
Design, Synthesis, and Evaluation of Novel 3-Carboranyl-1,8-Naphthalimide Derivatives as Potential Anticancer Agents

 , , , , , and
, , , , , and
Abstract
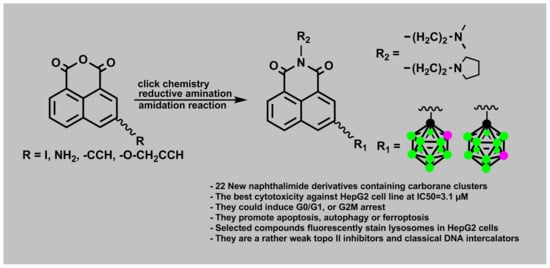
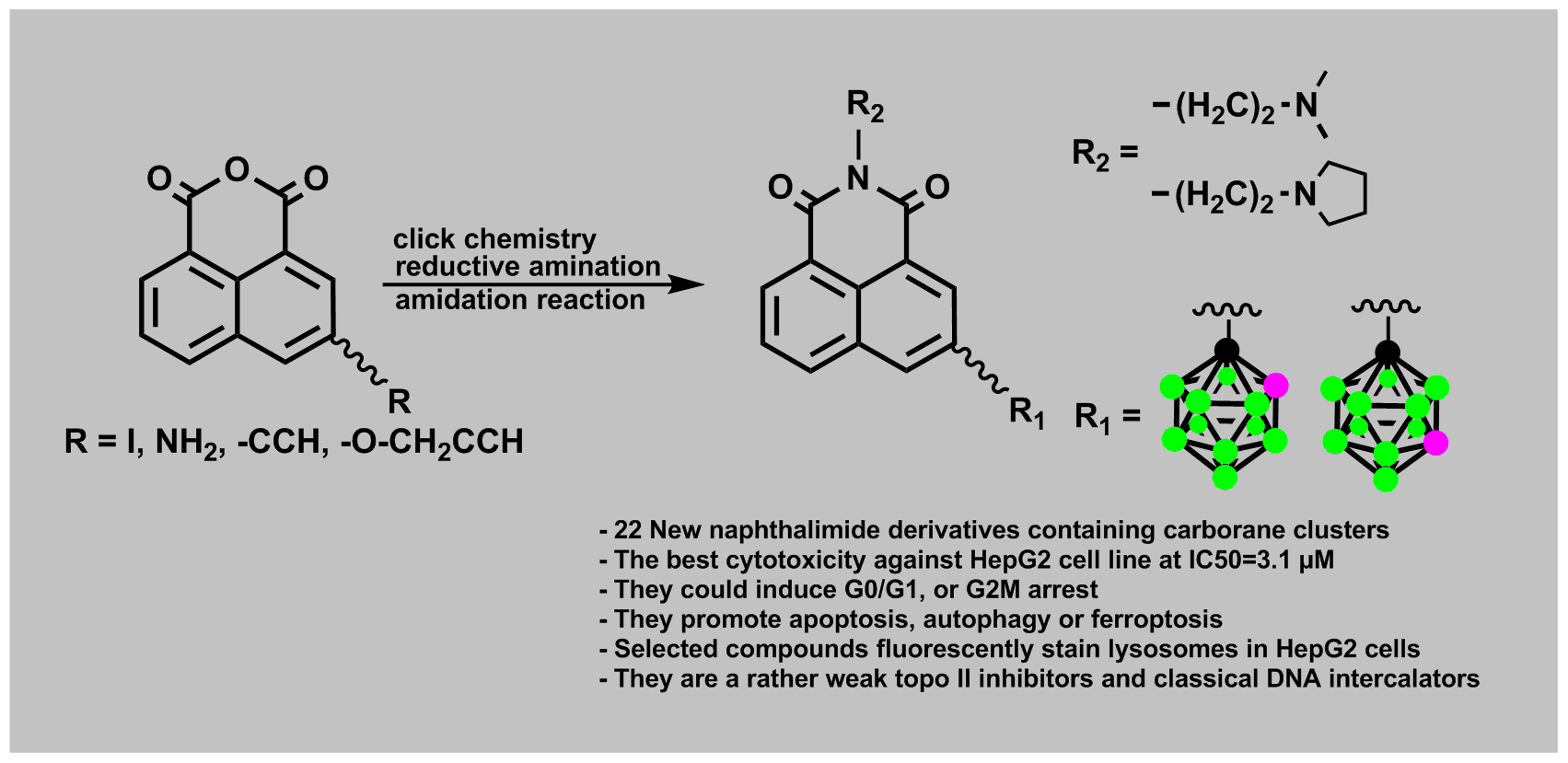
:
1. Introduction
2. Results and Discussion
2.1. Chemistry
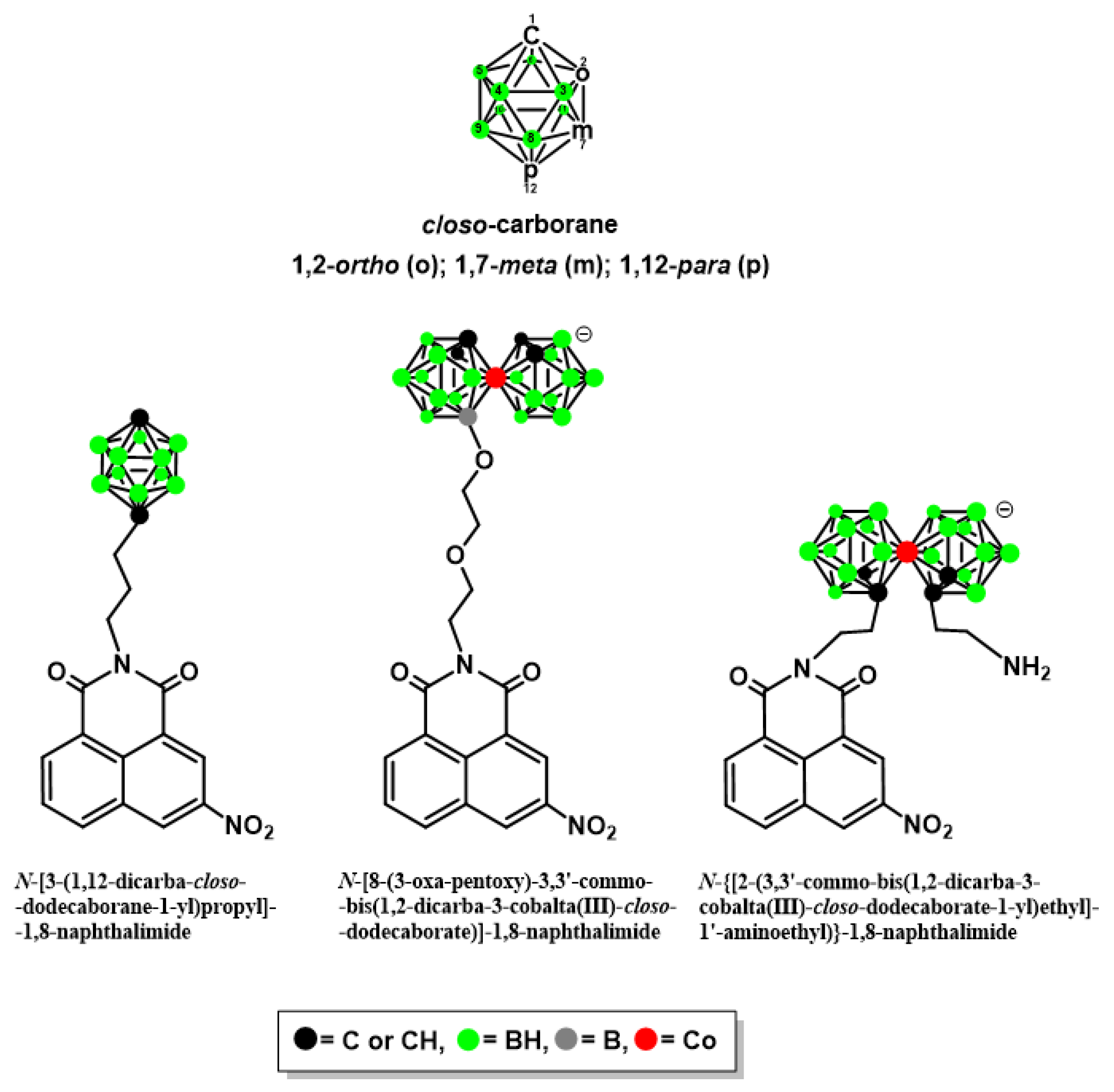
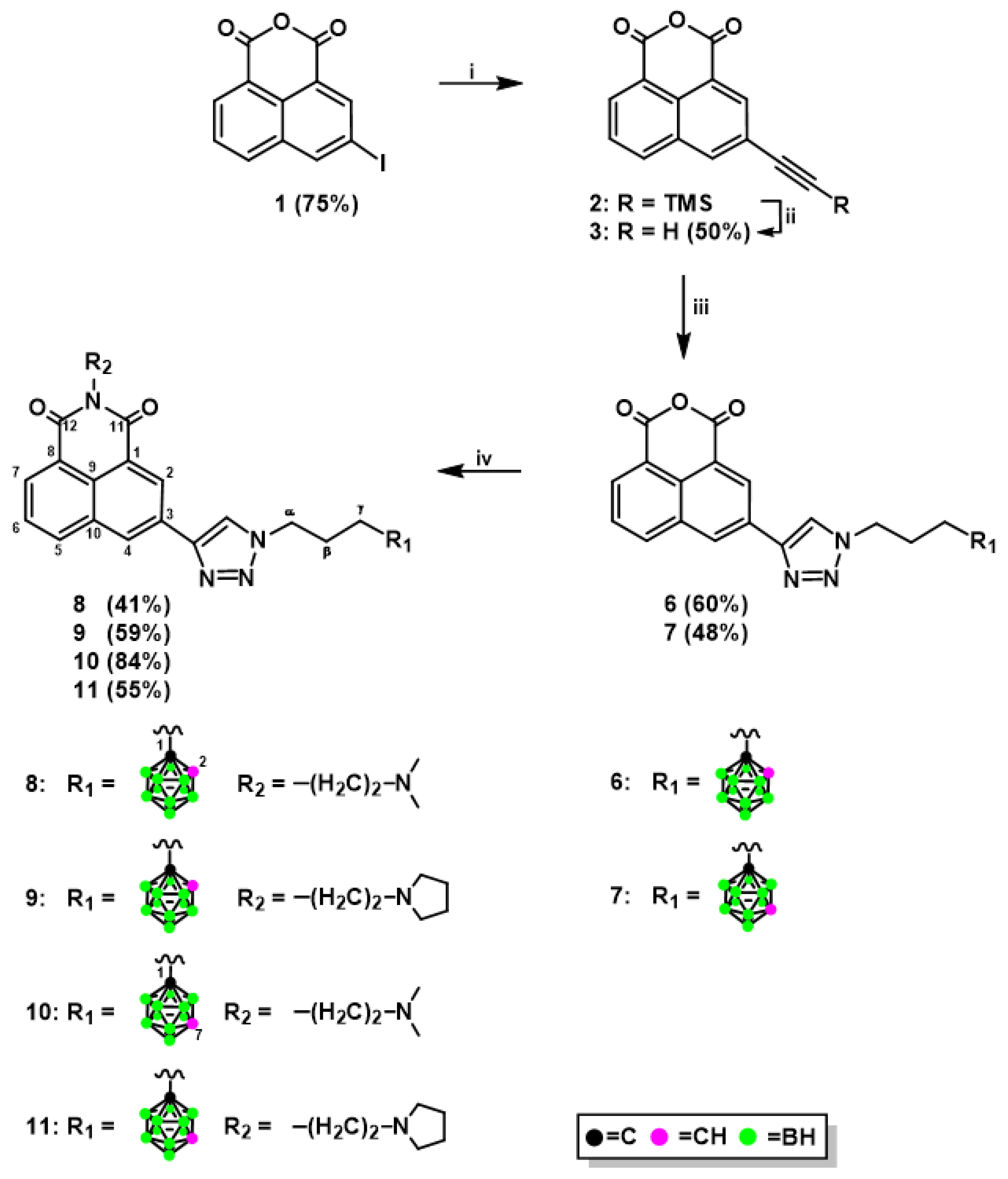
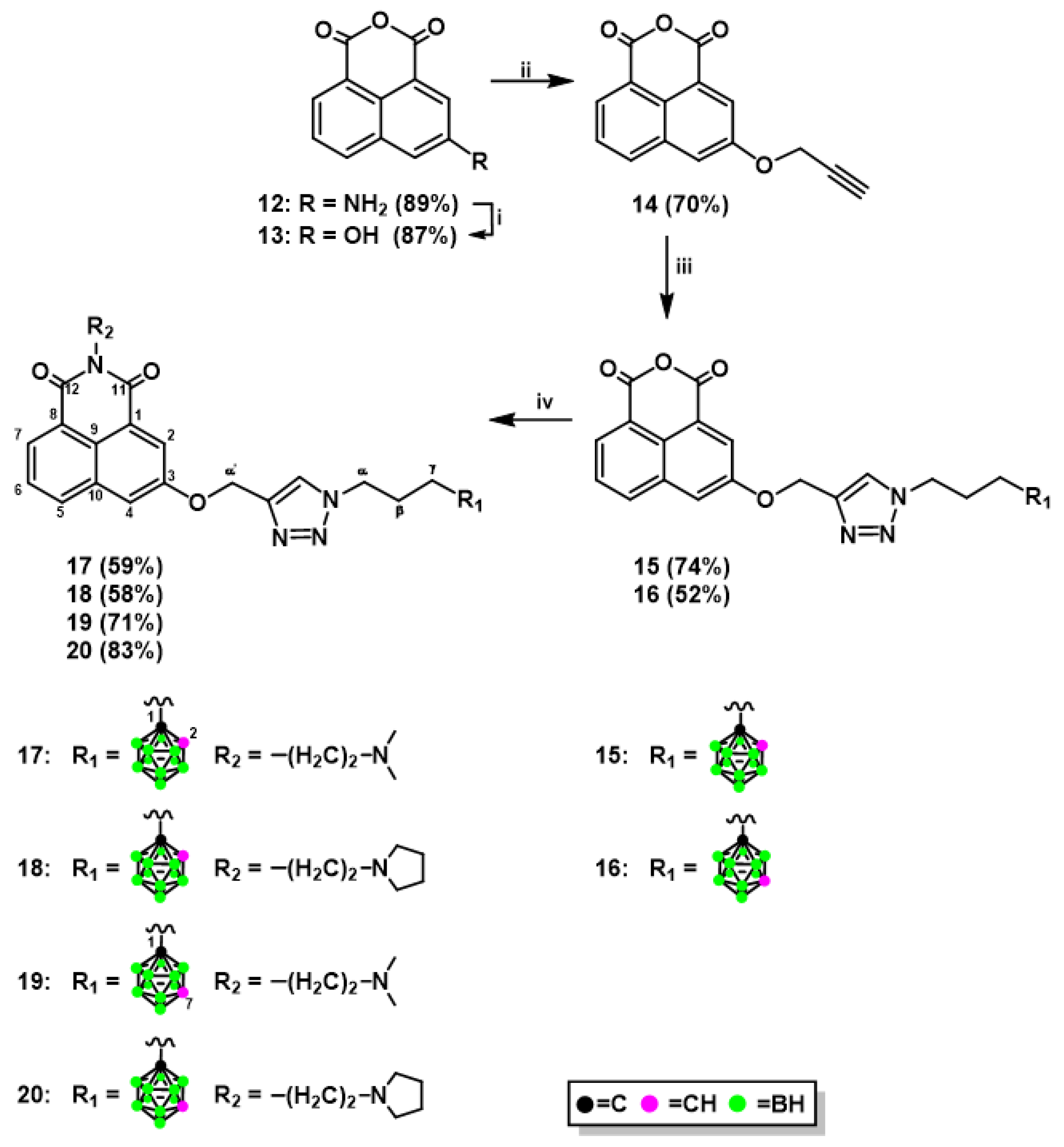
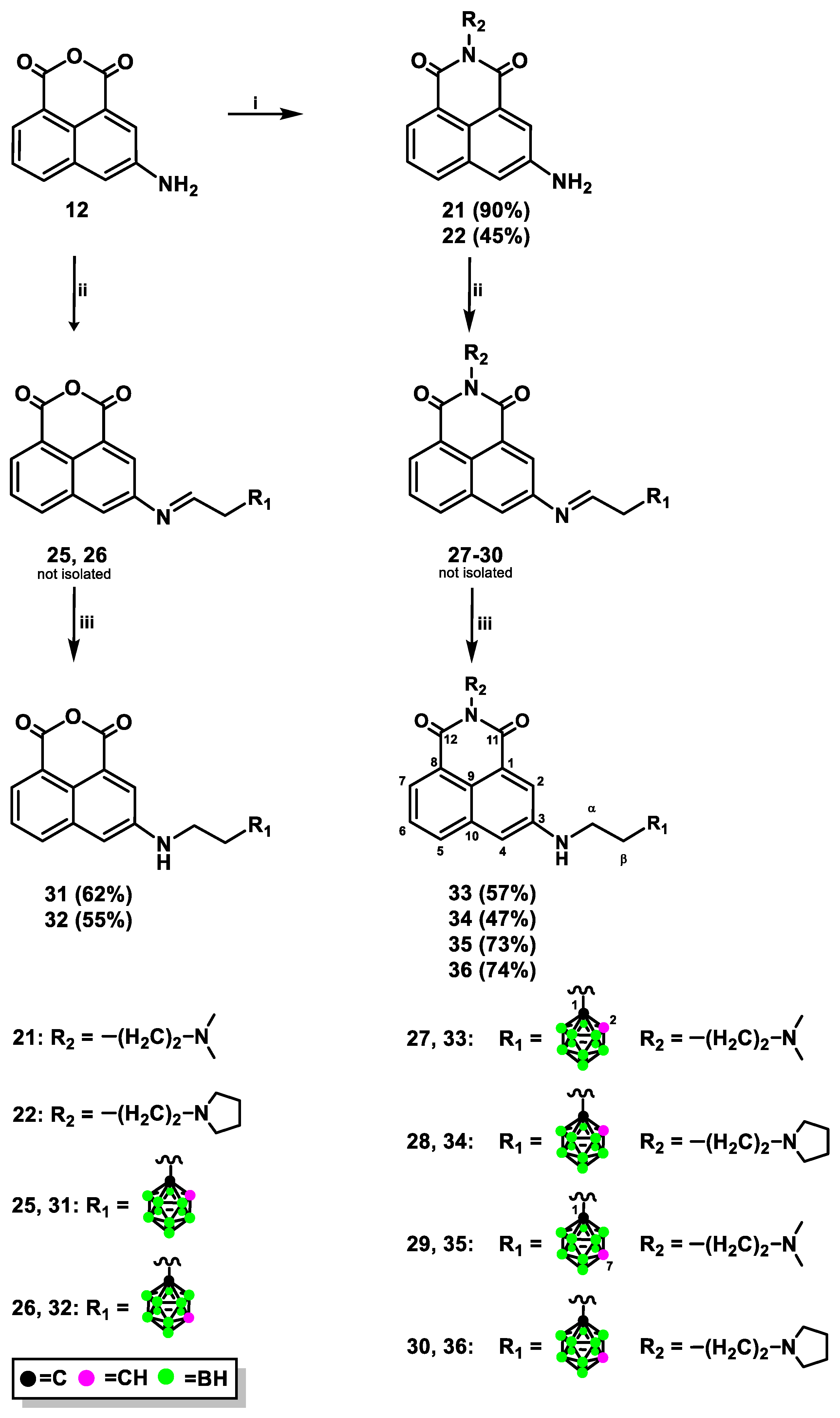
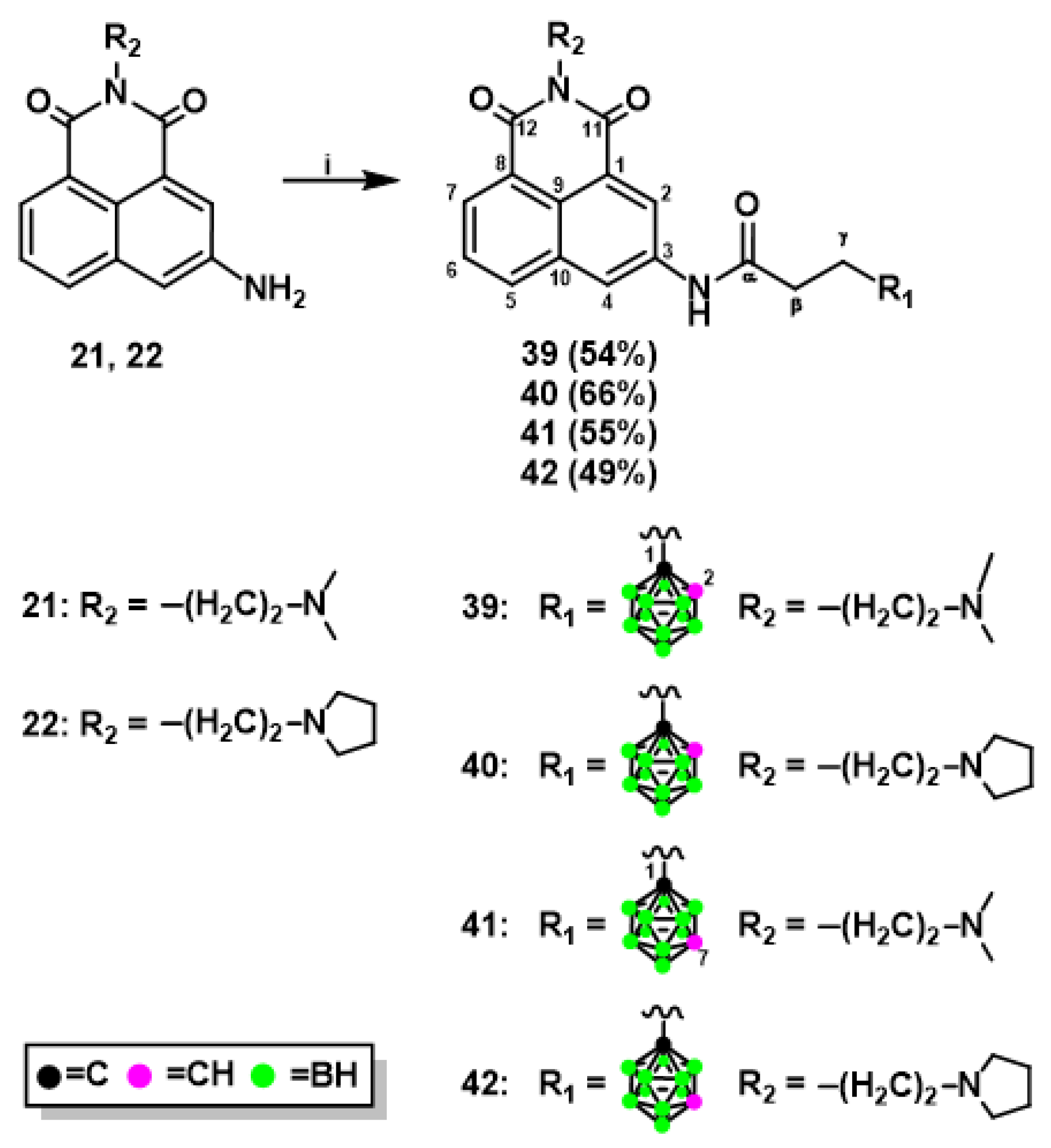
2.1.1. Synthesis of Mitonafide and Pinafide Analogs Containing Carborane Clusters
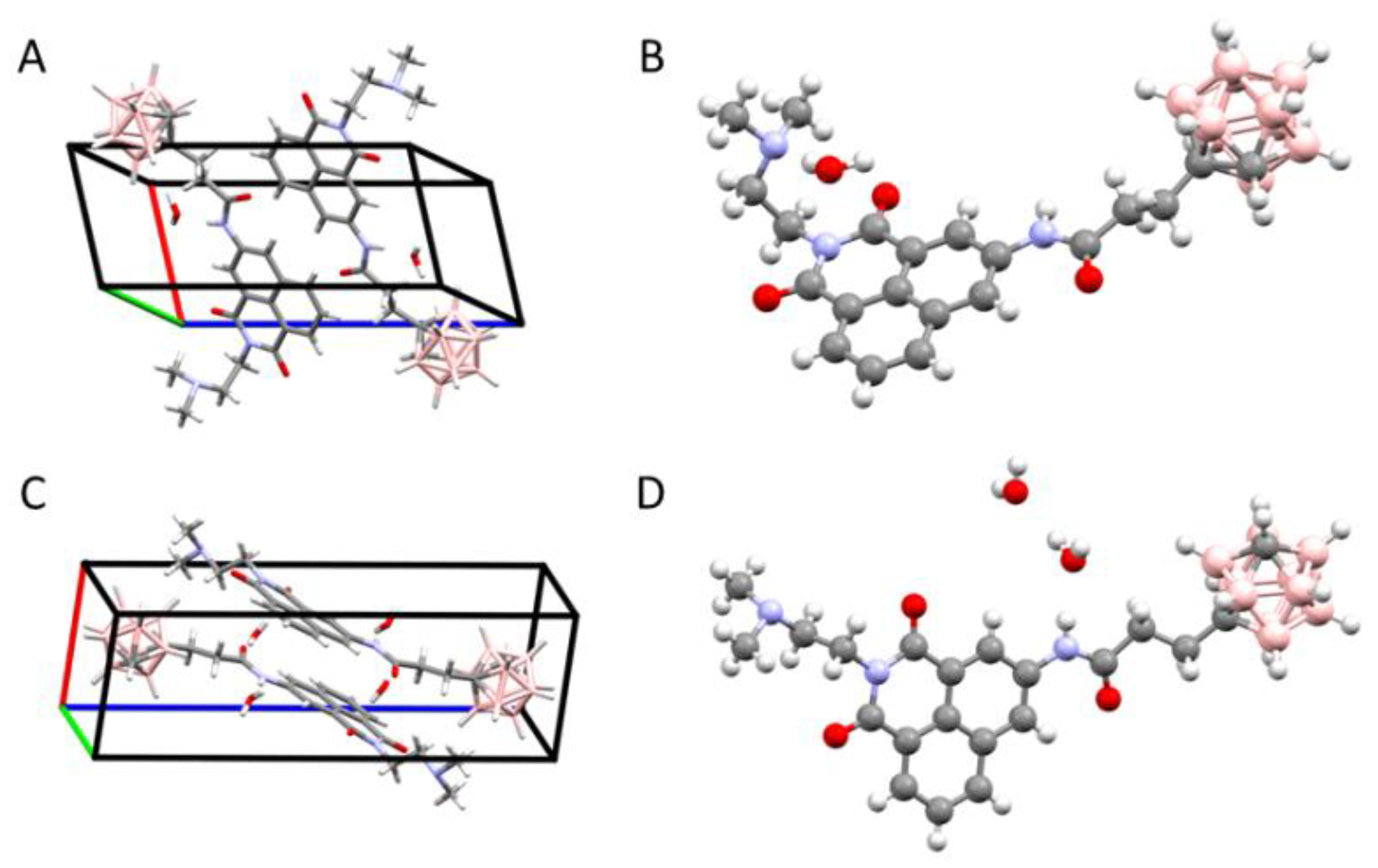
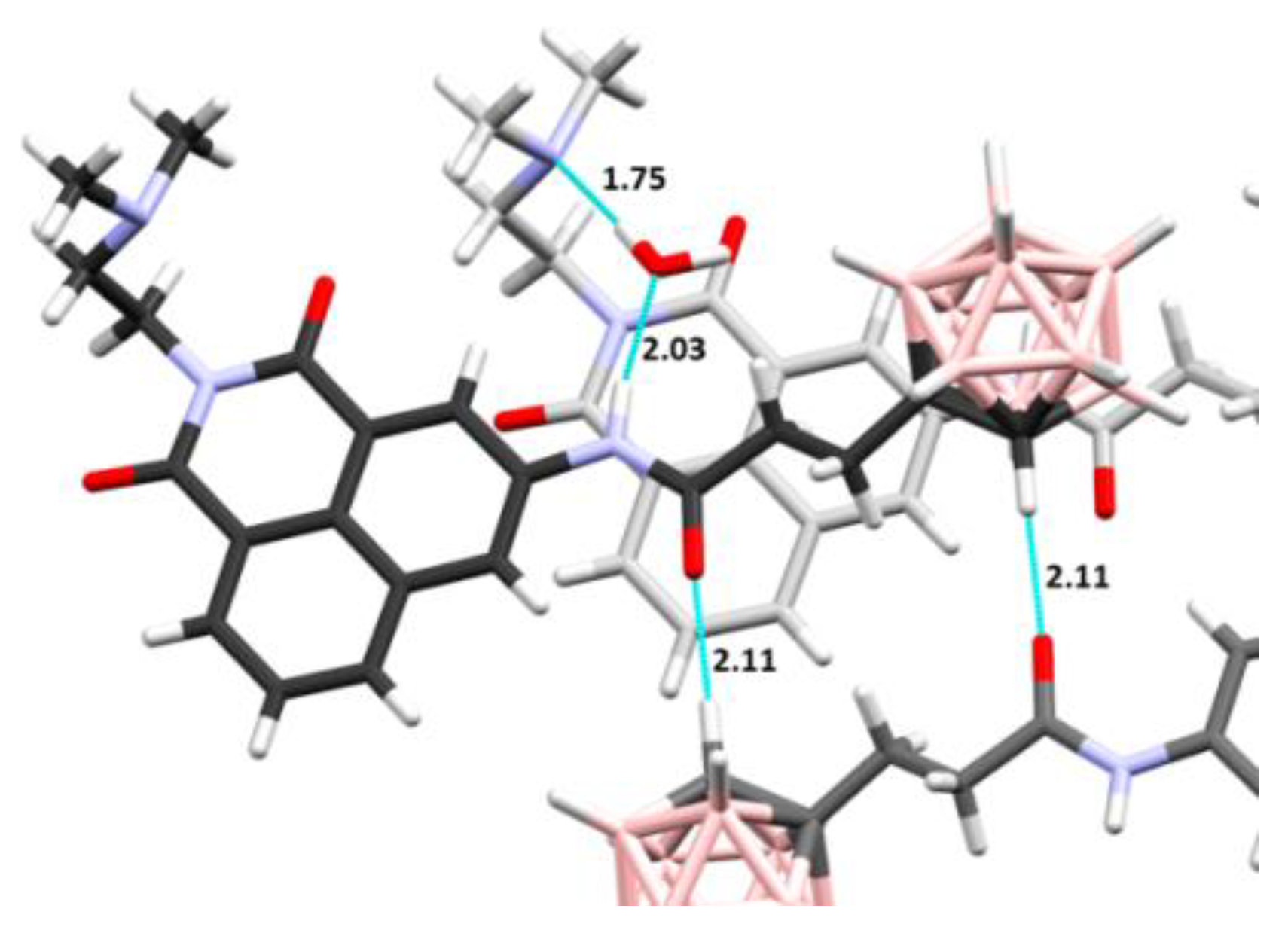
2.1.2. X-ray Structural Analysis
2.2. Biological Investigation
2.2.1. In Vitro Cytotoxic Activity
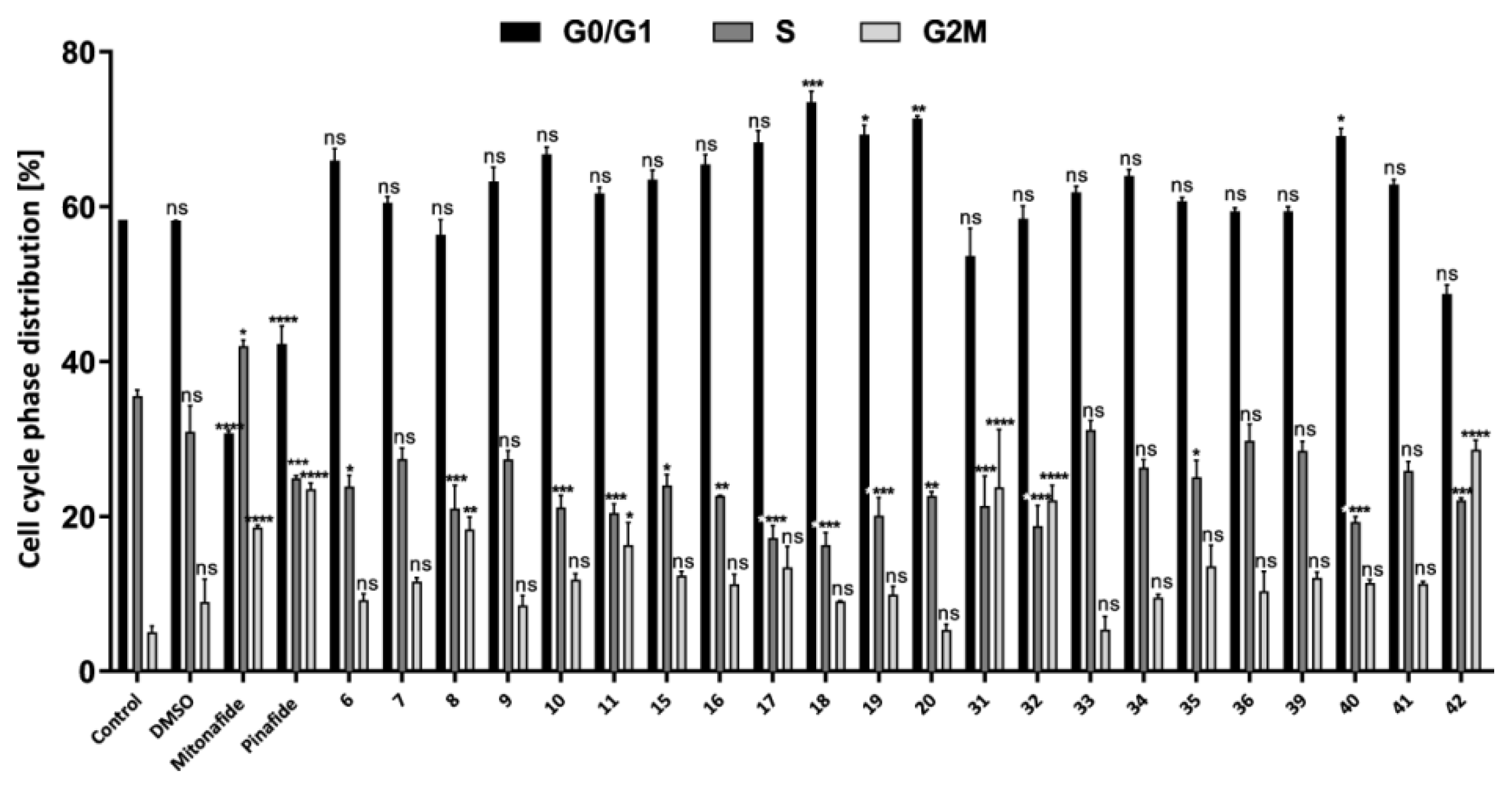
2.2.2. Cell Cycle Analysis by Flow Cytometry
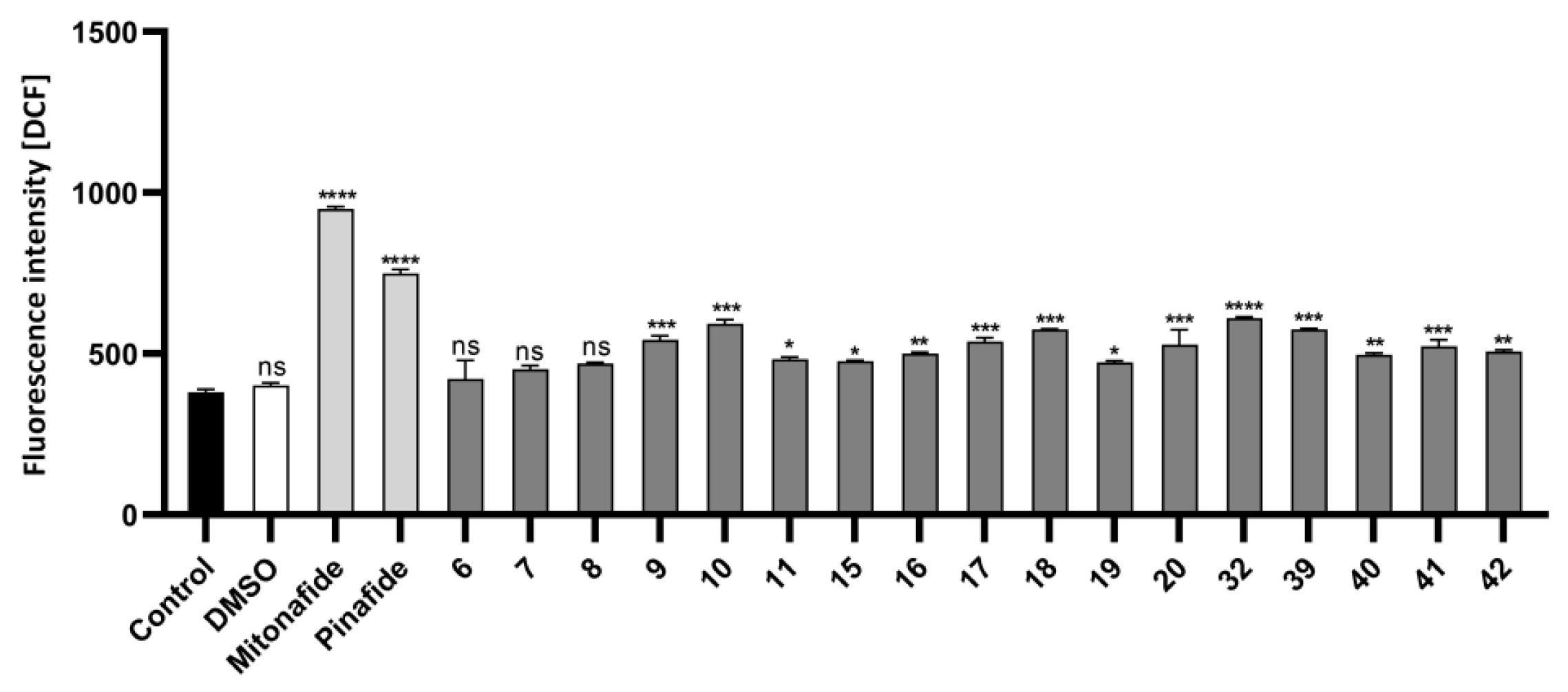
2.2.3. Oxidative Stress Measurement in HepG2 Cells by Flow Cytometry
2.2.4. Analysis of 8-oxo-dG in HepG2 Cells
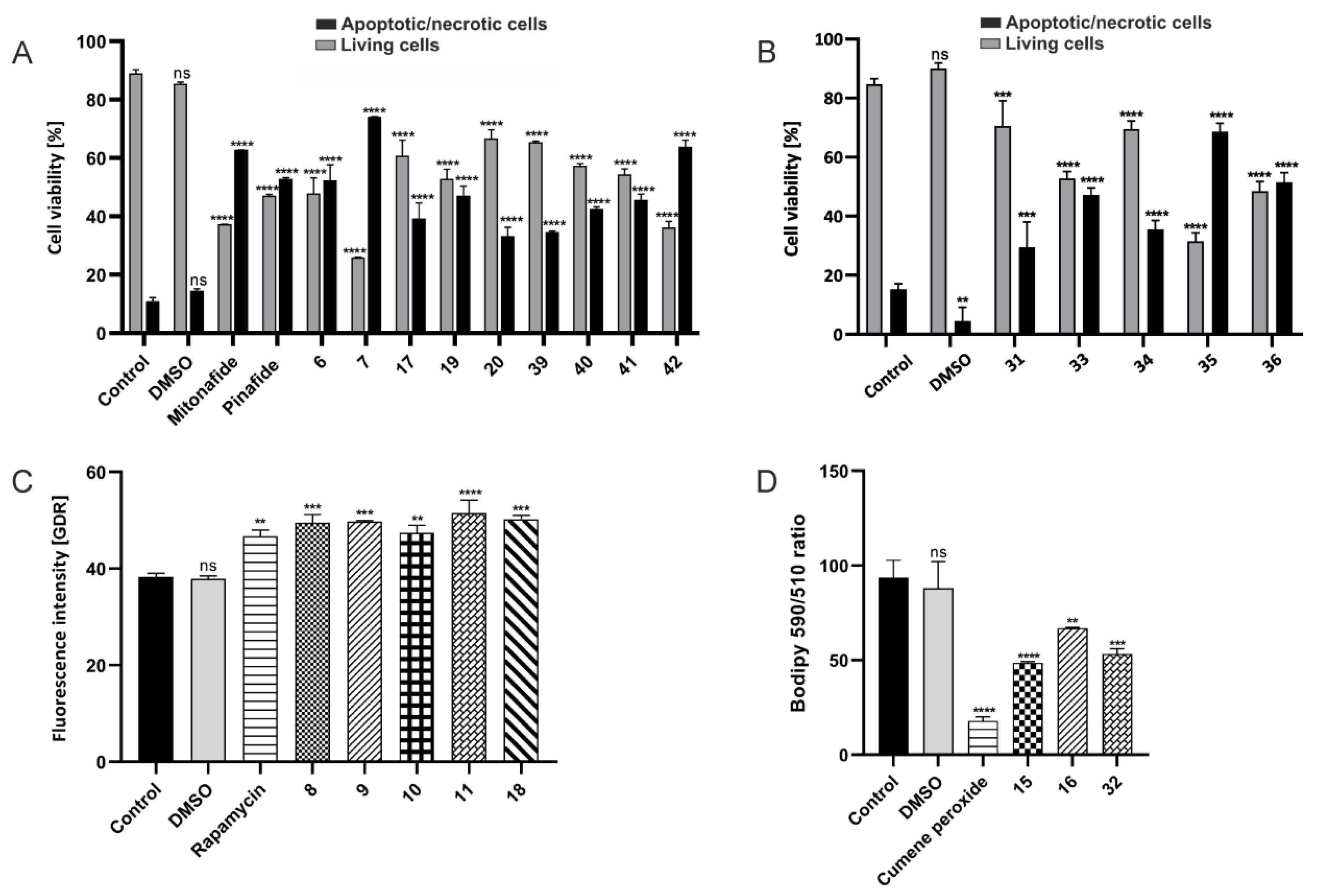
2.2.5. Apoptosis/Necrosis, Autophagy, and Ferroptosis Assays by Flow Cytometry
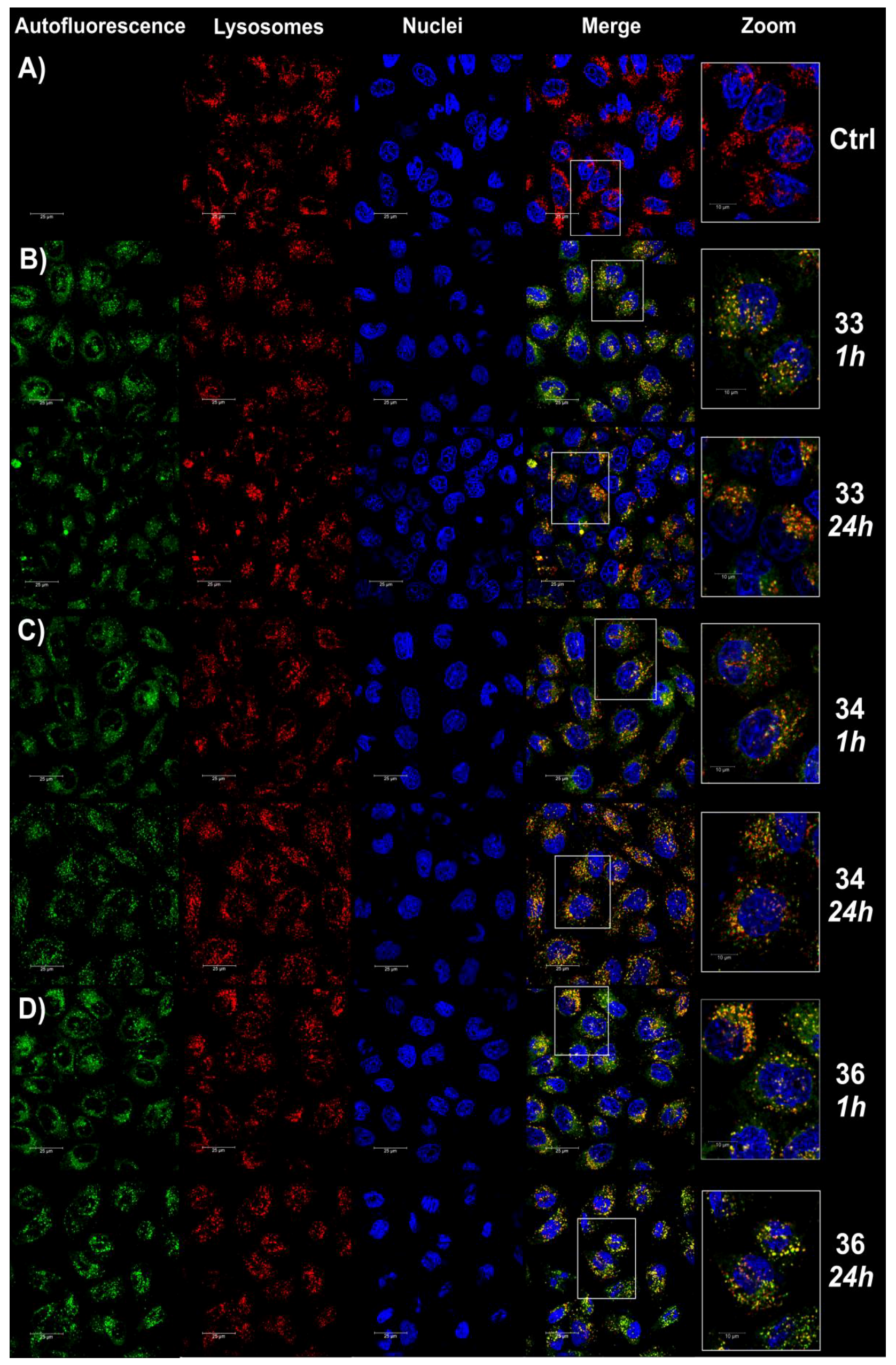
2.2.6. Fluorescence Imaging of Lysosomes
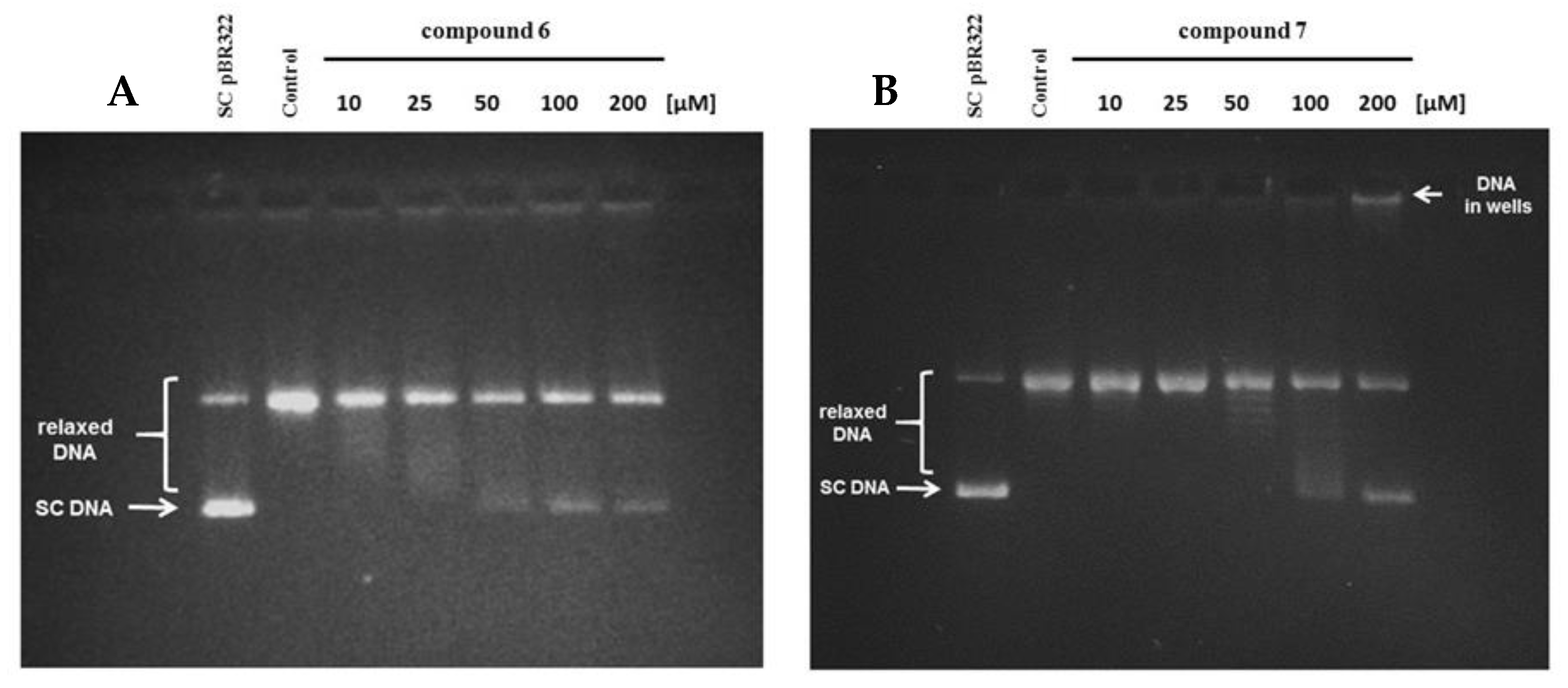
2.2.7. Human Topoisomerase IIα Relaxation Assay
2.3. Physicochemical Investigation with DNA
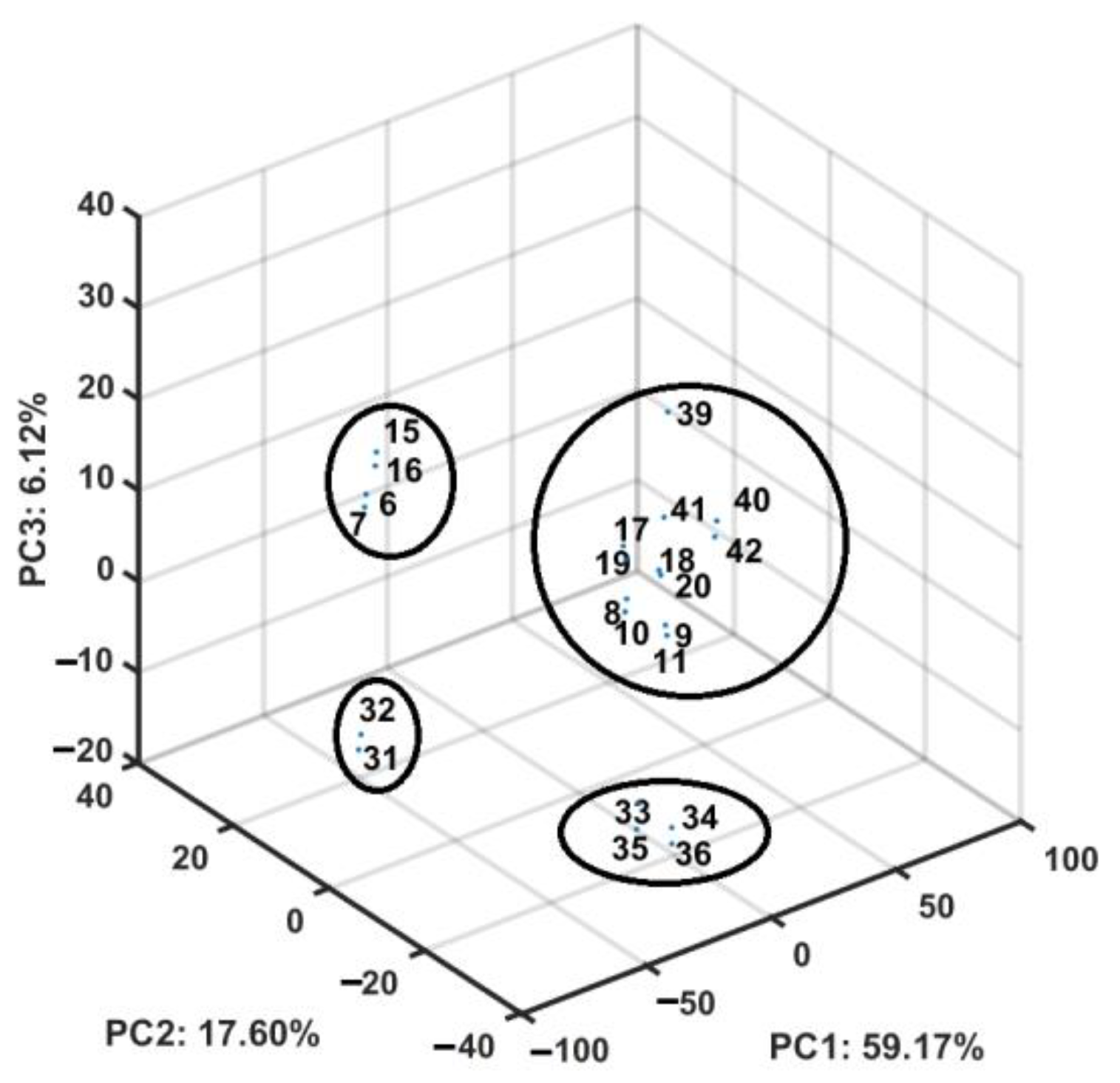
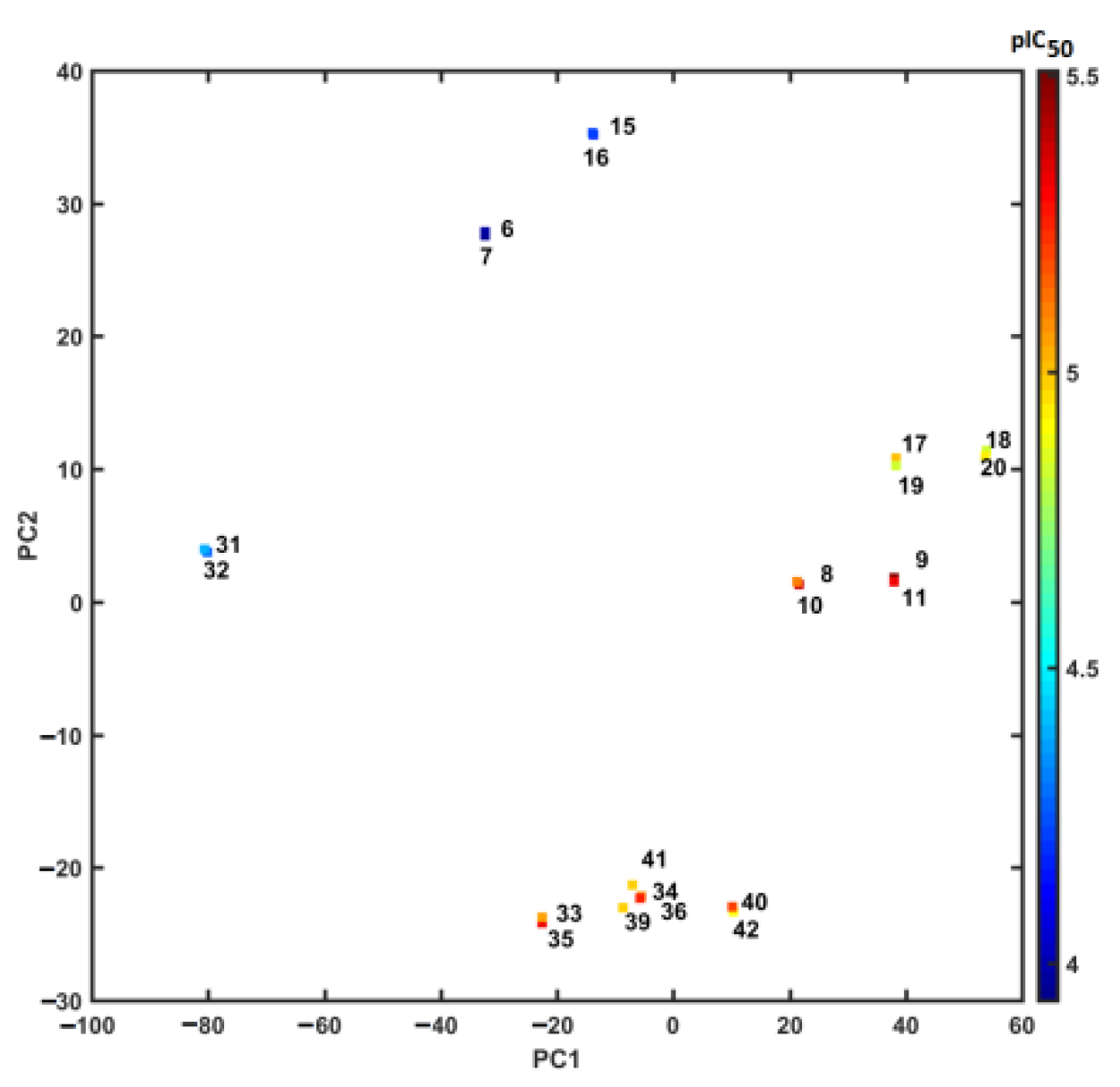
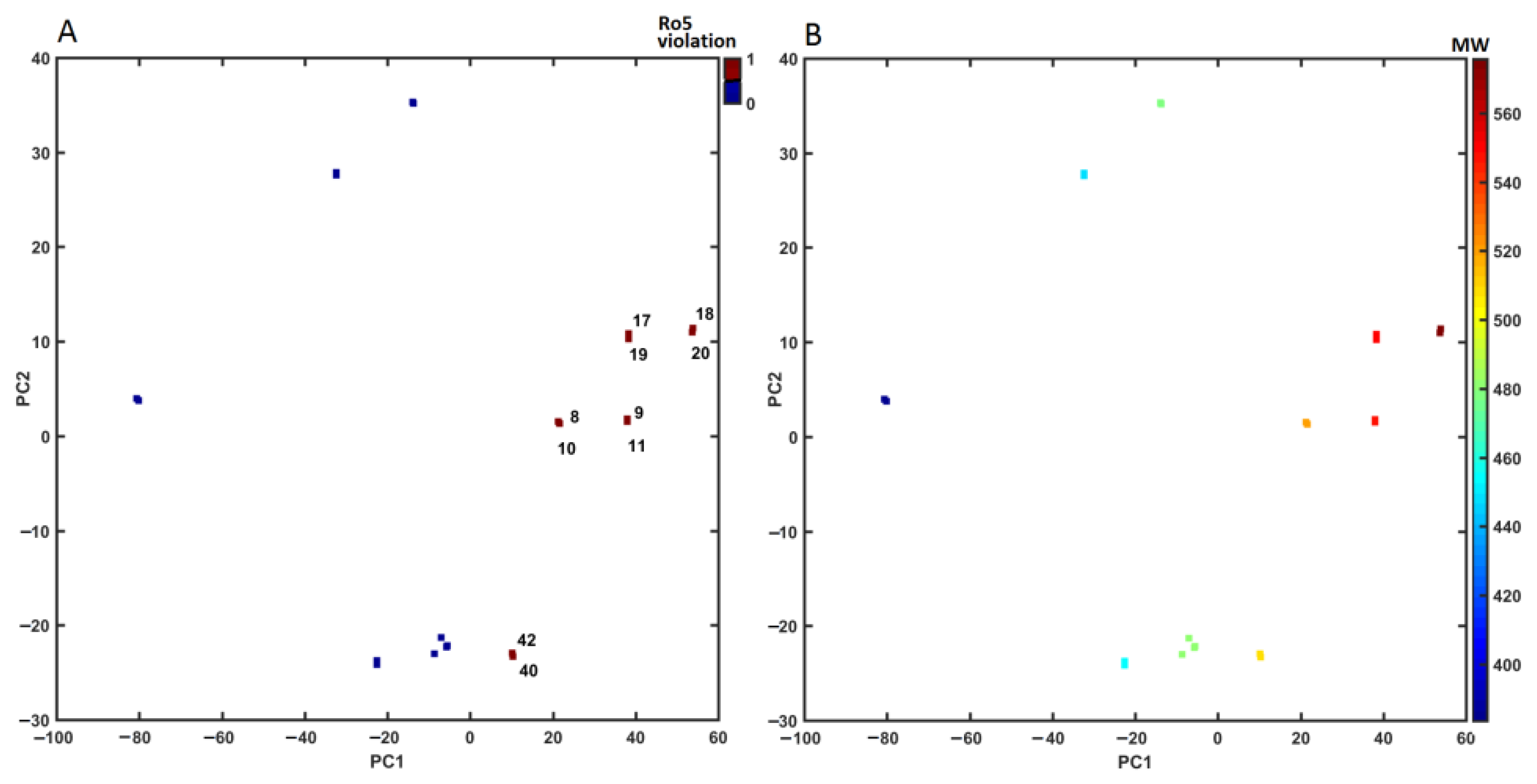
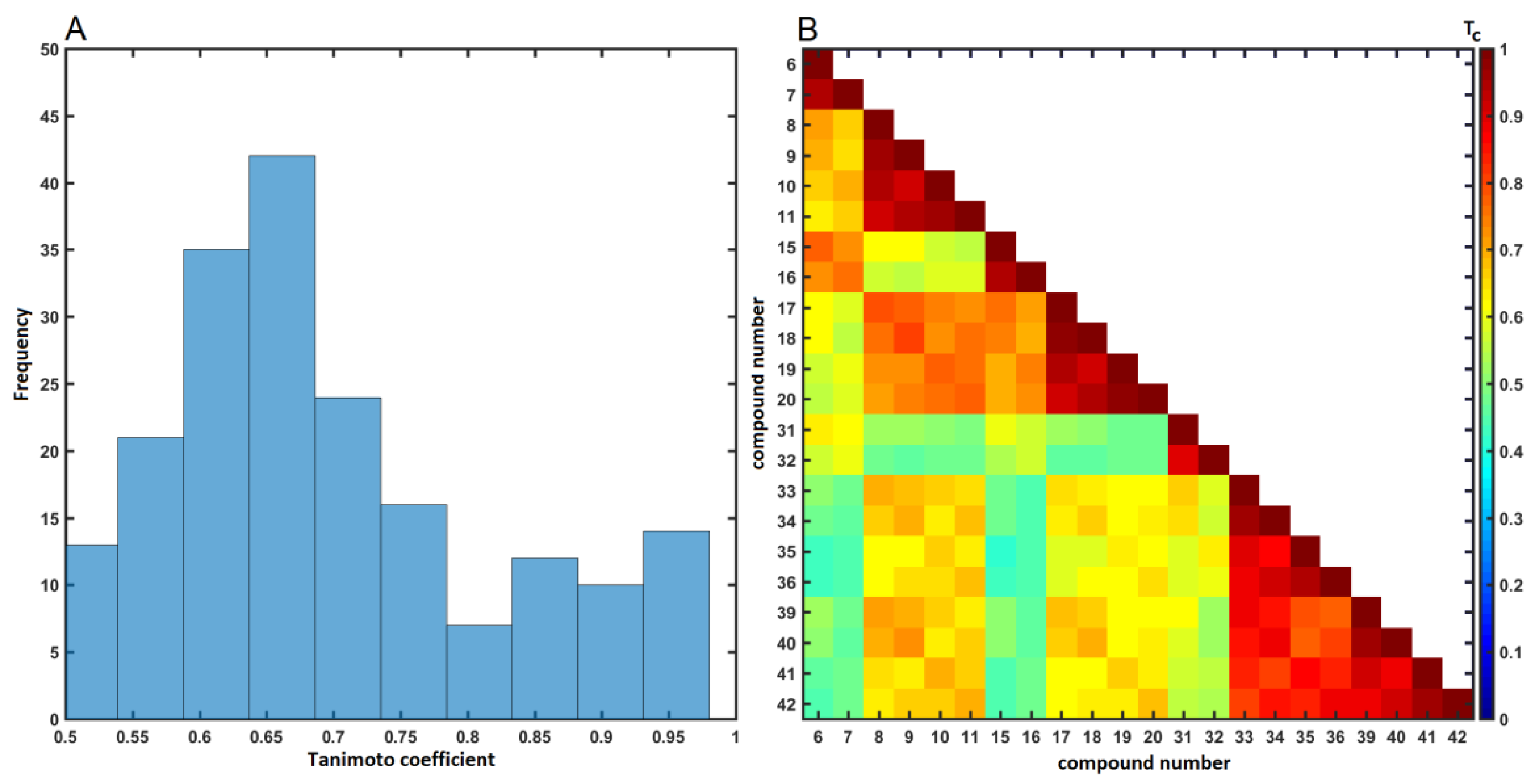
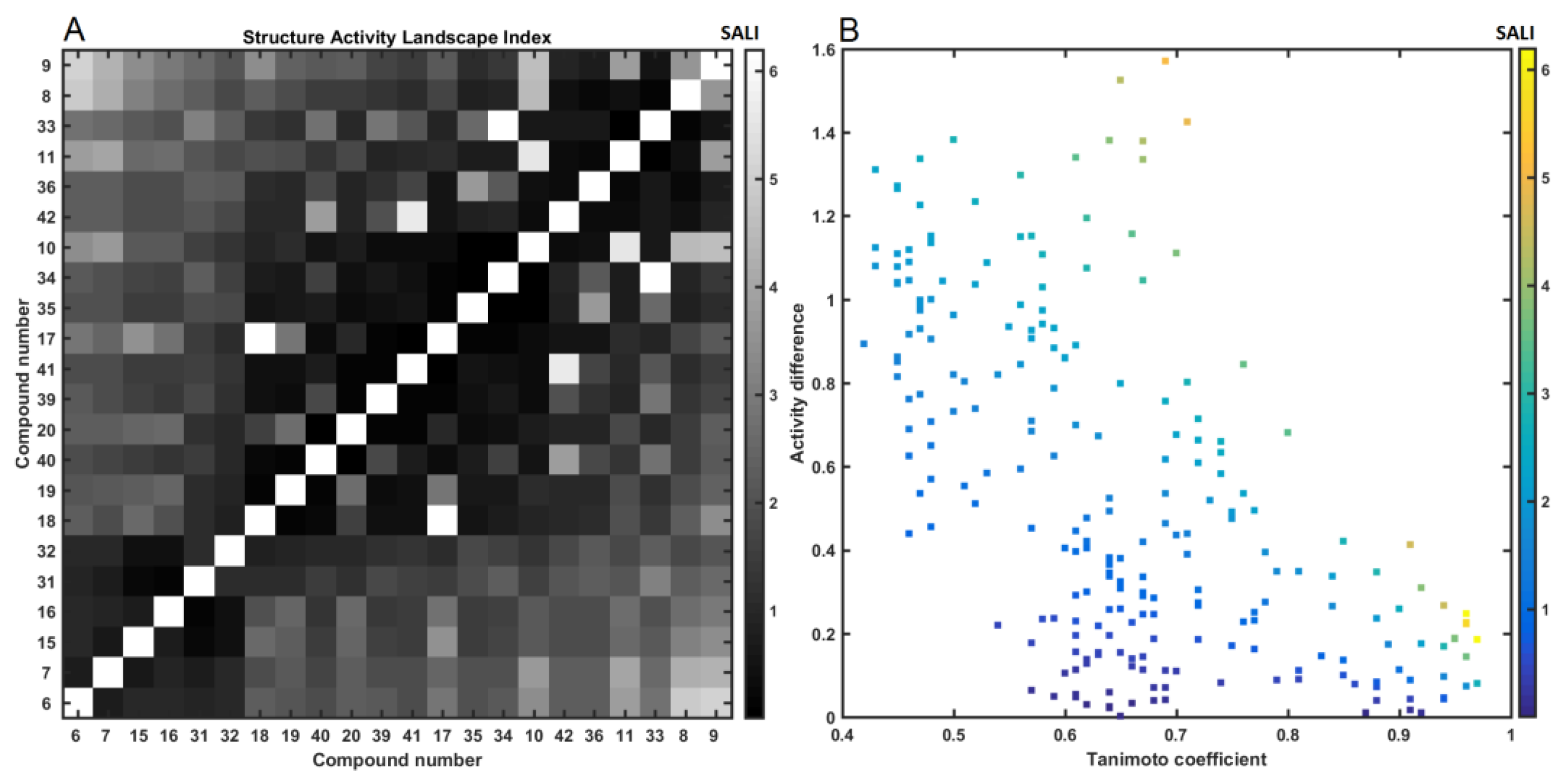
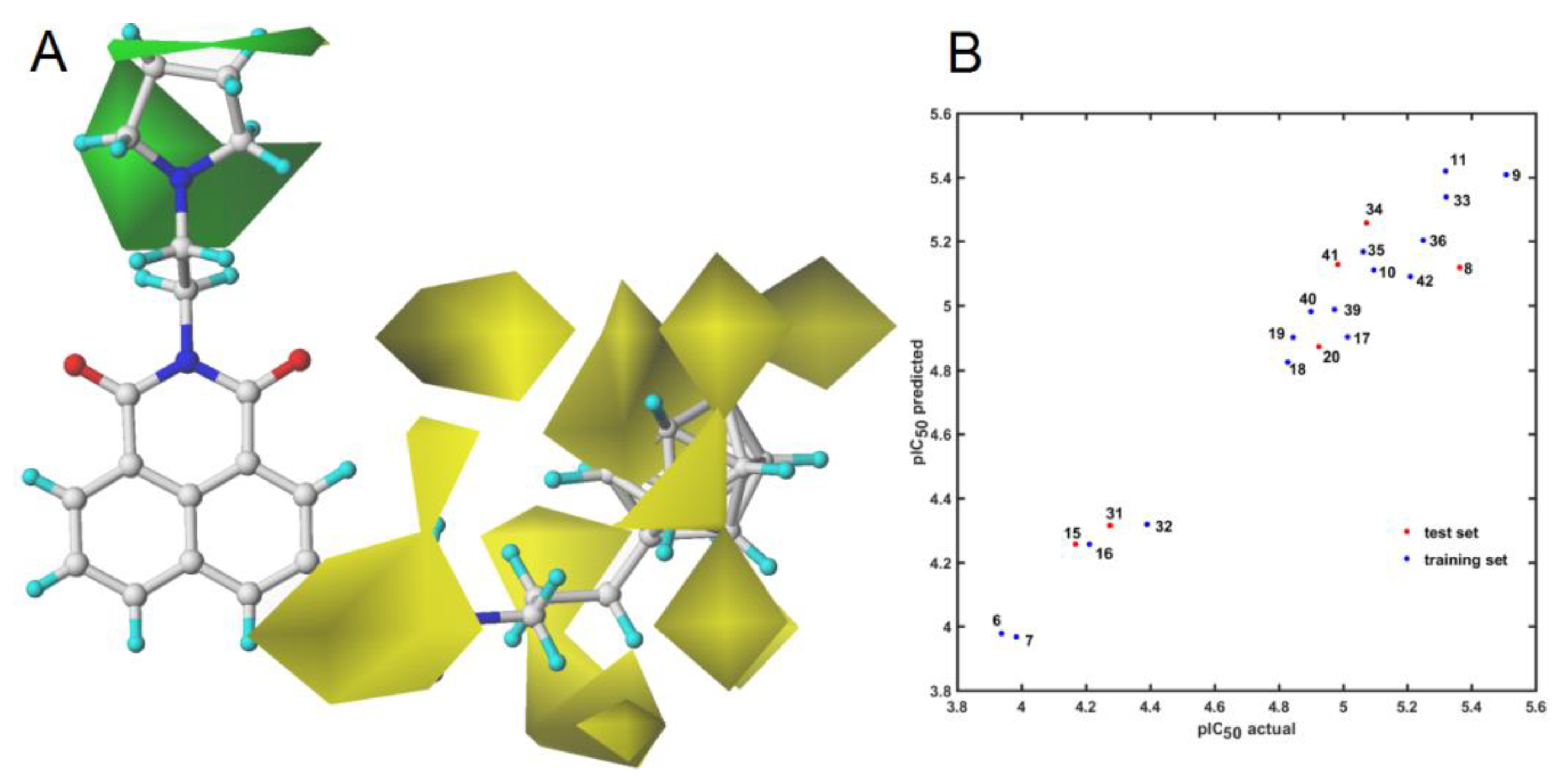
2.4. Similarity-Based Assessment of Property Profile
3. Materials and Methods
3.1. Chemistry
3.1.1. Synthesis of 3-Ethynyl-1,8-Naphthalic Anhydride (3)
3.1.2. Synthesis of 3-Prop-2-yn-1-yloxy-1,8-Naphthalic Anhydride (14)
3.1.3. Synthesis of 1,8-Naphthalic Anhydride Derivatives 6, 7, and Naphthalimide Derivatives 8–11 Modified with Carborane Clusters via Click Reactions
3.1.4. Synthesis of 1,8-Naphthalic Anhydride Derivatives 15, 16, and Naphthalimide Derivatives 17–20 Modified with Carborane Clusterc via Click Reactions
3.1.5. Synthesis of 1,8-Naphthalic Anhydride Derivatives 31, 32 Modified with Carborane Cluster via Reductive Amination
3.1.6. Synthesis of Naphthalimide Derivatives 33-36 Modified with Carborane Cluster via Reductive Amination
3.1.7. Synthesis of Naphthalimide Derivatives 39-42 Modified with Carborane Cluster via Amidation Reaction
3.2. Biological Investigation
3.2.1. Cytotoxicity Assay
3.2.2. Cell Cycle Analysis by Flow Cytometry
3.2.3. Oxidative Stress Measurements in HepG2 Cells by Flow Cytometry
3.2.4. Analysis of 8-Oxo-dG in HepG2 Cells by HPLC-UV-ED
3.2.5. Apoptosis/Necrosis Assay by Flow Cytometry
3.2.6. Apoptosis Detection Using Annexin V Conjugate Staining
3.2.7. Autophagy Assay by Flow Cytometry
3.2.8. Lipid Peroxidation Measurements by Flow Cytometry
3.2.9. Fluorescence Imaging Experiment
3.2.10. Human Topoisomerase IIα Relaxation Assay
3.2.11. Statistical Analysis
3.3. Physicochemical Investigation with DNA
