
Liver Disease: Induction, Progression, Immunological Mechanisms, and Therapeutic Interventions

 , ,
, ,  and
and
Abstract
:1. Liver Fibrosis Overview, Causes, and Burden
1.1. Alcoholic Liver Disease
1.2. Non-Alcoholic Fatty Liver Disease
1.3. Chronic Hepatitis C
1.4. Chronic Hepatitis B
2. Inflammation and Progression of Liver Fibrosis
2.1. Kupffer Cells and Liver Inflammation in Non-Alcoholic Fatty Liver Disease
2.2. T Cell-Mediated Inflammation in Autoimmune Hepatitis
2.3. Natural Killer and Natural Killer T Cell-Related Inflammation in Viral Hepatitis
2.4. Alternative Stem Cell Lines for the Treatment of Liver Cirrhosis: A Focus on Mesenchymal Stem Cells
3. Liver Fibrosis Therapies
3.1. Pre-Clinical Liver Fibrosis Treatments with MSCs
3.2. Clinical Translation of Liver Fibrosis Therapies with MSCs
3.3. Gene Delivery and Genetically Modified Stem Cells
3.4. Targeted Molecular Pathways and Small Molecule Drug Delivery
3.5. Monoclonal Antibodies
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Definition & Facts for Cirrhosis. Available online: www.niddk.nih.gov/health-information/liver-disease/cirrhosis/definition-facts (accessed on 18 August 2020).
- Mayo Clinic Staff Cirrhosis. Available online: https://www.mayoclinic.org/diseases-conditions/cirrhosis/symptoms-causes/syc-20351487 (accessed on 18 August 2020).
- Liver Biopsy and Liver Function Tests—Diagnosing Liver Disease. Available online: https://liverfoundation.org/for-patients/about-the-liver/diagnosing-liver-disease/ (accessed on 10 March 2020).
- Liver Function Tests. Available online: https://www.mayoclinic.org/tests-procedures/liver-function-tests/about/pac-20394595 (accessed on 10 March 2020).
- Baryah, A.N.S.; Midha, V.; Mahajan, R.; Sood, A. Impact of Corona Virus Disease-19 (COVID-19) pandemic on gastrointestinal disorders. Indian J. Gastroenterol. 2020, 39, 214–219. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R.; Kaneider, N.C. Pathways of liver injury in alcoholic liver disease. J. Hepatol. 2011, 55, 1159–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, G.; Bala, S. Alcoholic liver disease and the gut-liver axis. World J. Gastroenterol. 2010, 16, 1321–1329. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhao, Y.R.; Tian, Z. Roles of hepatic stellate cells in acute liver failure: From the perspective of inflammation and fibrosis. World J. Hepatol. 2019, 11, 412–420. [Google Scholar] [CrossRef]
- Yu, J.; Marsh, S.; Hu, J.; Feng, W.; Wu, C. The Pathogenesis of Nonalcoholic Fatty Liver Disease: Interplay between Diet, Gut Microbiota, and Genetic Background. Gastroenterol. Res. Pract. 2016, 2016, 2862173. [Google Scholar] [CrossRef] [Green Version]
- Tiniakos, D.G.; Vos, M.B.; Brunt, E.M. Nonalcoholic fatty liver disease: Pathology and pathogenesis. Annu. Rev. Pathol. 2010, 5, 145–171. [Google Scholar] [CrossRef] [Green Version]
- Johnson, A.; Milner, J.; Makowski, L. The inflammation highway: Metabolism accelerates inflammatory traffic in obesity. Immunol. Rev. 2012, 249, 218–238. [Google Scholar] [CrossRef] [Green Version]
- Carr, R.M.; Oranu, A.; Khungar, V. Nonalcoholic Fatty Liver Disease: Pathophysiology and Management. Gastroenterol. Clin. North Am. 2016, 45, 639–652. [Google Scholar] [CrossRef] [Green Version]
- Hepatitis C Questions and Answers for the Public. Available online: https://www.cdc.gov/hepatitis/hcv/cfaq.htm (accessed on 18 August 2020).
- Welzel, T.M.; Morgan, T.R.; Bonkovsky, H.L.; Naishadham, D.; Pfeiffer, R.M.; Wright, E.C.; Hutchinson, A.A.; Crenshaw, A.T.; Bashirova, A.; Carrington, M.; et al. Variants in Interferon-α Pathway Genes and Response to Pegylated-Interferon-α2a plus Ribavirin for Treatment of Chronic HCV Infection in the HALT-C Trial. Hepatology 2009, 49, 1847–1858. [Google Scholar] [CrossRef]
- Shah, N.; Pierce, T.; Kowdley, K.V. Review of direct-acting antiviral agents for the treatment of chronic hepatitis C. Expert Opin. Investig. Drugs 2013, 22, 1107–1121. [Google Scholar] [CrossRef]
- Kanwal, F.; Kramer, J.; Asch, S.M.; Chayanupatkul, M.; Cao, Y.; El-Serag, H.B. Risk of Hepatocellular Cancer in HCV Patients Treated With Direct-Acting Antiviral Agents. Gastroenterology 2017, 153, 996–1005.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phase 1 Study to Evaluate Antiviral Activity Of Small Molecule Direct Antiviral Agent At Multiple Doses In Subjects With Chronically Infected Hepatitis C Virus.—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT00671671?term=small+molecule%2C+drug&cond=liver+disease&draw=2&rank=1 (accessed on 11 June 2020).
