
Co-Incubation with PPARβ/δ Agonists and Antagonists Modeled Using Computational Chemistry: Effect on LPS Induced Inflammatory Markers in Pulmonary Artery



Abstract
:1. Introduction
2. Results
2.1. PPARβ/δ Expression and Basal NO Production Over Time in Pulmonary Artery
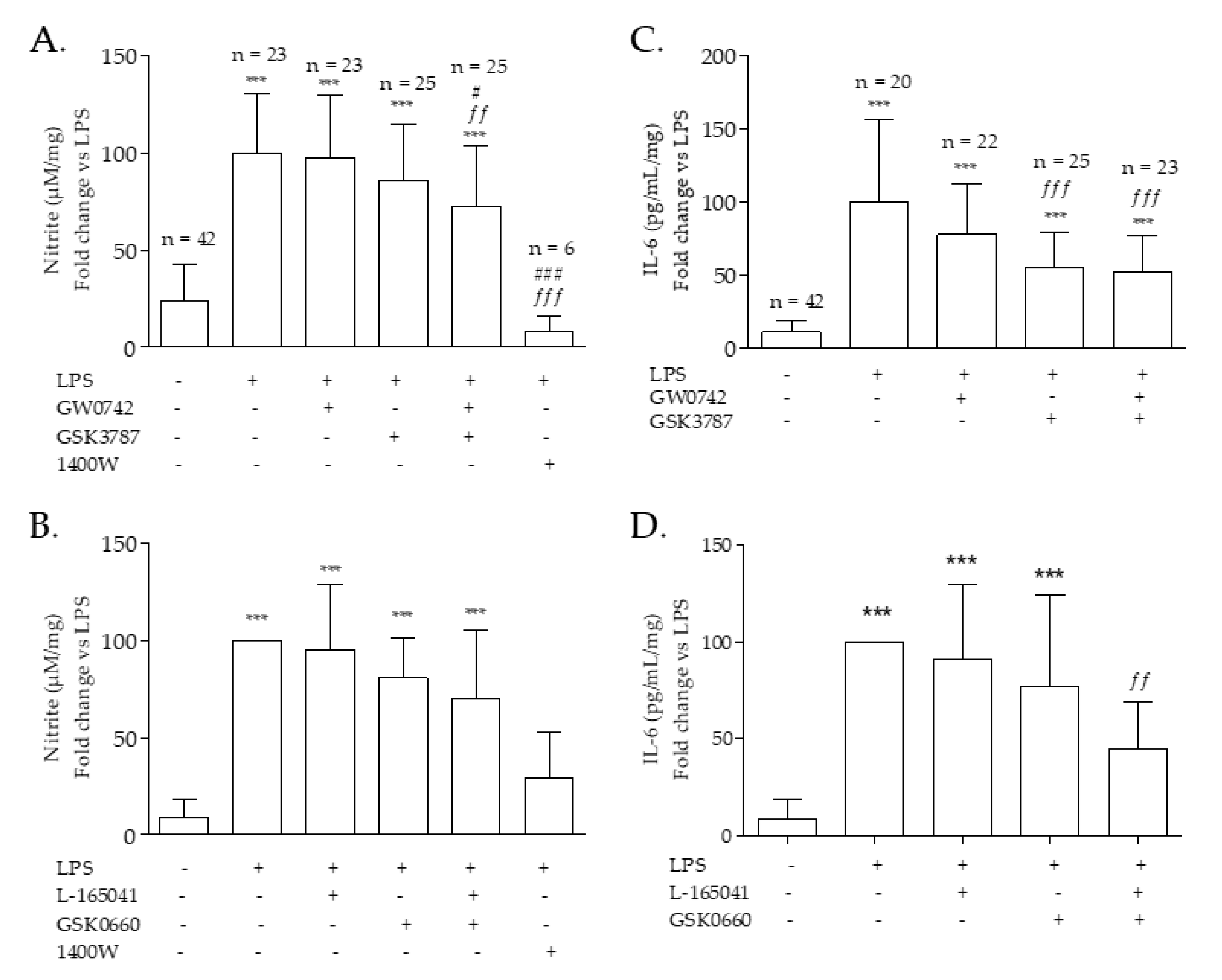
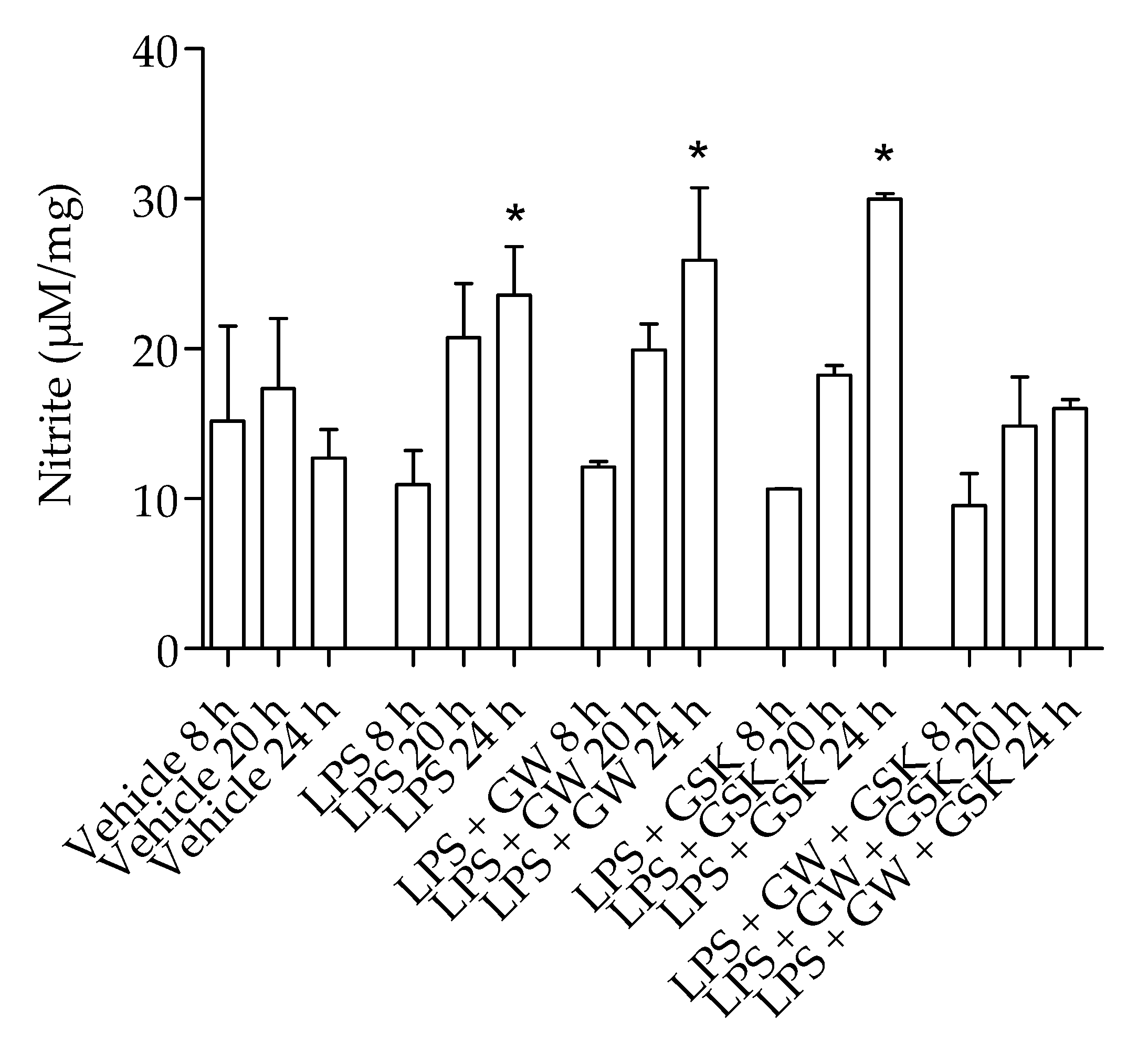
2.2. PPARβ/δ Regulation of LPS-Induced Inflammation
2.3. Marker Genes for PPARβ/δ Induction and Transrepression in Pulmonary Artery
2.4. Computational Chemistry: PPARβ/δ Docking Analysis
2.4.1. Docking of One PPARβ/δ Ligand
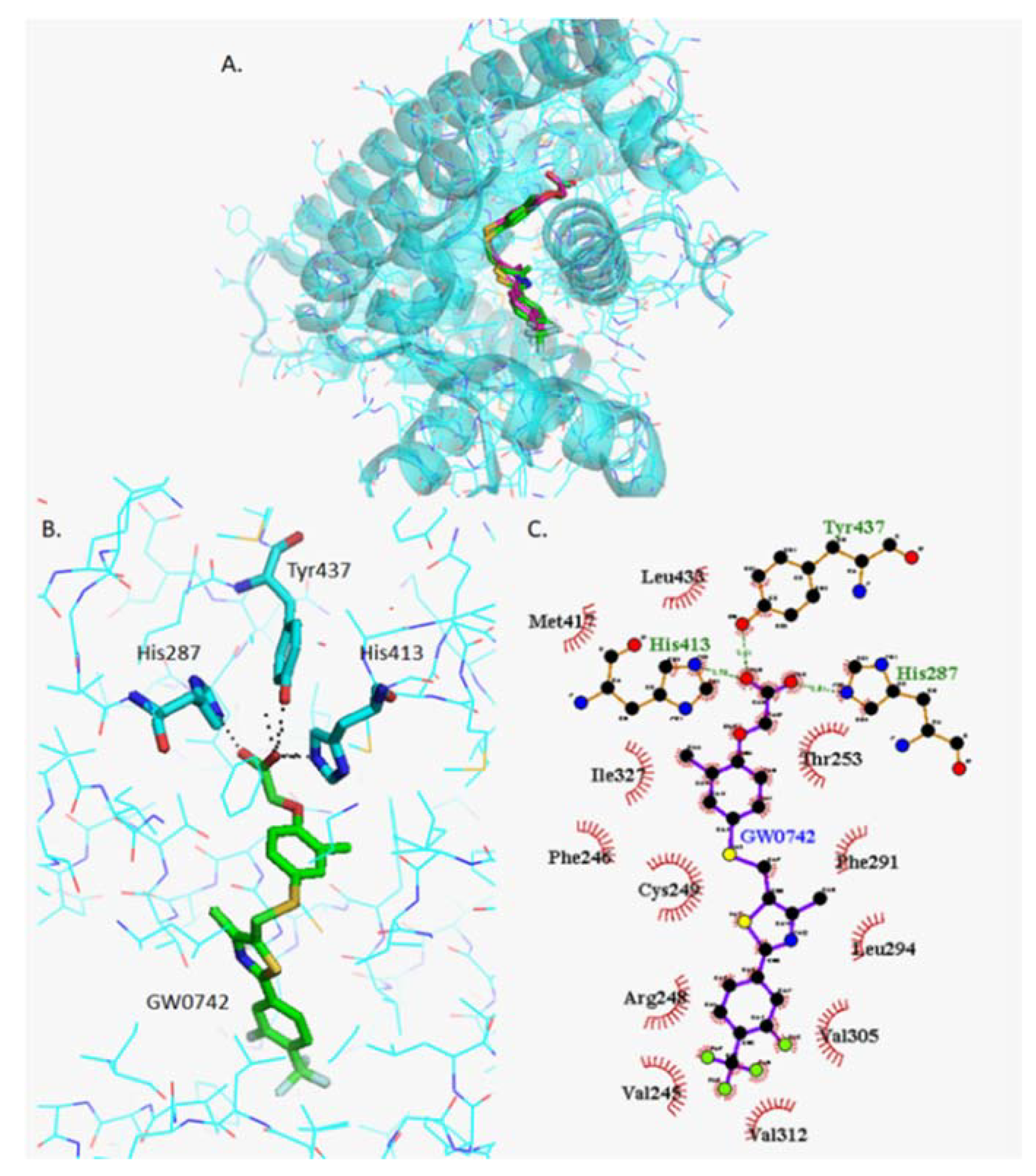
2.4.2. Docking of GW0742
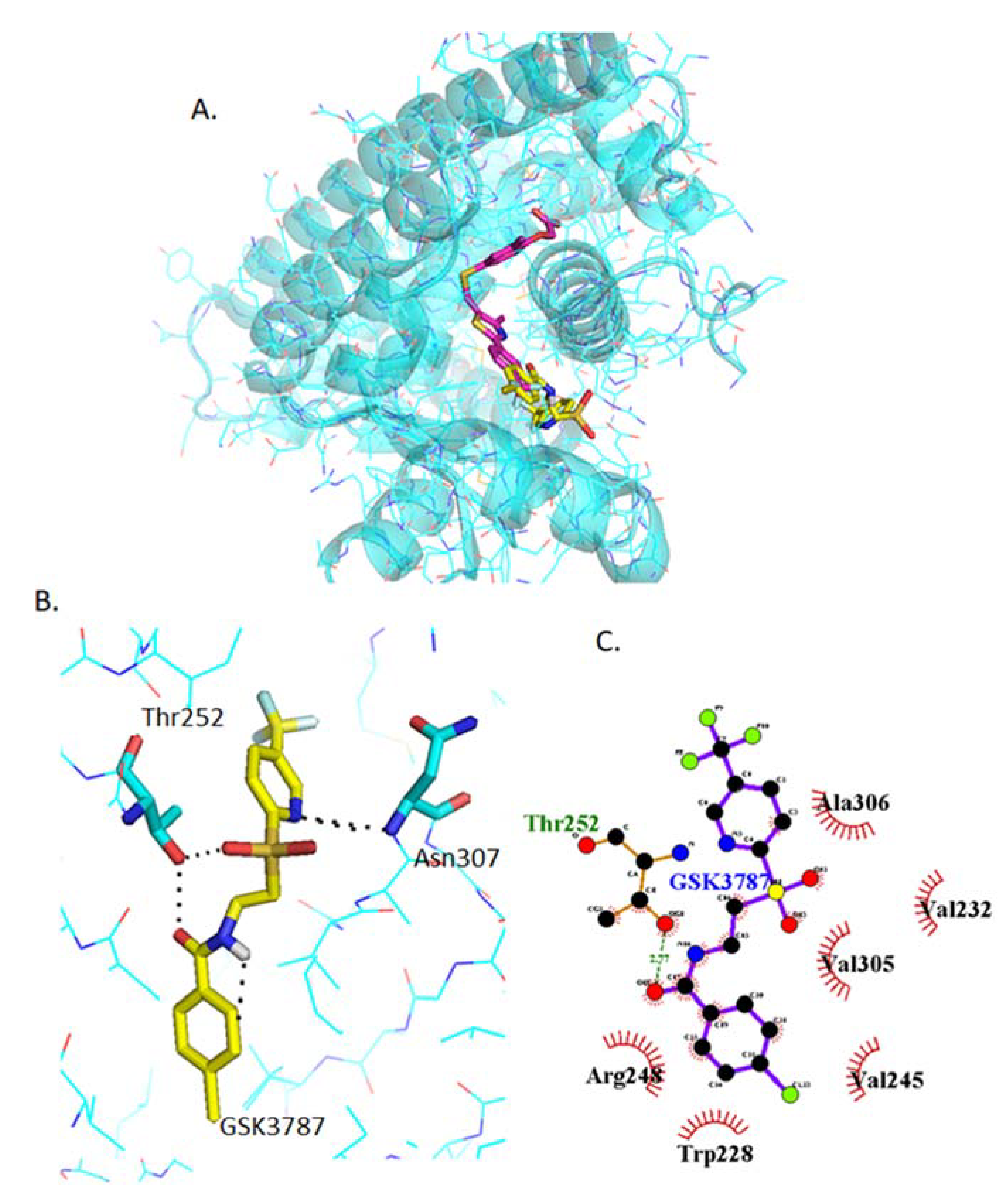
2.4.3. Docking of GSK3787
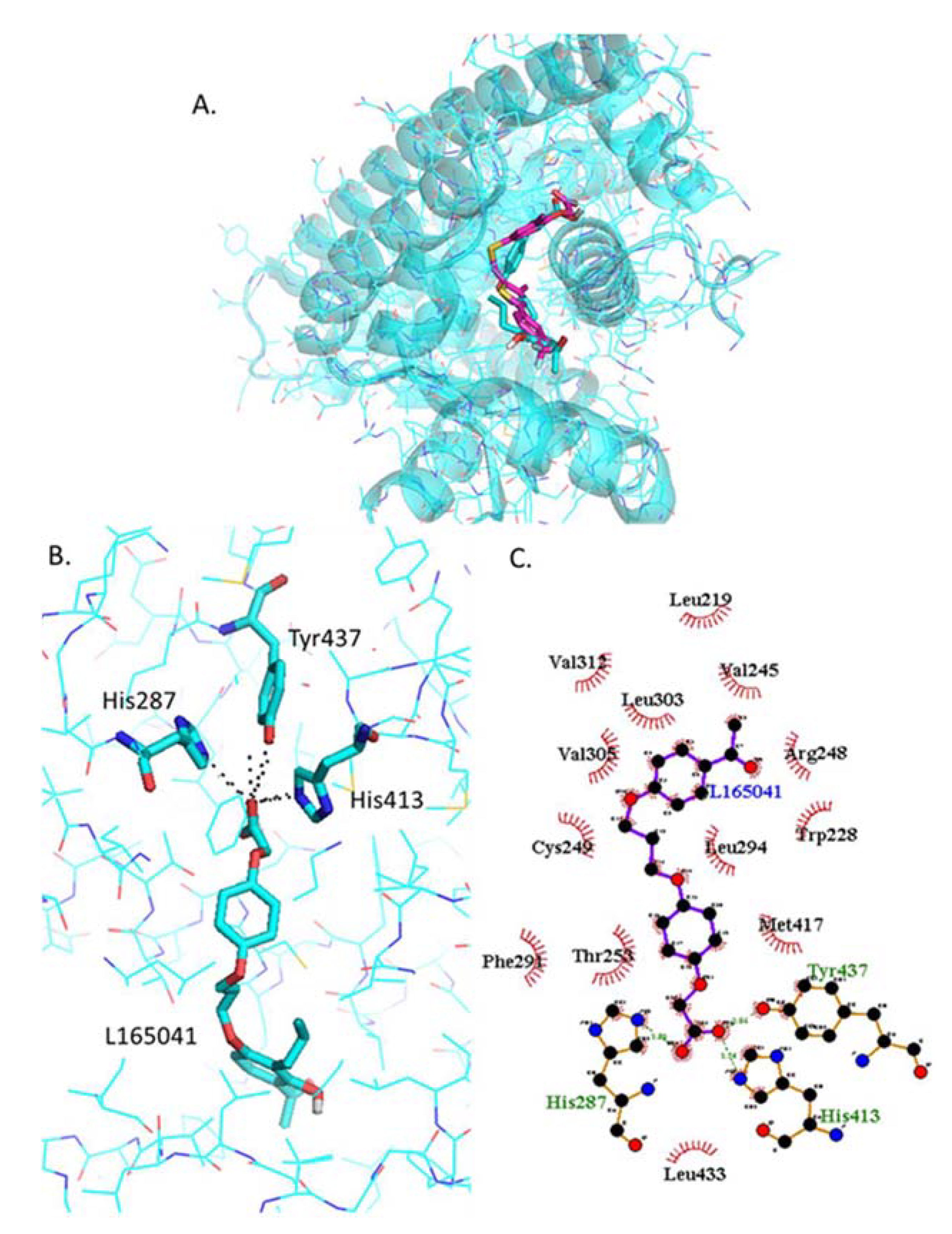
2.4.4. Docking of L−165042
2.4.5. Docking of GSK0660
2.4.6. Docking of Two PPARβ/δ Ligands Simultaneously
2.4.7. Analysis of GW0742 and GSK3787 Docked into GW0742-Bound PPARβ/δ
2.4.8. Analysis of GW0742 and GSK3787 Docked into GSK3787-Bound PPARβ/δ
2.4.9. Analysis of L−165041 and GSK0660 Docked into L−165041-Bound PPARβ/δ
2.4.10. Analysis of L−165041 and GSK0660 Docked into GSK0660-Bound PPARβ/δ
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Reagents
5.2. Animals
5.3. Quantification of PPARβ/δ Expression in Lung Tissues by Enzyme-Linked Immunosorbent Assay (ELISA)
5.4. Quantification of Nitric Oxide Released by Lung Tissues by the Griess Assay
5.5. Quantification of IL−6 Released by Pulmonary Artery by ELISA
5.6. Quantitative Real Time-Polymerase Chain Reaction (qRT-PCR)
5.7. Docking
5.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

References
- Issemann, I.; Green, S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 1990, 347, 645–650. [Google Scholar]
- Mandard, S.; Patsouris, D. Nuclear control of the inflammatory response in mammals by peroxisome proliferator-activated receptors. PPAR Res. 2013, 2013, 613864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliver, W.R., Jr.; Shenk, J.L.; Snaith, M.R.; Russell, C.S.; Plunket, K.D.; Bodkin, N.L.; Lewis, M.C.; Winegar, D.A.; Sznaidman, M.L.; Lambert, M.H.; et al. A selective peroxisome proliferator-activated receptor delta agonist promotes reverse