
Combined Transcriptome Analysis Reveals the Ovule Abortion Regulatory Mechanisms in the Female Sterile Line of Pinus tabuliformis Carr.

Abstract
:1. Introduction
2. Results
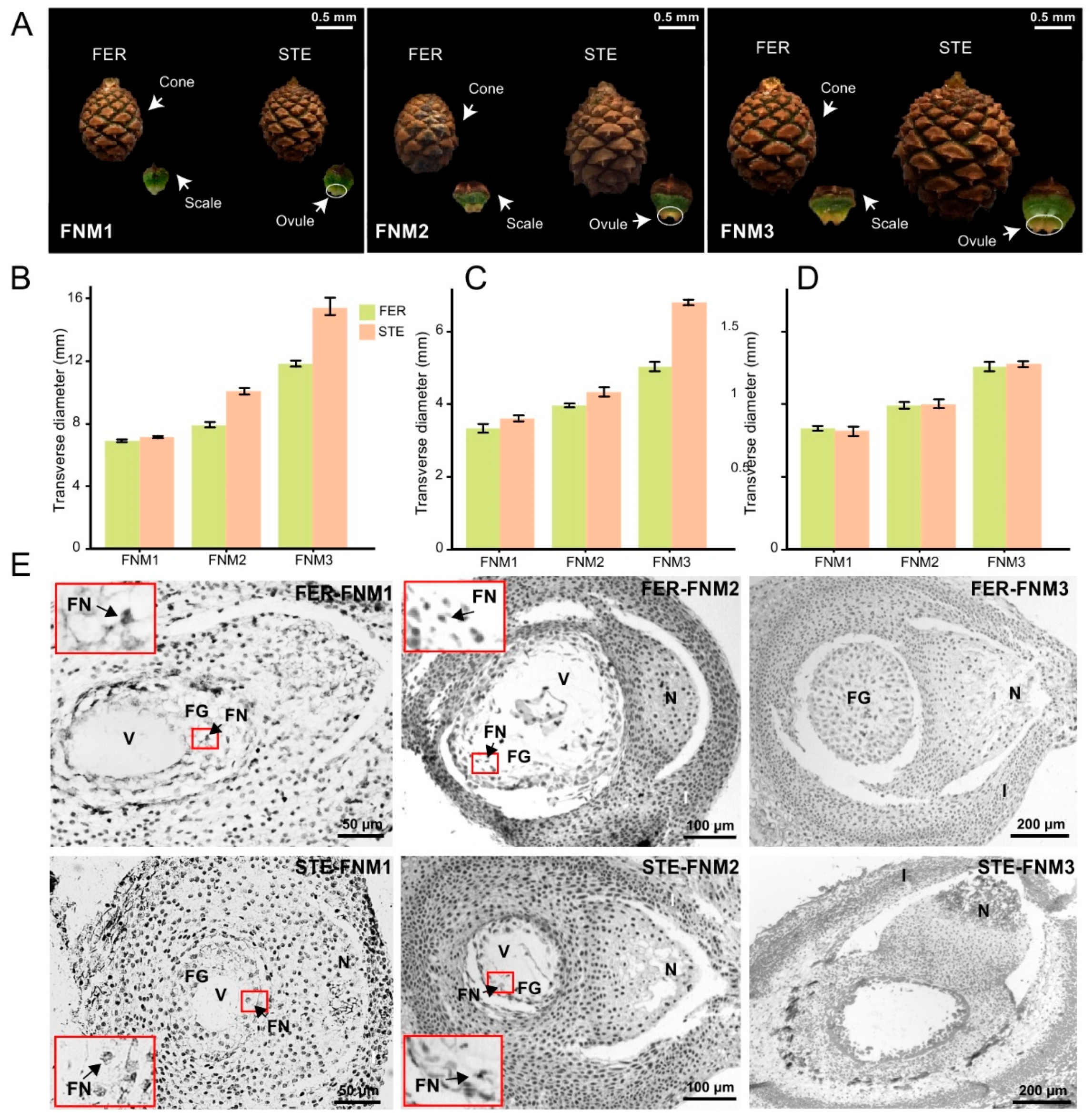
2.1. Cone and Ovule Phenotype Analysis of P. tabuliformis
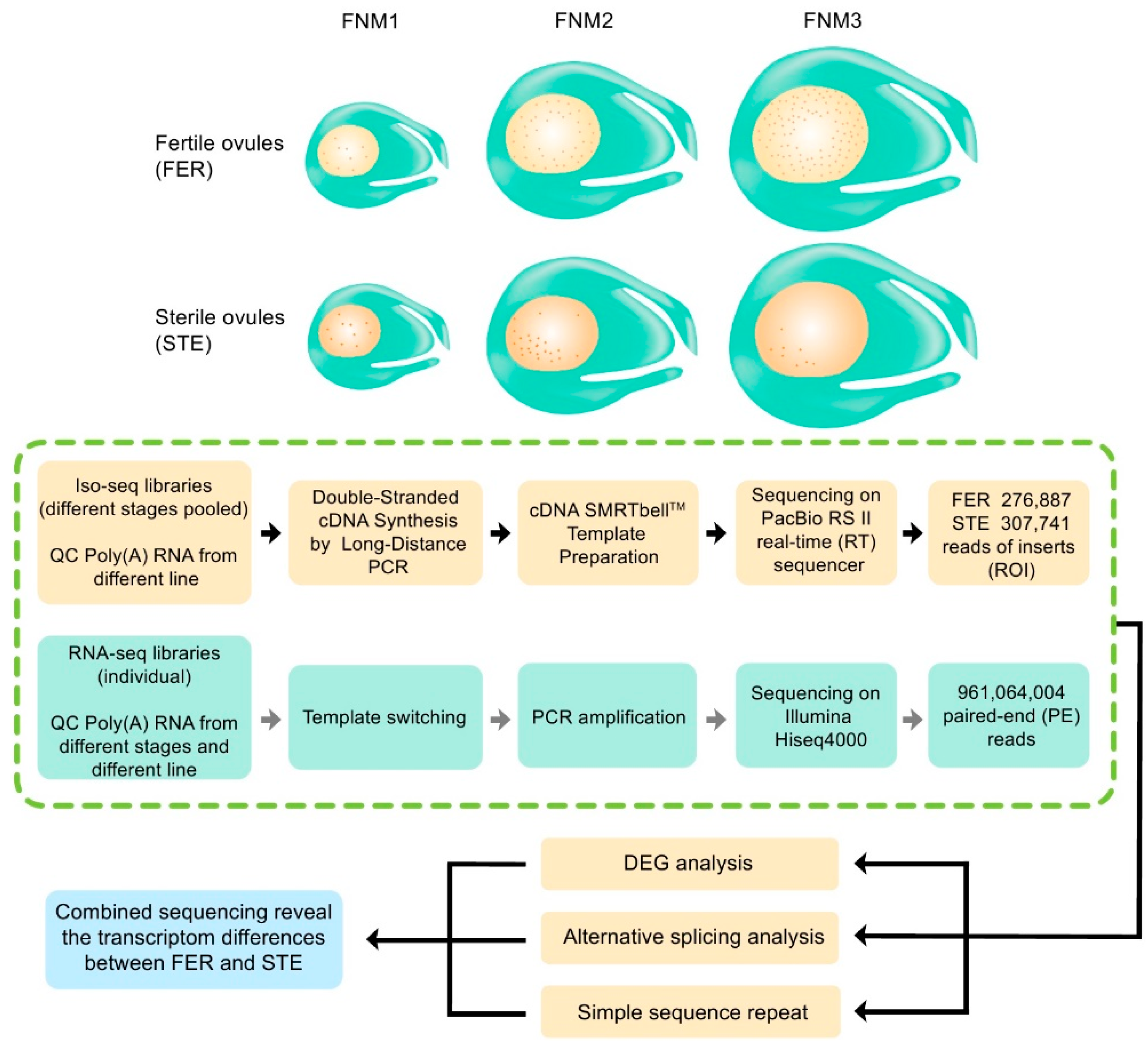
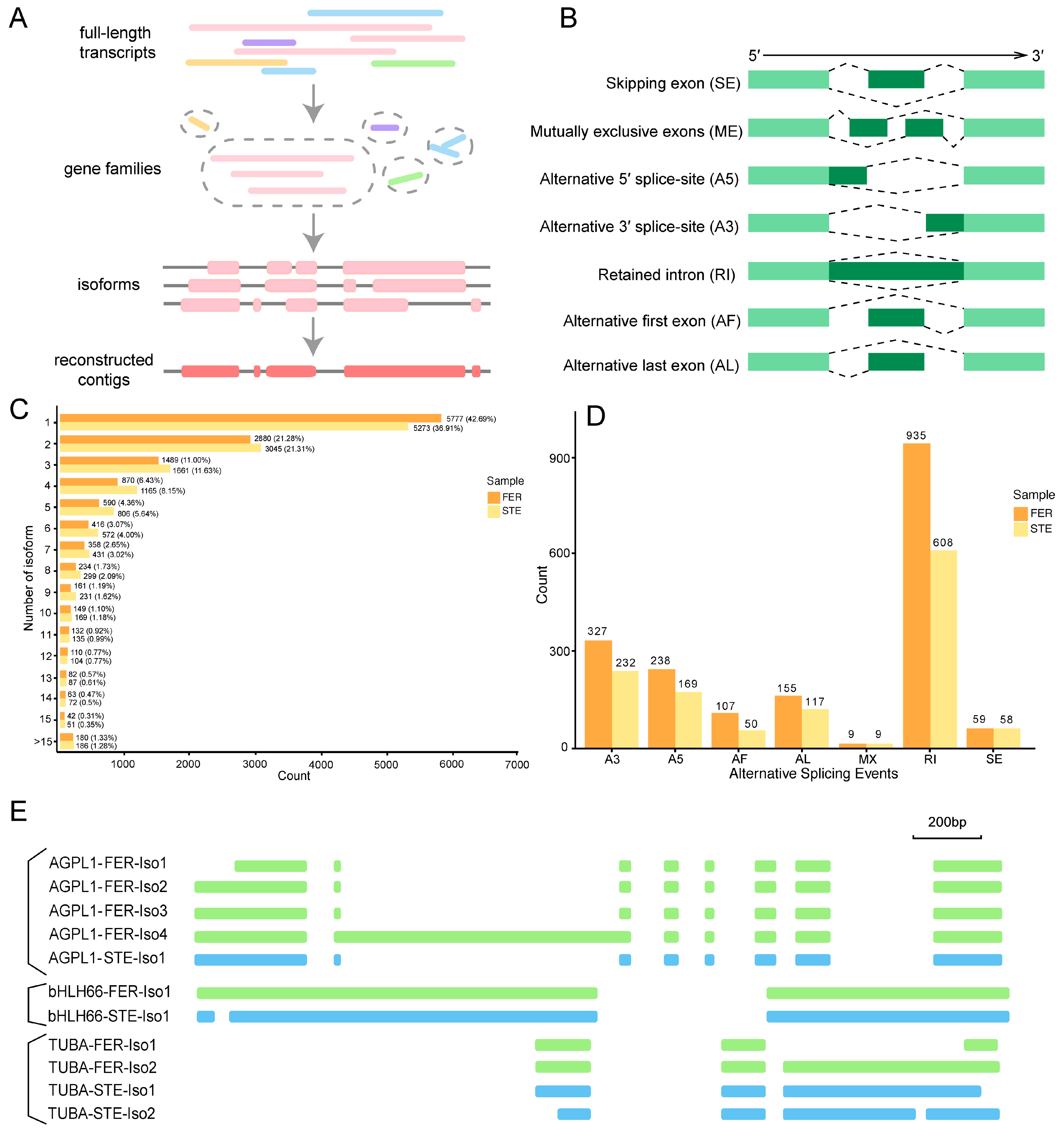
2.2. Combined Sequencing Approach to Analyze the Ovule of P. Tabuliformis
2.3. Functional Annotation of Full-Length Transcriptomes in P. Tabuliformis
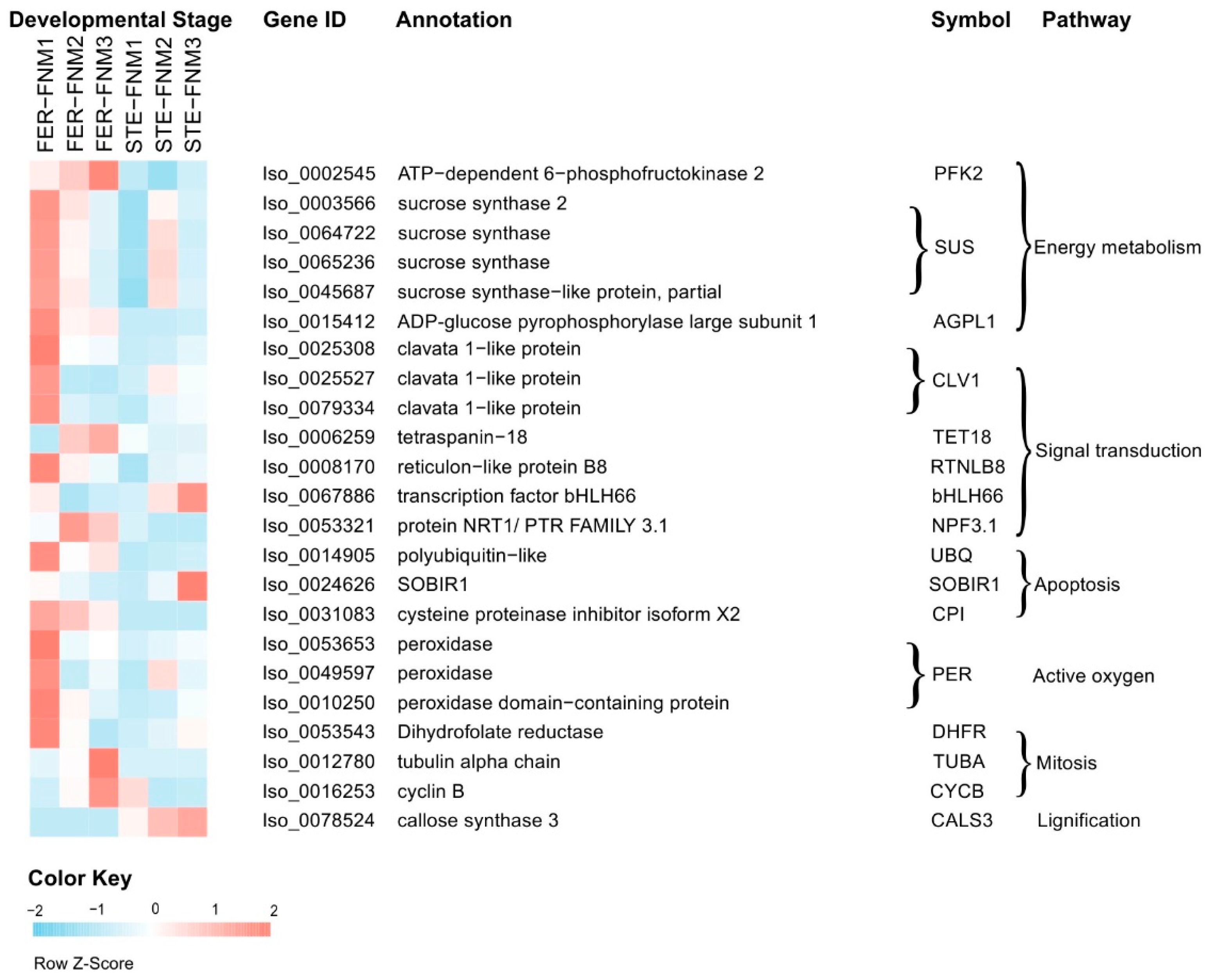
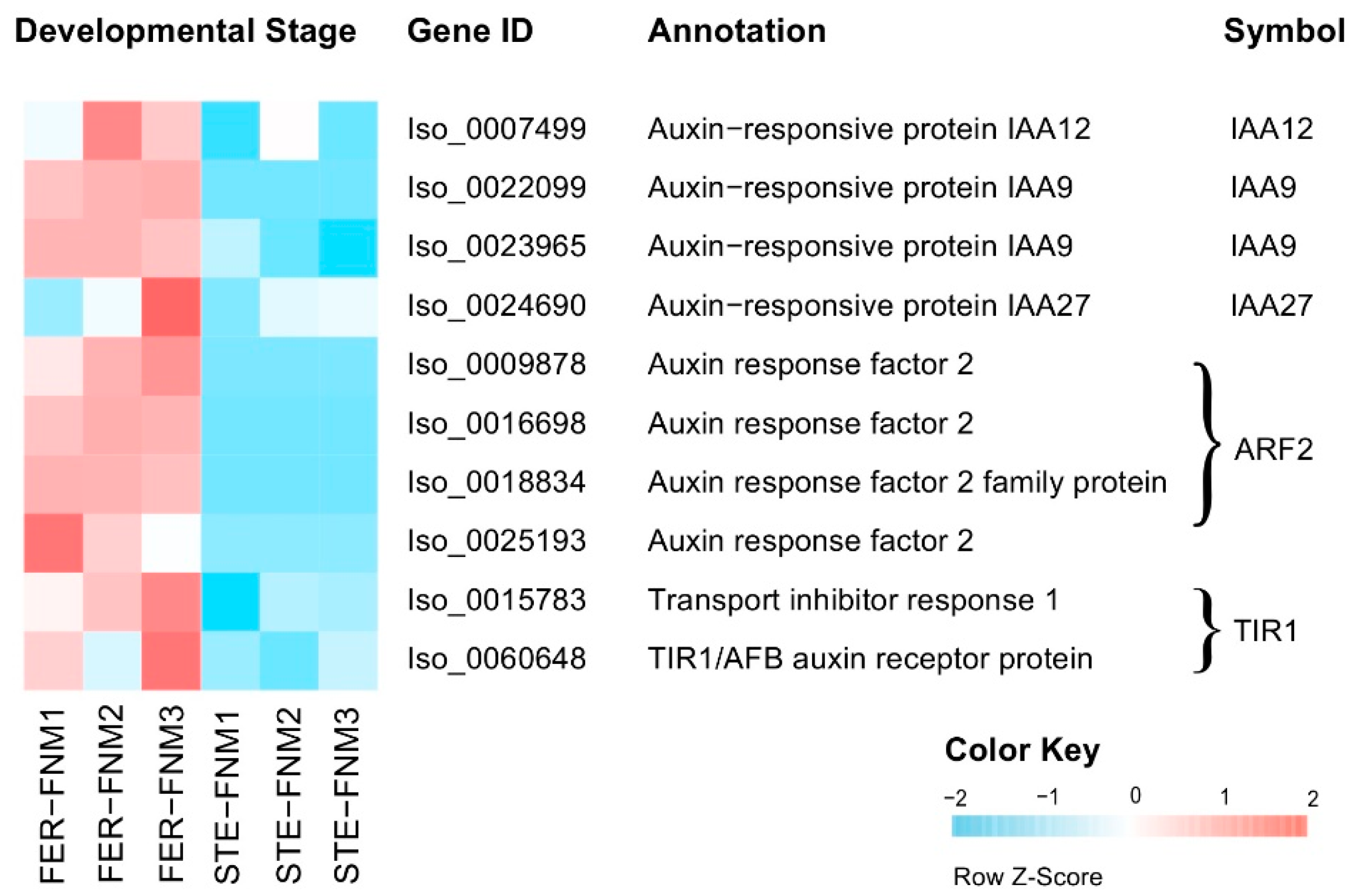
2.4. Candidate Genes with Opposite Expression Patterns during the FNM Process in STE and FER
2.5. AS Analysis of DEGs with Different Expression Patterns during the FNM Process in Two Lines
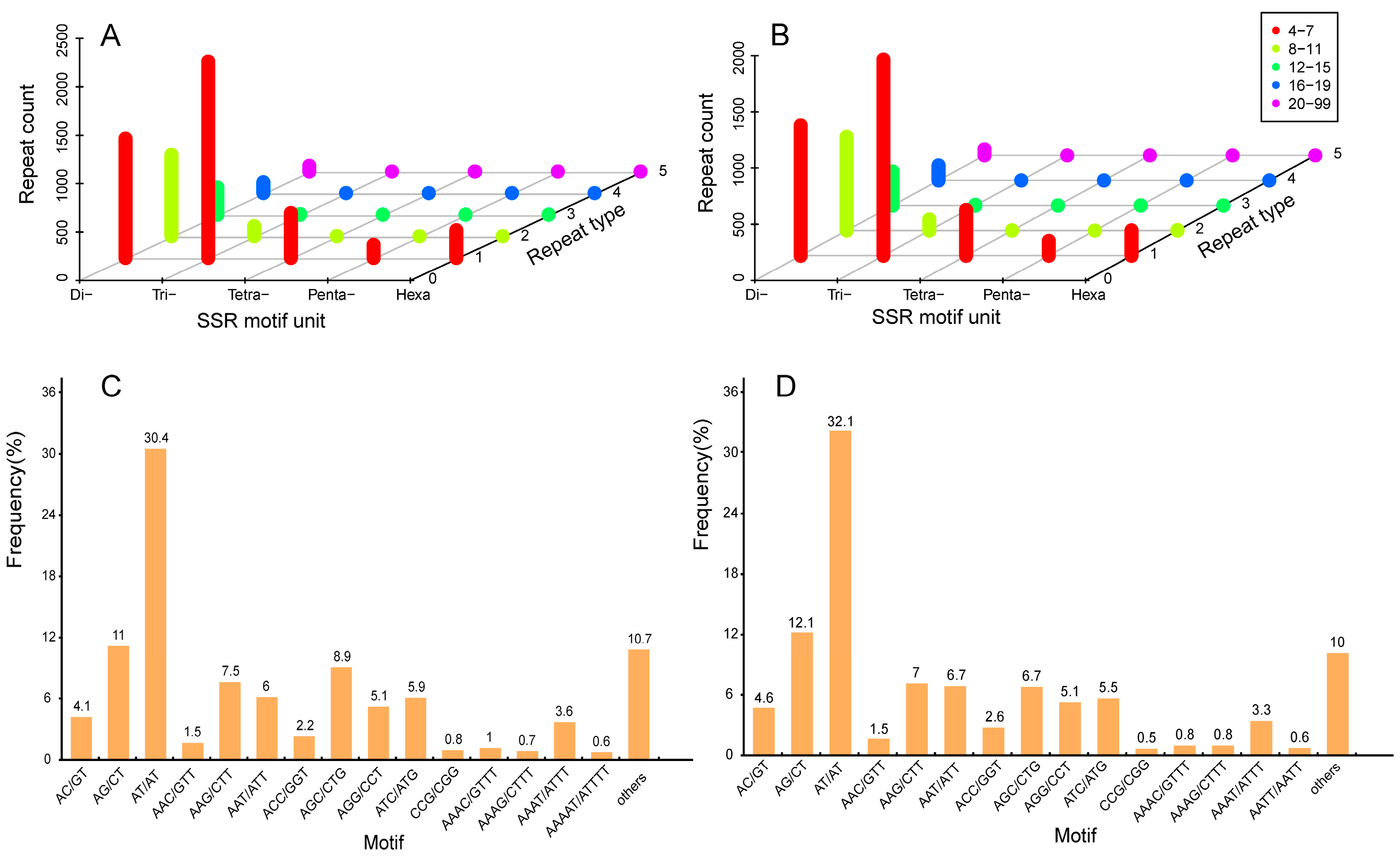
2.6. Development and Characterization of SSR Markers
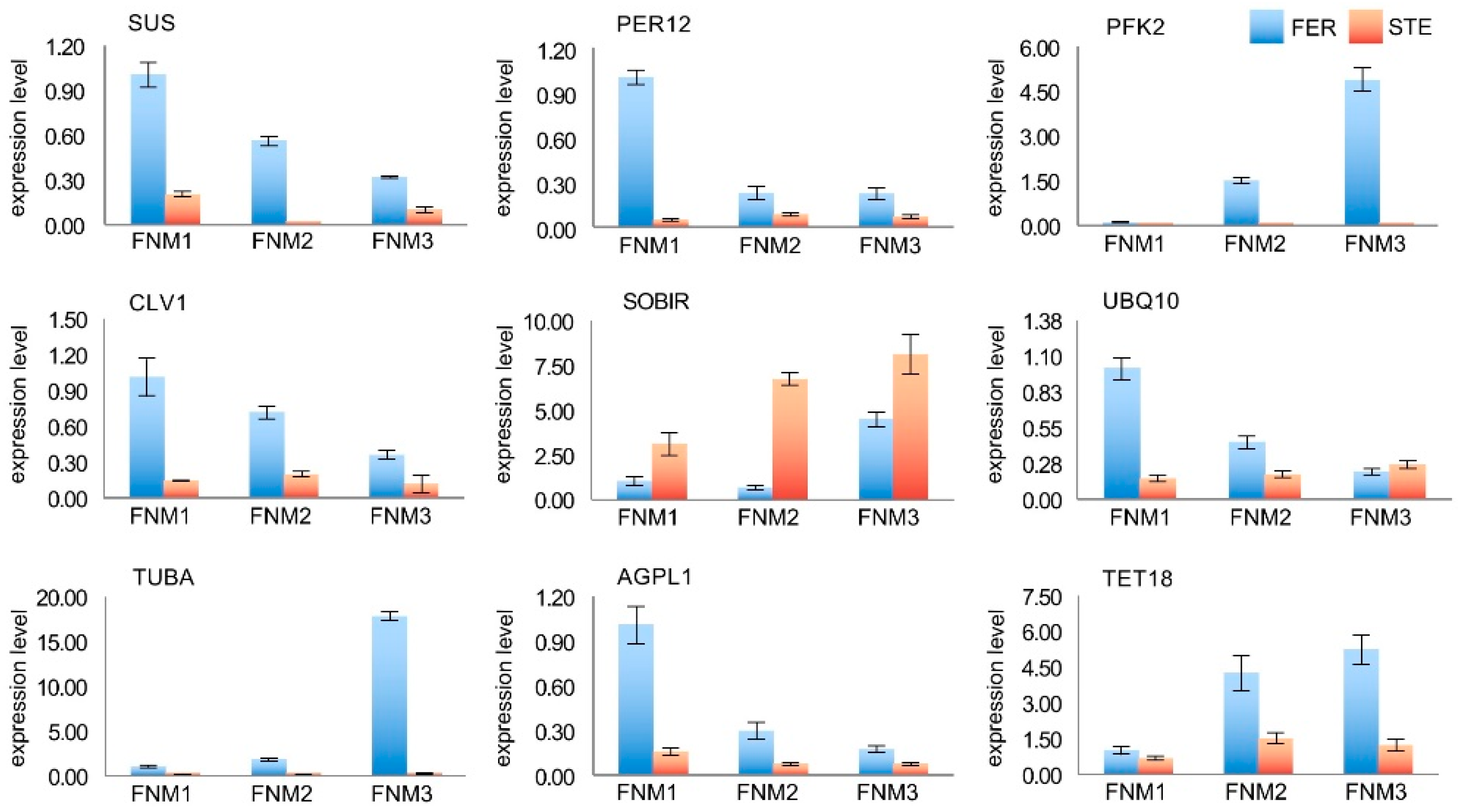
2.7. Verification of the Gene Expression Profile by qRT-PCR
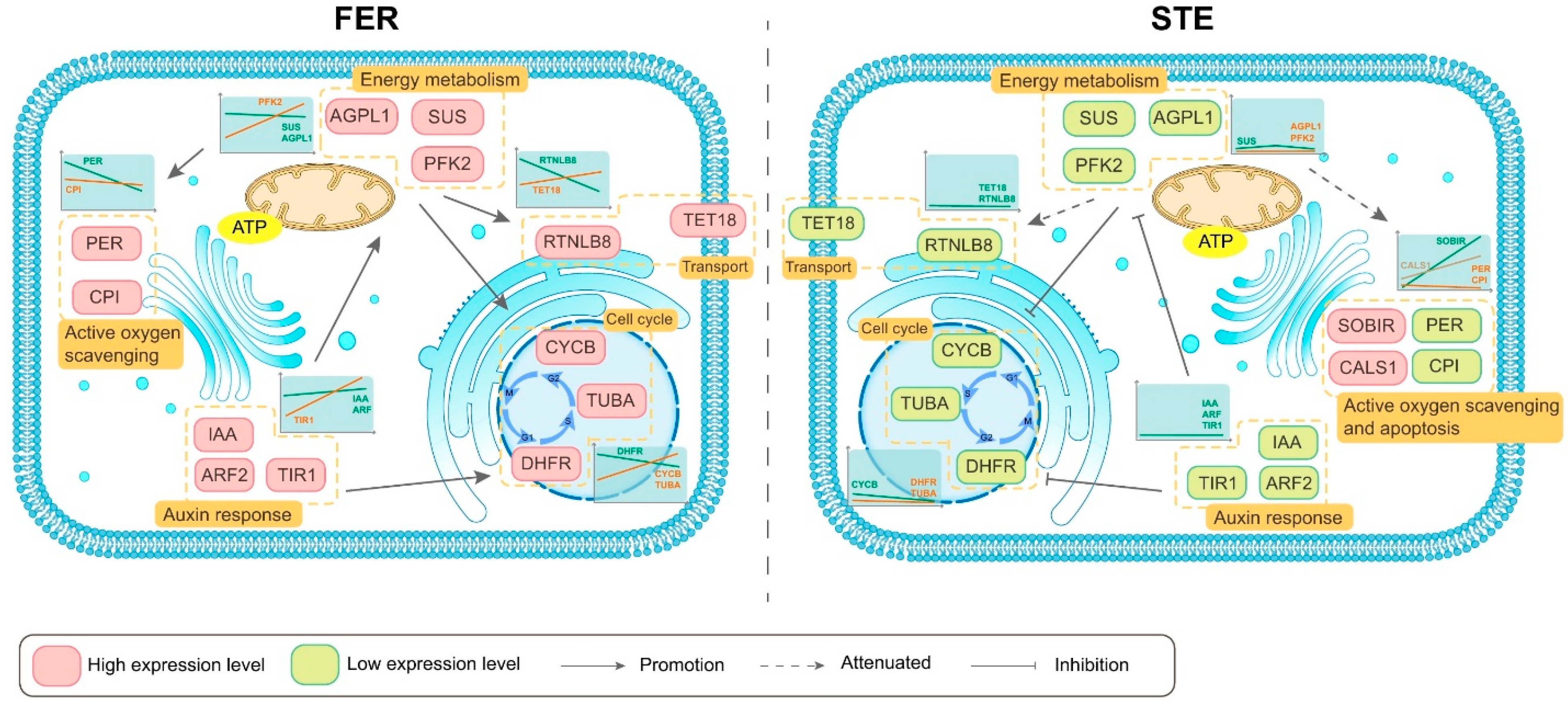
3. Discussion
4. Materials and Methods
4.1. Plant Materials and RNA Sample Preparation
4.2. Analysis of PacBio SMRT
4.3. Functional Annotation
4.4. Analysis of Differentially Expressed Genes (DEGs)
4.5. Alternative Splicing (AS) Detection
4.6. Simple Sequence Repeat (SSR) Marker Prediction
4.7. Real-Time Quantitative PCR Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Guo, A.; Zheng, C. Female gametophyte development. J. Plant Biol. 2013, 56, 345–356. [Google Scholar] [CrossRef]
- Hu, Q.; Gao, S.; Li, F. Advances of research on plant female sterility. J. Beijing For. Univ. 2004, 26, 87–91. [Google Scholar]
- Chen, L.; Zhang, J.; Li, H.; Niu, J.; Xue, H.; Liu, B.; Wang, Q.; Luo, X.; Zhang, F.; Zhao, D.; et al. Transcriptomic Analysis Reveals Candidate Genes for Female Sterility in Pomegranate Flowers. Front. Plant Sci. 2017, 8, 1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awasthi, A.; Paul, P.; Kumar, S.; Verma, S.K.; Prasad, R.; Dhaliwal, H.S. Abnormal endosperm development causes female sterility in rice insertional mutant OsAPC6. Plant Sci. 2012, 183, 167–174. [Google Scholar] [CrossRef]
- Rosellini, D.; Ferranti, F.; Barone, P.; Verones, F. Expression of female sterility in alfalfa (Medicago sativa L.). Sex. Plant Reprod. 2003, 15, 271–279. [Google Scholar] [CrossRef]
- Winiarczyk, K.; Kosmala, A. Development of the female gametophyte in the sterile ecotype of the bolting Allium sativum L. Sci. Hortic. 2009, 121, 353–360. [Google Scholar] [CrossRef]