3.3.1. Materials
3.3.2. Preparation of ct-DNA
3.3.3. Melting Temperature (Tm) Measurements
3.3.4. Circular Dichroism Measurements
3.3.5. Ultraviolet-Visible Spectra Titration
3.4. Theoretical Calculations
3.4.1. Model Building and CoMFA Modeling
3.4.2. Similarity-Based Activity Landscape (SALI)
3.4.3. Principal Component Analysis (PCA) and Partial Least Squares Method (PLS)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kamal, A.; Bolla, N.R.; Srikanth, P.S.; Sirvastava, A.K. Naphthalimide derivatives with therapeutic characteristics: A patent review. Expert Opin. Ther. Pat. 2013, 23, 299–317. [Google Scholar] [CrossRef]
- Shen, K.; Sun, L.; Zhang, H.; Xu, Y.; Qian, X.; Lu, Y.; Li, Q.; Ni, L.; Liu, J. A ROS-mediated lysosomal-mitochondrial pathways is induced by a novel amonafide analogue, 7c, in human HeLa cervix carcinoma cells. Cancer Lett. 2013, 333, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Li, Y.; Wang, Z.; Xiao, M.; Yin, P.; Lu, Y.; Qian, X.; Xu, Y.; Liu, J. 7b, a novel naphthalimide derivative, exhibited anti-inflammatory effects via targeted-inhibiting TAK-1 following down-regulation of ERK1/2- and p38 MAPK-mediated activation of NF-κB in LPS-stimulated RAW264.7 macrophages. Int. Immunopharmacol. 2013, 17, 216–228. [Google Scholar] [CrossRef] [PubMed]
- Pain, A.; Samanta, S.; Dutta, S.; Saxena, A.K.; Shanmugavel, M.; Kampasi, H.; Quazi, G.N.; Sanyula, U. Synthesis and evaluation of substituted naphthalimide nitrogen mustards as rationally designed anticancer compound. Acta Pol. Pharm. 2003, 60, 285–291. [Google Scholar] [PubMed]
- Jia, T.; Fu, C.; Huang, C.; Yang, H.; Jia, N. Highly sensitive naphthalimide-based fluorescence polarization probe for detecting cancer cells. ACS Appl. Mater. Inter. 2015, 7, 10013–10021. [Google Scholar] [CrossRef]
- Xu, Z.; Ren, Y.Y.; Fan, X.; Cheng, S.; Xu, Q.; Xu, L. A naphthalimide-based fluorescent probe for highly selective detection of pyrophosphate in aqueous solution and living cells. Tetrahedron 2015, 71, 5055–5058. [Google Scholar] [CrossRef]
- Lee, Y.H.; Tang, Y.; Verwilst, P.; Lin, W.; Kim, J.S. A biotin-guided formaldehyde sensor selectively detecting endogenous concentrations in cancerous cells and tissues. Chem. Commun. 2016, 52, 11247–11250. [Google Scholar] [CrossRef]
- Zhang, L.; Su, F.; Kong, X.; Lee, F.; Sher, S.; Day, K.; Tian, Y.; Meldrum, D.R. 1,8-Nahphtalimide derivative dyes targeting live cell mitochondria with large stokes shift. ChemBioChem 2016, 17, 1719–1724. [Google Scholar] [CrossRef] [Green Version]
- Tomczyk, M.D.; Walczak, K.Z. 1,8-Naphthalimide based DNA intercalators and anticancer agents. A systematic review from 2007 to 2017. Eur. J. Med. Chem. 2018, 159, 393–422. [Google Scholar] [CrossRef]
- Tandon, R.; Luxami, V.; Kaur, H.; Tandon, N.K. Paul, 1,8-Naphthalimide: A potent DNA intercalator and target for cancer therapy. Chem. Rec. 2017, 17, 1–39. [Google Scholar] [CrossRef]
