
The Mystery of Diabetic Cardiomyopathy: From Early Concepts and Underlying Mechanisms to Novel Therapeutic Possibilities

 , , and
, , and
Abstract
:1. Introduction
2. Definition
3. History
4. Evolution of the Disease and Functional Phenotype
5. Structural Features of the Diabetic Heart
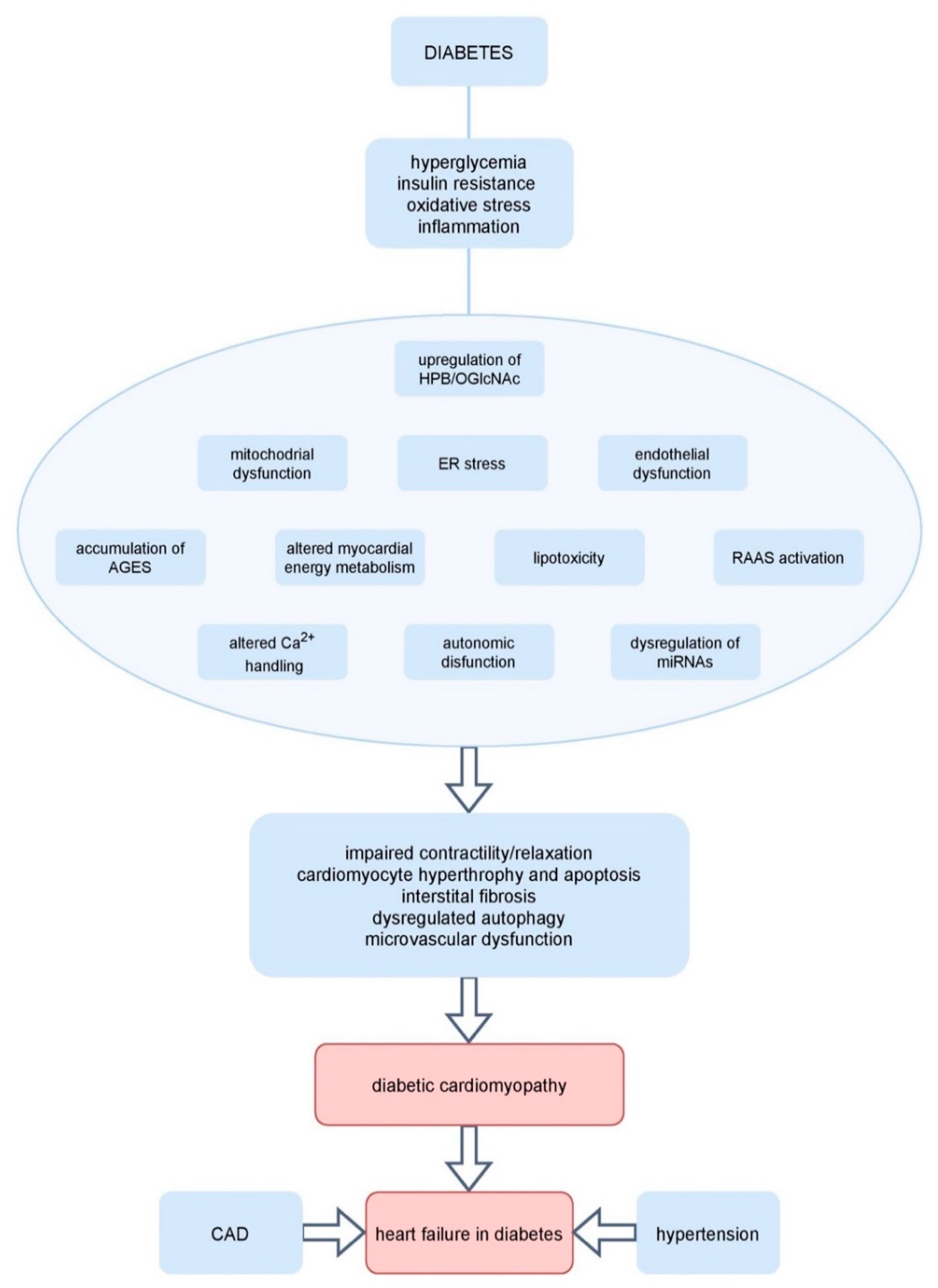
6. Pathophysiologic Mechanisms Underlying Diabetic Cardiomyopathy
6.1. Hyperglycemia
6.2. Insulin Resistance
6.3. Myocardial Energy Metabolism
6.4. Advanced Glycation End Products Modification
6.5. Neurohormonal Activation
6.6. Oxidative Stress
6.7. Inflammation
6.8. Mitochondrial Dysfunction
6.9. Altered Ca2+ Handling
6.10. O-Linked Beta-N-acetylglucosamine (O-GlcNAc) Protein Modification
6.11. microRNAs (miRNAs) Dysregulation
6.12. Endoplasmic Reticulum Stress
6.13. Epicardial Adipose Tissue
7. Signaling Pathways Contributing to Diabetic Cardiomyopathy
7.1. AMP-Activated Protein Kinase (AMPK)
7.2. Variations in Activations of Cardiac Peroxisome Proliferator-Activated Receptors (PPARs)
7.3. Cardiac Mitogen-Activated Protein Kinase (MAPK) Signaling
7.4. Protein Kinase C (PKC) Activation
7.5. NF-κB Activation
7.6. Transcription Factor Cyclic Adenosine 5-Monophosphate-Responsive Element Modulator (CREM) Activation
7.7. Exosomes Abnormalities
7.8. Abnormalities of Nuclear Factor Erythroid 2–Related Factor 2 (Nrf2)
7.9. SGLT2 Abnormalities
7.10. Mammalian Target of Rapamycin (mTOR) Signaling
8. Clinical Diagnosis
9. Therapeutic Possibilities
9.1. Heart Failure Prevention in Diabetes
9.1.1. Lifestyle Interventions
9.1.2. Metabolic Therapies
9.1.3. Targeting Oxidative Stress
9.1.4. SGLT2 Inhibitors
9.1.5. RAAS Inhibition
9.2. Conventional Therapies for Diabetes
9.2.1. Metformin
9.2.2. Sulfonylurea Agents
9.2.3. Insulin
9.2.4. Thiazolidinediones
9.2.5. Incretin-Based Therapies
9.2.6. SGLT2 Inhibitors
9.3. Conventional Therapies for Heart Failure
9.4. Metabolic Modulators
9.4.1. Trimetazidine
9.4.2. Perhexiline
9.4.3. Meldonium
9.4.4. Fenofibrate
9.4.5. GLP-1 Agonists and Mineralocorticoid Receptor Blockers
9.4.6. SGLT2 Inhibitors
9.5. Potential Therapies Targeting Signaling Pathways
9.5.1. p38 MAPK Inhibition
9.5.2. PKC β2 Inhibition
9.5.3. Phosphodiesterase Type 5 Inhibition
9.5.4. AMPK Activation
9.5.5. ET-1 Targeting
9.5.6. HBP and O-GlcNAc Targeting
9.5.7. NF-κB Inhibition
9.5.8. Nrf2 Activity
9.5.9. Heat Shock Protein20-Engineered Exosomes
9.5.10. microRNAs
9.6. Correction of Intestinal Dysbiosis
10. Gene Therapy
11. Cardiomyopathy in Type 1 and Type 2 Diabetes Mellitus
12. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lin, X.; Xu, Y.; Pan, X.; Xu, J.; Ding, Y.; Sun, X.; Song, X.; Ren, Y.; Shan, P.-F. Global, regional, and national burden and trend of diabetes in 195 countries and territories: An analysis from 1990 to 2025. Sci. Rep. 2020, 10, 14790. [Google Scholar] [CrossRef]
- Khan, M.A.B.; Hashim, M.J.; King, J.K.; Govender, R.D.; Mustafa, H.; Al Kaabi, J. Epidemiology of Type 2 Diabetes—Global Burden of Disease and Forecasted Trends. J. Epidemiol. Glob. Health 2020, 10, 107–111. [Google Scholar] [CrossRef] [Green Version]
- Forouzanfar, M.H.; Afshin, A.; Alexander, L.T.; Anderson, H.R.; Bhutta, Z.A.; Biryukov, S.; Brauer, M.; Burnett, R.; Cercy, K.; Charlson, F.J.; et al. GBD 2015 Risk Factors Collaborators. Global, regional, and national comparative risk assessment of 79 behavioural, environmental and occupational, and metabolic risks or clusters of risks, 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1659–1724. [Google Scholar] [CrossRef] [Green Version]
- De Rosa, S.; Arcidiacono, B.; Chiefari, E.; Brunetti, A.; Indolfi, C.; Foti, D.P. Type 2 Diabetes Mellitus and Cardiovascular Disease: Genetic and Epigenetic Links. Front. Endocrinol. 2018, 9, 2. [Google Scholar] [CrossRef] [PubMed]
- Huynh, K.; Bernardo, B.C.; McMullen, J.R.; Ritchie, R.H. Diabetic cardiomyopathy: Mechanisms and new treatment strategies targeting antioxidant signaling pathways. Pharmacol. Ther. 2014, 142, 375–415. [Google Scholar] [CrossRef] [PubMed]
- Kannel, W.B.; McGee, D.L. Diabetes and cardiovascular disease. The Framingham study. JAMA 1979, 241, 2035–2038. [Google Scholar] [CrossRef] [PubMed]
- Levelt, E.; Gulsin, G.; Neubauer, S.; McCann, G.P. MECHANISMS IN ENDOCRINOLOGY: Diabetic cardiomyopathy: Pathophysiology and potential metabolic interventions state of the art review. Eur. J. Endocrinol. 2018, 178, R127–R139. [Google Scholar] [CrossRef] [Green Version]
- Kannel, W.B.; Hjortland, M.; Castelli, W.P. Role of diabetes in congestive heart failure: The Framingham study. Am. J. Cardiol. 1974, 34, 29–34. [Google Scholar] [CrossRef]
- Dunlay, S.M.; Givertz, M.M.; Aguilar, D.; Allen, L.A.; Chan, M.; Desai, A.S.; Deswal, A.; Dickson, V.V.; Kosiborod, M.N.; Lekavich, C.L.; et al. Type 2 Diabetes Mellitus and Heart Failure: A Scientific Statement from the American Heart Association and the Heart Failure Society of America: This statement does not represent an update of the 2017 ACC/AHA/HFSA heart failure guideline. Circulation 2019, 140, e294–e324. [Google Scholar] [CrossRef]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; De Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef]
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circ Res. 2018, 122, 624–638. [Google Scholar] [CrossRef]
- Miki, T.; Yuda, S.; Kouzu, H.; Miura, T. Diabetic cardiomyopathy: Pathophysiology and clinical features. Heart Fail. Rev. 2013, 18, 149–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubler, S.; Dlugash, J.; Yuceoglu, Y.Z.; Kumral, T.; Branwood, A.W.; Grishman, A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am. J. Cardiol. 1972, 30, 595–602. [Google Scholar] [CrossRef]
- Jia, G.; DeMarco, V.G.; Sowers, J.R. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat. Rev. Endocrinol. 2016, 12, 144–153. [Google Scholar] [CrossRef]
- Talukder, M.A.; Kalyanasundaram, A.; Zuo, L.; Velayutham, M.; Nishijima, Y.; Periasamy, M.; Zweier, J.L. Is reduced SERCA2a expression detrimental or beneficial to postischemic cardiac function and injury? Evidence from heterozygous SERCA2a knockout mice. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H1426–H1434. [Google Scholar] [CrossRef]
- Pollack, P.S.; Malhotra, A.; Fein, F.S.; Scheuer, J. Effects of diabetes on cardiac contractile proteins in rabbits and reversal with insulin. Am. J. Physiol. 1986, 251, H448–H454. [Google Scholar] [CrossRef]
- Malhotra, A.; Sanghi, V. Regulation of contractile proteins in diabetic heart. Cardiovasc. Res. 1997, 34, 34–40. [Google Scholar] [CrossRef] [Green Version]
- Sundgren, N.C.; Giraud, G.D.; Schultz, J.M.; Lasarev, M.R.; Stork, P.J.; Thornburg, K.L. Extracellular signal-regulated kinase and phosphoinositol-3 kinase mediate IGF-1 induced proliferation of fetal sheep cardiomyocytes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 285, R1481–R1489. [Google Scholar] [CrossRef] [Green Version]