- Pagnussat, G.; Alandete-Saez, M.; Bowman, J.; Sundaresan, V. Auxin-Dependent Patterning and Gamete Specification in the Arabidopsis Female Gametophyte. Science 2009, 324, 1684–1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martín, M.; Distefano, A.; Zabaleta, E.; Pagnussat, G. New insights into the functional roles of reactive oxygen species during embryo sac development and fertilization in Arabidopsis thaliana. Plant Signal. Behav. 2014, 8, e25714. [Google Scholar] [CrossRef] [Green Version]
- Zsogon, A.; Szakonyi, D.; Shi, X.; Byrne, M. Ribosomal Protein RPL27a Promotes Female Gametophyte Development in a Dose-Dependent Manner. Plant Physiol. 2014, 165, 1133–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Mei, W.; Soltis, P.S.; Soltis, D.E.; Barbazuk, W.B. Detecting alternatively spliced transcript isoforms from single-molecule long-read sequences without a reference genome. Mol. Ecol. Resour. 2017, 17, 1243–1256. [Google Scholar] [CrossRef]
- Tilgner, H.; Grubert, F.; Sharon, D.; Snyder, M.P. Defining a personal, allele-specific, and single-molecule long-read transcriptome. Proc. Natl. Acad. Sci. USA 2014, 111, 9869–9874. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Li, Q.; Li, Y.; Qian, J.; Han, J. Chloroplast genome of Aconitum barbatum var. puberulum (Ranunculaceae) derived from CCS reads using the PacBio RS platform. Front. Plant Sci. 2015, 6, 42. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Tseng, E.; Regulski, M.; Clark, T.A.; Hon, T.; Jiao, Y.; Lu, Z.; Olson, A.; Stein, J.C.; Ware, D. Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nat. Commun. 2016, 7, 11708. [Google Scholar] [CrossRef] [Green Version]
- Blencowe, B.J. Alternative Splicing: New Insights from Global Analyses. Cell 2006, 126, 37–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, D.; Wang, Y.; Liu, Y.; Hu, J.; Guo, Y.; Gao, L.; Ma, R. SMRT sequencing of full-length transcriptome of flea beetle Agasicles hygrophila (Selman and Vogt). Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Dorothee, S.; John, B. Alternative Splicing at the Intersection of Biological Timing, Development, and Stress Responses. Plant Cell 2013, 25, 3640–3656. [Google Scholar]
- Zhu, F.Y.; Chen, M.X.; Ye, N.H.; Shi, L.; Ma, K.L.; Yang, J.F.; Cao, Y.Y.; Zhang, Y.; Yoshida, T.; Fernie, A.R.; et al. Proteogenomic analysis reveals alternative splicing and translation as part of the abscisic acid response in Arabidopsis seedlings. Plant J. 2017, 91, 518–533. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Peters, R.J.; Weirather, J.; Luo, H.; Liao, B.; Zhang, X.; Zhu, Y.; Ji, A.; Zhang, B.; Hu, S.; et al. Full-length transcriptome sequences and splice variants obtained by a combination of sequencing platforms applied to different root tissues of Salvia miltiorrhiza and tanshinone biosynthesis. Plant J. 2015, 82, 951–961. [Google Scholar] [CrossRef]
