
Heme Oxygenase 1: A Defensive Mediator in Kidney Diseases


1
Centre de Recherche des Cordeliers, INSERM, Sorbonne Université, Université de Paris, F-75006 Paris, France
2
U1167-RID-AGE, Institut Pasteur de Lille, Inserm, Univ. Lille, F-59000 Lille, France
3
Nephrology Department, CHU Lille, Univ. Lille, F-59000 Lille, France
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2021, 22(4), 2009; https://doi.org/10.3390/ijms22042009
Submission received: 18 January 2021
/
Revised: 4 February 2021
/
Accepted: 8 February 2021
/
Published: 18 February 2021
(This article belongs to the Section Molecular Biology)
Abstract
:The incidence of kidney disease is rising, constituting a significant burden on the healthcare system and making identification of new therapeutic targets increasingly urgent. The heme oxygenase (HO) system performs an important function in the regulation of oxidative stress and inflammation and, via these mechanisms, is thought to play a role in the prevention of non-specific injuries following acute renal failure or resulting from chronic kidney disease. The expression of HO-1 is strongly inducible by a wide range of stimuli in the kidney, consequent to the kidney’s filtration role which means HO-1 is exposed to a wide range of endogenous and exogenous molecules, and it has been shown to be protective in a variety of nephropathological animal models. Interestingly, the positive effect of HO-1 occurs in both hemolysis- and rhabdomyolysis-dominated diseases, where the kidney is extensively exposed to heme (a major HO-1 inducer), as well as in non-heme-dependent diseases such as hypertension, diabetic nephropathy or progression to end-stage renal disease. This highlights the complexity of HO-1’s functions, which is also illustrated by the fact that, despite the abundance of preclinical data, no drug targeting HO-1 has so far been translated into clinical use. The objective of this review is to assess current knowledge relating HO-1’s role in the kidney and its potential interest as a nephroprotection agent. The potential therapeutic openings will be presented, in particular through the identification of clinical trials targeting this enzyme or its products.
1. Introduction
The number of people worldwide with chronic kidney disease (CKD), acute kidney injury (AKI) or requiring renal replacement therapy exceeds 850 million [1]. The forecasted incidence is a cause for concern, with CKD projected to become the world’s fifth leading cause of death by 2040 [2]. It is generally accepted that kidney diseases are a significant public health issue and that the need for a multimodal approach to curb this presumed evolution is urgently needed [3]. Of key importance is the identification of novel therapeutic targets, both to limit the impact of acute renal events and to ameliorate poor outcomes in CKD, across the range of underlying etiologies. Several candidates for nephroprotection are currently being studied [4,5]. The heme oxygenase (HO) system is one of the targets that could be used to protect kidney structures from damage caused by oxidative stress, limiting inflammation and the consequent accelerated ageing.
HO was discovered in 1968 by Tenhunen and colleagues, who first described its ability to catalyze the breakdown of heme (Fe-protoporphyrin IX) to free iron, carbon monoxide (CO) and biliverdine, which is rapidly converted to bilirubin [6]. Two main isoforms of HO were described: inducible HO-1 and constitutive HO-2. Initially merely considered a recycling system for heme from aged red blood cells, HO has since been attributed a number of cytoprotective properties via its metabolites and downstream signaling, expanding its definition from that of a “molecular wrecking ball” to a “mesmerizing” trigger of cellular events, to quote Maines et al. [7].
Driven by its potential antioxidant and anti-inflammatory capacities, HO-1 is the subject of growing interest for its role in different human pathologies. Among the studied organs, the kidney has often been highlighted over the last 30 years, notably through the identification of a role for HO-1 in various renal, metabolic and vascular diseases of both an acute and chronic nature [8,9,10,11]. HO-1 is essential for efficient kidney function, as illustrated by kidney damage in humans with HO-1 deficiency and by numerous animal studies (described below). The kidney’s physiological functions specifically influence HO-1 regulation. Indeed, renal tissue is very susceptible to hypoxia [12], a major driver of HO-1 expression, and is particularly exposed to toxic molecules because of its filtration and reabsorption functions. This particularity of being a “detoxification” organ, shared with the liver, means the kidney is highly exposed to damage-associated molecular patterns (DAMPs) and stress mediators. Interestingly, the kidney possesses a capacity for the synthesis of albumin, alphaprotein and haptoglobin under ischemic or toxic ischemic damage, underlining the parallel with the liver [13,14]. Renal cells therefore require adaptation strategies to preserve their functions and integrity in such a harsh environment. The kidney is a major site of exposure to heme in the event of extravascular hemolysis or rhabdomyolysis, and hence also for the induction of HO-1 expression. Under these conditions, HO-1 overexpression is easily explained by the need to catabolize the free heme overload. However, HO-1’s involvement in kidney diseases goes beyond the framework of heme-related diseases, illustrating the complexity of its mechanisms of action which extend beyond heme catabolism alone [15].