- Levelt, E.; Mahmod, M.; Piechnik, S.K.; Ariga, R.; Francis, J.M.; Rodgers, C.T.; Clarke, W.T.; Sabharwal, N.; Schneider, J.E.; Karamitsos, T.D.; et al. Relationship Between Left Ventricular Structural and Metabolic Remodeling in Type 2 Diabetes. Diabetes 2016, 65, 44–52. [Google Scholar] [CrossRef] [Green Version]
- Finck, B.N.; Lehman, J.J.; Leone, T.C.; Welch, M.J.; Bennett, M.J.; Kovacs, A.; Han, X.; Gross, R.W.; Kozak, R.; Lopaschuk, G.D.; et al. The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J. Clin. Investig. 2002, 109, 121–130. [Google Scholar] [CrossRef]
- Wang, J.; Song, Y.; Wang, Q.; Kralik, P.M.; Epstein, P.N. Causes and Characteristics of Diabetic Cardiomyopathy. Rev. Diabet. Stud. 2006, 3, 108. [Google Scholar] [CrossRef] [Green Version]
- Van Linthout, S.; Seeland, U.; Riad, A.; Eckhardt, O.; Hohl, M.; Dhayat, N.; Richter, U.; Fischer, J.W.; Böhm, M.; Pauschinger, M.; et al. Reduced MMP-2 activity contributes to cardiac fibrosis in experimental diabetic cardiomyopathy. Basic Res. Cardiol. 2008, 103, 319–327. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, A.; Howarth, F.C.; Yanni, J.; Dobryznski, H.; Boyett, M.R.; Adeghate, E.; Bidasee, K.R.; Singh, J. Left ventricle structural remodelling in the prediabetic Goto-Kakizaki rat. Exp. Physiol. 2011, 96, 875–888. [Google Scholar] [CrossRef]
- Tokudome, T.; Horio, T.; Yoshihara, F.; Suga, S.-I.; Kawano, Y.; Kohno, M.; Kangawa, K. Direct effects of high glucose and insulin on protein synthesis in cultured cardiac myocytes and DNA and collagen synthesis in cardiac fibroblasts. Metabolism 2004, 53, 710–715. [Google Scholar] [CrossRef]
- Begum, N.; Ragolia, L. High glucose and insulin inhibit VSMC MKP-1 expression by blocking iNOS via p38 MAPK activation. Am. J. Physiol. Cell Physiol. 2000, 278, C81–C91. [Google Scholar] [CrossRef]
- Frustaci, A.; Kajstura, J.; Chimenti, C.; Jakoniuk, I.; Leri, A.; Maseri, A.; Nadal-Ginard, B.; Anversa, P. Myocardial Cell Death in Human Diabetes. Circ. Res. 2000, 87, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Fiordaliso, F.; Leri, A.; Cesselli, D.; Limana, F.; Safai, B.; Nadal-Ginard, B.; Anversa, P.; Kajstura, J. Hyperglycemia activates p53 and p53-regulated genes leading to myocyte cell death. Diabetes 2001, 50, 2363–2375. [Google Scholar] [CrossRef] [Green Version]
- Mellor, K.M.; Bell, J.R.; Young, M.J.; Ritchie, R.H.; Delbridge, L.M. Myocardial autophagy activation and suppressed survival signaling is associated with insulin resistance in fructose-fed mice. J. Mol. Cell. Cardiol. 2011, 50, 1035–1043. [Google Scholar] [CrossRef]
- Farhangkhoee, H.; Khan, Z.A.; Kaur, H.; Xin, X.; Chen, S.; Chakrabarti, S. Vascular endothelial dysfunction in diabetic cardiomyopathy: Pathogenesis and potential treatment targets. Pharmacol. Ther. 2006, 111, 384–399. [Google Scholar] [CrossRef]
- Mather, K.J.; Lteif, A.; Steinberg, H.O.; Baron, A.D. Interactions between endothelin and nitric oxide in the regulation of vascular tone in obesity and diabetes. Diabetes 2004, 53, 2060–2066. [Google Scholar] [CrossRef] [Green Version]
- Vinik, A.I.; Erbas, T.; Casellini, C.M. Diabetic cardiac autonomic neuropathy, inflammation and cardiovascular disease. J. Diabetes Investig. 2013, 4, 4–18. [Google Scholar] [CrossRef]
- Pappachan, J.M.; Sebastian, J.; Bino, B.C.; Jayaprakash, K.; Vijayakumar, K.; Sujathan, P.; Adinegara, L.A. Cardiac autonomic neuropathy in diabetes mellitus: Prevalence, risk factors and utility of corrected QT interval in the ECG for its diagnosis. Postgrad. Med. J. 2008, 84, 205–210. [Google Scholar] [CrossRef]
- Shanik, M.H.; Xu, Y.; Skrha, J.; Dankner, R.; Zick, Y.; Roth, J. Insulin resistance and hyperinsulinemia: Is hyperinsulinemia the cart or the horse? Diabetes Care 2008, 31 (Suppl. 2), S262–S268. [Google Scholar] [CrossRef] [Green Version]
- Garvey, W.T.; Maianu, L.; Huecksteadt, T.P.; Birnbaum, M.J.; Molina, J.M.; Ciaraldi, T.P. Pretranslational suppression of a glucose transporter protein causes insulin resistance in adipocytes from patients with non-insulin-dependent diabetes mellitus and obesity. J. Clin. Investig. 1991, 87, 1072–1081. [Google Scholar] [CrossRef] [Green Version]
- Robertson, R.P.; Harmon, J.; Tran, P.O.; Poitout, V. Beta-cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes. Diabetes 2004, 53 (Suppl. 1), S119–S124. [Google Scholar] [CrossRef] [Green Version]
- Yao, D.; Brownlee, M. Hyperglycemia-induced reactive oxygen species increase expression of the receptor for advanced glycation end products (RAGE) and RAGE ligands. Diabetes 2010, 59, 249–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ligeti, L.; Szenczi, O.; Prestia, C.; Szabó, C.; Horvath, K.; Marcsek, Z.; Van Stiphout, R.; Van Riel, N.; Buijs, J.D.; Van Der Vusse, G.; et al. Altered calcium handling is an early sign of streptozotocin-induced diabetic cardiomyopathy. Int. J. Mol. Med. 2006, 17, 1035–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavrentyev, E.N.; Estes, A.M.; Malik, K.U. Mechanism of High Glucose–Induced Angiotensin II Production in Rat Vascular Smooth Muscle Cells. Circ. Res. 2007, 101, 455–464. [Google Scholar] [CrossRef] [Green Version]
- Bugger, H.; Riehle, C.; Jaishy, B.; Wende, A.R.; Tuinei, J.; Chen, D.; Soto, J.; Pires, K.M.; Boudina, S.; Theobald, H.A.; et al. Genetic loss of insulin receptors worsens cardiac efficiency in diabetes. J. Mol. Cell. Cardiol. 2012, 52, 1019–1026. [Google Scholar] [CrossRef] [Green Version]
- Qi, Y.; Xu, Z.; Zhu, Q.; Thomas, C.; Kumar, R.; Feng, H.; Dostal, D.E.; White, M.F.; Baker, K.M.; Guo, S. Myocardial Loss of IRS1 and IRS2 Causes Heart Failure and Is Controlled by p38 MAPK During Insulin Resistance. Diabetes 2013, 62, 3887–3900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amato, L.; Paolisso, G.; Cacciatore, F.; Ferrara, N.; Ferrara, P.; Canonico, S.; Varricchio, M.; Rengo, F. Congestive heart failure predicts the development of non-insulin-dependent diabetes mellitus in the elderly. The Osservatorio Geriatrico Regione Campania Group. Diabetes Metab. 1997, 23, 213–218. [Google Scholar] [PubMed]
- Mazumder, P.K.; O’Neill, B.T.; Roberts, M.W.; Buchanan, J.; Yun, U.J.; Cooksey, R.C.; Boudina, S.; Abel, E.D. Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts. Diabetes 2004, 53, 2366–2374. [Google Scholar] [CrossRef] [Green Version]
- Lopaschuk, G.D. Metabolic abnormalities in the diabetic heart. Heart Fail. Rev. 2002, 7, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Taegtmeyer, H.; McNulty, P.; Young, M.E. Adaptation and maladaptation of the heart in diabetes: Part I: General concepts. Circulation 2002, 105, 1727–1733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawlak, M.; Baugé, E.; Lalloyer, F.; Lefebvre, P.; Staels, B. Ketone Body Therapy Protects from Lipotoxicity and Acute Liver Failure Upon Pparα Deficiency. Mol. Endocrinol. 2015, 29, 1134–1143. [Google Scholar] [CrossRef] [Green Version]