- Wang, K.-R.; Qian, F.; Sun, Q.; Ma, C.-I.; Rong, R.-X.; Cao, Z.-R.; Wang, X.-M.; Li, X.-L. Substituents effects on cytotoxic activity, spectroscopic property, and DNA binding property of naphthalimide derivatives. Chem. Biol. Drug Des. 2016, 87, 664–672. [Google Scholar] [CrossRef]
- Scholz, M.; Hey-Hawkins, E. Carbaboranes as pharmacophores: Properties, synthesis and application strategies. Chem. Rev. 2011, 111, 7035–7062. [Google Scholar] [CrossRef]
- Issa, F.; Kassiou, M.; Rendina, L.M. Boron in drug discovery: Carboranes as unique pharmacophores in biologically active compounds. Chem. Rev. 2011, 111, 5701–5722. [Google Scholar] [CrossRef]
- Leśnikowski, Z.J. Challenges and opportunities for the application of boron clusters in drug design. J. Med. Chem. 2016, 59, 7738–7758. [Google Scholar] [CrossRef]
- Leśnikowski, Z.J. Recent developments with boron as a platform for novel drug design. Expert Opin. Drug Dis. 2016, 11, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Goszczyński, T.M.; Fink, K.; Boratyński, J. Icosahedral boron clusters as modifying entities for biomolecules. Expert Opin. Drug Dis. 2018, 18, 205–213. [Google Scholar] [CrossRef]
- Hey-Hawkins, E.; Vinas Teixidor, C. Boron-based Compounds: Potential and Emerging Applications in Medicine, 1st ed.; John Wiley & Sons Ltd: Hoboken, NJ, USA, 2018. [Google Scholar]
- Ali, F.; Hosmane, N.S.; Zhu, Y. Boron chemistry for medical applications. Molecules 2020, 25, 828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leśnikowski, Z.J. What are the current challenges with the application of boron clusters to drug design. Expert Opin. Drug Dis. 2020. [Google Scholar] [CrossRef]
- Nekvinda, J.; Różycka, D.; Rykowski, S.; Wyszko, E.; Fedoruk-Wyszomirska, A.; Gurda, D.; Orlicka-Płocka, M.; Giel-Pietraszuk, M.; Kiliszek, A.; Rypniewski, W.; et al. Synthesis of naphthalimide-carborane and metallacarborane conjugates: Anticancer activity, DNA binding ability. Bioorg. Chem. 2020, 94, 103432. [Google Scholar] [CrossRef] [PubMed]
- Meldal, M.; Tornoe, C.W. Cu-catalyzed azide-alkyne cycloaddition. Chem. Rev. 2008, 108, 2952–3015. [Google Scholar] [CrossRef] [PubMed]
- Nwe, K.; Brechbiel, M.W. Growing applications of “click chemistry” for bioconjugation in contemporary biomedical research. Cancer Biother. Radio. 2009, 24, 289–302. [Google Scholar]
- Matuszewski, M.; Kiliszek, A.; Rypniewski, W.; Lesnikowski, Z.J.; Olejniczak, A.B. Nucleoside bearing clusters and their phosphoramidites–building blocks for modified oligonucleotide synthesis. New. J. Chem. 2015, 39, 1202–1221. [Google Scholar] [CrossRef]
- Chan, T.R.; Hilgraf, R.; Sharpless, K.B.; Fokin, V.V. Polytriazoles as Copper(I)-stabilizing ligands in calatysis. Org. Lett. 2004, 6, 2853–2855. [Google Scholar] [CrossRef] [PubMed]
- Afanasyev, O.I.; Kuchuk, E.; Usanov, D.L.; Chusov, D. Reductive amination in the synthesis of pharmaceuticals. Chem. Rev. 2019, 119, 11857–11911. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yang, L.; Houa, C.; Cao, H. A new N-imidazolyl-1,8-naphthalimide based fluorescence sensor for fluoride detection. Org. Biomol. Chem. 2012, 10, 6271–6274. [Google Scholar] [CrossRef]