cholesterol transport. Proc. Natl. Acad. Sci. USA 2001, 98, 5306–5311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.J.; Kim, M.Y.; Hwang, J.S.; Kim, H.J.; Lee, J.H.; Chang, K.C.; Kim, J.H.; Han, C.W.; Kim, J.H.; Seo, H.G. PPARdelta inhibits IL-1beta-stimulated proliferation and migration of vascular smooth muscle cells via up-regulation of IL-1Ra. Cell. Mol. Life Sci. CMLS 2010, 67, 2119–2130. [Google Scholar] [CrossRef] [PubMed]
- Quintela, A.M.; Jimenez, R.; Piqueras, L.; Gomez-Guzman, M.; Haro, J.; Zarzuelo, M.J.; Cogolludo, A.; Sanz, M.J.; Toral, M.; Romero, M.; et al. PPARbeta activation restores the high glucose-induced impairment of insulin signalling in endothelial cells. BJP 2014, 171, 3089–3102. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Yamamoto, J.; Iwasaki, S.; Asaba, H.; Hamura, H.; Ikeda, Y.; Watanabe, M.; Magoori, K.; Ioka, R.X.; Tachibana, K.; et al. Activation of peroxisome proliferator-activated receptor δ induces fatty acid β-oxidation in skeletal muscle and attenuates metabolic syndrome. Proc. Natl. Acad. Sci. USA 2003, 100, 15924–15929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.-X.; Lee, C.-H.; Tiep, S.; Yu, R.T.; Ham, J.; Kang, H.; Evans, R.M. Peroxisome-proliferator-activated receptor δ activates fat metabolism to prevent obesity. Cell 2003, 113, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Ooi, E.M.; Watts, G.F.; Sprecher, D.L.; Chan, D.C.; Barrett, P.H. Mechanism of action of a peroxisome proliferator-activated receptor (PPAR)-delta agonist on lipoprotein metabolism in dyslipidemic subjects with central obesity. J. Clin. Endocrinol. Metab. 2011, 96, E1568–E1576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, E.J.; Pearce, G.L.; Jones, N.P.; Sprecher, D.L. Lipid effects of peroxisome proliferator-activated receptor-delta agonist GW501516 in subjects with low high-density lipoprotein cholesterol: Characteristics of metabolic syndrome. ATVB 2012, 32, 2289–2294. [Google Scholar] [CrossRef] [Green Version]
- Geiger, L.N.; Dunsford, W.S.; Lewis, D.J.; Brennan, C.; Liu, K.C.; Newsholme, S.J. Rat carcinogenicity study with GW501516, a PPAR delta agonist. Toxicol. Sci. Off. J. Soc. Toxicol. 2009, 108, 895. [Google Scholar]