- Li, J.; Harata-Lee, Y.; Denton, M.D.; Feng, Q.; Rathjen, J.R.; Qu, Z.; Adelson, D.L. Long read reference genome-free reconstruction of a full-length transcriptome from Astragalus membranaceus reveals transcript variants involved in bioactive compound biosynthesis. Cell Discov. 2017, 3, 17031. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhang, L.; Zhang, P.; Yang, Z.; Zhan, X.; Shen, X.; Zhang, Z.; Hu, X.; Xuan, D.; Wu, W.; et al. Characterization and Gene Mapping of an Open Hul Male Sterile Mutant ohms1 Caused by Alternative Splicing in Rice. Chin. J. Rice Sci. 2015, 29, 457–466. [Google Scholar]
- Lawson, M.J.; Zhang, L. Distinct patterns of SSR distribution in the Arabidopsis thaliana and rice genomes. Genome Biol. 2006, 7, R14. [Google Scholar] [CrossRef] [Green Version]
- Dou, B.; Hou, B.; Xu, H.; Lou, X.; Chi, X.; Yang, J.; Wang, F.; Ni, Z.; Sun, Q. Efficient mapping of a female sterile gene in wheat (Triticum aestivum L.). Genet. Res. 2009, 91, 337–343. [Google Scholar] [CrossRef] [Green Version]
- Kato, K.K. Molecular Mapping of the Male-Sterile, Female-Sterile Mutant Gene (st8) in Soybean. J. Hered. 2003, 94, 425–428. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Y.; Taylor, A.; McManus, H. Evolution of the life cycle in land plants. J. Syst. Evol. 2012, 50, 171–194. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Zhang, W.; Gong, Z.; Zheng, C. Morphologic and anatomical observations in the process of ovulate strobilus generation and development in Pinus tabuliformis. J. Beijing For. Univ. 2017, 39, 1–12. [Google Scholar]
- Chen, K.; Abbott, R.J.; Milne, R.I.; Tian, X.M.; Liu, J. Phylogeography of Pinus tabulaeformis Carr. (Pinaceae), a dominant species of coniferous forest in northern China. Mol. Ecol. 2008, 17, 4276–4288. [Google Scholar] [CrossRef]
- Zhang, M.; Zheng, C.-X. Archegonium and fertilization in Coniferopsida. Trees 2016, 30, 75–86. [Google Scholar] [CrossRef]
- Li, F.; Zheng, C. A Preliminary Study on Ovule Abortion during Later Stage of Development in Pinus tabulaeformis. J. Beijing For. Univ. 1990, 12, 68–73. [Google Scholar]
- Yao, Y.; Han, R.; Gong, Z.; Zheng, C.; Zhao, Y. RNA-seq Analysis Reveals Gene Expression Profiling of Female Fertile and Sterile Ovules of PinusTabulaeformis Carr. during Free Nuclear Mitosis of the Female Gametophyte. Int. J. Mol. Sci. 2018, 19, 2246. [Google Scholar] [CrossRef] [Green Version]