- Brana, M.F.; Castellano, J.M.; Roldan, C.M.; Santos, A.; Vazquez, D.; Jimenez, A. Synthesis and mode(s) of action of a new series of imide derivatives of 3-nitro-1,8-naphthalic. Cancer Chemoth. Pharm. 1980, 4, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Dozzo, P.; Kasar, R.A.; Kahl, S.B. Simple, High-yield methods for the synthesis of aldehydes directly from o-, m-, and p-carborane and their further conversions. Inorg. Chem. 2005, 44, 8053–8057. [Google Scholar] [CrossRef]
- Bae, J.W.; Lee, S.H.; Jung, Y.J.; Yoon, C.-O.M.; Yoon, C.M. Reduction of ketones to alcohols using a decaborane/pyrrolidine/cerium(III) chloride system in methanol. Tetrahedron Lett. 2001, 42, 2137–2139. [Google Scholar] [CrossRef]
- Bae, J.W.; Lee, S.H.; Cho, Y.J.; Yoon, C.M. A reductive amination of carbonyls with amines using decaborane in methanol. J. Chem. Soc. Perkin Trans. 2000, 1, 145–146. [Google Scholar] [CrossRef]
- Lee, S.H.; Park, Y.J.; Yoon, C.M. Reductive etherification of aromatic aldehydes with decaborane. Tetrahedron Lett. 1999, 40, 6049–6050. [Google Scholar] [CrossRef]
- Bae, J.W.; Jung, Y.J.; Lee, S.H.; Yoon, C.-O.M.; Yoon, C.M. A one-pot synthesis of N-alkylaminobenzenes from nitroaromatics: Reduction followed by reductive amination of using B10H14. Chem. Commun. 2000, 19, 1857–1858. [Google Scholar] [CrossRef]
- Brown, D.G.; Boström, J. Analysis of past and present synthetic methodologies on medicinal chemistry: Where have all the new reaction gone? J. Med. Chem. 2016, 59, 4443–4458. [Google Scholar] [CrossRef] [Green Version]
- Malmquist, J.; Sjöberg, S. Asymmetric Synthesis of p-Carboranylalanine (p-Car) and 2-Methyl-o-Carboranylalanine (Me-o-Car). Tetrahedron 1996, 52, 9207–9218. [Google Scholar] [CrossRef]
- Naeslund, C.; Ghirmai, S.; Sjöberg, S. Enantioselective synthesis of m-carboranylalanine, a boron rich analogue of phenylalanine. Tetrahedron 2005, 61, 1181–1186. [Google Scholar] [CrossRef]
- Różycka, D.; Leśnikowski, Z.J.; Olejniczak, A.B. Synthesis of boron cluster analogs of penicillin and their antibacterial activity. J. Organomet. Chem. 2019, 881, 19–24. [Google Scholar] [CrossRef]
- Różycka, D.; Korycka-Machała, M.; Żaczek, A.; Dziadek, J.; Gurda, D.; Orlicka-Płocka, M.; Wyszko, E.; Biniek-Antosiak, K.; Rypniewski, W.; Olejniczak, A.B. Novel isoniazid-carborane hybrids active in vitro against Mycobacterium Tuberculosis. Pharm. Basel 2020, 13, 465. [Google Scholar]
- Hardie, M.J.; Raston, C.L. Crystalline hydrogen bonded complexes of o-carborane. CrystEngComm 2001, 3, 162–164. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Berg, K.; Hansen, M.B.; Nielsen, S.E. A new sensitive bioassay for precise quantification of interferon activity as measured via the mitochondrial dehydrogenase function in cells (MTT-method). APMIS 1990, 98, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Yang, S.; Li, S.; Liu, S.; Tang, S.; Liu, Z.; Meng, X.; Yu, S. A novel triazolonaphthalimide induces apoptosis and inhibits tumor growth by targeting DNA and DNA-associated processes. Oncotarget 2017, 8, 37394–37408. [Google Scholar] [CrossRef] [Green Version]
