Y.; Zhang, X.; Gao, B.; Chopyk, J.; Schwanemann, L.K.; Ventura-Cots, M.; Bataller, R.; Bosques-Padilla, F.; et al. Intestinal Virome in Patients with Alcoholic Hepatitis. Hepatology 2020, 72, 2182–2196. [Google Scholar] [CrossRef]
- Ponsioen, C.Y.; Defoer, J.; Kate, F.J.W.T.; Weverling, G.J.; Tytgat, G.N.J.; Pannekoek, Y.; Wertheim-Dillen, P.M.E. A survey of infectious agents as risk factors for primary sclerosing cholangitis: Are Chlamydia species involved? Eur. J. Gastroenterol. Hepatol. 2002, 14, 641–648. [Google Scholar] [CrossRef]
- Georgiadou, S.P.; Zachou, K.; Liaskos, C.; Gabeta, S.; Rigopoulou, E.I.; Dalekos, G.N. Occult hepatitis B virus infection in patients with autoimmune liver diseases. Liver Int. 2009, 29, 434–442. [Google Scholar] [CrossRef]
- Kim, S.R.; Imoto, S.; Taniguchi, M.; Kim, K.I.; Sasase, N.; Matsuoka, T.; Maekawa, Y.; Ninomiya, T.; Ando, K.; Mita, K.; et al. Primary Sclerosing Cholangitis and Hepatitis C Virus Infection. Intervirology 2005, 48, 268–272. [Google Scholar] [CrossRef]
- Nayudu, S.K.; Kumbum, K.; Balar, B.; Niazi, M.; Chilimuri, S. Small Duct Primary Sclerosing Cholangitis in Association With Hepatitis C Virus Infection: A Case Report. Gastroenterol. Res. 2011, 4, 39–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahimpour, S.; Nasiri-Toosi, M.; Khalili, H.; Daryani, N.E.; Taromlou, M.K.N.; Azizi, Z. A Triple Blinded, Randomized, Placebo-Controlled Clinical Trial to Evaluate the Efficacy and Safety of Oral Vancomycin in Primary Sclerosing Cholangitis: A Pilot Study. J. Gastrointest. Liver Dis. 2016, 25, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Tabibian, J.H.; Weeding, E.; Jorgensen, R.A.; Petz, J.L.; Keach, J.C.; Talwalkar, J.A.; Lindor, K.D. Randomised clinical trial: Vancomycin or metronidazole in patients with primary sclerosing cholangitis—A pilot study. Aliment. Pharmacol. Ther. 2013, 37, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Färkkilä, M.; Karvonen, A.-L.; Nurmi, H.; Nuutinen, H.; Taavitsainen, M.; Pikkarainen, P.; Kärkkäinen, P. Metronidazole and ursodeoxycholic acid for primary sclerosing cholangitis: A randomized placebo-controlled trial. Hepatology 2004, 40, 1379–1386. [Google Scholar] [CrossRef]
- Tabibian, J.H.; Gossard, A.; El-Youssef, M.; Eaton, J.E.; Petz, J.; Jorgensen, R.; Enders, F.B.; Tabibian, A.; Lindor, K.D. Prospective Clinical Trial of Rifaximin Therapy for Patients With Primary Sclerosing Cholangitis. Am. J. Ther. 2017, 24, e56–e63. [Google Scholar] [CrossRef] [Green Version]
- Silveira, M.G.; Torok, N.J.; Gossard, A.A.; Keach, J.C.; Jorgensen, R.R.A.; Petz, R.J.L.; Lindor, K.D. Minocycline in the Treatment of Patients With Primary Sclerosing Cholangitis: Results of a Pilot Study. Am. J. Gastroenterol. 2009, 104, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Allegretti, J.R.; Kassam, Z.; Carrellas, M.; Mullish, B.; Marchesi, J.R.; Pechlivanis, A.; Smith, M.; Gerardin, Y.; Timberlake, S.; Pratt, D.S.; et al. Fecal Microbiota Transplantation in Patients With Primary Sclerosing Cholangitis: A Pilot Clinical Trial. Am. J. Gastroenterol. 2019, 114, 1071–1079. [Google Scholar] [CrossRef]
- Shah, A.; Macdonald, G.A.; Morrison, M.; Holtmann, G. Targeting the Gut Microbiome as a Treatment for Primary Sclerosing Cholangitis: A Conceptional Framework. Am. J. Gastroenterol. 2020, 115, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Hilscher, M.; Enders, F.B.; Carey, E.J.; Lindor, K.D.; Tabibian, J.H. Alkaline phosphatase normalization is a biomarker of improved survival in primary sclerosing cholangitis. Ann. Hepatol. 2016, 15, 246–253. [Google Scholar] [CrossRef]
- Rupp, C.; Rössler, A.; Halibasic, E.; Sauer, P.; Weiss, K.-H.; Friedrich, K.; Wannhoff, A.; Stiehl, A.; Stremmel, W.; Trauner, M.; et al. Reduction in alkaline phosphatase is associated with longer survival in primary sclerosing cholangitis, independent of dominant stenosis. Aliment. Pharmacol. Ther. 2014, 40, 1292–1301. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Crawford, D.; Burger, D.; Martin, N.; Walker, M.; Talley, N.J.; Tallis, C.; Jones, M.; Stuart, K.; Keely, S.; et al. Effects of Antibiotic Therapy in Primary Sclerosing Cholangitis with and without Inflammatory Bowel Disease: A Systematic Review and Meta-Analysis. Semin. Liver Dis. 2019, 39, 432–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Begley, M.; Hill, C.; Gahan, C.G.M. Bile Salt Hydrolase Activity in Probiotics. Appl. Environ. Microbiol. 2006, 72, 1729–1738. [Google Scholar] [CrossRef] [Green Version]
- Kitahara, M.; Sakata, S.; Sakamoto, M.; Benno, Y. Comparison among Fecal Secondary Bile Acid Levels, Fecal Microbiota andClostridium scindensCell Numbers in Japanese. Microbiol. Immunol. 2004, 48, 367–375. [Google Scholar] [CrossRef]