- Ner-Gaon, H.; Halachmi, R.; Savaldi-Goldstein, S.; Rubin, E.; Ophir, R.; Fluhr, R. Intron retention is a major phenomenon in alternative splicing in Arabidopsis. Plant J. 2004, 39, 877–885. [Google Scholar] [CrossRef]
- Zeng, D.; Chen, X.; Peng, J.; Yang, C.; Peng, M.; Zhu, W.; Xie, D.; He, P.; Wei, P.; Lin, Y.; et al. Single-molecule long-read sequencing facilitates shrimp transcriptome research. Sci. Rep. 2018, 8, 16920. [Google Scholar] [CrossRef]
- Wang, H.; Liu, R.; Wang, J.; Wang, P.; Shen, Y.; Liu, G. The Arabidopsis kinesin gene AtKin-1 plays a role in the nuclear division process during megagametogenesis. Plant Cell Rep. 2014, 33, 819–828. [Google Scholar] [CrossRef] [PubMed]
- Lo’pez-Almansa, J.C.; Pannell, J.R.; Gil, L. Female sterility in Ulmus minor (Ulmaceae): A hypothesis invo king the cost of sex in a clonal plant. Am. J. Bot. 2003, 90, 603–609. [Google Scholar] [CrossRef]
- Yu, L.; Chunyu, Z.; Tong, C.; Huaiqing, H.; Peng, L.; Ray, A.B.; Paul, M.H.; Jingbo, J.; Jinxing, L. Mutation in SUMO E3 ligase, SIZ1, Disrupts the MatureFemale Gametophyte in Arabidopsis. PLoS ONE 2012, 7, e0126164. [Google Scholar]
- Anderson, S.N.; Johnson, C.S.; Jones, D.S.; Conrad, L.J.; Gou, X.; Russell, S.D.; Sundaresan, V. Transcriptomes of isolatedOryza sativagametes characterized by deep sequencing: Evidence for distinct sex-dependent chromatin and epigenetic states before fertilization. Plant J. 2013, 76, 729–741. [Google Scholar] [CrossRef]
- Yang, L.; Wu, Y.; Yu, M.; Mao, B.; Zhao, B.; Wang, J. Genome-wide transcriptome analysis of female-sterile rice ovule shed light on its abortive mechanism. Planta 2016, 244, 1011–1028. [Google Scholar] [CrossRef]
- Loraine, A.E.; McCormick, S.; Estrada, A.; Patel, K.; Qin, P. RNA-Seq of Arabidopsis Pollen Uncovers Novel Transcription and Alternative Splicing. Plant Physiol. 2013, 162, 1092–1109. [Google Scholar] [CrossRef] [Green Version]
- Forsthoefel, N.R.; Klag, K.A.; McNichol, S.R.; Arnold, C.E.; Vernon, C.R.; Wood, W.W.; Vernon, D.M. Arabidopsis PIRL6 Is Essential for Male and Female Gametogenesis and Is Regulated by Alternative Splicing. Plant Physiol. 2018, 178, 1154–1169. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.H.; Yang, Z.Q.; Wang, J.; Luo, Q.F.; Li, H.G. Development and characterization of SSR markers from Pinus massoniana and their transferability to P. elliottii, P. caribaea and P. yunnanensis. Genet. Mol. Res. 2014, 13, 1508–1513. [Google Scholar] [CrossRef]