- Pollock, C.B.; Yin, Y.; Yuan, H.; Zeng, X.; King, S.; Li, X.; Kopelovich, L.; Albanese, C.; Glazer, R.I. PPARdelta activation acts cooperatively with 3-phosphoinositide-dependent protein kinase-1 to enhance mammary tumorigenesis. PLoS ONE 2011, 6, e16215. [Google Scholar] [CrossRef]
- Jones, D.; Boudes, P.F.; Swain, M.G.; Bowlus, C.L.; Galambos, M.R.; Bacon, B.R.; Doerffel, Y.; Gitlin, N.; Gordon, S.C.; Odin, J.A.; et al. Seladelpar (MBX-8025), a selective PPAR-δ agonist, in patients with primary biliary cholangitis with an inadequate response to ursodeoxycholic acid: A double-blind, randomised, placebo-controlled, phase 2, proof-of-concept study. Lancet Gastroenterol. Hepatol. 2017, 2, 716–726. [Google Scholar] [CrossRef] [Green Version]
- Yu, K.; Bayona, W.; Kallen, C.B.; Harding, H.P.; Ravera, C.P.; McMahon, G.; Brown, M.; Lazar, M.A. Differential activation of peroxisome proliferator-activated receptors by eicosanoids. J. Biol. Chem. 1995, 270, 23975–23983. [Google Scholar] [CrossRef] [Green Version]
- Forman, B.M.; Chen, J.; Evans, R.M. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors alpha and delta. Proc. Natl. Acad. Sci. USA 1997, 94, 4312–4317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moraes, L.A.; Piqueras, L.; Bishop-Bailey, D. Peroxisome proliferator-activated receptors and inflammation. Pharmacol. Ther. 2006, 110, 371–385. [Google Scholar] [CrossRef] [PubMed]
- Nandhikonda, P.; Yasgar, A.; Baranowski, A.M.; Sidhu, P.S.; McCallum, M.M.; Pawlak, A.J.; Teske, K.; Feleke, B.; Yuan, N.Y.; Kevin, C.; et al. Peroxisome proliferation-activated receptor delta agonist GW0742 interacts weakly with multiple nuclear receptors, including the vitamin D receptor. Biochemistry 2013, 52, 4193–4203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, L.; Busch, D.; Qiao, Z.; van Griensven, M.; Teuben, M.; Hildebrand, F.; Pape, H.C.; Pfeifer, R. Dose-dependent effects of peroxisome proliferator-activated receptors beta/delta agonist on systemic inflammation after haemorrhagic shock. Cytokine 2018, 103, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Neels, J.G.; Grimaldi, P.A. Physiological functions of peroxisome proliferator-activated receptor beta. Physiol. Rev. 2014, 94, 795–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cronet, P.; Petersen, J.F.W.; Folmer, R.; Blomberg, N.; Sjöblom, K.; Karlsson, U.; Lindstedt, E.