- Park, J.M.; Shin, Y.J.; Riew, T.R.; Lee, M.Y. The indilinone MAZ51 induces cell rounding and G2M cell cycle arrest in glioma cells without the inhibition of VEGFR-3 phoshorylation: Involvment of the RhoA and Akt/GSK3beta signaling pathways. PLoS ONE 2014, 9, e109055. [Google Scholar]
- Kasai, H.; Nishimura, S. Hydroxylation of deoxyguanosine at the C-8 position by ascorbic acid and other reducing agents. Nucleic Acids Res. 1984, 12, 2137–2145. [Google Scholar] [CrossRef] [Green Version]
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef]
- Xie, S.Q.; Li, Q.; Zhang, Y.H.; Wang, J.H.; Mei, Z.H.; Zhao, J.; Wang, C.J. NPC-16, a novel naphthalimide-polyamine conjugate, induced apoptosis and autophagy in human hepatoma HepG2 cells and Bel-7402 cells. Apoptosis 2011, 16, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Ling, J.; Kim, S.G.; Blenis, J. Rapamycin: One drug, many effects. Cell Metab. 2014, 19, 373–379. [Google Scholar]
- Li, M.; Yang, Z.; Vollmer, L.L.; Gao, Y.; Fu, Y.; Liu, C.; Chen, X.; Liu, P.; Vogt, A.; Yin, X.M. AMDE-1 is a dual function chemical for autophagy activation and inhibition. PLoS ONE 2015, 10, e0122083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Cao, F.; Yin, H.; Huang, Z.; Lin, Z.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 888. [Google Scholar] [CrossRef] [PubMed]
- Bonam, S.R.; Wang, F.; Muller, S. Lysosomes as a therapeutic target. Nat. Rev. Drug Discov. 2019, 18, 923–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, W.; Jiang, H.; Tang, X.; Liu, W. A reversible ratiometric two-photon lysosome-targeted probe for real-time monitoring of pH changes in living cells. J. Mater. Chem. B 2017, 5, 4768–4773. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Liang, X.; Zhang, H.; Xie, H.; Liu, J.; Xu, Y.; Zhu, W.; Wang, Y.; Wang, X.; Tan, S.; et al. A new class of naphthalimide-based antitumor agents that inhibit Topoisomarese II and induce lysosomal membrane permeabilization and apoptosis. J. Med. Chem. 2010, 53, 2589–2600. [Google Scholar] [CrossRef] [PubMed]
- Rong, R.-X.; Wang, S.-S.; Liu, X.; Li, R.-F.; Wang, K.-R.; Cao, Z.-R.; Li, X.-L. Lysosomes-targeting imaging and anitcancer properties of novel bis-naphthalimide derivatives. Bioorg. Med. Chem. Lett. 2018, 28, 742–747. [Google Scholar] [CrossRef]
- Tomczyk, M.D.; Byczek-Wyrostek, A.; Strama, K.; Wawszków, M.; Kasprzycki, P.; Walczak, K.Z. Anticancer activity and Topoisomerase II inhibition of naphthalimides with ω-Hydroxylalkylamine Side-Chains of Different Lengths. Med. Chem. 2019, 15, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, L.R.; Baguley, B.C. Topoisomerase II enzymes and mutagenicity. Environ. Mol. Mutagen. 1994, 24, 245–261. [Google Scholar] [CrossRef]
- Wijeratne, S.S.; Patel, J.M.; Kiang, C.H. Melting Transitions of DNA-Capped Gold Nanoparticle Assemblies; Springer: New York, NY, USA, 2012; pp. 269–282. [Google Scholar]