The aim of this review is to summarize the large amount of experimental and clinical data describing the importance of HO-1 in the kidney, without forgetting that HO-2 is also significant: constitutively expressed in the kidney under homeostatic conditions, it is in the first line of defense against ischemia and other insults, and its renal cytoprotective effects have been demonstrated in heme protein- and ischemic-induced AKI [16]. Compared to the literature about HO-1, however, there are very few studies examining the functional significance of HO-2 in kidney diseases; therefore, this review will focus upon HO-1. After a general introduction on the characteristics of HO-1 in the kidney, we will describe the role of this enzyme in different renal cell populations and in kidney diseases. Given its primary function of recycling heme, we will approach these pathological states according to whether or not there is a massive release of free heme. Finally, current clinical applications will be presented, revealing the disparity between the profusion of available experimental data and the relative paucity of therapeutic applications.
2. About Heme Oxygenase-1 in Kidney
2.1. Why Is HO-1 Particularly Important for the Kidney?
Over the past few decades, the key role of the HO-1 system in the pathophysiology of kidney diseases has been supported by several studies. In the two HO-1-deficient patients reported up to date, the kidney was among the damaged organs. Both patients had hematuria and proteinuria. Kidney biopsies revealed increased mesangial proliferation and focal thickening of the capillary loops by light microscopy, and endothelial detachment in the glomerular capillary by electron microscopy [17,18]. Renal injuries were also present in both animal models invalidated for HMOX1, with some differences apparent between the mouse [19] and rat models [20]. In Hmox1−/− mice, lack of functional heme oxygenase was responsible for enhanced susceptibility to oxidative stress [19], decreased hemophagocytosis of senescent red blood cells by tissue macrophages, increased hemolysis and redistribution of iron from splenic and hepatic macrophages to hepatocytes and renal proximal tubule cells [21]. Similarly, Hmox1−/− rats exhibited anemia, splenomegaly, and prominent interstitial inflammatory cell infiltrates and fibrosis scarring in their kidneys. While glomerular lesions were anecdotal in the mouse model, Atsaves et al. found an increase in mesangial matrix and focal and segmental glomerulosclerotic lesions in Hmox1−/− rats. These morphological findings were associated with increased blood urea nitrogen, serum creatinine and albuminuria, but interestingly there was no increase in iron deposits in the glomeruli, tubules or interstitium [20].
Spontaneous renal damage in both human and animal models invalidated for HMOX suggests that HO-1 is important for efficient renal function. The renal protective role of this enzyme has been confirmed in multiple models of renal challenge in HO-1 invalidation/inhibition or overexpression conditions [22]. However, the histological differences cited above illustrate the difficulties of transposing results from one species to another. Differences in the regulation of HO-1 between humans and mice are well described, motivating the generation of human hHO-1 BAC transgenic mice [23]. In this model, HO-1 is overexpressed, and it remains unknown as to whether this higher background expression may obscure subtle damage over longer periods of time, or even have adverse effects itself.
A better understanding of how HO-1 functions in the kidney, and how its expression or function is controlled, is essential for our comprehension of kidney physiology and pathology but it is (as always) important to remember the limits of any study in its interpretation and translation: we will attempt to sustain this critical viewpoint throughout this review.
2.2. Regulation of HO-1 Expression in Kidneys
HO-1 protein is ubiquitous, being anchored in the membrane of the endoplasmic reticulum and also localized in mitochondrial nuclei and caveoli, while the HO-1 gene (HMOX1), which encodes HO-1, is located on Chromosome 22. Many stimuli can modulate the transcription of HMOX1, reflecting the broad spectrum of DNA-binding motifs within its promoter and the large number of signaling pathways which lead to its transcription. The main regulator of the HMOX1 gene is nuclear erythroid factor 2 (Nrf2), whose activation depends on its interaction with the Kelch-type, ECH-associated protein (Keap1). The Keap–Nrf2 interaction promotes the degradation of Nrf2 (healthy state), while destabilization of this complex releases Nrf2 which translocates into the nucleus and upregulates certain genes, including HO-1 (stressed state). There are many other activators or repressors of HO-1 forming a complex biomolecular network (detailed in previous reviews [7,10]). Details on these are beyond the scope of this review and we will here focus upon the renal-specific features of HO-1 expression.