- Wagner, F.; Thompson, R.; Kantaridis, C.; Simpson, P.; Troke, P.J.F.; Jagannatha, S.; Neelakantan, S.; Purohit, V.S.; Hammond, J.L. Antiviral activity of the hepatitis C virus polymerase inhibitor filibuvir in genotype 1-infected patients. Hepatology 2011, 54, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Baumert, T.F.; Thimme, R.; von Weizsäcker, F. Pathogenesis of hepatitis B virus infection. World J. Gastroenterol. 2007, 13, 82–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann-Haefelin, C.; Thimme, R. Entering the spotlight: Hepatitis B surface antigen-specific B cells. J. Clin. Investig. 2018, 128, 4257–4259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunn, C.; Brunetto, M.; Reynolds, G.; Christophides, T.; Kennedy, P.T.; Lampertico, P.; Das, A.; Lopes, A.R.; Borrow, P.; Williams, K.; et al. Cytokines induced during chronic hepatitis B virus infection promote a pathway for NK cell-mediated liver damage. J. Exp. Med. 2007, 204, 667–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakimi, K.; Lane, T.E.; Wieland, S.; Asensio, V.C.; Campbell, I.L.; Chisari, F.V.; Guidotti, L.G. Blocking chemokine responsive to γ-2/interferon (IFN)-γ inducible protein and monokine induced by IFN-γ activity in vivo reduces the pathogenetic but not the antiviral potential of hepatitis B virus-specific cytotoxic T lymphocytes. J. Exp. Med. 2001, 194, 1755–1766. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.W.; Harmon, C.; O’Farrelly, C. Liver immunology and its role in inflammation and homeostasis. Cell. Mol. Immunol. 2016, 13, 267–276. [Google Scholar] [CrossRef] [Green Version]
- Gao, B. Basic liver immunology. Cell. Mol. Immunol. 2016, 13, 265–266. [Google Scholar] [CrossRef] [Green Version]
- Konturek, P.; Harsch, I.; Konturek, K.; Schink, M.; Konturek, T.; Neurath, M.; Zopf, Y. Gut–Liver Axis: How Do Gut Bacteria Influence the Liver? Med. Sci. 2018, 6, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamataki, Z.; Swadling, L. The liver as an immunological barrier redefined by single-cell analysis. Immunology 2020, 160, 157–170. [Google Scholar] [CrossRef] [Green Version]
- Béland, K.; Lapierre, P.; Djilali-Saiah, I.; Alvarez, F. Liver Restores Immune Homeostasis after Local Inflammation despite the Presence of Autoreactive T Cells. PLoS ONE 2012, 7, e48192. [Google Scholar] [CrossRef]
- Elpek, G.Ö. Cellular and molecular mechanisms in the pathogenesis of liver fibrosis: An update. World J. Gastroenterol. 2014, 20, 7260–7276. [Google Scholar] [CrossRef]
- Noor, M.T.; Manoria, P. Immune Dysfunction in Cirrhosis. J. Clin. Transl. Hepatol. 2017, 20, 2564. [Google Scholar] [CrossRef] [Green Version]
- Zadorozhna, M.; Di Gioia, S.; Conese, M.; Mangieri, D. Neovascularization is a key feature of liver fibrosis progression: Anti-angiogenesis as an innovative way of liver fibrosis treatment. Mol. Biol. Rep. 2020, 47, 2279–2288. [Google Scholar] [CrossRef] [PubMed]
- Naim, A.; Pan, Q.; Baig, M.S. Matrix Metalloproteinases (MMPs) in Liver Diseases. J. Clin. Exp. Hepatol. 2017, 7, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Brenner, D.A.; Kisseleva, T. Combatting Fibrosis: Exosome-Based Therapies in the Regression of Liver Fibrosis. Hepatol. Commun. 2019, 3, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Evolving challenges in hepatic fibrosis. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Joo, Y.E.; Seo, Y.H.; Lee, W.S.; Kim, H.S.; Choi, S.K.; Rew, J.S.; Park, C.S.; Kim, S.J. Expression of tissue inhibitors of metalloproteinases (TIMPs) in hepatocellular carcinoma. Korean J. Intern. Med. 2000, 15, 171. [Google Scholar] [CrossRef]
- Arpino, V.; Brock, M.; Gill, S.E. The role of TIMPs in regulation of extracellular matrix proteolysis. Matrix Biol. 2015, 44–46, 247–254. [Google Scholar] [CrossRef]
- Xu, R.; Zhang, Z.; Wang, F. Liver fibrosis: Mechanisms of immune-mediated liver injury. Cell. Mol. Immunol. 2012, 9, 296–301. [Google Scholar] [CrossRef]
- Knolle, P.A.; Wohlleber, D. Immunological functions of liver sinusoidal endothelial cells. Cell. Mol. Immunol. 2016, 13, 347–353. [Google Scholar] [CrossRef] [Green Version]
- Land, W.G. Role of Damage-Associated Molecular Patterns in Light of Modern Environmental Research: A Tautological Approach. Int. J. Environ. Res. 2020, 14, 583–604. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Lefebvre, A.T.; Horuzsko, A. Kupffer Cell Metabolism and Function. J. Enzymol. Metab. 2015, 1, 101. [Google Scholar] [PubMed]
- Chen, J.; Deng, X.; Liu, Y.; Tan, Q.; Huang, G.; Che, Q.; Guo, J.; Su, Z. Kupffer cells in non-alcoholic fatty liver disease: Friend or foe? Int. J. Biol. Sci. 2020, 16, 2367–2378. [Google Scholar] [CrossRef]