- Fu, W.E.I.; Du, Z.-X.; Ren, H.E.; Buahom, N.; Liu, H.-W.; Huang, X.I.N. Polymorphic microsatellite markers in Taxus chinensis var. mairei (Taxaceae). J. Genet. 2013, 93, 81–84. [Google Scholar] [CrossRef]
- Han, S.; Wu, Z.; Jin, Y.; Yang, W.; Shi, H. RNA-Seq analysis for transcriptome assembly, gene identification, and SSR mining in ginkgo (Ginkgo biloba L.). Tree Genet. Genomes 2015, 11, 37. [Google Scholar] [CrossRef]
- Jiri, F.; Vieten, A.; Sauer, M.; Weijers, D.; Schwarz, H.; Hamann, T.; Offringa, R.; Jürgens, G. Efflux-dependent auxin gradients establish the apical–basal axis of Arabidopsis. Nature 2003, 426, 147–153. [Google Scholar]
- Fu, W.; Zhao, Z.; Ge, X.; Ding, L.; Li, Z. Anatomy and transcript profiling of gynoecium development in female sterile Brassica napus mediated by one alien chromosome from Orychophragmus violaceus. BMC Genom. 2014, 19, 2–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, H.; Park, C.; Gutierrez, C.; Wojcikowska, B.; Pencik, A.; Novak, O.; Chen, J.; Grunewald, W.; Dresselhaus, T.; Friml, J.; et al. Maternal auxin supply contributes to early embryo patterning in Arabidopsis. Nat. Plants 2018, 4, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Candela, H.; Panoli, A.; Martin, M.V.; Alandete-Saez, M.; Simon, M.; Neff, C.; Swarup, R.; Bellido, A.; Yuan, L.; Pagnussat, G.C.; et al. Auxin Import and Local Auxin Biosynthesis Are Required for Mitotic Divisions, Cell Expansion and Cell Specification during Female Gametophyte Development in Arabidopsis thaliana. PLoS ONE 2015, 10, e0126164. [Google Scholar] [CrossRef]
- Wu, M.F.; Tian, Q.; Reed, J.W. Arabidopsis microRNA167 controls patterns of ARF6 and ARF8 expression, and regulates both female and male reproduction. Development 2006, 133, 4211–4218. [Google Scholar] [CrossRef] [Green Version]
- Dharmasiri, N.; Dharmasiri, S.; Estelle, M. The F-box protein TIR1 is an auxin receptor. Nature 2005, 435, 441–445. [Google Scholar] [CrossRef]
- Ito, A.; Hayama, H.; Kashimura, Y. Sugar metabolism in buds during flower bud formation: A comparison of two Japanese pear [Pyrus pyrifolia (Burm.) Nak.] cultivars possessing different flowering habits. Sci. Hortic. 2002, 96, 163–175. [Google Scholar] [CrossRef]
- Kong, J.; Li, Z.; Tan, Y.-P.; Wan, C.-X.; Li, S.-Q.; Zhu, Y.-G. Different gene expression patterns of sucrose-starch metabolism during pollen maturation in cytoplasmic male-sterile and male-fertile lines of rice. Physiol. Plant. 2007, 130, 136–147. [Google Scholar] [CrossRef]