In the kidney, HO-1 protein levels are undetectable under homeostatic conditions, except in the tubules where it nevertheless remains low [24]. Many stress conditions are known to up-regulate the renal transcription HO-1, including oxidative stress, heat shock, hypoxia, heavy metals and toxins (reviewed in Bolisetty et al. [8]). Under these conditions, HO-1 expression remains heterogenous among the different renal compartments. The kidney consists of two main areas: the output cortex and the medulla in the innermost region. The medulla contains most of the length of the nephrons, the functional components of the kidney that filter fluid from the blood. The renal cortex includes the glomeruli, those “tufts” of capillaries where plasma is filtered through the glomerular basement membrane. The filtered fluid then flows along the convoluted proximal tubule to the loop of Henle, and then to the convoluted distal tubule and collecting ducts, which drain into the ureter (Figure 1). Glomerular filtration and reabsorption of solutes (the kidney’s two major functions) are supported by renal blood flow, which is among the highest in the body, with kidneys receiving about 20 to 25% of cardiac output. Compared to the cortex, blood flow to the renal medulla is relatively low. This disparity is required to enable an effective urine concentrating mechanism and also explains the particular susceptibility of the medulla to decreased blood flow. Under stress conditions, HO-1 is thus more highly expressed in the medulla than the cortex [25]. Its expression is very strong in tubules (especially proximal tubules), but minimal or absent in glomeruli, as illustrated in Figure 2 in both hemolytic patients and heme-injected mice. However, it has been shown that prior induction of HO-1 within the glomeruli prevented subsequent development of nephrotoxic glomerulonephritis in a Lewis rat model, but the major producer of HO-1 in the glomeruli was the infiltrating macrophages not the intrinsic glomerular cells [26]. This lower capacity of glomeruli to express HO-1, which is a key cytoprotective mechanism, could explain glomeruli’s particular sensitivity to stressors in certain pathologies [27].
Taken together, these data show that HO-1 levels are normally undetectable in the kidney but that HO-1 is highly inducible in tubules during pathologies, notably via its main transcription factor Nrf2. The capacity of glomeruli to induce HO-1 is lower, however.
2.3. HO-1 and Nephroprotection
Like HO-2, HO-1 drives the NADPH-dependent addition of an oxygen molecule to the porphyrin ring of heme, thus catalyzing the oxidation of heme and the equimolar release of biliverdin, free iron, and carbon oxide (CO). All these events are cytoprotective by removing free heme, regulating iron efflux, and increasing bilirubin and CO levels. Indeed, the noxious effects of free heme are well described, particularly at the vascular site via direct toxicity, but also through the stimulation of pro-inflammatory signaling pathways (such as DAMPs) and the activation of the complement system [28]. Similar to endothelial cells, such adverse effects are reported with other cell types when massively exposed to heme. This is the case for human skeletal muscle fibers, in which exposure to heme induces contractile dysfunction [29], and renal tubular cells [30]. In addition, iron is notably toxic even at low concentrations. It participates in the Fenton reaction, producing hydroxyl, and can result in cell death by ferroptosis, a cell-death pathway characterized by an iron-dependent decrease in glutathione (GSH) levels and the accumulation of lipid hydroperoxides to lethal levels [31]. Thus, it has been reported that the cytoprotection of HO-1 was due, at least in part, to an increased iron flux [32], which following its release is either stored in ferritin or exported from macrophages to plasma for re-use via the transmembrane ferrous exporter ferroportin. CO counteracts vasoconstriction and has powerful anti-oxidant properties, as does biliverdin, which is enzymatically converted to bilirubin via biliverdin reductase [33].