- Ferrannini, E.; Baldi, S.; Frascerra, S.; Astiarraga, B.; Heise, T.; Bizzotto, R.; Mari, A.; Pieber, T.R.; Muscelli. Shift to Fatty Substrate Utilization in Response to Sodium-Glucose Cotransporter 2 Inhibition in Subjects Without Diabetes and Patients with Type 2 Diabetes. Diabetes 2016, 65, 1190–1195. [Google Scholar] [CrossRef] [Green Version]
- Mudaliar, S.; Alloju, S.; Henry, R.R. Can a Shift in Fuel Energetics Explain the Beneficial Cardiorenal Outcomes in the EMPA-REG OUTCOME Study? A Unifying Hypothesis. Diabetes Care 2016, 39, 1115–1122. [Google Scholar] [CrossRef] [Green Version]
- Daniele, G.; Xiong, J.; Solis-Herrera, C.; Merovci, A.; Eldor, R.; Tripathy, D.; DeFronzo, R.A.; Norton, L.; Abdul-Ghani, M. Dapagliflozin Enhances Fat Oxidation and Ketone Production in Patients with Type 2 Diabetes. Diabetes Care 2016, 39, 2036–2041. [Google Scholar] [CrossRef] [Green Version]
- Yamagishi, S. Potential clinical utility of advanced glycation end product cross-link breakers in age- and diabetes-associated disorders. Rejuvenation Res. 2012, 15, 564–572. [Google Scholar] [CrossRef]
- Goh, S.Y.; Cooper, M.E. Clinical review: The role of advanced glycation end products in progression and complications of diabetes. J. Clin. Endocrinol. Metab. 2008, 93, 1143–1152. [Google Scholar] [CrossRef] [Green Version]
- Willemsen, S.; Hartog, J.W.; van Veldhuisen, D.J.; van der Meer, P.; Roze, J.F.; Jaarsma, T.; Schalkwijk, C.; van der Horst, I.C.; Hillege, H.L.; Voors, A.A. The role of advanced glycation end-products and their receptor on outcome in heart failure patients with preserved and reduced ejection fraction. Am. Heart J. 2012, 164, 742–749.e3. [Google Scholar] [CrossRef]
- Jia, G.; Habibi, J.; DeMarco, V.G.; Martinez-Lemus, L.A.; Ma, L.; Whaley-Connell, A.T.; Aroor, A.R.; Domeier, T.L.; Zhu, Y.; Meininger, G.A.; et al. Endothelial Mineralocorticoid Receptor Deletion Prevents Diet-Induced Cardiac Diastolic Dysfunction in Females. Hypertension 2015, 66, 1159–1167. [Google Scholar] [CrossRef] [Green Version]
- Singh, V.P.; Le, B.; Khode, R.; Baker, K.M.; Kumar, R. Intracellular angiotensin II production in diabetic rats is correlated with cardiomyocyte apoptosis, oxidative stress, and cardiac fibrosis. Diabetes 2008, 57, 3297–3306. [Google Scholar] [CrossRef] [Green Version]
- Schneider, J.G.; Tilly, N.; Hierl, T.; Sommer, U.; Hamann, A.; Dugi, K.; Leidig-Bruckner, G.; Kasperk, C. Elevated plasma endothelin-1 levels in diabetes mellitus. Am. J. Hypertens. 2002, 15, 967–972. [Google Scholar] [CrossRef] [Green Version]
- Davidson, K.W.; Burg, M.; Shimbo, D. Endothelin-1 release and stimulation of the inflammatory cascade: Is acute coronary syndrome triggered by watching spectator sports? J. Am. Coll. Cardiol. 2010, 55, 643–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widyantoro, B.; Emoto, N.; Nakayama, K.; Anggrahini, D.W.; Adiarto, S.; Iwasa, N.; Yagi, K.; Miyagawa, K.; Rikitake, Y.; Suzuki, T.; et al. Endothelial cell-derived endothelin-1 promotes cardiac fibrosis in diabetic hearts through stimulation of endothelial-to-mesenchymal transition. Circulation 2010, 121, 2407–2418. [Google Scholar] [CrossRef] [Green Version]
- Bisognano, J.D.; Weinberger, H.D.; Bohlmeyer, T.J.; Pende, A.; Raynolds, M.V.; Sastravaha, A.; Roden, R.; Asano, K.; Blaxall, B.C.; Wu, S.C.; et al. Myocardial-directed overexpression of the human β1-adrenergic receptor in transgenic mice. J. Mol. Cell. Cardiol. 2000, 32, 817–830. [Google Scholar] [CrossRef] [PubMed]
- Olshansky, B.; Sabbah, H.N.; Hauptman, P.J.; Colucci, W.S. Parasympathetic Nervous System and Heart Failure. Circulation 2008, 118, 863–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahn, J.K.; Zola, B.; Juni, J.E.; Vinik, A.I. Radionuclide assessment of left ventricular diastolic filling in diabetes mellitus with and without cardiac autonomic neuropathy. J. Am. Coll. Cardiol. 1986, 7, 1303–1309. [Google Scholar] [CrossRef] [Green Version]
- Marwick, T.H.; Ritchie, R.; Shaw, J.E.; Kaye, D. Implications of Underlying Mechanisms for the Recognition and Management of Diabetic Cardiomyopathy. J. Am. Coll. Cardiol. 2018, 71, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Bugger, H.; Abel, E.D. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia 2014, 57, 660–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seddon, M.; Looi, Y.H.; Shah, A.M. Oxidative stress and redox signalling in cardiac hypertrophy and heart failure. Heart 2007, 93, 903–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiranandani, N.; Bupha-Intr, T.; Janssen, P.M.L. SERCA overexpression reduces hydroxyl radical injury in murine myocardium. Am. J. Physiol. Circ. Physiol. 2006, 291, H3130–H3135. [Google Scholar] [CrossRef] [Green Version]
- Ho, F.M.; Lin, W.W.; Chen, B.C.; Chao, C.M.; Yang, C.R.; Lin, L.Y.; Lai, C.C.; Liu, S.H.; Liau, C.S. High glucose-induced apoptosis in human vascular endothelial cells is mediated through NF-kappaB and c-Jun NH2-terminal kinase pathway and prevented by PI3K/Akt/eNOS pathway. Cell Signal. 2006, 18, 391–399. [Google Scholar] [CrossRef]
- Murdoch, C.E.; Grieve, D.J.; Cave, A.C.; Looi, Y.H.; Shah, A.M. NADPH oxidase and heart failure. Curr. Opin. Pharmacol. 2006, 6, 148–153. [Google Scholar] [CrossRef]
- Wold, L.E.; Ceylan-Isik, A.F.; Ren, J. Oxidative stress and stress signaling: Menace of diabetic cardiomyopathy. Acta Pharmacol. Sin. 2005, 26, 908–917. [Google Scholar] [CrossRef]
- Sugamura, K.; Keaney, J.F., Jr. Reactive oxygen species in cardiovascular disease. Free Radic. Biol. Med. 2011, 51, 978–992. [Google Scholar] [CrossRef] [Green Version]
- Haffner, S.M. The metabolic syndrome: Inflammation, diabetes mellitus, and cardiovascular disease. Am. J. Cardiol. 2006, 97, 3A–11A. [Google Scholar] [CrossRef]
- Pal, P.B.; Sonowal, H.; Shukla, K.; Srivastava, S.K.; Ramana, K.V. Aldose Reductase Mediates NLRP3 Inflammasome-Initiated Innate Immune Response in Hyperglycemia-Induced Thp1 Monocytes and Male Mice. Endocrinology 2017, 158, 3661–3675. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Westermann, D.; Rutschow, S.; Van Linthout, S.; Linderer, A.; Bücker-Gärtner, C.; Sobirey, M.; Riad, A.; Pauschinger, M.; Schultheiss, H.P.; Tschöpe, C. Inhibition of p38 mitogen-activated protein kinase attenuates left ventricular dysfunction by mediating pro-inflammatory cardiac cytokine levels in a mouse model of diabetes mellitus. Diabetologia 2006, 49, 2507–2513. [Google Scholar] [CrossRef]