- Mehal, W.Z.; Azzaroli, F.; Crispe, I.N. Antigen Presentation by Liver Cells Controls Intrahepatic T Cell Trapping, Whereas Bone Marrow-Derived Cells Preferentially Promote Intrahepatic T Cell Apoptosis. J. Immunol. 2001, 167, 667–673. [Google Scholar] [CrossRef] [Green Version]
- Scoazec, J.-Y.; Feldmann, G. The cell adhesion molecules of hepatic sinusoidal endothelial cells. J. Hepatol. 1994, 20, 296–300. [Google Scholar] [CrossRef]
- Lin, R.; Zhang, J.; Zhou, L.; Wang, B. Altered function of monocytes/macrophages in patients with autoimmune hepatitis. Mol. Med. Rep. 2016, 13, 3874–3880. [Google Scholar] [CrossRef] [Green Version]
- Crux, N.B.; Elahi, S. Human Leukocyte Antigen (HLA) and immune regulation: How do classical and non-classical HLA alleles modulate immune response to human immunodeficiency virus and hepatitis C virus infections? Front. Immunol. 2017, 8, 1–26. [Google Scholar] [CrossRef]
- Christen, U.; Hintermann, E. Immunopathogenic mechanisms of autoimmune hepatitis: How much do we know from animal models? Int. J. Mol. Sci. 2016, 17, 2007. [Google Scholar] [CrossRef]
- Zheng, M.; Tian, Z. Liver-Mediated Adaptive Immune Tolerance. Front. Immunol. 2019, 10, 2525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006, 439, 682–687. [Google Scholar] [CrossRef]
- Saltzman, W.M. Tissue Engineering: Principles for the Design of Replacement Organs and Tissues, 1st ed.; Oxford University Press: New York, NY, USA, 2004. [Google Scholar]
- Power, C.; Rasko, J.E.J. Will cell reprogramming resolve the embryonic stem cell controversy? a narrative review. Ann. Intern. Med. 2011, 155, 114–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbett, J.L.; Duncan, S.A. iPSC-Derived Hepatocytes as a Platform for Disease Modeling and Drug Discovery. Front. Med. 2019, 6, 1–12. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Y.; Zhang, L.; Li, J.; Zhu, C. Mesenchymal stem cells: Potential application for the treatment of hepatic cirrhosis. Stem Cell Res. Ther. 2018, 9, 59. [Google Scholar] [CrossRef] [Green Version]
- Miyajima, A.; Tanaka, M.; Itoh, T. Stem/progenitor cells in liver development, homeostasis, regeneration, and reprogramming. Cell Stem Cell 2014, 14, 561–574. [Google Scholar] [CrossRef] [Green Version]
- Ko, S.; Russell, J.O.; Molina, L.M.; Monga, S.P. Liver Progenitors and Adult Cell Plasticity in Hepatic Injury and Repair: Knowns and Unknowns. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 23–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christ, B.; Pelz, S. Implication of hepatic stem cells in functional liver repopulation. Cytom. Part A 2013, 83 A, 90–102. [Google Scholar] [CrossRef]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Krause, D.S.; Deans, R.J.; Keating, A.; Prockop, D.J.; Horwitz, E.M. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Friedenstein, A.J.; Piatetzky-Shapiro, I.I.; Petrakova, K.V. Osteogenesis in transplants of bone marrow cells. J. Embryol. Exp. Morphol. 1966, 16, 381–390. [Google Scholar]
- Melief, S.M.; Zwaginga, J.J.; Fibbe, W.E.; Roelofs, H. Adipose Tissue-Derived Multipotent Stromal Cells Have a Higher Immunomodulatory Capacity Than Their Bone Marrow-Derived Counterparts. Stem Cells Transl. Med. 2013, 2, 455–463. [Google Scholar] [CrossRef]
- Jia, Z.; Wang, S.; Liu, Q. Identification of differentially expressed genes by single-cell transcriptional profiling of umbilical cord and synovial fluid mesenchymal stem cells. J. Cell. Mol. Med. 2020, 24, 1945–1957. [Google Scholar] [CrossRef] [Green Version]
- Battula, V.L.; Treml, S.; Bareiss, P.M.; Gieseke, F.; Roelofs, H.; De Zwart, P.; Müller, I.; Schewe, B.; Skutella, T.; Fibbe, W.E.; et al. Isolation of functionally distinct mesenchymal stem cell subsets using antibodies against CD56, CD271, and mesenchymal stem cell antigen-1. Haematologica 2009, 94, 173–184. [Google Scholar] [CrossRef] [Green Version]
- Phinney, D.G.; Sensebé, L. Mesenchymal stromal cells: Misconceptions and evolving concepts. Cytotherapy 2013, 15, 140–145. [Google Scholar] [CrossRef]
- Anzalone, R.; Lo Iacono, M.; Corrao, S.; Magno, F.; Loria, T.; Cappello, F.; Zummo, G.; Farina, F.; La Rocca, G. New emerging potentials for human wharton’s jelly mesenchymal stem cells: Immunological features and hepatocyte-like differentiative capacity. Stem Cells Dev. 2010, 19, 423–438. [Google Scholar] [CrossRef]
- Hu, L.; Hu, J.; Zhao, J.; Liu, J.; Ouyang, W.; Yang, C.; Gong, N.; Du, L.; Khanal, A.; Chen, L. Side-by-side comparison of the biological characteristics of human umbilical cord and adipose tissue-derived mesenchymal stem cells. Biomed Res. Int. 2013, 2013, 438248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, H.J.; Bae, Y.K.; Kim, M.; Kwon, S.J.; Jeon, H.B.; Choi, S.J.; Kim, S.W.; Yang, Y.S.; Oh, W.; Chang, J.W. Comparative analysis of human mesenchymal stem cells from bone marrow, adipose tissue, and umbilical cord blood as sources of cell therapy. Int. J. Mol. Sci. 2013, 14, 17986–18001. [Google Scholar] [CrossRef]