- Baez, M.; Cabrera, R.; Guixe, V.; Babul, J. Unfolding Pathway of the Dimeric and Tetrameric Forms of Phosphofructokinase-2 from Escherichia coli. Biochemistry 2007, 46, 6141–6148. [Google Scholar] [CrossRef]
- Kotlarz, D.; Buc, H. Regulatory Properties of Phosphofructokinase 2 from Escherichia coli. Eur. J. Biochem. 1981, 117, 569–574. [Google Scholar] [CrossRef]
- Huck, N. The Arabidopsis mutant feronia disrupts the female gametophytic control of pollen tube reception. Development 2003, 130, 2149–2159. [Google Scholar] [CrossRef] [Green Version]
- Reimann, R.; Kost, B.; Dettmer, J. TETRASPANINs in Plants. Front. Plant Sci. 2017, 8, 545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Muto, A.; Van de Velde, J.; Neyt, P.; Himanen, K.; Vandepoele, K.; Van Lijsebettens, M. Functional Analysis of the Arabidopsis TETRASPANIN Gene Family in Plant Growth and Development. Plant Physiol. 2015, 169, 2200–2214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kriechbaumer, V.; Botchway, S.W.; Slade, S.E.; Knox, K.; Frigerio, L.; Oparka, K.; Hawes, C. Reticulomics: Protein-Protein Interaction Studies with Two Plasmodesmata-Localized Reticulon Family Proteins Identify Binding Partners Enriched at Plasmodesmata, Endoplasmic Reticulum, and the Plasma Membrane. Plant Physiol. 2015, 169, 1933–1945. [Google Scholar] [CrossRef]
- Tolley, N.; Sparkes, I.; Craddock, C.P.; Eastmond, P.J.; Runions, J.; Hawes, C.; Frigerio, L. Transmembrane domain length is responsible for the ability of a plant reticulon to shape endoplasmic reticulum tubules in vivo. Plant J. 2010, 64, 411–418. [Google Scholar] [CrossRef] [Green Version]
- Kayes, J.M.; Clark, S.E. CLAVATA2, a regulator of meristem and organ development in Arabidopsis. Development 1998, 125, 3843–3851. [Google Scholar]
- Ferrándiz, C.; Pelaz, S.; Yanofsky, M.F. Control of carpel and fruit development in Arabidopsis. Annu. Rev. Biochem. 1999, 68, 321–354. [Google Scholar] [CrossRef] [PubMed]
- Trehin, C.; Ahn, I.-O.; Perennes, C.; Couteau, F.; Lalanne, E.; Bergounioux, C. Cloning of upstream sequences responsible for cell cycle regulation of the Nicotiana sylvestris CycB1;1 gene. Plant Mol. Biol. 1997, 35, 667–672. [Google Scholar] [CrossRef]
- Gambino, J.; Bergen, L.G.; Morris, N.R. Effects of mitotic and tubulin mutations on microtubule architecture in actively growing protoplasts of Aspergillus nidulans. J. Cell Biol. 1984, 99, 830–838. [Google Scholar] [CrossRef] [Green Version]