- Taylor, M.R.; Flannigan, K.L.; Rahim, H.; Mohamud, A.; Lewis, I.A.; Hirota, S.A.; Greenway, S.C. Vancomycin relieves mycophenolate mofetil–induced gastrointestinal toxicity by eliminating gut bacterial β-glucuronidase activity. Sci. Adv. 2019, 5, eaax2358. [Google Scholar] [CrossRef] [Green Version]
- Abarbanel, D.N.; Seki, S.M.; Davies, Y.; Marlen, N.; Benavides, J.A.; Cox, K.; Nadeau, K.C.; Cox, K.L. Immunomodulatory Effect of Vancomycin on Treg in Pediatric Inflammatory Bowel Disease and Primary Sclerosing Cholangitis. J. Clin. Immunol. 2013, 33, 397–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deneau, M.R.; Mack, C.; Mogul, D.; Perito, E.R.; Valentino, P.L.; Amir, A.Z.; DiGuglielmo, M.; Draijer, L.G.; El-Matary, W.; Furuya, K.N.; et al. Oral Vancomycin, Ursodeoxycholic Acid, or No Therapy for Pediatric Primary Sclerosing Cholangitis: A Matched Analysis. Hepatology 2021, 73, 1061–1073. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.-Z.; Reilly, C.R.; Steward-Harrison, L.C.; Balouch, F.; Muir, R.; Lewindon, P.J. Oral vancomycin induces clinical and mucosal remission of colitis in children with primary sclerosing cholangitis–ulcerative colitis. Gut 2019, 68, 1533–1535. [Google Scholar] [CrossRef]
- Bakker, G.J.; Nieuwdorp, M. Fecal Microbiota Transplantation: Therapeutic Potential for a Multitude of Diseases beyond Clostridium difficile. Microbiol. Spectr. 2017, 5, 5. [Google Scholar] [CrossRef]
- Wortelboer, K.; Nieuwdorp, M.; Herrema, H. Fecal microbiota transplantation beyond Clostridioides difficile infections. EBioMedicine 2019, 44, 716–729. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.-W.; Kuo, C.-H.; Kuo, F.-C.; Wang, Y.-K.; Hsu, W.-H.; Yu, F.-J.; Hu, H.-M.; Hsu, P.-I.; Wang, J.-Y.; Wu, D.-C. Fecal microbiota transplantation: Review and update. J. Formos. Med. Assoc. 2019, 118, S23–S31. [Google Scholar] [CrossRef]
- Cammarota, G.; Masucci, L.; Ianiro, G.; Bibbò, S.; Dinoi, G.; Costamagna, G.; Sanguinetti, M.; Gasbarrini, A. Randomised clinical trial: Faecal microbiota transplantation by colonoscopy vs. vancomycin for the treatment of recurrentClostridium difficileinfection. Aliment. Pharmacol. Ther. 2015, 41, 835–843. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.R.; Khoruts, A.; Staley, C.; Sadowsky, M.J.; Abd, M.; Alani, M.; Bakow, B.; Curran, P.; McKenney, J.; Tisch, A.; et al. Effect of Fecal Microbiota Transplantation on Recurrence in Multiply Recurrent Clostridium difficile Infection. Ann. Intern. Med. 2016, 165, 609–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hvas, C.L.; Dahl Jørgensen, S.M.; Jørgensen, S.P.; Storgaard, M.; Lemming, L.; Hansen, M.M.; Erikstrup, C.; Dahlerup, J.F. Fecal Microbiota Transplantation Is Superior to Fidaxomicin for Treatment of Recurrent Clostridium difficile Infection. Gastroenterology 2019, 156, 1324–1332.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, J.; Grinspan, A. Fecal Microbiota Transplantation for Inflammatory Bowel Disease. Gastroenterol. Hepatol. 2016, 12, 374–379. [Google Scholar]
- Tan, P.; Li, X.; Shen, J.; Feng, Q. Fecal Microbiota Transplantation for the Treatment of Inflammatory Bowel Disease: An Update. Front. Pharmacol. 2020, 11, 574533. [Google Scholar] [CrossRef] [PubMed]
- DeFilipp, Z.; Bloom, P.P.; Soto, M.T.; Mansour, M.K.; Sater, M.; Huntley, M.H.; Turbett, S.; Chung, R.T.; Chen, Y.-B.; Hohmann, E.L. Drug-Resistant E. coli Bacteremia Transmitted by Fecal Microbiota Transplant. N. Engl. J. Med. 2019, 381, 2043–2050. [Google Scholar] [CrossRef] [PubMed]
- Beuers, U.; Hohenester, S.; Wenniger, L.J.M.D.B.; Kremer, A.; Jansen, P.L.M.; Elferink, R.P.J.O. The biliary HCO3− umbrella: A unifying hypothesis on pathogenetic and therapeutic aspects of fibrosing cholangiopathies. Hepatology 2010, 52, 1489–1496. [Google Scholar] [CrossRef]
- Trampert, D.C.; van de Graaf, S.F.; Jongejan, A.; Elferink, R.P.O.; Beuers, U. Hepatobiliary acid-base homeostasis: Insights from analogous secretory epithelia. J. Hepatol. 2021, 74, 428–441. [Google Scholar] [CrossRef]
- Schmucker, D.L.; Ohta, M.; Kanai, S.; Sato, Y.; Kitani, K. Hepatic injury induced by bile salts: Correlation between biochemical and morphological events. Hepatology 1990, 12, 1216–1221. [Google Scholar] [CrossRef]
- Verdier, J.; Luedde, T.; Sellge, G. Biliary Mucosal Barrier and Microbiome. Visc. Med. 2015, 31, 156–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, T.; Zhang, Z.; Liu, B.; Hou, D.; Liang, Y.; Zhang, J.; Shi, P. Gut microbiota dysbiosis and bacterial community assembly associated with cholesterol gallstones in large-scale study. BMC Genom. 2013, 14, 669. [Google Scholar] [CrossRef] [Green Version]