- Der Lee, K.; Kuo, T.K.C.; Whang-Peng, J.; Chung, Y.F.; Lin, C.T.; Chou, S.H.; Chen, J.R.; Chen, Y.P.; Lee, O.K.S. In vitro hepatic differentiation of human mesenchymal stem cells. Hepatology 2004, 40, 1275–1284. [Google Scholar]
- Lysy, P.A.; Campard, D.; Smets, F.; Malaise, J.; Mourad, M.; Najimi, M.; Sokal, E.M. Persistence of a chimerical phenotype after hepatocyte differentiation of human bone marrow mesenchymal stem cells. Cell Prolif. 2008, 41, 36–58. [Google Scholar] [CrossRef] [PubMed]
- Kazemnejad, S.; Allameh, A.; Soleimani, M.; Gharehbaghian, A.; Mohammadi, Y.; Amirizadeh, N.; Jazayery, M. Biochemical and molecular characterization of hepatocyte-like cells derived from human bone marrow mesenchymal stem cells on a novel three-dimensional biocompatible nanofibrous scaffold. J. Gastroenterol. Hepatol. 2009, 24, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Yin, H.; Liu, M.; Xu, G.; Zhou, X.; Ge, P.; Yang, H.; Mao, Y. Impaired albumin function: A novel potential indicator for liver function damage? Ann. Med. 2019, 51, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Nasef, A.; Chapel, A.; Mazurier, C.; Bouchet, S.; Lopez, M.; Mathieu, N.; Sensebé, L.; Zhang, Y.; Gorin, N.-C.; Thierry, D.; et al. MSC’s effect on T-lymphocytes. Gene Expr. 2018, 13, 217–226. [Google Scholar] [CrossRef]
- Li, N.; Hua, J. Interactions between mesenchymal stem cells and the immune system. Cell. Mol. Life Sci. 2017, 74, 2345–2360. [Google Scholar] [CrossRef] [PubMed]
- Sotiropoulou, P.A.; Papamichail, M. Immune properties of mesenchymal stem cells. Methods Mol. Biol. 2007, 407, 225–243. [Google Scholar] [PubMed]
- Galleu, A.; Riffo-Vasquez, Y.; Trento, C.; Lomas, C.; Dolcetti, L.; Cheung, T.S.; Von Bonin, M.; Barbieri, L.; Halai, K.; Ward, S.; et al. Apoptosis in mesenchymal stromal cells induces in vivo recipient-mediated immunomodulation. Sci. Transl. Med. 2017, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, A.R.R.; Dahlke, M.H. Immunomodulation by Mesenchymal Stem Cells (MSCs): Mechanisms of action of living, apoptotic, and dead MSCs. Front. Immunol. 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, S.; Chen, Q.L.; Song, Y.N.; Sun, Y.; Wei, B.; Li, X.Y.; Hu, Y.Y.; Liu, P.; Su, S.B. Mechanisms of CCl4-induced liver fibrosis with combined transcriptomic and proteomic analysis. J. Toxicol. Sci. 2016, 41, 561–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.; Li, J.J.; Cao, D.Y.; Li, X.; Zhang, L.Y.; He, Y.; Yue, S.Q.; Wang, D.S.; Dou, K.F. Intravenous injection of mesenchymal stem cells is effective in treating liver fibrosis. World J. Gastroenterol. 2012, 18, 1048–1058. [Google Scholar] [CrossRef]
- Sziksz, E.; Pap, D.; Lippai, R.; Béres, N.J.; Fekete, A.; Szabó, A.J.; Vannay, Á. Fibrosis Related Inflammatory Mediators: Role of the IL-10 Cytokine Family. Mediat. Inflamm. 2015, 2015, 764641. [Google Scholar] [CrossRef] [PubMed]
- Abdel Aziz, M.T.; Atta, H.M.; Mahfouz, S.; Fouad, H.H.; Roshdy, N.K.; Ahmed, H.H.; Rashed, L.A.; Sabry, D.; Hassouna, A.A.; Hasan, N.M. Therapeutic potential of bone marrow-derived mesenchymal stem cells on experimental liver fibrosis. Clin. Biochem. 2007, 40, 893–899. [Google Scholar] [CrossRef]
- Meier, R.P.H.; Mahou, R.; Morel, P.; Meyer, J.; Montanari, E.; Muller, Y.D.; Christofilopoulos, P.; Wandrey, C.; Gonelle-Gispert, C.; Bühler, L.H. Microencapsulated human mesenchymal stem cells decrease liver fibrosis in mice. J. Hepatol. 2015, 62, 634–641. [Google Scholar] [CrossRef]
- Jung, K.H.; Shin, H.P.; Lee, S.; Lim, Y.J.; Hwang, S.H.; Han, H.; Park, H.K.; Chung, J.H.; Yim, S.V. Effect of human umbilical cord blood-derived mesenchymal stem cells in a cirrhotic rat model. Liver Int. 2009, 29, 898–909. [Google Scholar] [CrossRef]
- Wang, Y.; Han, Z.B.; Ma, J.; Zuo, C.; Geng, J.; Gong, W.; Sun, Y.; Li, H.; Wang, B.; Zhang, L.; et al. A toxicity study of multiple-administration human umbilical cord mesenchymal stem cells in cynomolgus monkeys. Stem Cells Dev. 2012, 21, 1401–1408. [Google Scholar] [CrossRef]
- Guo, G.; Zhuang, X.; Xu, Q.; Wu, Z.; Zhu, Y.; Zhou, Y.; Li, Y.; Lu, Y.; Zhang, B.; Talbot, P.; et al. Peripheral infusion of human umbilical cord mesenchymal stem cells rescues acute liver failure lethality in monkeys. Stem Cell Res. Ther. 2019, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Ji, R.; Zhang, N.; You, N.; Li, Q.; Liu, W.; Jiang, N.; Liu, J.; Zhang, H.; Wang, D.; Tao, K.; et al. The differentiation of MSCs into functional hepatocyte-like cells in a liver biomatrix scaffold and their transplantation into liver-fibrotic mice. Biomaterials 2012, 33, 8995–9008. [Google Scholar] [CrossRef]