- Mergny, J.L.; Duval-Valentin, G.; Nguyen, C.H.; Perrouault, L.; Faucon, B.; Rougee, M.; Montenay Garestier, T.; Bisagni, E.; Helene, C. Triple helix-specific ligands. Science 1992, 256, 1681–1684. [Google Scholar] [CrossRef]
- Lenglet, G.; David-Cordonnier, M.-H. DNA-destabilizing agents as an alternative approach for targeting DNA: Mechanism of action and cellular consequence. J. Nucleic. Acids 2010. [Google Scholar] [CrossRef] [Green Version]
- Lesnik, E.A.; Guinosso, C.J.; Kawasaka, A.M.; Sasmor, H.; Zounes, M.; Cummins, L.L.; Ecker, D.J.; Cook, P.D.; Freier, S.M. Oligonucleotides containing 2′-O-modified adenosine: Synthesis and effects on stability of DNA:RNA duplex. Biochem. US 1993, 32, 7832–7838. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, V.I.; Minchenkova, L.E.; Schyolkina, A.K.; Poletayev, A.I. Different conformations of double-stranded nucleic acids in solution as revealed by circular dichroism. Biopolymers 1973, 12, 89–110. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Shang, L.; Wang, Z.X.; Dong, S.J. Spectrometric and voltammetric investigation of interaction of neutral red with calf thymus DNA: pH effect. Biophys. Chem. 2005, 118, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Mati, S.S.; Roy, S.S.; Chall, S.; Bhattacharya, S.; Bhattacharya, S.C. Unveiling the groove binding mechanism of a biocompatible naphthalimide-based organoselenocyanate with calf thymus DNA: An “ex vivo” fluorescence imaging application appended by biophysical experiments and molecular docking simulations. J. Phys. Chem. B 2013, 117, 14655–16665. [Google Scholar] [CrossRef]
- Vardevanyan, P.O.; Antonyan, A.P.; Parsadanyan, M.A.; Davtyan, H.G.; Karapetyan, A.T. The binding of ethidium bromide with DNA: Interaction with single- and double-stranded structures. Exp. Mol. Med. 2003, 35, 527–533. [Google Scholar] [CrossRef] [Green Version]
- Peltason, L.; Bajorath, J. Systematic computational analysis of structure-activity relationships: Concepts, challenges and recent advances. Future Med. Chem. 2009, 1, 451–466. [Google Scholar] [CrossRef]
- Bak, A.; Pizova, H.; Kozik, V.; Vorcakova, K.; Kos, J.; Treml, J.; Odehnalova, K.; Oravec, M.; Imramovsky, A.; Bobal, P.; et al. SAR-mediated similarity assessment of the property profile for new, silicon-based AChE/BChE inhibitors. Int. J. Mol. Sci. 2019, 20, 5385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colquhoun, D. The quantitative analysis of drug–receptor interactions: A short history. Trends Pharmacol. Sci. 2006, 27, 149–157. [Google Scholar] [CrossRef]
- Bak, A.; Kozik, V.; Malik, I.; Jampilek, J.; Smolinski, A. Probability-driven 3D pharmacophore mapping of antimycobacterial potential of hybrid molecules combining phenylcarbamoyloxy and N-arylpiperazine fragments. SAR QSAR Environ. Res. 2018, 29, 801–821. [Google Scholar] [CrossRef] [PubMed]
- Grammatica, P. Principles of QSAR models validation: Internal and external. Qsar Comb. Sci. 2007, 26, 694–701. [Google Scholar] [CrossRef]