- Westermann, D.; Rutschow, S.; Jäger, S.; Linderer, A.; Anker, S.; Riad, A.; Unger, T.; Schultheiss, H.P.; Pauschinger, M.; Tschöpe, C. Contributions of inflammation and cardiac matrix metalloproteinase activity to cardiac failure in diabetic cardiomyopathy: The role of angiotensin type 1 receptor antagonism. Diabetes 2007, 56, 641–646. [Google Scholar] [CrossRef] [Green Version]
- Van Linthout, S.; Tschöpe, C. Inflammation—Cause or Consequence of Heart Failure or Both? Curr. Heart Fail. Rep. 2017, 14, 251–265. [Google Scholar] [CrossRef] [Green Version]
- Anderson, E.J.; Kypson, A.P.; Rodriguez, E.; Anderson, C.A.; Lehr, E.J.; Neufer, P.D. Substrate-Specific Derangements in Mitochondrial Metabolism and Redox Balance in the Atrium of the Type 2 Diabetic Human Heart. J. Am. Coll. Cardiol. 2009, 54, 1891–1898. [Google Scholar] [CrossRef] [Green Version]
- Anderson, E.J.; Rodriguez, E.; Anderson, C.A.; Thayne, K.; Chitwood, W.R.; Kypson, A.P. Increased propensity for cell death in diabetic human heart is mediated by mitochondrial-dependent pathways. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H118–H124. [Google Scholar] [CrossRef] [Green Version]
- Sena, L.A.; Chandel, N.S. Physiological Roles of Mitochondrial Reactive Oxygen Species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [Green Version]
- Kaludercic, N.; Di Lisa, F. Mitochondrial ROS Formation in the Pathogenesis of Diabetic Cardiomyopathy. Front. Cardiovasc. Med. 2020, 7, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisner, D.A.; Caldwell, J.L.; Kistamás, K.; Trafford, A.W. Calcium and Excitation-Contraction Coupling in the Heart. Circ. Res. 2017, 121, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Chen, P.; Deng, J.; Lv, J.; Liu, J. Resveratrol and polydatin as modulators of Ca2+mobilization in the cardiovascular system. Ann. N. Y. Acad. Sci. 2017, 1403, 82–91. [Google Scholar] [CrossRef]
- Kanaporis, G.; Blatter, L.A. Membrane potential determines calcium alternans through modulation of SR Ca2+ load and L-type Ca2+current. J. Mol. Cell. Cardiol. 2017, 105, 49–58. [Google Scholar] [CrossRef] [Green Version]
- Bidasee, K.R.; Zhang, Y.; Shao, C.H.; Wang, M.; Patel, K.P.; Dincer, U.D.; Besch, H.R., Jr. Diabetes increases formation of advanced glycation end products on Sarco(endo)plasmic reticulum Ca2+-ATPase. Diabetes 2004, 53, 463–473. [Google Scholar] [CrossRef] [Green Version]
- Belke, D.D.; Swanson, E.A.; Dillmann, W.H. Decreased sarcoplasmic reticulum activity and contractility in diabetic db/db mouse heart. Diabetes 2004, 53, 3201–3208. [Google Scholar] [CrossRef] [Green Version]
- Van den Bergh, A.; Vanderper, A.; Vangheluwe, P.; Desjardins, F.; Nevelsteen, I.; Verreth, W.; Wuytack, F.; Holvoet, P.; Flameng, W.; Balligand, J.L.; et al. Dyslipidaemia in type II diabetic mice does not aggravate contractile impairment but increases ventricular stiffness. Cardiovasc. Res. 2008, 77, 371–379. [Google Scholar] [CrossRef] [Green Version]
- Ye, G.; Metreveli, N.S.; Donthi, R.V.; Xia, S.; Xu, M.; Carlson, E.C.; Epstein, P.N. Catalase protects cardiomyocyte function in models of type 1 and type 2 diabetes. Diabetes 2004, 53, 1336–1343. [Google Scholar] [CrossRef] [Green Version]
- Zeidan, Q.; Hart, G.W. The intersections between O-GlcNAcylation and phosphorylation: Implications for multiple signaling pathways. J. Cell Sci. 2010, 123 Pt 1, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Hart, G.W.; Housley, M.P.; Slawson, C. Cycling of O-linked beta-N-acetylglucosamine on nucleocytoplasmic proteins. Nature 2007, 446, 1017–1022. [Google Scholar] [CrossRef] [PubMed]
- Makino, A.; Dai, A.; Han, Y.; Youssef, K.D.; Wang, W.; Donthamsetty, R.; Scott, B.T.; Wang, H.; Dillmann, W.H. O-GlcNAcase overexpression reverses coronary endothelial cell dysfunction in type 1 diabetic mice. Am. J. Physiol. Physiol. 2015, 309, C593–C599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darley-Usmar, V.M.; Ball, L.E.; Chatham, J.C. Protein O-linked β-N-acetylglucosamine: A novel effector of cardiomyocyte metabolism and function. J. Mol. Cell. Cardiol. 2012, 52, 538–549. [Google Scholar] [CrossRef] [Green Version]
- Rajamani, U.; Essop, M.F.; Essop, F. Hyperglycemia-mediated activation of the hexosamine biosynthetic pathway results in myocardial apoptosis. Am. J. Physiol. Physiol. 2010, 299, C139–C147. [Google Scholar] [CrossRef]
- Barwari, T.; Joshi, A.; Mayr, M. MicroRNAs in Cardiovascular Disease. J. Am. Coll. Cardiol. 2016, 68, 2577–2584. [Google Scholar] [CrossRef] [Green Version]
- Zampetaki, A.; Kiechl, S.; Drozdov, I.; Willeit, P.; Mayr, U.; Prokopi, M.; Mayr, A.; Weger, S.; Oberhollenzer, F.; Bonora, E.; et al. Plasma microRNA profiling reveals loss of endothelial miR-126 and other microRNAs in type 2 diabetes. Circ. Res. 2010, 107, 810–817. [Google Scholar] [CrossRef] [PubMed]
- Kuethe, F.; Sigusch, H.H.; Bornstein, S.R.; Hilbig, K.; Kamvissi, V.; Figulla, H.R. Apoptosis in Patients with Dilated Cardiomyopathy and Diabetes: A Feature of Diabetic Cardiomyopathy? Horm. Metab. Res. 2007, 39, 672–676. [Google Scholar] [CrossRef]
- Yang, L.; Zhao, D.; Ren, J.; Yang, J. Endoplasmic reticulum stress and protein quality control in diabetic cardiomyopathy. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 209–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González, N.; Moreno-Villegas, Z.; González-Bris, A.; Egido, J.; Lorenzo, Ó. Regulation of visceral and epicardial adipose tissue for preventing cardiovascular injuries associated to obesity and diabetes. Cardiovasc. Diabetol. 2017, 16, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.H.; Xie, Z. Regulation of interplay between autophagy and apoptosis in the diabetic heart: New role of AMPK. Autophagy 2013, 9, 624–625. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, K.; Fujii, H.; Takahashi, T.; Kodama, M.; Aizawa, Y.; Ohta, Y.; Ono, T.; Hasegawa, G.; Naito, M.; Nakajima, T.; et al. Constitutive Regulation of Cardiac Fatty Acid Metabolism through Peroxisome Proliferator-activated Receptor α Associated with Age-dependent Cardiac Toxicity. J. Biol. Chem. 2000, 275, 22293–22299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.T.; Grayburn, P.; Karim, A.; Shimabukuro, M.; Higa, M.; Baetens, D.; Orci, L.; Unger, R.H. Lipotoxic heart disease in obese rats: Implications for human obesity. Proc. Natl. Acad. Sci. USA 2000, 97, 1784–1789. [Google Scholar] [CrossRef] [Green Version]
- Young, M.E.; Patil, S.; Ying, J.; Depre, C.; Ahuja, H.S.; Shipley, G.L.; Stepkowski, S.M.; Davies, P.J.; Taegtmeyer, H. Uncoupling protein 3 transcription is regulated by peroxisome proliferator-activated receptor α in the adult rodent heart. FASEB J. 2001, 15, 833–845. [Google Scholar] [CrossRef]
- Burkart, E.M.; Sambandam, N.; Han, X.; Gross, R.W.; Courtois, M.; Gierasch, C.M.; Shoghi, K.; Welch, M.J.; Kelly, D.P. Nuclear receptors PPARβ/δ and PPARα direct distinct metabolic regulatory programs in the mouse heart. J. Clin. Investig. 2007, 117, 3930–3939. [Google Scholar] [CrossRef]