- Jiménez, E.; Sánchez, B.; Farina, A.; Margolles, A.; Rodríguez, J.M. Characterization of the bile and gall bladder microbiota of healthy pigs. Microbiology 2014, 3, 937–949. [Google Scholar] [CrossRef]
- Steck, N.; Hoffmann, M.; Sava, I.G.; Kim, S.C.; Hahne, H.; Tonkonogy, S.L.; Mair, K.; Krueger, D.; Pruteanu, M.; Shanahan, F.; et al. Enterococcus faecalis Metalloprotease Compromises Epithelial Barrier and Contributes to Intestinal Inflammation. Gastroenterology 2011, 141, 959–971. [Google Scholar] [CrossRef] [Green Version]
- Katt, J.; Schwinge, D.; Schoknecht, T.; Quaas, A.; Sobottka, I.; Burandt, E.; Becker, C.; Neurath, M.F.; Lohse, A.W.; Herkel, J.; et al. Increased T helper type 17 response to pathogen stimulation in patients with primary sclerosing cholangitis. Hepatology 2013, 58, 1084–1093. [Google Scholar] [CrossRef]
- Kwon, W.; Jang, J.-Y.; Kim, E.-C.; Park, J.W.; Han, I.W.; Kang, M.J.; Kim, S.-W. Changing trend in bile microbiology and antibiotic susceptibilities: Over 12 years of experience. Infection 2013, 41, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Litvak, Y.; Byndloss, M.; Tsolis, R.M.; Bäumler, A.J. Dysbiotic Proteobacteria expansion: A microbial signature of epithelial dysfunction. Curr. Opin. Microbiol. 2017, 39, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Beuers, U.; Spengler, U.; Kruis, W.; Aydemir, Ü.; Wiebecke, B.; Heldwein, W.; Weinzierl, M.; Pape, G.R.; Sauerbruch, T.; Paumgartner, G. Ursodeoxycholic acid for treatment of primary sclerosing cholangitis: A placebo-controlled trial. Hepatology 1992, 16, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Olsson, R.; Boberg, K.M.; de Muckadell, O.B.S.; Lindgren, S.; Hultcrantz, R.; Folvik, G.; Bell, H.; Gangsøy–Kristiansen, M.; Matre, J.; Rydning, A.; et al. High-Dose Ursodeoxycholic Acid in Primary Sclerosing Cholangitis: A 5-Year Multicenter, Randomized, Controlled Study. Gastroenterology 2005, 129, 1464–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindor, K.D.; Kowdley, K.V.; Luketic, V.A.C.; Harrison, M.E.; McCashland, T.; Befeler, A.S.; Harnois, D.; Jorgensen, R.; Petz, J.; Keach, J.; et al. High-dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis. Hepatology 2009, 50, 808–814. [Google Scholar] [CrossRef]
- Fickert, P.; Wagner, M. Biliary bile acids in hepatobiliary injury—What is the link? J. Hepatol. 2017, 67, 619–631. [Google Scholar] [CrossRef] [Green Version]
- Kowdley, K.V.; Vuppalanchi, R.; Levy, C.; Floreani, A.; Andreone, P.; LaRusso, N.F.; Shrestha, R.; Trotter, J.; Goldberg, D.; Rushbrook, S.; et al. A randomized, placebo-controlled, phase II study of obeticholic acid for primary sclerosing cholangitis. J. Hepatol. 2020, 73, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Trauner, M.; Gulamhusein, A.; Hameed, B.; Caldwell, S.; Shiffman, M.L.; Landis, C.; Eksteen, B.; Agarwal, K.; Muir, A.; Rushbrook, S.; et al. The Nonsteroidal Farnesoid X Receptor Agonist Cilofexor (GS-9674) Improves Markers of Cholestasis and Liver Injury in Patients With Primary Sclerosing Cholangitis. Hepatology 2019, 70, 788–801. [Google Scholar] [CrossRef] [Green Version]
- Mizuno, S.; Hirano, K.; Tada, M.; Yamamoto, K.; Yashima, Y.; Yagioka, H.; Kawakubo, K.; Ito, Y.; Kogure, H.; Sasaki, T.; et al. Bezafibrate for the treatment of primary sclerosing cholangitis. J. Gastroenterol. 2010, 45, 758–762. [Google Scholar] [CrossRef]
- Mizuno, S.; Hirano, K.; Isayama, H.; Watanabe, T.; Yamamoto, N.; Nakai, Y.; Sasahira, N.; Tada, M.; Omata, M.; Koike, K. Prospective study of bezafibrate for the treatment of primary sclerosing cholangitis. J. Hepato-Biliary-Pancreatic Sci. 2015, 22, 766–770. [Google Scholar] [CrossRef]
- Fickert, P.; Hirschfield, G.; Denk, G.; Marschall, H.-U.; Altorjay, I.; Färkkilä, M.; Schramm, C.; Spengler, U.; Chapman, R.; Bergquist, A.; et al. norUrsodeoxycholic acid improves cholestasis in primary sclerosing cholangitis. J. Hepatol. 2017, 67, 549–558. [Google Scholar] [CrossRef] [Green Version]
- Shoda, J.; Okada, K.; Inada, Y.; Kusama, H.; Utsunomiya, H.; Oda, K.; Yokoi, T.; Yoshizato, K.; Suzuki, H. Bezafibrate induces multidrug-resistance P-Glycoprotein 3 expression in cultured human hepatocytes and humanized livers of chimeric mice. Hepatol. Res. 2007, 37, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Inoue, I.; Noji, S.; Awata, T.; Takahashi, K.; Nakajima, T.; Sonoda, M.; Komoda, T.; Katayama, S. Bezafibrate has an antioxidant effect: Peroxisome proliferator-activated receptor α is associated with Cu2+, Zn2+-superoxide dismutase in the liver. Life Sci. 1998, 63, 135–144. [Google Scholar] [CrossRef]