- Bak, A.; Kozik, V.; Smolinski, A.; Jampilek, J. Multidimensional (3D/4D-QSAR) probability-guided pharmacophore mapping: Investigation of activity profile for a series of drug absorption promoters. RSC Adv. 2016, 6, 76183–76205. [Google Scholar] [CrossRef]
- Maggiora, G.M.; Shanmugasundaram, V. Molecular similarity measures. Methods Mol. Biol. 2011, 672, 39–100. [Google Scholar]
- Lopez-Lopez, E.; Prieto-Martínez, F.D.; Medina-Franco, J.L. Activity landscape and molecular modeling to explore the SAR of dual epigenetic inhibitors: A focus on G9a and DNMT1. Molecules 2018, 23, 3282. [Google Scholar] [CrossRef] [Green Version]
- Gieleciak, R.; Magdziarz, T.; Bak, A.; Polanski, J. Modeling robust QSAR. 1. Coding molecules in 3D-QSAR-from a point to surface sectors and molecular volumes. J. Chem. Inf. Model. 2005, 45, 1447–1455. [Google Scholar] [CrossRef]
- Polanski, J.; Bak, A.; Gieleciak, R.; Magdziarz, T. Modeling robust QSAR. J. Chem. Inf. Model. 2003, 46, 2310–2318. [Google Scholar] [CrossRef]
- Golbraikh, A.; Tropsha, A. Beware of q2! J. Mol. Graph. Mod. 2002, 20, 269–276. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, A71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. App. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Quintana-Espinoza, P.; García-Luis, J.; Amesty, Á.; Martín-Rodríguez, P.; Lorenzo-Castrillejo, I.; Ravelo, A.G.; Fernández-Pérez, L.; Machín, F.; Estévez-Braun, A. Synthesis and study of antiproliferative, antitopoisomerase II, DNA-intercalating and DNA-damaging activities of arylnaphthalimides. Bioorgan. Med. Chem. 2013, 21, 6484–6495. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, K.; Shimabukuro, S.; Mabuchi, M.; Maeda, H. A Naphthalimide-based Cd2+ fluorescent probe with carbamoylmethyl groups working as chelators and PET-promoters under neutral conditions. Chem. Eur. J. 2016, 22, 8579–8585. [Google Scholar] [CrossRef]
- Barciszewska, A.-M.; Giel-Pietraszuk, M.; Perrigue, P.M.; Naskręt-Barciszewska, M. Total DNA methylation changes reflect random oxidative DNA damage in gliomas. Cells 2019, 8, 1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egawa, Y.; Hayashida, R.; Seki, T.; Anzai, J. Fluorometric determination of heparyn based on self-quenching of fluorescein-labeled protamine. Talanta 2008, 76, 736–741. [Google Scholar] [CrossRef] [PubMed]
- Nie, M.Y.; Wang, Y.; Li, H.L. Electrochemical and spectral properties of phenylhydrazine in the presence of ß-cyclodextrin. Pol. J. Chem. 1997, 71, 816–822. [Google Scholar]
- Bajorath, J.; Peltason, L.; Wawer, M.; Guha, R.; Lajiness, M.S.; Van Drie, J.H. Navigating structure–activity landscapes. Drug Discov. Today 2009, 14, 698–705. [Google Scholar] [CrossRef]
- Guha, R.; Van Drie, J.H. Assessing how well a modeling protocol captures a structure–activity landscape. J. Chem. Inf. Model. 2008, 48, 1716–1728. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.; Scheiber, J.; Jenkins, J.L.; Sukuru, S.C. How similar are similarity searching methods? A principal component analysis of molecular descriptor space. J. Chem. Inf. Model. 2009, 49, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Bak, A.; Polanski, J. Modeling robust QSAR 3: SOM-4D-QSAR with iterative variable elimination IVE-PLS: Application to steroid, azo dye, and benzoic acid series. J. Chem. Inf. Model. 2007, 47, 1469–1480. [Google Scholar] [CrossRef] [PubMed]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 a [µM] |