- Gorelova, V.; De Lepeleire, J.; Van Daele, J.; Pluim, D.; Mei, C.; Cuypers, A.; Leroux, O.; Rebeille, F.; Schellens, J.H.M.; Blancquaert, D.; et al. Dihydrofolate Reductase/Thymidylate Synthase Fine-Tunes the Folate Status and Controls Redox Homeostasis in Plants. Plant Cell 2017, 29, 2831–2853. [Google Scholar] [CrossRef] [Green Version]
- Slansky, J.E.; Farnham, P.J. Transcriptional regulation of the dihydrofolate reductase gene. Bioessays 1996, 18, 55–62. [Google Scholar] [CrossRef]
- Liu, J.; Ding, P.; Sun, T.; Nitta, Y.; Dong, O.; Huang, X.; Yang, W.; Li, X.; Botella, J.R.; Zhang, Y. Heterotrimeric G Proteins Serve as a Converging Point in Plant Defense Signaling Activated by Multiple Receptor-Like Kinases. Plant Physiol. 2013, 161, 2146–2158. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Wang, X.; Wang, D.; Xu, F.; Ding, X.; Zhang, Z.; Bi, D.; Cheng, Y.T.; Chen, S.; Li, X.; et al. Regulation of cell death and innate immunity by two receptor-like kinases in Arabidopsis. Cell Host Microbe 2009, 6, 34–44. [Google Scholar] [CrossRef] [Green Version]
- Leslie, M.E.; Lewis, M.W.; Youn, J.Y.; Daniels, M.J.; Liljegren, S.J. The EVERSHED receptor-like kinase modulates floral organ shedding in Arabidopsis. Development 2010, 137, 467–476. [Google Scholar] [CrossRef] [Green Version]
- Milosevic, N.; Slusarenko, A.J. Active oxygen metabolism and lignification in the hypersensitive response in bean. Physiol. Mol. Plant Pathol. 1996, 49, 143–158. [Google Scholar] [CrossRef]
- Kawano, T. Roles of the reactive oxygen species-generating peroxidase reactions in plant defense and growth induction. Plant Cell Rep. 2003, 21, 829–837. [Google Scholar] [CrossRef]
- Luna, E.; Pastor, V.; Robert, J.; Flors, V.; Mauch-Mani, B.; Ton, J. Callose deposition: A multifaceted plant defense response. Mol. Plant Microbe Interact. 2011, 24, 183–193. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.; Hong, Z.; Sivaramakrishnan, M.; Mahfouz, M.; Verma, D.P. Callose synthase (CalS5) is required for exine formation during microgametogenesis and for pollen viability in Arabidopsis. Plant J. 2005, 42, 315–328. [Google Scholar] [CrossRef]
- Tiwari, B.S.; Belenghi, B.; Levine, A. Oxidative stress increased respiration and generation of reactive oxygen species, resulting in ATP depletion, opening of mitochondrial permeability transition, and programmed cell death. Plant Physiol. 2002, 128, 1271–1281. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Zheng, C. RAPD analysis in female sterility clone 28 of Pinus tabulae- formis Carr. J. Beijing For. Univ. 2002, 24, 35–38. [Google Scholar]