- Lemoinne, S.; Pares, A.; Reig, A.; Ben Belkacem, K.; Fankem, A.D.K.; Gaouar, F.; Poupon, R.; Housset, C.; Corpechot, C.; Chazouillères, O. Primary sclerosing cholangitis response to the combination of fibrates with ursodeoxycholic acid: French–Spanish experience. Clin. Res. Hepatol. Gastroenterol. 2018, 42, 521–528. [Google Scholar] [CrossRef]
- Callea, F.; Sergi, C.; Fabbretti, G.; Brisigotti, M.; Cozzutto, C.; Medicina, D. Precancerous lesions of the biliary tree. J. Surg. Oncol. 1993, 53, 131–133. [Google Scholar] [CrossRef]
- Bouvier, A.-M.; Remontet, L.; Jougla, E.; Launoy, G.; Grosclaude, P.; Buémi, A.; Tretarre, B.; Velten, M.; Dancourt, V.; Menegoz, F.; et al. Incidence of gastrointestinal cancers in France. Gastroentérologie Clinique et Biologique 2004, 28, 877–881. [Google Scholar] [CrossRef]
- Song, J.; Li, Y.; Bowlus, C.L.; Yang, G.; Leung, P.S.C.; Gershwin, M.E. Cholangiocarcinoma in Patients with Primary Sclerosing Cholangitis (PSC): A Comprehensive Review. Clin. Rev. Allergy Immunol. 2019, 58, 134–149. [Google Scholar] [CrossRef]
- Chung, B.K.; Karlsen, T.H.; Folseraas, T. Cholangiocytes in the pathogenesis of primary sclerosing cholangitis and development of cholangiocarcinoma. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1390–1400. [Google Scholar] [CrossRef]
- Ehlken, H.; Schramm, C. Primary Sclerosing Cholangitis and Cholangiocarcinoma: Pathogenesis and Modes of Diagnostics. Dig. Dis. 2013, 31, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Timmer, M.R.; Beuers, U.; Fockens, P.; Ponsioen, C.Y.; Rauws, E.A.; Wang, K.K.; Krishnadath, K.K. Genetic and Epigenetic Abnormalities in Primary Sclerosing Cholangitis-associated Cholangiocarcinoma. Inflamm. Bowel Dis. 2013, 19, 1789–1797. [Google Scholar] [CrossRef]
- Burak, K.W.; Angulo, P.; Pasha, T.M.; Egan, K.M.; Petz, J.; Lindor, K.D. Incidence and Risk Factors for Cholangiocarcinoma in Primary Sclerosing Cholangitis. Am. J. Gastroenterol. 2004, 99, 523–526. [Google Scholar] [CrossRef]
- Yamada, D.; Rizvi, S.; Razumilava, N.; Bronk, S.F.; Davila, J.I.; Champion, M.D.; Borad, M.J.; Bezerra, J.A.; Chen, X.; Gores, G.J. IL-33 facilitates oncogene-induced cholangiocarcinoma in mice by an interleukin-6-sensitive mechanism. Hepatology 2015, 61, 1627–1642. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Baluyut, A.; Ismail, A.; Zaman, A.; Sood, G.; Ghalib, R.; McCashland, T.M.; Reddy, K.R.; Zervos, X.; Anbari, M.A.; et al. Cholangiocarcinoma in patients with primary sclerosing cholangitis: A multicenter case-control study. Hepatology 2000, 31, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Saab, M.; Mestivier, D.; Sohrabi, M.; Rodriguez, C.; Khonsari, M.R.; Faraji, A.; Sobhani, I. Characterization of biliary microbiota dysbiosis in extrahepatic cholangiocarcinoma. PLoS ONE 2021, 16, e0247798. [Google Scholar] [CrossRef] [PubMed]
- Carpino, G.; Cardinale, V.; Renzi, A.; Hov, J.R.; Berloco, P.B.; Rossi, M.; Karlsen, T.H.; Alvaro, D.; Gaudio, E. Activation of biliary tree stem cells within peribiliary glands in primary sclerosing cholangitis. J. Hepatol. 2015, 63, 1220–1228. [Google Scholar] [CrossRef] [PubMed]
- Moeini, A.; Haber, P.K.; Sia, D. Cell of origin in biliary tract cancers and clinical implications. JHEP Rep. 2021, 3, 100226. [Google Scholar] [CrossRef] [PubMed]
- Boyd, S.; Tenca, A.; Jokelainen, K.; Mustonen, H.; Krogerus, L.; Arola, J.; Färkkilä, M. Screening primary sclerosing cholangitis and biliary dysplasia with endoscopic retrograde cholangiography and brush cytology: Risk factors for biliary neoplasia. Endoscopy 2016, 48, 432–439. [Google Scholar] [CrossRef]
- Fevery, J.; Verslype, C.; Lai, G.; Aerts, R.; Van Steenbergen, W. Incidence, Diagnosis, and Therapy of Cholangiocarcinoma in Patients with Primary Sclerosing Cholangitis. Dig. Dis. Sci. 2007, 52, 3123–3135. [Google Scholar] [CrossRef]
- Thanan, R.; Pairojkul, C.; Pinlaor, S.; Khuntikeo, N.; Wongkham, C.; Sripa, B.; Ma, N.; Vaeteewoottacharn, K.; Furukawa, A.; Kobayashi, H.; et al. Inflammation-related DNA damage and expression of CD133 and Oct3/4 in cholangiocarcinoma patients with poor prognosis. Free. Radic. Biol. Med. 2013, 65, 1464–1472. [Google Scholar] [CrossRef]
- Kerr, S.E.; Fritcher, E.G.B.; Campion, M.B.; Voss, J.S.; Kipp, B.R.; Halling, K.C.; Lewis, J.T. Biliary dysplasia in primary sclerosing cholangitis harbors cytogenetic abnormalities similar to cholangiocarcinoma. Hum. Pathol. 2014, 45, 1797–1804. [Google Scholar] [CrossRef] [PubMed]