-L.; Bamberg, K. Structure of the PPARα and -γ ligand binding domain in complex with AZ 242; ligand selectivity and agonist activation in the PPAR family. Structure 2001, 9, 699–706. [Google Scholar] [CrossRef] [Green Version]
- Helsen, C.; Claessens, F. Looking at nuclear receptors from a new angle. Mol. Cell. Endocrinol. 2014, 382, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Calvo, R.; Serrano, L.; Coll, T.; Moullan, N.; Sanchez, R.M.; Merlos, M.; Palomer, X.; Laguna, J.C.; Michalik, L.; Wahli, W.; et al. Activation of peroxisome proliferator-activated receptor beta/delta inhibits lipopolysaccharide-induced cytokine production in adipocytes by lowering nuclear factor-kappaB activity via extracellular signal-related kinase 1/2. Diabetes 2008, 57, 2149–2157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnegg, C.I.; Kooshki, M.; Hsu, F.-C.; Sui, G.; Robbins, M.E. PPARδ prevents radiation-induced proinflammatory responses in microglia via transrepression of NF-κB and inhibition of the PKCα/MEK1/2/ERK1/2/AP-1 pathway. Free Radic. Biol. Med. 2012, 52, 1734–1743. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.; Wang, Y.; Tang, Z.; Zhang, H.; Qin, X.; Zhu, Y.; Guan, Y.; Wang, X.; Staels, B.; Chien, S.; et al. Suppression of pro-inflammatory adhesion molecules by PPAR-delta in human vascular endothelial cells. ATVB 2008, 28, 315–321. [Google Scholar] [CrossRef]
- Di Paola, R.; Crisafulli, C.; Mazzon, E.; Esposito, E.; Paterniti, I.; Galuppo, M.; Genovese, T.; Thiemermann, C.; Cuzzocrea, S. GW0742, a high-affinity PPAR-beta/delta agonist, inhibits acute lung injury in mice. Shock 2010, 33, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Fu, L.; Ning, W.; Guo, L.; Sun, X.; Dey, S.K.; Chaturvedi, R.; Wilson, K.T.; DuBois, R.N. Peroxisome proliferator-activated receptor delta promotes colonic inflammation and tumor growth. Proc. Natl. Acad. Sci. USA 2014, 111, 7084–7089. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Fu, M.; Zhu, X.; Xiao, Y.; Mou, Y.; Zheng, H.; Akinbami, M.A.; Wang, Q.; Chen, Y.E. Peroxisome proliferator-activated receptor delta is up-regulated during vascular lesion formation and promotes post-confluent cell proliferation in vascular smooth muscle cells. JBC 2002, 277, 11505–11512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Wang, H.; Guo, Y.; Ning, W.; Katkuri, S.; Wahli, W.; Desvergne, B.; Dey, S.K.; DuBois, R.N. Crosstalk between peroxisome proliferator-activated receptor delta and VEGF stimulates cancer progression. Proc. Natl. Acad. Sci. USA 2006, 103, 19069–19074. [Google Scholar] [CrossRef] [Green Version]
- Lim, H.J.; Lee, S.; Park, J.H.; Lee, K.S.; Choi, H.E.; Chung, K.S.; Lee, H.H.; Park, H.Y. PPAR delta agonist L-165041 inhibits rat vascular smooth muscle cell proliferation and migration via inhibition of cell cycle. Atherosclerosis 2009, 202, 446–454. [Google Scholar] [CrossRef]
- Ham, S.A.; Yoo, T.; Hwang, J.S.; Kang, E.S.; Lee, W.J.; Paek, K.S.; Park, C.; Kim, J.H.; Do, J.T.; Lim, D.S.; et al. Ligand-activated PPARdelta modulates the migration and invasion of melanoma cells by regulating Snail expression. Am. J. Cancer Res. 2014, 4, 674–682. [Google Scholar]
- Bishop-Bailey, D.; Hla, T. Endothelial cell apoptosis induced by the peroxisome proliferator-activated receptor (PPAR) ligand 15-deoxy-Delta12, 14-prostaglandin J2. JBC 1999, 274, 17042–17048. [Google Scholar] [CrossRef] [Green Version]
- Haskova, Z.; Hoang, B.; Luo, G.; Morgan, L.A.; Billin, A.N.; Barone, F.C.; Shearer, B.G.; Barton, M.E.; Kilgore, K.S. Modulation of LPS-induced pulmonary neutrophil infiltration and cytokine production by the selective PPARbeta/delta ligand GW0742. Inflamm. Res. Off. J. Eur. Histamine Res. Soc. 2008, 57, 314–321. [Google Scholar]
- Bao, X.C.; Fang, Y.Q.; You, P.; Zhang, S.; Ma, J. Protective role of peroxisome proliferator-activated receptor beta/delta in acute lung injury induced by prolonged hyperbaric hyperoxia in rats. Respir. Physiol. Neurobiol. 2014, 199, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Shintani, Y.; Collino, M.; Osuchowski, M.F.; Busch, D.; Patel, N.S.; Sepodes, B.; Castiglia, S.; Fantozzi, R.; Bishop-Bailey, D.; et al. Protective role of peroxisome proliferator-activated receptor-beta/delta in septic shock. Am. J. Respir. Crit. Care Med. 2010, 182, 1506–1515. [Google Scholar] [CrossRef]