- Zhang, W.; Zheng, C.; Zhao, N.; Li, F. Observation on the formation of ovule tapetum in Pinus tabulaeformis Carr. J. Beijing For. Univ. 2015, 37, 53–57. [Google Scholar]
- Gordon, S.P.; Tseng, E.; Salamov, A.; Zhang, J.; Meng, X.; Zhao, Z.; Kang, D.; Underwood, J.; Grigoriev, I.V.; Figueroa, M.; et al. Widespread Polycistronic Transcripts in Fungi Revealed by Single-Molecule mRNA Sequencing. PLoS ONE 2015, 10, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3675–3676. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L.; et al. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef]
- Audic, S.; Claverie, J.M. The significance of digital gene expression profiles. Genome Res. 1997, 7, 986–995. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Alamancos, G.P.; Pagès, A.; Trincado, J.L.; Bellora, N.; Eyras, E. Leveraging transcript quantification for fast computation of alternative splicing profiles. RNA 2015, 21, 1521–1531. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Searching Item | Numbers | |
|---|---|---|
| FER | STE | |
| Total number of sequences examined | 40,828 | 44,195 |
| Total size of examined sequences (bp) | 112,127,427 | 98,231,600 |
| Total number of identified SSRs | 5530 | 5096 |
| Number of SSR-containing sequences | 4276 | 3948 |
| Number of sequences containing more than one SSR | 868 | 796 |
| Number of SSRs present in compound formation | 475 | 487 |
| Dinucleotide | 2516 | 2485 |
| Trinucleotide | 2132 | 1846 |
| Tetranucleotide | 463 | 407 |
| Pentanucleotide | 134 | 130 |
| Hexanucleotide | 285 | 228 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gong, Z.; Han, R.; Xu, L.; Hu, H.; Zhang, M.; Yang, Q.; Zeng, M.; Zhao, Y.; Zheng, C. Combined Transcriptome Analysis Reveals the Ovule Abortion Regulatory Mechanisms in the Female Sterile Line of Pinus tabuliformis Carr. Int. J. Mol. Sci. 2021, 22, 3138. https://doi.org/10.3390/ijms22063138
Gong Z, Han R, Xu L, Hu H, Zhang M, Yang Q, Zeng M, Zhao Y, Zheng C. Combined Transcriptome Analysis Reveals the Ovule Abortion Regulatory Mechanisms in the Female Sterile Line of Pinus tabuliformis Carr. International Journal of Molecular Sciences. 2021; 22(6):3138. https://doi.org/10.3390/ijms22063138
Chicago/Turabian StyleGong, Zaixin, Rui Han, Li Xu, Hailin Hu, Min Zhang, Qianquan Yang, Ming Zeng, Yuanyuan Zhao, and Caixia Zheng. 2021. "Combined Transcriptome Analysis Reveals the Ovule Abortion Regulatory Mechanisms in the Female Sterile Line of Pinus tabuliformis Carr." International Journal of Molecular Sciences 22, no. 6: 3138. https://doi.org/10.3390/ijms22063138