- Rose, B.A.; Force, T.; Wang, Y. Mitogen-Activated Protein Kinase Signaling in the Heart: Angels Versus Demons in a Heart-Breaking Tale. Physiol. Rev. 2010, 90, 1507–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marber, M.S.; Rose, B.; Wang, Y. The p38 mitogen-activated protein kinase pathway—A potential target for intervention in infarction, hypertrophy, and heart failure. J. Mol. Cell. Cardiol. 2011, 51, 485–490. [Google Scholar] [CrossRef] [Green Version]
- Strniskova, M.; Barancík, M.; Neckář, J.; Ravingerova, T. Mitogen-activated protein kinases in the acute diabetic myocardium. Mol. Cell. Biochem. 2003, 249, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Lei, S.; Li, H.; Xu, J.; Liu, Y.; Gao, X.; Wang, J.; Ng, K.F.; Lau, W.B.; Ma, X.L.; Rodrigues, B.; et al. Hyperglycemia-Induced Protein Kinase C β2 Activation Induces Diastolic Cardiac Dysfunction in Diabetic Rats by Impairing Caveolin-3 Expression and Akt/eNOS Signaling. Diabetes 2013, 62, 2318–2328. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Park, Y.; Zhang, H.; Xu, X.; Laine, G.A.; Dellsperger, K.C.; Zhang, C. Feed-forward signaling of TNF-α and NF-κB via IKK-β pathway contributes to insulin resistance and coronary arteriolar dysfunction in type 2 diabetic mice. Am. J. Physiol. Circ. Physiol. 2009, 296, H1850–H1858. [Google Scholar] [CrossRef] [PubMed]
- Barbati, S.A.; Colussi, C.; Bacci, L.; Aiello, A.; Re, A.; Stigliano, E.; Isidori, A.M.; Grassi, C.; Pontecorvi, A.; Farsetti, A.; et al. Transcription Factor CREM Mediates High Glucose Response in Cardiomyocytes and in a Male Mouse Model of Prolonged Hyperglycemia. Endocrinology 2017, 158, 2391–2405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.P.; Marlen, K.; Palma, J.F.; Schweitzer, A.; Reilly, L.; Gregoire, F.M.; Xu, G.G.; Blume, J.E.; Johnson, J.D. Overexpression of repressive cAMP response element modulators in high glucose and fatty acid-treated rat islets. A common mechanism for glucose toxicity and lipotoxicity? J. Biol. Chem. 2003, 278, 51316–51323. [Google Scholar] [CrossRef] [Green Version]
- Garcia, N.A.; Moncayo-Arlandi, J.; Sepulveda, P.; Diez-Juan, A. Cardiomyocyte exosomes regulate glycolytic flux in endothelium by direct transfer of GLUT transporters and glycolytic enzymes. Cardiovasc. Res. 2015, 109, 397–408. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Huang, W.; Liu, G.; Cai, W.; Millard, R.W.; Wang, Y.; Chang, J.; Peng, T.; Fan, G.C. Cardiomyocytes mediate anti-angiogenesis in type 2 diabetic rats through the exosomal transfer of miR-320 into endothelial cells. J. Mol. Cell. Cardiol. 2014, 74, 139–150. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.; Ichikawa, T.; Li, J.; Si, Q.; Yang, H.; Chen, X.; Goldblatt, C.S.; Meyer, C.J.; Li, X.; Cai, L.; et al. Diabetic downregulation of Nrf2 activity via ERK contributes to oxidative stress-induced insulin resistance in cardiac cells in vitro and in vivo. Diabetes 2011, 60, 625–633. [Google Scholar] [CrossRef] [Green Version]
- Lahnwong, S.; Chattipakorn, S.C.; Chattipakorn, N. Potential mechanisms responsible for cardioprotective effects of sodium–glucose co-transporter 2 inhibitors. Cardiovasc. Diabetol. 2018, 17, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdul-Ghani, M.A.; Norton, L.; DeFronzo, R.A. Role of Sodium-Glucose Cotransporter 2 (SGLT 2) Inhibitors in the Treatment of Type 2 Diabetes. Endocr. Rev. 2011, 32, 515–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahmoune, H.; Thompson, P.W.; Ward, J.M.; Smith, C.D.; Hong, G.; Brown, J. Glucose Transporters in Human Renal Proximal Tubular Cells Isolated from the Urine of Patients with Non-Insulin-Dependent Diabetes. Diabetes 2005, 54, 3427–3434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heerspink, H.J.; Perkins, B.A.; Fitchett, D.H.; Husain, M.; Cherney, D.Z. Sodium Glucose Cotransporter 2 Inhibitors in the Treatment of Diabetes Mellitus: Cardiovascular and Kidney Effects, Potential Mechanisms, and Clinical Applications. Circulation 2016, 134, 752–772. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef] [Green Version]
- Verma, S.; McMurray, J.J.V.; Cherney, D.Z.I. The Metabolodiuretic Promise of Sodium-Dependent Glucose Cotransporter 2 Inhibition: The Search for the Sweet Spot in Heart Failure. JAMA Cardiol. 2017, 2, 939–940. [Google Scholar] [CrossRef]
- Lam, C.S.P.; Chandramouli, C.; Ahooja, V.; Verma, S. SGLT-2 Inhibitors in Heart Failure: Current Management, Unmet Needs, and Therapeutic Prospects. J. Am. Hearth Assoc. 2019, 8, e013389. [Google Scholar] [CrossRef]
- Abdul-Ghani, M.; Del Prato, S.; Chilton, R.; De Fronzo, R.A. SGLT2 Inhibitors and Cardiovascular Risk: Lessons Learned from the EMPA-REG OUTCOME Study. Diabetes Care 2016, 39, 717–725. [Google Scholar] [CrossRef] [Green Version]
- Lin, B.; Koibuchi, N.; Hasegawa, Y.; Sueta, D.; Toyama, K.; Uekawa, K.; Ma, M.; Nakagawa, T.; Kusaka, H.; Kim-Mitsuyama, S. Glycemic control with empagliflozin, a novel selective SGLT2 inhibitor, ameliorates cardiovascular injury and cognitive dysfunction in obese and type 2 diabetic mice. Cardiovasc. Diabetol. 2014, 13, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Ptaszynska, A.; Hardy, E.; Johnsson, E.; Parikh, S.; List, J. Effects of dapagliflozin on cardiovascular risk factors. Postgrad. Med. 2013, 125, 181–189. [Google Scholar] [CrossRef]
- Sato, T.; Aizawa, Y.; Yuasa, S.; Kishi, S.; Fuse, K.; Fujita, S.; Ikeda, Y.; Kitazawa, H.; Takahashi, M.; Sato, M.; et al. The effect of dapagliflozin treatment on epicardial adipose tissue volume. Cardiovasc. Diabetol. 2018, 17, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrannini, E.; Muscelli, E.; Frascerra, S.; Baldi, S.; Mari, A.; Heise, T.; Broedl, U.C.; Woerle, H.J. Metabolic response to sodium-glucose cotransporter 2 inhibition in type 2 diabetic patients. J. Clin. Investig. 2014, 124, 499–508. [Google Scholar] [CrossRef] [Green Version]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Zannad, F. Effects of Sodium-Glucose Cotransporter 2 Inhibitors for the Treatment of Patients with Heart Failure. JAMA Cardiol. 2017, 2, 1025–1029. [Google Scholar] [CrossRef]
- Baartscheer, A.; Schumacher, C.A.; Wüst, R.C.; Fiolet, J.W.; Stienen, G.J.; Coronel, R.; Zuurbier, C.J. Empagliflozin decreases myocardial cytoplasmic Na+ through inhibition of the cardiac Na+/H+ exchanger in rats and rabbits. Diabetologia 2017, 60, 568–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sano, M. A new class of drugs for heart failure: SGLT2 inhibitors reduce sympathetic overactivity. J. Cardiol. 2018, 71, 471–476. [Google Scholar] [CrossRef] [Green Version]