- Khozoie, C.; Borland, M.G.; Zhu, B.; Baek, S.; John, S.; Hager, G.L.; Shah, Y.M.; Gonzalez, F.J.; Peters, J.M. Analysis of the peroxisome proliferator-activated receptor-beta/delta (PPARbeta/delta) cistrome reveals novel co-regulatory role of ATF4. BMC Genom. 2012, 13, 665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adhikary, T.; Kaddatz, K.; Finkernagel, F.; Schonbauer, A.; Meissner, W.; Scharfe, M.; Jarek, M.; Blocker, H.; Muller-Brusselbach, S.; Muller, R. Genomewide analyses define different modes of transcriptional regulation by peroxisome proliferator-activated receptor-beta/delta (PPARbeta/delta). PLoS ONE 2011, 6, e16344. [Google Scholar] [CrossRef] [PubMed]
- Di Paola, R.; Briguglio, F.; Paterniti, I.; Mazzon, E.; Oteri, G.; Militi, D.; Cordasco, G.; Cuzzocrea, S. Emerging role of PPAR-beta/delta in inflammatory process associated to experimental periodontitis. Mediat. Inflamm. 2011, 2011, 787159. [Google Scholar] [CrossRef] [PubMed]
- Bojic, L.A.; Burke, A.C.; Chhoker, S.S.; Telford, D.E.; Sutherland, B.G.; Edwards, J.Y.; Sawyez, C.G.; Tirona, R.G.; Yin, H.; Pickering, J.G.; et al. Peroxisome proliferator-activated receptor delta agonist GW1516 attenuates diet-induced aortic inflammation, insulin resistance, and atherosclerosis in low-density lipoprotein receptor knockout mice. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 52–60. [Google Scholar] [CrossRef] [Green Version]
- Perez-Diaz, N.; Zloh, M.; Patel, P.; Mackenzie, L.S. In silico modelling of prostacyclin and other lipid mediators to nuclear receptors reveal novel thyroid hormone receptor antagonist properties. Prostaglandins Other Lipid Mediat. 2016, 122, 18–27. [Google Scholar] [CrossRef]
- Jin, L.; Lin, S.; Rong, H.; Zheng, S.; Jin, S.; Wang, R.; Li, Y. Structural basis for iloprost as a dual peroxisome proliferator-activated receptor alpha/delta agonist. JBC 2011, 286, 31473–31479. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.E.; Lambert, M.H.; Montana, V.G.; Parks, D.J.; Blanchard, S.G.; Brown, P.J.; Sternbach, D.D.; Lehmann, J.M.; Wisely, G.B.; Willson, T.M.; et al. Molecular recognition of fatty acids by peroxisome proliferator-activated receptors. Mol. Cell 1999, 3, 397–403. [Google Scholar] [CrossRef]