- Kharaziha, P.; Hellström, P.M.; Noorinayer, B.; Farzaneh, F.; Aghajani, K.; Jafari, F.; Telkabadi, M.; Atashi, A.; Honardoost, M.; Zali, M.R.; et al. Improvement of liver function in liver cirrhosis patients after autologous mesenchymal stem cell injection: A phase I-II clinical trial. Eur. J. Gastroenterol. Hepatol. 2009, 21, 1199–1205. [Google Scholar] [CrossRef] [PubMed]
- Transplantation of Autologous Mesenchymal Stem Cell in Decompensate Cirrhotic Patients with Pioglitazone. Available online: https://clinicaltrials.gov/ct2/show/NCT01454336?term=Hepatic+portal+vein&type=Intr&cond=Liver+Fibrosis&draw=2&rank=1 (accessed on 16 April 2020).
- Vosough, M.; Moossavi, S.; Mardpour, S.; Akhlaghpoor, S.; Azimian, V.; Jarughi, N.; Hosseini, S.E.; Ashrafi, M.; Nikfam, S.; Aghdami, N. Repeated intraportal injection of mesenchymal stem cells in combination with pioglitazone in patients with compensated cirrhosis: A clinical report of two cases. Arch. Iran. Med. 2016, 19, 131–136. [Google Scholar] [PubMed]
- ABMSC Infusion Through Hepatic Artery in Portal Hypertension Surgery for the Treatment of Liver Cirrhosis. Available online: https://clinicaltrials.gov/ct2/show/NCT01560845 (accessed on 16 April 2020).
- Xu, L.; Gong, Y.; Wang, B.; Shi, K.; Hou, Y.; Wang, L.; Lin, Z.; Han, Y.; Lu, L.; Chen, D.; et al. Randomized trial of autologous bone marrow mesenchymal stem cells transplantation for hepatitis B virus cirrhosis: Regulation of Treg/Th17 cells. J. Gastroenterol. Hepatol. 2014, 29, 1620–1628. [Google Scholar] [CrossRef] [PubMed]
- Bone Mesenchymal Stem Cell (BMSC) Transplantation in Liver Cirrhosis Via Portal Vein—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT00993941 (accessed on 16 April 2020).
- Sakai, Y.; Takamura, M.; Seki, A.; Sunagozaka, H.; Terashima, T.; Komura, T.; Yamato, M.; Miyazawa, M.; Kawaguchi, K.; Nasti, A.; et al. Phase I clinical study of liver regenerative therapy for cirrhosis by intrahepatic arterial infusion of freshly isolated autologous adipose tissue-derived stromal/stem (regenerative) cell. Regen. Ther. 2017, 6, 52–64. [Google Scholar] [CrossRef]
- A Phase I/II Safety and Tolerability Dose Escalation Study of Autologous Stem Cells to Patients with Liver Insufficiency. Available online: https://www.clinicaltrials.gov/ct2/show/NCT00655707 (accessed on 16 April 2020).
- Yang, X.; Meng, Y.; Han, Z.; Ye, F.; Wei, L.; Zong, C. Mesenchymal Stem Cells Transplantation for Liver Cirrhosis Due to HCV Hepatitis N. Cell Biosci. 2020, 10, 123. [Google Scholar] [CrossRef]
- Kantarcıoğlu, M.; Demirci, H.; Avcu, F.; Karslioğlu, Y.; Babayiğit, M.A.; Karaman, B.; Öztürk, K.; Gürel, H.; Kayhan, M.A.; Kaçar, S.; et al. Efficacy of autologous mesenchymal stem cell transplantation in patients with liver cirrhosis. Turkish J. Gastroenterol. 2015, 26, 244–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steingen, C.; Brenig, F.; Baumgartner, L.; Schmidt, J.; Schmidt, A.; Bloch, W. Characterization of key mechanisms in transmigration and invasion of mesenchymal stem cells. J. Mol. Cell. Cardiol. 2008, 44, 1072–1084. [Google Scholar] [CrossRef] [PubMed]
- Marquez-Curtis, L.A.; Gul-Uludag, H.; Xu, P.; Chen, J.; Janowska-Wieczorek, A. CXCR4 transfection of cord blood mesenchymal stromal cells with the use of cationic liposome enhances their migration toward stromal cell-derived factor-1. Cytotherapy 2013, 15, 840–849. [Google Scholar] [CrossRef]
- Cheng, K.; Yang, N.; Mahato, R.I. TGF-β1 gene silencing for treating liver fibrosis. Mol. Pharm. 2009, 6, 772–779. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Mondal, G.; Dutta, R.; Mahato, R.I. Co-delivery of small molecule hedgehog inhibitor and miRNA for treating liver fibrosis. Biomaterials 2016, 76, 144–156. [Google Scholar] [CrossRef]
- Hyun, J.; Wang, S.; Kim, J.; Rao, K.M.; Park, S.Y.; Chung, I.; Ha, C.S.; Kim, S.W.; Yun, Y.H.; Jung, Y. MicroRNA-378 limits activation of hepatic stellate cells and liver fibrosis by suppressing Gli3 expression. Nat. Commun. 2016, 7, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Z.; Froh, M.; Wheeler, M.D.; Smutney, O.; Lehmann, T.G.; Thurman, R.G. Viral gene delivery of superoxide dismutase attenuates experimental cholestasis-induced liver fibrosis in the rat. Gene Ther. 2002, 9, 183–191. [Google Scholar] [CrossRef] [Green Version]
- Improvement of Liver Function in Liver Cirrhosis Patients After Autologous Mesenchymal Stem Cell Injection:a Phase I-II Clinical Trial—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT00420134 (accessed on 15 April 2020).