- Cadamuro, M.; Nardo, G.; Indraccolo, S.; Dall’Olmo, L.; Sambado, L.; Moserle, L.; Franceschet, I.; Colledan, M.; Massani, M.; Stecca, T.; et al. Platelet-derived growth factor-D and Rho GTPases regulate recruitment of cancer-associated fibroblasts in cholangiocarcinoma. Hepatology 2013, 58, 1042–1053. [Google Scholar] [CrossRef]
- Shen, H.; Ye, F.; Xie, L.; Yang, J.; Li, Z.; Xu, P.; Meng, F.; Li, L.; Xiaochen, B.; Bo, X.; et al. Metagenomic sequencing of bile from gallstone patients to identify different microbial community patterns and novel biliary bacteria. Sci. Rep. 2015, 5, 17450. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Shen, H.; Li, Z.; Meng, F.; Li, L.; Yang, J.; Chen, Y.; Bo, X.; Zhang, X.; Ni, M. Influence of the Biliary System on Biliary Bacteria Revealed by Bacterial Communities of the Human Biliary and Upper Digestive Tracts. PLoS ONE 2016, 11, e0150519. [Google Scholar] [CrossRef]
- Hiramatsu, K.; Harada, K.; Tsuneyama, K.; Sasaki, M.; Fujita, S.; Hashimoto, T.; Kaneko, S.; Kobayashi, K.; Nakanuma, Y. Amplification and sequence analysis of partial bacterial 16S ribosomal RNA gene in gallbladder bile from patients with primary biliary cirrhosis. J. Hepatol. 2000, 33, 9–18. [Google Scholar] [CrossRef]
- Jia, X.; Lu, S.; Zeng, Z.; Liu, Q.; Dong, Z.; Chen, Y.; Zhu, Z.; Hong, Z.; Zhang, T.; Du, G.; et al. Characterization of Gut Microbiota, Bile Acid Metabolism, and Cytokines in Intrahepatic Cholangiocarcinoma. Hepatology 2020, 71, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Jusakul, A.; Khuntikeo, N.; Haigh, W.G.; Kuver, R.; Ioannou, G.N.; Loilome, W.; Namwat, N.; Bhudhisawasdi, V.; Pugkhem, A.; Pairojkul, C.; et al. Identification of biliary bile acids in patients with benign biliary diseases, hepatocellular carcinoma and cholangiocarcinoma. Asian Pac. J. Cancer Prev. 2012, 13. [Google Scholar]
- Jiménez, F.A.; Guitron, A.; Segura-Lopez, F.K.; Méndez-Tenorio, A.; Iwai, S.; Hernández-Guerrero, A.; Torres, J. Microbiota studies in the bile duct strongly suggest a role for Helicobacter pylori in extrahepatic cholangiocarcinoma. Clin. Microbiol. Infect. 2016, 22, 178-e11–178-e22. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Dhakan, D.B.; Maji, A.; Saxena, R.; Prasoodanan, V.P.K.; Mahajan, S.; Pulikkan, J.; Kurian, J.; Gomez, A.M.; Scaria, J.; et al. Association of Flavonifractor plautii, a Flavonoid-Degrading Bacterium, with the Gut Microbiome of Colorectal Cancer Patients in India. MSystems 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Sobhani, I.; Tap, J.; Roudot-Thoraval, F.; Roperch, J.P.; Letulle, S.; Langella, P.; Corthier, G.; Van Nhieu, J.T.; Furet, J.-P. Microbial Dysbiosis in Colorectal Cancer (CRC) Patients. PLoS ONE 2011, 6, e16393. [Google Scholar] [CrossRef]
- Molinero, N.; Ruiz, L.; Milani, C.; Gutiérrez-Díaz, I.; Sánchez, B.; Mangifesta, M.; Segura, J.; Cambero, I.; Campelo, A.B.; Garcia, J.I.R.; et al. The human gallbladder microbiome is related to the physiological state and the biliary metabolic profile. Microbiome 2019, 7, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Rath, H.C.; Herfarth, H.H.; Ikeda, J.S.; Grenther, W.B.; Hamm, T.E.; Balish, E.; Taurog, J.D.; Hammer, R.E.; Wilson, K.H.; Sartor, R.B. Normal luminal bacteria, especially Bacteroides species, mediate chronic colitis, gastritis, and arthritis in HLA-B27/human beta2 microglobulin transgenic rats. J. Clin. Investig. 1996, 98, 945–953. [Google Scholar] [CrossRef] [Green Version]
- Nanjundappa, R.H.; Ronchi, F.; Wang, J.; Clemente-Casares, X.; Yamanouchi, J.; Umeshappa, C.S.; Yang, Y.; Blanco, J.; Bassolas-Molina, H.; Salas, A.; et al. A Gut Microbial Mimic that Hijacks Diabetogenic Autoreactivity to Suppress Colitis. Cell 2017, 171, 655–667.e17. [Google Scholar] [CrossRef] [Green Version]
- Lazaridis, K.N.; Gores, G.J. Primary Sclerosing Cholangitis and Cholangiocarcinoma. Semin. Liver Dis. 2006, 26, 042–051. [Google Scholar] [CrossRef] [PubMed]
- Randi, G.; Franceschi, S.; La Vecchia, C. Gallbladder cancer worldwide: Geographical distribution and risk factors. Int. J. Cancer 2006, 118, 1591–1602. [Google Scholar] [CrossRef] [PubMed]
- Crawford, R.W.; Gibson, D.L.; Kay, W.W.; Gunn, J.S. Identification of a Bile-Induced Exopolysaccharide Required for Salmonella Biofilm Formation on Gallstone Surfaces. Infect. Immun. 2008, 76, 5341–5349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawford, R.W.; Rosales-Reyes, R.; Ramirez-Aguilar, M.D.L.L.; Chapa-Azuela, O.; Alpuche-Aranda, C.; Gunn, J.S. Gallstones play a significant role in Salmonella spp. gallbladder colonization and carriage. Proc. Natl. Acad. Sci. USA 2010, 107, 4353–4358. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Escobedo, G.; Marshall, J.M.; Gunn, J.S. Chronic and acute infection of the gall bladder by Salmonella Typhi: Understanding the carrier state. Nat. Rev. Genet. 2010, 9, 9–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagaraja, V.; Eslick, G.D. Systematic review with meta-analysis: The relationship between chronicSalmonella typhicarrier status and gall-bladder cancer. Aliment. Pharmacol. Ther. 2014, 39, 745–750. [Google Scholar] [CrossRef] [PubMed]