- Ceriello, A.; Genovese, S.; Mannucci, E.; Gronda, E. Glucagon and heart in type 2 diabetes: New perspectives. Cardiovasc. Diabetol. 2016, 15, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eickhoff, M.K.; Dekkers, C.; Kramers, B.J.; Laverman, G.D.; Frimodt-Møller, M.; Jørgensen, N.R.; Faber, J.; Danser, A.; Gansevoort, R.T.; Rossing, P.; et al. Effects of Dapagliflozin on Volume Status When Added to Renin–Angiotensin System Inhibitors. J. Clin. Med. 2019, 8, 779. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zheng, Y.; Zhou, Y.; Liu, Y.; Xie, M.; Meng, W.; An, M. The expression and significance of mTORC1 in diabetic retinopathy. BMC Ophthalmol. 2020, 20, 1–7. [Google Scholar] [CrossRef]
- Xie, Z.; He, C.; Zou, M.H. AMP-activated protein kinase modulates cardiac autophagy in diabetic cardiomyopathy. Autophagy 2011, 7, 1254–1255. [Google Scholar] [CrossRef] [Green Version]
- Lorenzo-Almorós, A.; Tuñón, J.; Orejas, M.; Cortés, M.; Egido, J.; Lorenzo, Ó. Diagnostic approaches for diabetic cardiomyopathy. Cardiovasc. Diabetol. 2017, 16, 1–14. [Google Scholar] [CrossRef] [Green Version]
- McGavock, J.M.; Lingvay, I.; Zib, I.; Tillery, T.; Salas, N.; Unger, R.; Levine, B.D.; Raskin, P.; Victor, R.G.; Szczepaniak, L.S. Cardiac steatosis in diabetes mellitus: A 1H-magnetic resonance spectroscopy study. Circulation 2007, 116, 1170–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rijzewijk, L.J.; van der Meer, R.W.; Smit, J.W.; Diamant, M.; Bax, J.J.; Hammer, S.; Romijn, J.A.; de Roos, A.; Lamb, H.J. Myocardial Steatosis Is an Independent Predictor of Diastolic Dysfunction in Type 2 Diabetes Mellitus. J. Am. Coll. Cardiol. 2008, 52, 1793–1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welch, M.J.; Lewis, J.S.; Kim, J.; Sharp, T.L.; Dence, C.S.; Gropler, R.J.; Herrero, P. Assessment of myocardial metabolism in diabetic rats using small-animal PET: A feasibility study. J. Nucl. Med. 2006, 47, 689–697. [Google Scholar] [PubMed]
- Sasso, F.C.; Rambaldi, P.F.; Carbonara, O.; Nasti, R.; Torella, M.; Rotondo, A.; Torella, R.; Mansi, L. Perspectives of nuclear diagnostic imaging in diabetic cardiomyopathy. Nutr. Metab. Cardiovasc. Dis. 2010, 20, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, M.; Ishizu, T.; Seo, Y.; Yamamoto, M.; Suzuki, H.; Shimano, H.; Kawakami, Y.; Aonuma, K. Subendocardial Systolic Dysfunction in Asymptomatic Normotensive Diabetic Patients. Circ. J. 2015, 79, 1749–1755. [Google Scholar] [CrossRef] [Green Version]
- Natali, A.; Nesti, L.; Fabiani, I.; Calogero, E.; Di Bello, V. Impact of empagliflozin on subclinical left ventricular dysfunctions and on the mechanisms involved in myocardial disease progression in type 2 diabetes: Rationale and design of the EMPA-HEART trial. Cardiovasc. Diabetol. 2017, 16, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loganathan, R.; Novikova, L.; Boulatnikov, I.G.; Smirnova, I.V. Exercise-induced cardiac performance in autoimmune (Type 1) diabetes is associated with a decrease in myocardial diacylglycerol. J. Appl. Physiol. 2012, 113, 817–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Searls, Y.M.; Smirnova, I.V.; Fegley, B.R.; Stehno-Bittel, L. Exercise Attenuates Diabetes-Induced Ultrastructural Changes in Rat Cardiac Tissue. Med. Sci. Sports Exerc. 2004, 36, 1863–1870. [Google Scholar] [CrossRef] [Green Version]
- Epp, R.A.; Susser, S.E.; Morissette, M.P.; Kehler, D.S.; Jassal, D.S.; Duhamel, T.A. Exercise training prevents the development of cardiac dysfunction in the low-dose streptozotocin diabetic rats fed a high-fat diet. Can. J. Physiol. Pharmacol. 2013, 91, 80–89. [Google Scholar] [CrossRef]
- Diabetes Control and Complications Trial (DCCT)/Epidemiology of Diabetes Interventions and Complications (EDIC) Study Research Group. Intensive Diabetes Treatment and Cardiovascular Outcomes in Type 1 Diabetes: The DCCT/EDIC Study 30-Year Follow-up. Diabetes Care 2016, 39, 686–693. [Google Scholar] [CrossRef] [Green Version]
- Peter, P.S.; Malhotra, A.; Sadoshima, J. Thioredoxin Prevents Oxidative Stress Induced by Diabetes Mellitus. Circulation 2006, 114, 329. [Google Scholar]
- Turdi, S.; Li, Q.; Lopez, F.L.; Ren, J. Catalase alleviates cardiomyocyte dysfunction in diabetes: Role of Akt, Forkhead transcriptional factor and silent information regulator 2. Life Sci. 2007, 81, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, S.; Kinugawa, S.; Ide, T.; Matsusaka, H.; Inoue, N.; Ohta, Y.; Yokota, T.; Sunagawa, K.; Tsutsui, H. Overexpression of glutathione peroxidase attenuates myocardial remodeling and preserves diastolic function in diabetic heart. Am. J. Physiol. Circ. Physiol. 2006, 291, H2237–H2245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsushima, S.; Zablocki, D.; Sadoshima, J. Application of recombinant thioredoxin1 for treatment of heart disease. J. Mol. Cell. Cardiol. 2011, 51, 570–573. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.; Zheng, S.; Thongboonkerd, V.; Xu, M.; Pierce, W.M., Jr.; Klein, J.B.; Epstein, P.N. Cardiac mitochondrial damage and biogenesis in a chronic model of type 1 diabetes. Am. J. Physiol. Metab. 2004, 287, E896–E905. [Google Scholar] [CrossRef] [Green Version]
- Mortensen, S.A.; Rosenfeldt, F.; Kumar, A.; Dolliner, P.; Filipiak, K.J.; Pella, D.; Alehagen, U.; Steurer, G.; Littarru, G.P. The effect of coenzyme Q10 on morbidity and mortality in chronic heart failure: Results from Q-SYMBIO: A randomized double-blind trial. JACC Heart Fail. 2014, 2, 641–649. [Google Scholar] [CrossRef]
- Huynh, K.; Kiriazis, H.; Du, X.J.; Love, J.E.; Jandeleit-Dahm, K.A.; Forbes, J.M.; McMullen, J.R.; Ritchie, R.H. Coenzyme Q10 attenuates diastolic dysfunction, cardiomyocyte hypertrophy and cardiac fibrosis in the db/db mouse model of type 2 diabetes. Diabetologia 2012, 55, 1544–1553. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.A.; Murphy, M.P. Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann. N. Y. Acad. Sci. 2010, 1201, 96–103. [Google Scholar] [CrossRef]
- Gray, S.P.; Jha, J.C.; Kennedy, K.; van Bommel, E.; Chew, P.; Szyndralewiez, C.; Touyz, R.M.; Schmidt, H.; Cooper, M.E.; Jandeleit-Dahm, K. Combined NOX1/4 inhibition with GKT137831 in mice provides dose-dependent reno- and atheroprotection even in established micro- and macrovascular disease. Diabetologia 2017, 60, 927–937. [Google Scholar] [CrossRef] [Green Version]
- Verma, S.; McMurray, J.J.V. SGLT2 inhibitors and mechanisms of cardiovascular benefit: A state-of-the-art review. Diabetologia 2018, 61, 2108–2117. [Google Scholar] [CrossRef] [Green Version]
- Paolisso, G.; Gambardella, A.; Verza, M.; D’Amore, A.; Sgambato, S.; Varricchio, M. ACE inhibition improves insulin-sensitivity in aged insulin-resistant hypertensive patients. J. Hum. Hypertens. 1992, 6, 175–179. [Google Scholar]