- Safety and Efficacy Study of Co-transfering of Mesenchymal Stem Cell and Regulatory T Cells in Treating End-stage Liver Disease—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03460795?term=stem+cell%2C+MSC&cond=Liver+Diseases&draw=2&rank=1 (accessed on 11 June 2020).
- Safety of UC-MSC Transfusion for ACLF Patients—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04822922?term=stem+cell%2C+MSC&recrs=abdef&cond=Liver+Diseases&draw=2&rank=2 (accessed on 15 April 2020).
- Sakai, Y.; Seki, A.; Kaneko, S. Liver Regeneration Therapy by Intrahepatic Arterial Administration of Autologous Adipose Tissue Derived Stromal Cells. Jpn. J. Clin. Med. 2015, 73, 492–498. [Google Scholar]
- A Trial of IDN-6556 in Post Orthotopic Liver Transplant for Chronic HCV (POLT-HCV-SVR). Available online: https://clinicaltrials.gov/ct2/show/NCT02138253 (accessed on 16 April 2020).
- Pirfenidone and Advanced Liver Fibrosis.—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04099407 (accessed on 15 April 2020).
- Evaluation of Irbesartan on Hepatic Fibrosis in Chronic Hepatitis C—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT00265642 (accessed on 15 April 2020).
- Effects of Losartan on Hepatic Fibrogenesis in Chronic Hepatitis C. Available online: https://clinicaltrials.gov/ct2/show/NCT00298714?term=gene&type=Intr&cond=Liver+Fibrosis&draw=2&rank=1 (accessed on 11 June 2020).
- Guselkumab (Anti-IL 23 Monoclonal Antibody) for Alcohol Associated Liver Disease—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04736966?term=antibodies&cond=Liver+Diseases&draw=2&rank=2 (accessed on 11 June 2020).
- Anti-LPS Antibody Treatment for Pediatric NAFLD—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/study/NCT03042767?term=antibodies&cond=Liver+Diseases&draw=3&rank=5 (accessed on 11 June 2020).
- Development of Novel MRI Methods for Detecting, Discriminating, and Measuring Liver Fibrosis and Congestion in Fontan Patients. Available online: https://clinicaltrials.gov/ct2/show/NCT03539757 (accessed on 16 April 2020).
- Liver Fibrosis Evaluation Using Ultrasound Shear Wave Imaging. Available online: https://clinicaltrials.gov/ct2/show/NCT03637959 (accessed on 16 April 2020).
- Evaluation of Innovative Ultrasonic Techniques for Non-invasive Diagnosis of Liver Fibrosis in Patients with Chronic Viral Hepatitis B or C (FIBRECHO). Available online: https://clinicaltrials.gov/ct2/show/NCT01537965?term=device&cond=liver+fibrosis&draw=3&rank=12%0A (accessed on 16 April 2020).
- Teixeira-Clerc, F.; Julien, B.; Grenard, P.; Van Nhieu, J.T.; Deveaux, V.; Li, L.; Serriere-Lanneau, V.; Ledent, C.; Mallat, A.; Lotersztajn, S. CB1 cannabinoid receptor antagonism: A new strategy for the treatment of liver fibrosis. Nat. Med. 2006, 12, 671–676. [Google Scholar] [CrossRef]
- Moreno, M.; Gonzalo, T.; Kok, R.J.; Sancho-Bru, P.; van Beuge, M.; Swart, J.; Prakash, J.; Temming, K.; Fondevila, C.; Beljaars, L.; et al. Reduction of advanced liver fibrosis by short-term targeted delivery of an angiotensin receptor blocker to hepatic stellate cells in rats. Hepatology 2010, 51, 942–952. [Google Scholar] [CrossRef]
- Rockey, D.C. Current and Future Anti-Fibrotic Therapies for Chronic Liver Disease. Clin. Liver Dis. 2008, 12, 939–962. [Google Scholar] [CrossRef] [Green Version]
- Colmenero, J.; Bataller, R.; Sancho-Bru, P.; Domínguez, M.; Moreno, M.; Forns, X.; Bruguera, M.; Arroyo, V.; Brenner, D.A.; Ginès, P. Effects of losartan on hepatic expression of nonphagocytic NADPH oxidase and fibrogenic genes in patients with chronic hepatitis C. Am. J. Physiol.—Gastrointest. Liver Physiol. 2009, 297, G726. [Google Scholar] [CrossRef] [Green Version]
- Evaluation of Irbesartan on Hepatic Fibrosis in Chronic Hepatitis C (Fibrosar). Available online: https://clinicaltrials.gov/ct2/show/NCT00265642 (accessed on 16 April 2020).