- Sepe, L.P.; Hartl, K.; Iftekhar, A.; Berger, H.; Kumar, N.; Goosmann, C.; Chopra, S.; Schmidt, S.C.; Gurumurthy, R.K.; Meyer, T.F.; et al. Genotoxic Effect of Salmonella Paratyphi A Infection on Human Primary Gallblader Cells. MBio. 2020, 11. [Google Scholar] [CrossRef]
- Chumduri, C.; Gurumurthy, R.K.; Zietlow, R.; Meyer, T.F. Subversion of host genome integrity by bacterial pathogens. Nat. Rev. Mol. Cell Biol. 2016, 17, 659–673. [Google Scholar] [CrossRef]
- Koeppel, M.; Garcia-Alcalde, F.; Glowinski, F.; Schlaermann, P.; Meyer, T.F. Helicobacter pylori Infection Causes Characteristic DNA Damage Patterns in Human Cells. Cell Rep. 2015, 11, 1703–1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toller, I.M.; Neelsen, K.J.; Steger, M.; Hartung, M.L.; Hottiger, M.; Stucki, M.; Kalali, B.; Gerhard, M.; Sartori, A.; Lopes, M.; et al. Carcinogenic bacterial pathogen Helicobacter pylori triggers DNA double-strand breaks and a DNA damage response in its host cells. Proc. Natl. Acad. Sci. USA 2011, 108, 14944–14949. [Google Scholar] [CrossRef] [Green Version]
- Hartl, K.; Sigal, M. Microbe-Driven Genotoxicity in Gastrointestinal Carcinogenesis. Int. J. Mol. Sci. 2020, 21, 7439. [Google Scholar] [CrossRef] [PubMed]
- Sigal, M.; Meyer, T.F. Coevolution between the Human Microbiota and the Epithelial Immune System. Dig. Dis. 2016, 34, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Faïs, T.; Delmas, J.; Barnich, N.; Bonnet, R.; Dalmasso, G. Colibactin: More Than a New Bacterial Toxin. Toxins 2018, 10, 151. [Google Scholar] [CrossRef] [Green Version]
- Dziubańska-Kusibab, P.J.; Berger, H.; Battistini, F.; Bouwman, B.A.M.; Iftekhar, A.; Katainen, R.; Cajuso, T.; Crosetto, N.; Orozco, M.; Aaltonen, L.A.; et al. Colibactin DNA-damage signature indicates mutational impact in colorectal cancer. Nat. Med. 2020, 26, 1063–1069. [Google Scholar] [CrossRef]
- Pleguezuelos-Manzano, C.; Puschhof, J.; Huber, A.R.; Van Hoeck, A.; Wood, H.M.; Nomburg, J.; Gurjao, C.; Manders, F.; Dalmasso, G.; Stege, P.B.; et al. Mutational signature in colorectal cancer caused by genotoxic pks+ E. coli. Nature 2020, 580, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Iftekhar, A.; Berger, H.; Bouznad, N.; Heuberger, J.; Boccellato, F.; Dobrindt, U.; Hermeking, H.; Sigal, M.; Meyer, T.F. Genomic aberrations after short-term exposure to colibactin-producing E. coli transform primary colon epithelial cells. Nat. Commun. 2021, 12, 1–15. [Google Scholar] [CrossRef]
Figure 1.
Bidirectional communication between gut and liver through the biliary tract, portal vein, and systemic circulation. The liver communicates with the intestine by transportation of bile salts and antimicrobial molecules (e.g., immunoglobulin A) to the intestinal lumen through the biliary tract, maintaining gut eubiosis by controlling unrestricted bacterial overgrowth. Additionally, endogenous and exogenous metabolites from the liver such as free fatty acids (FFA), ethanol or choline metabolites are transported to the intestine via the capillary system [32,33]. Pathogen-associated molecular patterns (PAMPs).
Figure 1.
Bidirectional communication between gut and liver through the biliary tract, portal vein, and systemic circulation. The liver communicates with the intestine by transportation of bile salts and antimicrobial molecules (e.g., immunoglobulin A) to the intestinal lumen through the biliary tract, maintaining gut eubiosis by controlling unrestricted bacterial overgrowth. Additionally, endogenous and exogenous metabolites from the liver such as free fatty acids (FFA), ethanol or choline metabolites are transported to the intestine via the capillary system [32,33]. Pathogen-associated molecular patterns (PAMPs).

Figure 2.
Altered gut microbiota in patients with PSC compared to healthy individuals. Several clinical studies analyzing fecal and mucosal samples have demonstrated that the gut microbiome of individuals with PSC is distinct from that of healthy controls. PSC, primary sclerosing cholangitis.
Figure 2.
Altered gut microbiota in patients with PSC compared to healthy individuals. Several clinical studies analyzing fecal and mucosal samples have demonstrated that the gut microbiome of individuals with PSC is distinct from that of healthy controls. PSC, primary sclerosing cholangitis.