- Hiller, K.-H.; Ruile, P.; Kraus, G.; Bauer, W.R.; Waller, C. Tissue ACE inhibition improves microcirculation in remote myocardium after coronary stenosis: MR imaging study in rats. Microvasc. Res. 2010, 80, 484–490. [Google Scholar] [CrossRef]
- Huynh, K.; Kiriazis, H.; Du, X.J.; Love, J.E.; Gray, S.P.; Jandeleit-Dahm, K.A.; McMullen, J.R.; Ritchie, R.H. Targeting the upregulation of reactive oxygen species subsequent to hyperglycemia prevents type 1 diabetic cardiomyopathy in mice. Free Radic. Biol. Med. 2013, 60, 307–317. [Google Scholar] [CrossRef]
- American Diabetes Association. 9. Pharmacologic Approaches to Glycemic Treatment: Standards of Medical Care in Diabetes—2021. Diabetes Care 2021, 44 (Suppl. 1), S111–S124. [Google Scholar] [CrossRef]
- Eurich, D.T.; Weir, D.L.; Majumdar, S.R.; Tsuyuki, R.T.; Johnson, J.A.; Tjosvold, L.; Vanderloo, S.E.; McAlister, F.A. Comparative Safety and Effectiveness of Metformin in Patients with Diabetes Mellitus and Heart Failure. Circ. Hearth Fail. 2013, 6, 395–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Wang, M.; Zhang, Y.; Cai, F.; Jiang, B.; Zha, W.; Yu, W. Metformin Ameliorates Diabetic Cardiomyopathy by Activating the PK2/PKR Pathway. Front. Physiol. 2020, 11, 425. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Asanuma, H.; Fujita, M.; Takahama, H.; Wakeno, M.; Ito, S.; Ogai, A.; Asakura, M.; Kim, J.; Minamino, T.; et al. Metformin prevents progression of heart failure in dogs: Role of AMP-activated protein kinase. Circulation 2009, 119, 2568–2577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzoulaki, I.; Molokhia, M.; Curcin, V.; Little, M.P.; Millett, C.J.; Ng, A.; Hughes, R.I.; Khunti, K.; Wilkins, M.R.; Majeed, A.; et al. Risk of cardiovascular disease and all cause mortality among patients with type 2 diabetes prescribed oral antidiabetes drugs: Retrospective cohort study using UK general practice research database. BMJ 2009, 339, b4731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.G.; Yoon, D.; Park, S.; Han, S.J.; Kim, D.J.; Lee, K.W.; Park, R.W.; Kim, H.J. Dipeptidyl Peptidase-4 Inhibitors and Risk of Heart Failure in Patients with Type 2 Diabetes Mellitus. Circ. Hearth Fail. 2017, 10, e003957. [Google Scholar] [CrossRef] [PubMed]
- Cooper, L.B.; Mi, X.; Mentz, R.J.; Green, J.B.; Anstrom, K.J.; Hernandez, A.F.; Curtis, L.H. Management of newly treated diabetes in Medicare beneficiaries with and without heart failure. Clin. Cardiol. 2016, 40, 38–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Home, P.D.; Pocock, S.J.; Beck-Nielsen, H.; Curtis, P.S.; Gomis, R.; Hanefeld, M.; Jones, N.P.; Komajda, M.; McMurray, J.J.; RECORD Study Team. Rosiglitazone evaluated for cardiovascular outcomes in oral agent combination therapy for type 2 diabetes (RECORD): A multicentre, randomised, open-label trial. Lancet 2009, 373, 2125–2135. [Google Scholar] [CrossRef]
- Komajda, M.; McMurray, J.J.; Beck-Nielsen, H.; Gomis, R.; Hanefeld, M.; Pocock, S.J.; Curtis, P.S.; Jones, N.P.; Home, P.D. Heart failure events with rosiglitazone in type 2 diabetes: Data from the RECORD clinical trial. Eur. Hearth J. 2010, 31, 824–831. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, A.V.; Usmani, A.; Rajamanickam, A.; Moheet, A. Thiazolidinediones and risk of heart failure in patients with or at high risk of type 2 diabetes mellitus: A meta-analysis and meta-regression analysis of placebo-controlled randomized clinical trials. Am. J. Cardiovasc. Drugs 2011, 11, 115–128. [Google Scholar] [CrossRef]
- Guan, Y.; Hao, C.; Cha, D.R.; Rao, R.; Lu, W.; Kohan, D.E.; Magnuson, M.A.; Redha, R.; Zhang, Y.; Breyer, M.D. Thiazolidinediones expand body fluid volume through PPARgamma stimulation of ENaC-mediated renal salt absorption. Nat. Med. 2005, 11, 861–866. [Google Scholar] [CrossRef]
- Nikolaidis, L.A.; Elahi, D.; Hentosz, T.; Doverspike, A.; Huerbin, R.; Zourelias, L.; Stolarski, C.; Shen, Y.T.; Shannon, R.P. Recombinant glucagon-like peptide-1 increases myocardial glucose uptake and improves left ventricular performance in conscious dogs with pacing-induced dilated cardiomyopathy. Circulation 2004, 110, 955–961. [Google Scholar] [CrossRef] [Green Version]
- Monji, A.; Mitsui, T.; Bando, Y.K.; Aoyama, M.; Shigeta, T.; Murohara, T. Glucagon-like peptide-1 receptor activation reverses cardiac remodeling via normalizing cardiac steatosis and oxidative stress in type 2 diabetes. Am. J. Physiol. Circ. Physiol. 2013, 305, H295–H304. [Google Scholar] [CrossRef]
- Nikolaidis, L.A.; Mankad, S.; Sokos, G.G.; Miske, G.; Shah, A.; Elahi, D.; Shannon, R.P. Effects of glucagon-like peptide-1 in patients with acute myocardial infarction and left ventricular dysfunction after successful reperfusion. Circulation 2004, 109, 962–965. [Google Scholar] [CrossRef] [Green Version]
- Margulies, K.B.; Hernandez, A.F.; Redfield, M.M.; Givertz, M.M.; Oliveira, G.H.; Cole, R.; Mann, D.L.; Whellan, D.J.; Kiernan, M.S.; Felker, G.M.; et al. Effects of Liraglutide on Clinical Stability Among Patients with Advanced Heart Failure and Reduced Ejection Fraction: A Randomized Clinical Trial. JAMA 2016, 316, 500–508. [Google Scholar] [CrossRef]
- Borghetti, G.; von Lewinski, D.; Eaton, D.M.; Sourij, H.; Houser, S.R.; Wallner, M. Diabetic Cardiomyopathy: Current and Future Therapies. Beyond Glycemic Control. Front. Physiol. 2018, 9, 1514. [Google Scholar] [CrossRef] [PubMed]
- Figtree, G.A.; Rådholm, K.; Barrett, T.D.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Matthews, D.R.; Shaw, W.; Neal, B. Effects of Canagliflozin on Heart Failure Outcomes Associated with Preserved and Reduced Ejection Fraction in Type 2 Diabetes Mellitus. Circulation 2019, 139, 2591–2593. [Google Scholar] [CrossRef] [Green Version]
- Grubić Rotkvić, P.; Cigrovski Berković, M.; Bulj, N.; Rotkvić, L.; Ćelap, I. Sodium-glucose cotransporter 2 inhibitors’ mechanisms of action in heart failure. World J. Diabetes 2020, 11, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Majowicz, M.P.; Gonzalez Bosc, L.V.; Albertoni Borghese, M.F.; Delgado, M.F.; Ortiz, M.C.; Sterin Speziale, N.; Vidal, N.A. Atrial natriuretic peptide and endothelin-3 target renal sodium-glucose cotransporter. Peptides 2003, 24, 1971–1976. [Google Scholar] [CrossRef] [PubMed]
- Volpe, M.; Carnovali, M.; Mastromarino, V. The natriuretic peptides system in the pathophysiology of heart failure: From molecular basis to treatment. Clin. Sci. 2015, 130, 57–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E., Jr.; Colvin, M.M.; Drazner, M.H.; Filippatos, G.S.; Fonarow, G.C.; Givertz, M.M.; et al. 2017 ACC/AHA/HFSA Focused Update of the 2013 ACCF/AHA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. Circulation 2017, 136, e137–e161. [Google Scholar] [CrossRef] [PubMed]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.; Coats, A.; Falk, V.; González-Juanatey, J.R.; Harjola, V.P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar] [CrossRef]