- Poo, J.L.; Torre, A.; Aguilar-Ramírez, J.R.; Cruz, M.; Mejía-Cuán, L.; Cerda, E.; Velázquez, A.; Patiño, A.; Ramírez-Castillo, C.; Cisneros, L.; et al. Benefits of prolonged-release pirfenidone plus standard of care treatment in patients with advanced liver fibrosis: PROMETEO study. Hepatol. Int. 2020, 14, 817–827. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S.; Ochi, T.; Shimada, H.; Inagaki, K.; Fujita, I.; Nii, A.; Moffat, M.A.; Katragadda, M.; Violand, B.N.; Arch, R.H.; et al. Anti-PDGF-B monoclonal antibody reduces liver fibrosis development. Hepatol. Res. 2010, 40, 1128–1141. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Abdelmalek, M.F.; Caldwell, S.; Shiffman, M.L.; Diehl, A.M.; Ghalib, R.; Lawitz, E.J.; Rockey, D.C.; Schall, R.A.; Jia, C.; et al. Simtuzumab Is Ineffective for Patients with Bridging Fibrosis or Compensated Cirrhosis Caused by Nonalcoholic Steatohepatitis. Gastroenterology 2018, 155, 1140–1153. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Jalan, R.; Kaser, A.; Davies, N.A.; Offner, F.A.; Hodges, S.J.; Ludwiczek, O.; Shawcross, D.; Zoller, H.; Alisa, A.; et al. Anti-tumor necrosis factor-alpha monoclonal antibody therapy in severe alcoholic hepatitis. J. Hepatol. 2003, 38, 419–425. [Google Scholar] [CrossRef]
- Sano, S.; Kubo, H.; Morishima, H.; Goto, R.; Zheng, R.; Nakagawa, H. Guselkumab, a human interleukin-23 monoclonal antibody in Japanese patients with generalized pustular psoriasis and erythrodermic psoriasis: Efficacy and safety analyses of a 52-week, phase 3, multicenter, open-label study. J. Dermatol. 2018, 45, 529–539. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
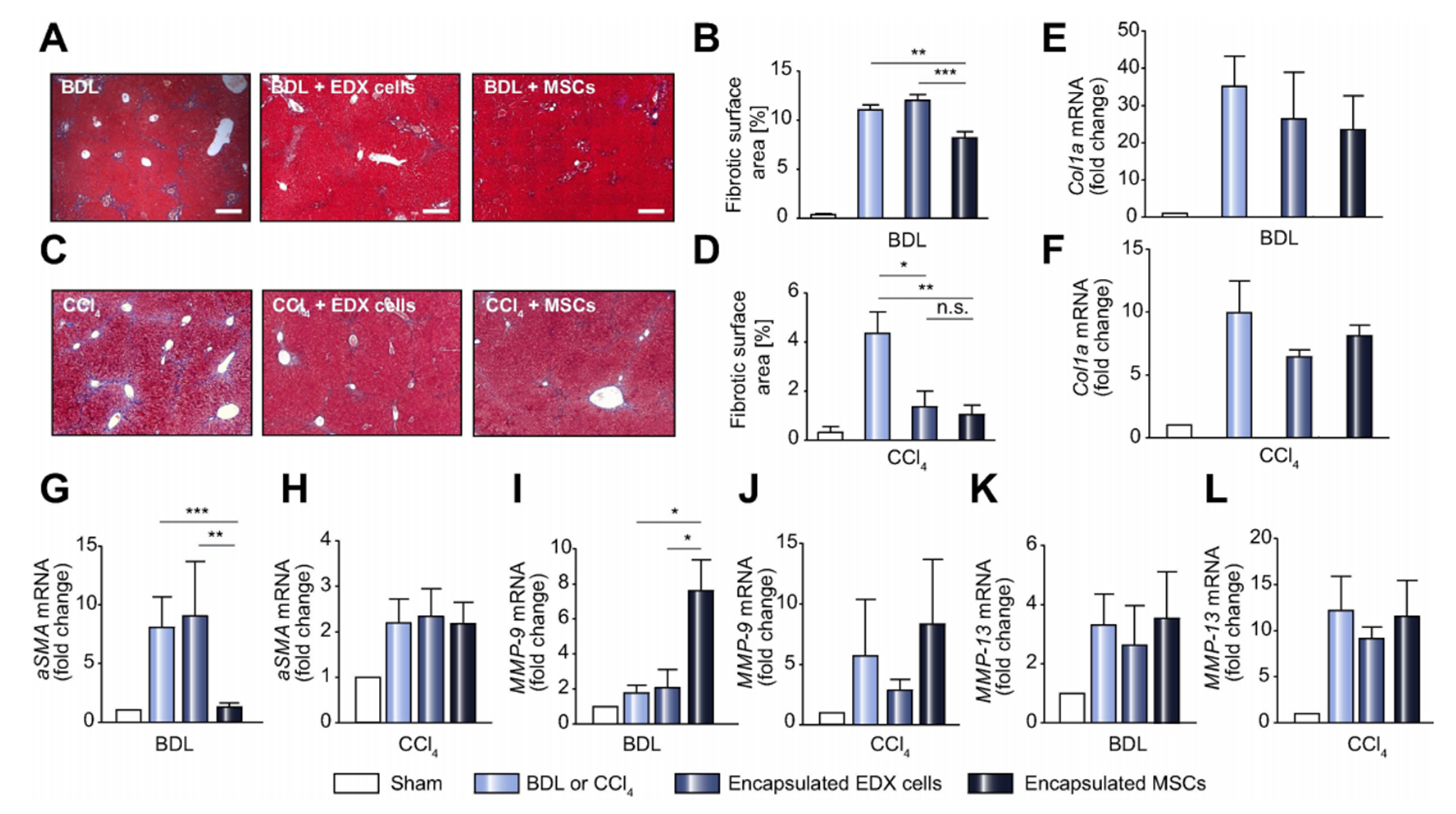{kind=link}
| General Category | Target Disease Condition | Therapeutic Agent | Responsible Company/Lab | Phase | ClinicalTrials.gov Identifier |
|---|---|---|---|---|---|
| Stem Cells | Compensated Liver Cirrhosis | Autologous Mesenchymal stem cells (MSCs) and Pioglitazone | Royan Institute | Phase I | NCT01454336 [84] |
| Hepatitis C Virus (HCV) Infection | Autologous Mesenchymal stem cells (MSCs) | Saglik Bilimleri Universitesi Gulhane Tip Fakultesi | Phase I/II | NCT02705742 [91] | |
| End-stage Liver Cirrhosis | Autograft MSCs Differentiated Into Progenitor of Hepatocytes | Shahid Beheshti University of Medical Sciences and Tarbiat Modarres University | Phase I/II | NCT00420134 [99] | |
| Chronic liver insufficiency | Autologous Expanded CD34+ Haemopoietic cells | Imperial College London | Phase I/II | NCT00655707 [90] | |
| Liver Cirrhosis | Co-transferring of MSCs and Tregs | Nanjing Medical University | Phase I/II | NCT03460795 [100] | |
| Liver Cirrhosis | Autologous Bone Mesenchymal Stem Cells (bMSCs) via Portal Vein | Sun Yat-sen University | Phase II | NCT00993941 [88] | |
| Acute-On-Chronic Liver Failure | Human umbilical cord-derived mesenchyme stem cells (hUC-MSCs) | Hai Li, Shanghai Jiao Tong University School of Medicine | Phase II | NCT04822922 [101] | |