- Wu, C.C.; Baiga, T.J.; Downes, M.; La Clair, J.J.; Atkins, A.R.; Richard, S.B.; Fan, W.; Stockley-Noel, T.A.; Bowman, M.E.; Noel, J.P.; et al. Structural basis for specific ligation of the peroxisome proliferator-activated receptor delta. Proc. Natl. Acad. Sci. USA 2017, 114, E2563–E2570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maltarollo, V.G.; Togashi, M.; Nascimento, A.S.; Honorio, K.M. Structure-based virtual screening and discovery of new PPARdelta/gamma dual agonist and PPARdelta and gamma agonists. PLoS ONE 2015, 10, e0118790. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, L.S.; Lymn, J.S.; Hughes, A.D. Linking phospholipase C isoforms with differentiation function in human vascular smooth muscle cells. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2013, 1833, 3006–3012. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
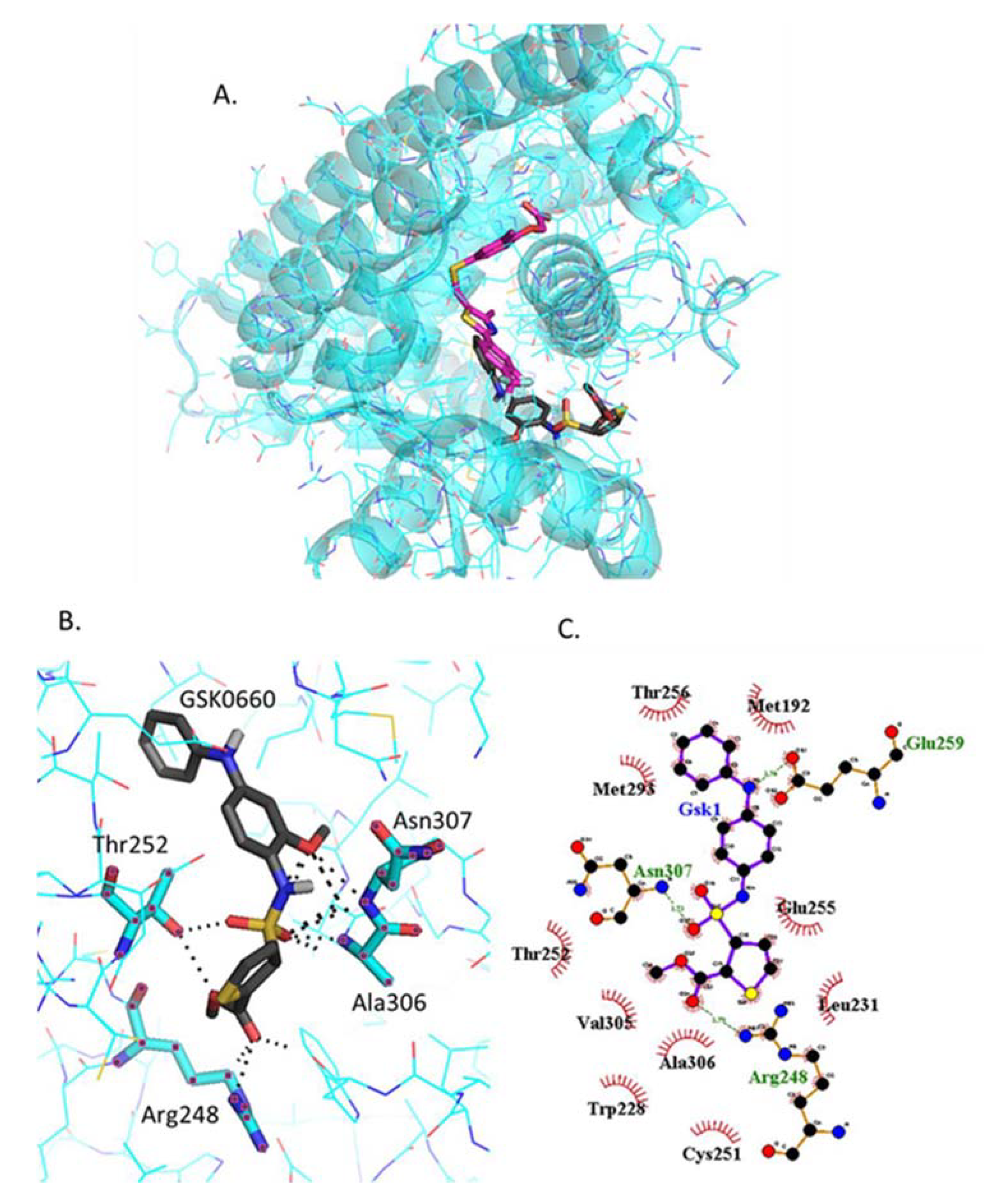{kind=link}
{kind=link}
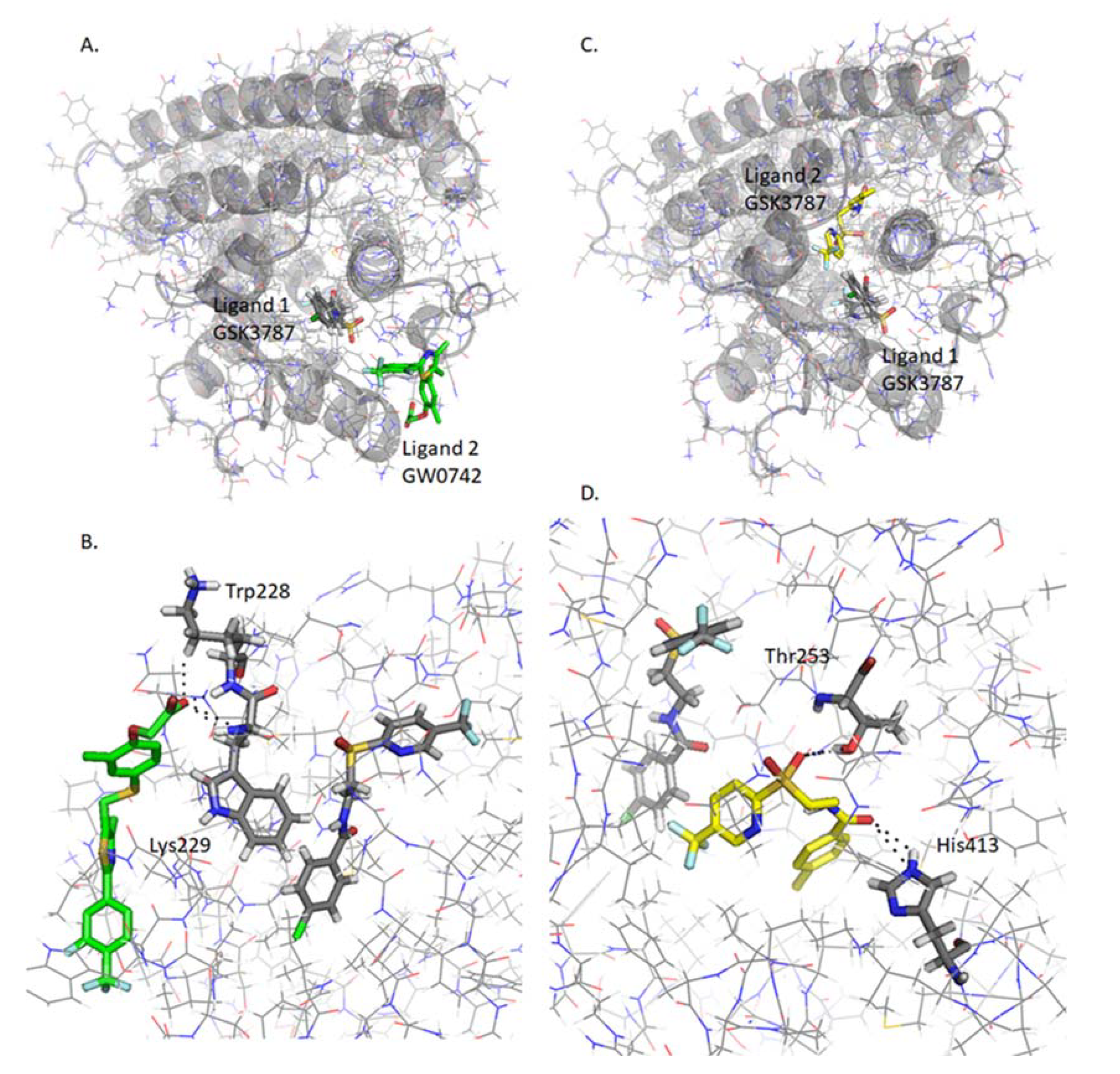{kind=link}
{kind=link}
| Best Fit | Agonists | Antagonists | ||||||
|---|---|---|---|---|---|---|---|---|
| GW0742 | L-165041 | GSK3787 | GSK0660 | |||||
| Affinity (Kcal/mol) | Aa with Polar Interactions | Affinity (Kcal/mol) | Aa with Polar Interactions | Affinity (Kcal/mol) | Aa with Polar Interactions | Affinity (Kcal/mol) | Aa with Polar Interactions | |
| 1 | −11.1 | His287 His413 Tyr437 | −8.7 | His287 His413 Tyr437 | −9.1 | Thr252 Asn307 | −8.6 | Arg248 Thr252 Ala306 Asn307 |
| 2 | −10.8 | Thr253 His287 His413 Tyr437 | −8 | Met192 Thr252 Thr256 Ile290 Ala306 | −8.9 | Thr252 | −8.3 | Arg248 Thr252 Ala306 |
| 3 | −9.9 | Thr253 His413 | −7.6 | Thr252 Arg258 Glu259 | −8.6 | Thr252 | −8.1 | Thr256 Asn307 |
| 4 | −9.6 | Thr256 | −7.5 | Thr252 Thr253 Ala306 | −8.5 | Thr256 | −8.1 | Asn307 |
| 5 | −9.3 | No bonds | −7.5 | Trp228 Thr252 Thr256 Ile290 | −8.4 | No bonds | −7.9 | Thr256 Ala306 Asn307 |
| 6 | −8.8 | Thr253 | −7.1 | Thr252 Thr256 Ala306 | −8.3 | Thr252 Thr253 | −7.6 | Asn307 |