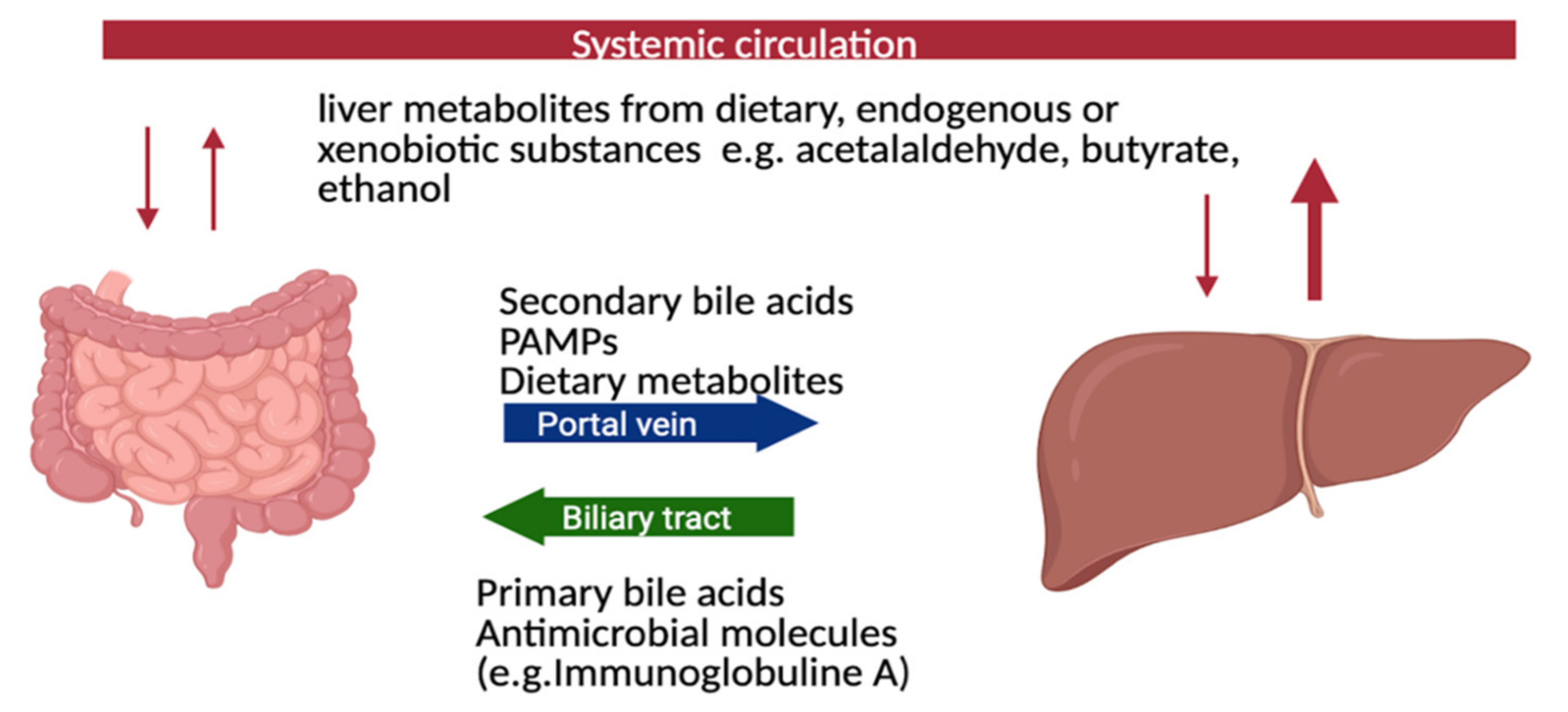{kind=link}
{kind=link}
Table 1.
Summary of studies analyzing fecal, mucosal, and bile samples of patients with PSC.
| Reference | Sample Origin | No. of PSC Patients with and without IBD (n=) | Microbiology Assessment | Major Findings with Regard to the Gut Microbiome |
|---|---|---|---|---|
| Sabino et al., 2016 [59] | Stool | 66 | 16sRNA gene sequencing | Significant over-representation of Enterococcus, Fusobacterium, and Lactobacillus genera |
| Kummen et al., 2017 [27] | Stool | 85 | 16sRNA gene sequencing | Enrichment of the Veillonella genus |
| Bajer et al., 2017 [25] | Stool | 43 | 16sRNA gene sequencing | Rothia, Enterococcus, Streptococcus, Veillonella were overrepresented in PSC regardless of concomitant IBD Decreased abundance of Adlercreutzia equolifaciens and Prevotella copri |
| Iwasawa et al., 2017 [60] | Stool | 27 | 16sRNA gene sequencing | Abundance of Enterococcus (E. faecalis especially), Streptococcus (with prevalence of Streptococcus parasanguinis) and Veillonella species Parabacteroides overrepresented |
| Rühlemann et al., 2019 [12] | Stool | 137 | 16sRNA gene sequencing | Increased abundance in eight genera Veillonella, Streptococcus, Lactobacillus, Enterococcus; Proteobacteria, Lactobacillales (Bacilli), Parabacteroides, and Bacteroides |
| Lemoinne et al., 2020 [61] | Stool | 49 | 16sRNA gene sequencing | Fungal dysbiosis: increased proportion of Exophiala, decreased proportion of Saccharomyces cerevisiae Bacterial dysbiosis: Firmicutes decreased (except Veillonella increased) and Proteobacteria increased |
| Kummen et al., 2021 [62] | Stool | 136 | metagenomic shotgun sequencing | Increased abundance of Clostridiales species Depletion of Eubacterium, Ruminococcus obeum |
| Rossen et al., 2015 [63] | Ileum mucosa | 12 | 16sRNA gene sequencing | Abundance of uncultured Clostridiales II |
| Kevans et al., 2016 [26] | Colon mucosa | 31 | 16sRNA gene sequencing | No changes |
| Torres et al., 2016 [64] | Ileum, colon mucosa | 20 | 16sRNA gene sequencing | significant increase in Barnesiellaceae and Blautia |
| Quraishi et al., 2017 [24] | Colon mucosa | 11 | 16sRNA gene sequencing | significant increase in Escherichia, Megasphera and Lachnospiraceae |
| Quraishi et al., 2020 [65] | Sigmoid mucosa | 20 | 16sRNA gene sequencing | significantly higher abundance of the genera Bacteroides fragilis, Roseburia, Shewanella, and Clostridium ramosum in patients with PSC-IBD compared to patients with UC |
| Liwinski et al., 2020 [66] | Duodenal and oral fluids, duodenal mucosa, bile | 46 | 16sRNA gene sequencing | Duodenal mucosa biopsy: overrepresentation of Escherichia coli and Veillonella dispar Bile fluid: overrepresentation of Enterococcus (especially Enterococcus faecalis), Proteobacteria Staphylococcus, Neisseria, Veillonella dispar |
PSC, primary sclerosing cholangitis; IBD, inflammatory bowel disease; UC, ulcerative colitis.