| Liver Cirrhosis, portal hypertension, hepatic Decompensation | Autologous bone marrow stem cells infusion (ABMSCi) and abdominal portal hypertension surgery | Wenzhou Medical University | Phase II/III | NCT01560845 [86] | |
| Liver Cirrhosis | Intrahepatic Arterial Administration of Autologous Adipose Tissue Derived Stromal Cells | Kanazawa University | N/A | NCT01062750 [102] | |
| Small Molecules | Chronic Hepatitis C Virus (HCV) | Small Molecule Agent (PF-868554), direct antiviral agent | Pfizer | Phase I | NCT00671671 [17] |
| Chronic Hepatitis C Virus (HCV) Infection | Emricasan (IDN-6556) | Conatus Pharmaceuticals Inc. | Phase II | NCT02138253 [103] | |
| Chronic Liver Fibrosis | Prolonged-Release Pirfenidone Formulation | Grupo Mexicano para el Estudios de las Enfermedades Hepaticas | Phase II | NCT04099407 [104] | |
| Hepatic Fibrosis in Chronic Hepatitis C | Irbesartan | French National Institute for Health and Medical Research-French National Agency for Research on AIDS and Viral Hepatitis (Inserm-ANRS) and Sanofi | Phase III | NCT00265642 [105] | |
| Hepatic Fibrogenesis in Chronic Hepatitis C | Losartan | Hospital Clinic of Barcelona | Phase IV | NCT00298714 [106] | |
| Antibody Therapy | Alcoholic Liver Disease | Guselkumab, a humanized anti-IL23 monoclonal antibody | University of California, San Diego | Phase I | NCT04736966 [107] |
| Non-alcoholic Fatty Acid Liver Disease | IMM-124E, polyclonal antibody against endotoxin lipopolysaccharide (LPS) | Immuron Ltd., Emory University, and Advanced MR Analytics AB | Phase II | NCT03042767 [108] | |
| Diagnostic Tools | Liver Fibrosis and Congestion in Fontan Patients | Non-Contrast Magnetic Resonance Imaging, Device | Children’s Hospital Medical Center, Cincinnati | N/A | NCT03539757 [109] |
| Liver Fibrosis | Mechanical Vibrations with Ultrasound Shear Wave Imaging, Device | Mayo Clinic | N/A | NCT03637959 [110] | |
| Liver Fibrosis | Fibroscan® of Echosens, Aixplorer® of Supersonic Imagine, Aplio XG of Toshiba, QRS software developed by Pr I.Bricault, Acuson S2000 of Siemens, Device | University Hospital, Grenoble & Clinical Investigation Centre for Innovative Technology Network | N/A | NCT01537965 [111] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neshat, S.Y.; Quiroz, V.M.; Wang, Y.; Tamayo, S.; Doloff, J.C. Liver Disease: Induction, Progression, Immunological Mechanisms, and Therapeutic Interventions. Int. J. Mol. Sci. 2021, 22, 6777. https://doi.org/10.3390/ijms22136777
Neshat SY, Quiroz VM, Wang Y, Tamayo S, Doloff JC. Liver Disease: Induction, Progression, Immunological Mechanisms, and Therapeutic Interventions. International Journal of Molecular Sciences. 2021; 22(13):6777. https://doi.org/10.3390/ijms22136777
Chicago/Turabian StyleNeshat, Sarah Y., Victor M. Quiroz, Yuanjia Wang, Sebastian Tamayo, and Joshua C. Doloff. 2021. "Liver Disease: Induction, Progression, Immunological Mechanisms, and Therapeutic Interventions" International Journal of Molecular Sciences 22, no. 13: 6777. https://doi.org/10.3390/ijms22136777