| 7 | −8.7 | Thr252 | −6.9 | Tyr284 Arg361 | −8.3 | Thr252 | −7.5 | Ala306 Asn307 |
| 8 | −8.4 | Arg258 | −6.8 | Glu255 Asn307 | −8.2 | Met192 | −7.4 | Thr256 Asn307 |
| Ligand 1 | Ligand 2 | ||||
|---|---|---|---|---|---|
| Molecule | Affinity (Kcal/mol) | Amino Acid with Polar Interactions | Molecule | Affinity (Kcal/mol) | Amino Acid with Polar Interactions |
| GW0742 | −11.1 | His287 His413 Tyr437 | GW0742 | −8.5 | Arg258 |
| GSK3787 | −7.7 | Trp228 Thr252 | |||
| GSK3787 | −9.1 | Thr252 Asn307 | GW0742 | −8.1 | Trp228 Lys229 |
| GSK3787 | −7.4 | Thr253 His413 | |||
| L−165041 | −8.7 | His287 His413 Tyr437 | L-165041 | −8.3 | Met192 Cys251 Thr252 Thr256 Ile290 Ala306 |
| GSK0660 | −6.5 | Arg198 Asn339 | |||
| GSK0660 | −8.6 | Arg248 Thr252 Ala306 Asn307 | L-165041 | −8.1 | Thr253 His413 |
| GSK0660 | −8.9 | Thr252 Thr253 | |||
| Combination 1 | Combination 2 | |
|---|---|---|
| Vehicle | 0.01% DMSO | 0.01% DMSO |
| LPS | LPS 1 μg/mL | LPS 1 μg/mL |
| LPS + agonist | LPS + GW0742 | LPS + L-165041 |
| LPS + antagonist | LPS + GSK3787 | LPS + GSK0660 |
| LPS + agonist + antagonist | LPS + GW0742 + GSK3787 | LPS + L-165041 + GSK0660 |
| LPS + 1400 W | LPS + 1400 W | LPS + 1400 W |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perez Diaz, N.; Lione, L.A.; Hutter, V.; Mackenzie, L.S. Co-Incubation with PPARβ/δ Agonists and Antagonists Modeled Using Computational Chemistry: Effect on LPS Induced Inflammatory Markers in Pulmonary Artery. Int. J. Mol. Sci. 2021, 22, 3158. https://doi.org/10.3390/ijms22063158
Perez Diaz N, Lione LA, Hutter V, Mackenzie LS. Co-Incubation with PPARβ/δ Agonists and Antagonists Modeled Using Computational Chemistry: Effect on LPS Induced Inflammatory Markers in Pulmonary Artery. International Journal of Molecular Sciences. 2021; 22(6):3158. https://doi.org/10.3390/ijms22063158
Chicago/Turabian StylePerez Diaz, Noelia, Lisa A. Lione, Victoria Hutter, and Louise S. Mackenzie. 2021. "Co-Incubation with PPARβ/δ Agonists and Antagonists Modeled Using Computational Chemistry: Effect on LPS Induced Inflammatory Markers in Pulmonary Artery" International Journal of Molecular Sciences 22, no. 6: 3158. https://doi.org/10.3390/ijms22063158