Table 2.
Overview of therapeutic agents targeting gut microbiota and their study characteristics.
| References | Therapeutic Agents | Study Type | PSC Patients (N=) | Treatment Duration | Primary Endpoint | Results |
|---|---|---|---|---|---|---|
| Rahimpour et al., 2016 [106] | Vancomycin | RCT | 8 | 12 weeks | Decrease in MRS at week 12 | Significant reduction in mean MRS and ALP levels |
| Tabibian et al., 2013 [107] | Vancomycin vs.Metronidazole | RCT | 9 9 | 12 weeks | Decrease in ALP levels at week 12 | Non-dose dependent significant ALP reduction (for low and high dose vancomycin) |
| Färkkilä et al., 2004 [108] | Metronidazole (and UDCA) | RCT | 39 | 36 months | Decrease in ALP or other liver enzymes, MRS, symptoms or histology at week 36 | Significant ALP level and MRS reduction, improvement (of stage and grade) in histology |
| Tabibian et al., 2017 [109] | Rifaximin | Open-label study | 16 | 12 weeks | Decrease in ALP levels at months 3 | No significant reduction |
| Silveira et al., 2009 [110] | Minocycline | Open-label study | 16 | 1 year | Decrease in ALP levels at month 12 | Significant ALP reduction and mean MRS |
| Allegretti et al., 2019 [111] | Fecal transplantation | Open-label pilot | 10 | 24 weeks | ≥50% ALP reduction at week 24 | Significant reduction in ALP levels in 3/10 |
Mayo risk score (MRS); number of treated PSC patients (N); ursodeoxycholic acid (UDCA); randomized controlled trial (RCT); alkaline phosphatase (ALP).
Table 3.
Overview of a selection of therapeutic agents targeting bile acid pathways and their study characteristics.
Table 3.
Overview of a selection of therapeutic agents targeting bile acid pathways and their study characteristics.
| References | Therapeutic Agents | Target Receptor/ Mechanism | Study Type | N | Treatment Duration | Primary Endpoint | Results |
|---|---|---|---|---|---|---|---|
| Beuers et al., 1992 [141] | UDCA 13–15 mg/kgper day | Choleretic effect, unknown receptor | RCT | 5 | 12 months | Significant ALP reduction | |
| Olsson et al., 2005 [142] | UDCA 17–23 mg/kgper day | Choleretic effect, unknown receptor | RCT | 110 | 5 years | Occurrence of liver transplantation | Significant improvement in ALP levels, no impact on survival |
| Lindor et al., 2009 [143] | UDCA 28–30 mg/kgper day | Choleretic effect, unknown receptor | RCT | 76 | 6 years | Changes in ALP and other liver enzymes | Significant improvement in ALP levels, no impact on survival, higher doses are associated with a higher rate of serious adverse events |
| Fickert et al., 2017 [144] | NorUDCA | Choleretic effect, unknown receptor | RCT | 161 | 12 weeks | Mean relative change in ALP levels | Significant dose-dependent ALP reductions |
| Kowdley et al., 2020 [145] | OCA | Endogenous FXR agonist | RCT | 76 | 24 weeks | Change in ALP at week 24 | Significant reduction in ALP levels (only for use of OCA 5–10 mg) |
| Trauner et al., 2019 [146] | Cilofexor | Nonsteroidal FXR agonist | RCT | 42 | 12 weeks | Change in ALP and other liver enzymes at week 12 | Significant reduction in ALP levels and other liver enzymes |
| Mizuno et al., 2010 [147] | Bezafibrate | PPARa-agonist | Open-label pilot | 7 | 6 months | Change in ALP levels at month 6 | Significant ALP reduction |
| Mizuno et al., 2015 [148] | Bezafibrate | PPARa-agonist | Open-label pilot | 11 | 12 weeks | Improvement in liver function tests | Significant ALP reduction |
| NCT04024813 | Seladelpar | Selective PPARδ agonist | RCT | 24 weeks | Change in ALP at week 24 | Ongoing |
Ursodeoxycholic acid (UDCA); norursodeoxycholic acid (norUDCA); obeticholic acid (OCA); farnesoid X receptor (FXR); number of treated PSC patients (N); peroxisome-proliferator-activated receptor delta (PPARδ); peroxisome-proliferator-activated receptor alpha (PPARα).
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Özdirik, B.; Müller, T.; Wree, A.; Tacke, F.; Sigal, M. The Role of Microbiota in Primary Sclerosing Cholangitis and Related Biliary Malignancies. Int. J. Mol. Sci. 2021, 22, 6975. https://doi.org/10.3390/ijms22136975
AMA Style
Özdirik B, Müller T, Wree A, Tacke F, Sigal M. The Role of Microbiota in Primary Sclerosing Cholangitis and Related Biliary Malignancies. International Journal of Molecular Sciences. 2021; 22(13):6975. https://doi.org/10.3390/ijms22136975
Chicago/Turabian StyleÖzdirik, Burcin, Tobias Müller, Alexander Wree, Frank Tacke, and Michael Sigal. 2021. "The Role of Microbiota in Primary Sclerosing Cholangitis and Related Biliary Malignancies" International Journal of Molecular Sciences 22, no. 13: 6975. https://doi.org/10.3390/ijms22136975
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.