|---|---|
| 6 | 115.38 ± 1.84 |
| 7 | 103.78 ± 1.16 |
| 8 | 4.33 ± 0.82 |
| 9 | 3.10 ± 0.42 |
| 10 | 8.02 ± 0.46 |
| 11 | 4.79 ± 1.36 |
| 15 | 67.78 ± 2.32 |
| 16 | 61.47 ± 2.33 |
| 17 | 9.68 ± 2.00 |
| 18 | 14.85 ± 0.86 |
| 19 | 14.30 ± 2.88 |
| 20 | 11.89 ± 0.48 |
| 31 | 53.09 ± 3.68 |
| 32 | 40.81 ± 1.67 |
| 33 | 4.77 ± 1.89 |
| 34 | 8.44 ± 1.70 |
| 35 | 8.65 ± 0.10 |
| 36 | 5.63 ± 0.16 |
| 39 | 10.63 ± 2.69 |
| 40 | 12.59 ± 0.68 |
| 41 | 10.38 ± 0.57 |
| 42 | 6.17 ± 1.21 |
| Compound | 8-oxo-dG/106dG ± SD |
|---|---|
| Control | 8.77 ± 0.02 |
| 31 | 31.77 ± 0.09 |
| 33 | 24.43 ± 0.02 |
| 34 | 29.43 ± 0.11 |
| 35 | 107.34 ± 0.57 |
| 36 | 25.33 ± 0.13 |
| Compound | ΔTm [°C] | Kb [M–1] |
|---|---|---|
| ct-DNA | ||
| 6 | 1.50 | 2.00 ± 1.90 × 105 |
| 7 | –2.17 | 7.88 ± 3.26 × 105 |
| 8 | 1.50 | 1.40 ± 1.04 × 105 |
| 9 | 0.83 | 3.58 ± 1.50 × 105 |
| 10 | 0.50 | 2.48 ± 1.44 × 105 |
| 11 | 0.50 | 7.20 ± 6.40 × 104 |
| 15 | 0.50 | 2.40 ± 1.0 × 104 |
| 16 | 0.50 | 1.92 ± 1.58 × 105 |
| 17 | 0.83 | 2.17 ± 1.10 × 105 |
| 18 | –0.17 | 2.48 ± 0.62 × 105 |
| 19 | 0.17 | 3.88 ± 0.38 × 105 |
| 20 | –0.17 | 3.12 ± 1.22 × 105 |
| 31 | –1.17 | nd |
| 32 | –0.83 | nd |
| 33 | 0.50 | 2.06 ± 1.80 × 105 |
| 34 | 0.50 | 1.62 ± 1.40 × 105 |
| 35 | 0.17 | 1.98 ± 1.58 × 105 |
| 36 | 0.17 | 2.20 ± 2.14 × 105 |
| 39 | 1.50 | 3.62 ± 1.10 × 105 |
| 40 | 1.17 | 6.60 ± 2.80 × 104 |
| 41 | 0.83 | nd |
| 42 | –0.17 | nd |
| Mitonafide | 5.17 | 2.54 × 105 |
| Pinafide | 6.50 | 6.60 × 104 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rykowski, S.; Gurda-Woźna, D.; Orlicka-Płocka, M.; Fedoruk-Wyszomirska, A.; Giel-Pietraszuk, M.; Wyszko, E.; Kowalczyk, A.; Stączek, P.; Bak, A.; Kiliszek, A.; et al. Design, Synthesis, and Evaluation of Novel 3-Carboranyl-1,8-Naphthalimide Derivatives as Potential Anticancer Agents. Int. J. Mol. Sci. 2021, 22, 2772. https://doi.org/10.3390/ijms22052772
Rykowski S, Gurda-Woźna D, Orlicka-Płocka M, Fedoruk-Wyszomirska A, Giel-Pietraszuk M, Wyszko E, Kowalczyk A, Stączek P, Bak A, Kiliszek A, et al. Design, Synthesis, and Evaluation of Novel 3-Carboranyl-1,8-Naphthalimide Derivatives as Potential Anticancer Agents. International Journal of Molecular Sciences. 2021; 22(5):2772. https://doi.org/10.3390/ijms22052772
Chicago/Turabian StyleRykowski, Sebastian, Dorota Gurda-Woźna, Marta Orlicka-Płocka, Agnieszka Fedoruk-Wyszomirska, Małgorzata Giel-Pietraszuk, Eliza Wyszko, Aleksandra Kowalczyk, Paweł Stączek, Andrzej Bak, Agnieszka Kiliszek, and et al. 2021. "Design, Synthesis, and Evaluation of Novel 3-Carboranyl-1,8-Naphthalimide Derivatives as Potential Anticancer Agents" International Journal of Molecular Sciences 22, no. 5: 2772. https://doi.org/10.3390/ijms22052772