- Zhang, L.; Ding, W.Y.; Wang, Z.H.; Tang, M.X.; Wang, F.; Li, Y.; Zhong, M.; Zhang, Y.; Zhang, W. Early administration of trimetazidine attenuates diabetic cardiomyopathy in rats by alleviating fibrosis, reducing apoptosis and enhancing autophagy. J. Transl. Med. 2016, 14, 109. [Google Scholar] [CrossRef] [Green Version]
- Lee, L.; Campbell, R.; Scheuermann-Freestone, M.; Taylor, R.; Gunaruwan, P.; Williams, L.; Ashrafian, H.; Horowitz, J.; Fraser, A.G.; Clarke, K.; et al. Metabolic modulation with perhexiline in chronic heart failure: A randomized, controlled trial of short-term use of a novel treatment. Circulation 2005, 112, 3280–3288. [Google Scholar] [CrossRef] [Green Version]
- Liepinsh, E.; Skapare, E.; Svalbe, B.; Makrecka, M.; Cirule, H.; Dambrova, M. Anti-diabetic effects of mildronate alone or in combination with metformin in obese Zucker rats. Eur. J. Pharmacol. 2011, 658, 277–283. [Google Scholar] [CrossRef]
- Kassiotis, C.; Rajabi, M.; Taegtmeyer, H. Metabolic Reserve of the Heart: The Forgotten Link Between Contraction and Coronary Flow. Prog. Cardiovasc. Dis. 2008, 51, 74–88. [Google Scholar] [CrossRef] [Green Version]
- Ramírez, E.; Klett-Mingo, M.; Ares-Carrasco, S.; Picatoste, B.; Ferrarini, A.; Rupérez, F.J.; Caro-Vadillo, A.; Barbas, C.; Egido, J.; Tuñón, J.; et al. Eplerenone attenuated cardiac steatosis, apoptosis and diastolic dysfunction in experimental type-II diabetes. Cardiovasc. Diabetol. 2013, 12, 172. [Google Scholar] [CrossRef] [Green Version]
- Patel, V.B.; Shah, S.; Verma, S.; Oudit, G.Y. Epicardial adipose tissue as a metabolic transducer: Role in heart failure and coronary artery disease. Heart Fail. Rev. 2017, 22, 889–902. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Lau, K.; Eby, B.; Lozano, P.; He, C.; Pennington, B.; Li, H.; Rathi, S.; Dong, Y.; Tian, R.; et al. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes 2011, 60, 1770–1778. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Belke, D.; Suarez, J.; Swanson, E.; Clark, R.; Hoshijima, M.; Dillmann, W.H. Adenovirus-mediated overexpression of O-GlcNAcase improves contractile function in the diabetic heart. Circ. Res. 2005, 96, 1006–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariappan, N.; Elks, C.M.; Sriramula, S.; Guggilam, A.; Liu, Z.; Borkhsenious, O.; Francis, J. NF-kappaB-induced oxidative stress contributes to mitochondrial and cardiac dysfunction in type II diabetes. Cardiovasc. Res. 2010, 85, 473–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Wang, S.; Zhou, S.; Yan, X.; Wang, Y.; Chen, J.; Mellen, N.; Kong, M.; Gu, J.; Tan, Y.; et al. Sulforaphane prevents the development of cardiomyopathy in type 2 diabetic mice probably by reversing oxidative stress-induced inhibition of LKB1/AMPK pathway. J. Mol. Cell. Cardiol. 2014, 77, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Gu, H.; Huang, W.; Peng, J.; Li, Y.; Yang, L.; Qin, D.; Essandoh, K.; Wang, Y.; Peng, T.; et al. Hsp20-Mediated Activation of Exosome Biogenesis in Cardiomyocytes Improves Cardiac Function and Angiogenesis in Diabetic Mice. Diabetes 2016, 65, 3111–3128. [Google Scholar] [CrossRef] [Green Version]
- Katare, R.; Caporali, A.; Zentilin, L.; Avolio, E.; Sala-Newby, G.; Oikawa, A.; Cesselli, D.; Beltrami, A.P.; Giacca, M.; Emanueli, C.; et al. Intravenous Gene Therapy With PIM-1 Via a Cardiotropic Viral Vector Halts the Progression of Diabetic Cardiomyopathy Through Promotion of Prosurvival Signaling. Circ. Res. 2011, 108, 1238–1251. [Google Scholar] [CrossRef]
- Bastin, M.; Andreelli, F. The gut microbiota and diabetic cardiomyopathy in humans. Diabetes Metab. 2020, 46, 197–202. [Google Scholar] [CrossRef]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef]
- Tan, Y.; Zhang, Z.; Zheng, C.; Wintergerst, K.A.; Keller, B.B.; Cai, L. Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: Preclinical and clinical evidence. Nat. Rev. Cardiol. 2020, 17, 585–607. [Google Scholar] [CrossRef] [PubMed]
- Costantino, S.; Paneni, F.; Mitchell, K.; Mohammed, S.A.; Hussain, S.; Gkolfos, C.; Berrino, L.; Volpe, M.; Schwarzwald, C.; Lüscher, T.F.; et al. Hyperglycaemia-induced epigenetic changes drive persistent cardiac dysfunction via the adaptor p66 Shc. Int. J. Cardiol. 2018, 268, 179–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diabetes Control and Complications Trial Research Group; Nathan, D.M.; Genuth, S.; Lachin, J.; Cleary, P.; Crofford, O.; Davis, M.; Rand, L.; Siebert, C. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N. Engl. J. Med. 1993, 329, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.A.; Zhang, E.; Natarajan, R. Epigenetic mechanisms in diabetic complications and metabolic memory. Diabetologia 2015, 58, 443–455. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| PARAMETERS | CANVAS Program | DECLARE-TIMI 58 | EMPA-REG OUTCOME |
|---|---|---|---|
| Intervention | Canagliflozin/placebo | Dapagliflozin/placebo | Empagliflozin/placebo |
| Median follow-up(years) | 3.6 | 4.2 | 3.1 |
| Number of patients | 10142 | 17160 | 7020 |
| Prior cardiovascular disease/heart failure (%) | 65.6/14.4 | 40/10 | 99/10 |
| Primary outcome (3-point MACE) | 0.86 (95% CI 0.75–0.97) Noninferiority, p < 0.001 Superiority, p = 0.02 | 0.93 (95%CI 0.84–1.03) Noninferiority, p < 0.001 Superiority, p = 0.17 | 0.86 (95% CI 0.74–0.99) Noninferiority, p < 0.001 Superiority, p = 0.04 |
| Cardiovascular death | 0.87 (0.72–1.06) | 0.98 (0.81–1.17) | 0.62 (0.49–0.77) * |
| Myocardial infarction | 0.89 (0.73–1.09) | 0.89 (0.77–1.01) | 0.87 (0.70–1.09) |
| Stroke | 0.87 (0.69–1.09) | 1.01 (0.84–1.21) | 1.18 (0.89–1.56) |
| Heart failure hospitalization | 0.67 (0.52–0.87) * | 0.73 (0.61–0.88) * | 0.65 (0.50–0.85) * |
| All-cause mortality | 0.87 (0.74–1.01) | 0.93 (0.82–1.04) | 0.68 (0.57–0.82) * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grubić Rotkvić, P.; Planinić, Z.; Liberati Pršo, A.-M.; Šikić, J.; Galić, E.; Rotkvić, L. The Mystery of Diabetic Cardiomyopathy: From Early Concepts and Underlying Mechanisms to Novel Therapeutic Possibilities. Int. J. Mol. Sci. 2021, 22, 5973. https://doi.org/10.3390/ijms22115973
Grubić Rotkvić P, Planinić Z, Liberati Pršo A-M, Šikić J, Galić E, Rotkvić L. The Mystery of Diabetic Cardiomyopathy: From Early Concepts and Underlying Mechanisms to Novel Therapeutic Possibilities. International Journal of Molecular Sciences. 2021; 22(11):5973. https://doi.org/10.3390/ijms22115973
Chicago/Turabian StyleGrubić Rotkvić, Petra, Zrinka Planinić, Ana-Marija Liberati Pršo, Jozica Šikić, Edvard Galić, and Luka Rotkvić. 2021. "The Mystery of Diabetic Cardiomyopathy: From Early Concepts and Underlying Mechanisms to Novel Therapeutic Possibilities" International Journal of Molecular Sciences 22, no. 11: 5973. https://doi.org/10.3390/ijms22115973