Transposing these properties to the renal level, it is easy to understand research interest in harnessing this molecule in order to treat kidney disease. The positive effects have been described for almost 30 years, following the work of Nath et al., who reported that HO-1 conferred protection in a rat model of glycerol-induced, acute renal failure. In their model, a single prior injection of hemoglobin rapidly induced HO-1 messenger RNA and protein within the kidney, which prevented progression to renal failure [34]. The nephroprotective effects of HO-1 and its metabolites have since been reported in different models of renal disease [8,35]. The potential importance of HO-1 in the pathogenesis of kidney diseases is also suggested by the link between polymorphisms in the HO-1 gene and renal outcome. Individuals with shorter (GT)n repeats in the HO-1 promoter region have a higher transcriptional activity and thus higher HO-1 levels than individuals with longer (GT)n repeats [36]. Interestingly, long (GT)n repeats have been associated with worse prognosis in several contexts, namely, a higher risk for AKI after cardiac surgery or decreased renal function after kidney transplantation [37,38]. However, there is no evidence of a protective effect of short repeat (GT)n for graft or recipient survival following renal transplant [39], underlining the complexity of the mechanisms involved. The levels of HO-1 expression may also vary with age. The renal expression of gene HO-1 in control C57Bl6 mice was significantly decreased at 18 compared with 3 months of age [40]. In another study, protein HO-1 expression was not significantly different at baseline between 6–8-week-old mice and 1-year-old mice, but the older animals’ ability to upregulate HO-1 in response to ischemia reperfusion injuries (IRI) was impaired (especially in the medulla), and they exhibited worse renal function compared with young animals [41]. These results echo the renal sensitivity of elderly humans to nephrotoxic agents.
To summarize, the preventive or genetic induction of HO-1 expression has been shown in many studies to confer renal protection. A decrease in renal HO-1 expression may contribute to the kidney’s sensitivity to certain pathologies.
3. HO-1 in Renal Cell Populations
Comparative analyses have revealed that HO-1 is not expressed to the same extent in the distinct cell populations of the kidney for a given stimulus (Figure 1). During intravascular hemolysis or in the presence of HO-1-inducing stress stimuli or drugs in the circulation, the endothelial cells are the first cells to be challenged by these systemic stressors. If the stressor is filtered in the glomeruli, then the podocytes are exposed and may be damaged, as seen in diabetic nephropathy. Further, the stressor could encounter mesangial cells which have a secretory and immunomodulatory role and proliferate during HO-1 deficiency. Finally, the stressor may reach the tubular epithelium which are the most-studied cell type in relation to HO-1 (the proximal and distal tubules must be considered separately with respect to the epithelium). Tubular epithelial cells have the most potent capacity to overexpress HO-1 and suffer the most severe injury in case of its deficiency or during intravascular hemolysis and rhabdomyolysis. This section describes the key functions of the main kidney cell types and the specific role of HO-1 within them.
3.1. Endothelial Cells
Endothelium is comprised of a monolayer of endothelial cells (EC) which accomplish critical functions in the blood vessels. They cover the interior of blood vessels and perform barrier functions, but they also serve as an exchange interface with adjacent tissues, a sensor for tissue and intravascular stress, and as a source of defense mediators [42]. As such, EC are the first line of cells to encounter circulating stressors and HO-1 expression is essential for their survival, with HO-1-deficient patients and murine models exhibiting extensive endothelial damage [19,43]. The pro-survival effects of HO-1 in EC occur in every organ and there exists a large body of work regarding its induction by various molecules [44,45]. According to its localization (in different organs, in vessels or capillaries), EC structure, specialization and functions vary. Thus, EC from different vascular beds have dynamic expression profiles (both spatially and temporally) and this might explain their selective involvement in different disease processes [42].
In the kidney, three main types of endothelium can be distinguished: glomerular, peritubular capillary, and medium/large vessel endothelium [46], each with different levels of HO-1 expression or function. Glomerular EC are “fenestrated” and covered by a glycocalyx [47] responsible for restricting macromolecule passage to the urinary chamber [48]. Despite the glycocalix, small hydrophobic molecules (e.g., heme) can enter cell membranes and activate proinflammatory and prothrombotic pathways, including TLR-4 signaling, thus promoting blood cell adhesion and vaso-occlusion (Schaer et al., 2013). Peritubular capillaries’ EC are also fenestrated and lay upon a thin stroma. They transport reabsorbed components, sustaining tubular epithelial cell function [46]. Finally, the endothelia of both medium and large vessels are contiguous, with interconnected EC. The structural and functional diversity of EC in the kidney as a function of their localization makes them differentially sensitive to specific stressors, such as toxic or proinflammatory filtrated molecules for glomerular EC, hypoxia for peritubular capillaries, or changes in shear stress, with correspondingly specific responses to such assaults [46].
The main factors affecting HO-1 expression in EC are hypoxia, shear stress, the presence of heme and other circulating stressors (ammonia [49], S-adenosyl methionine [50], etc.), circulating substances (statins [51], resveratrol [52], etc.) or drugs. Oxygen partial pressure decreases along the peritubular capillary, notably under stress conditions with excessive O2 consumption. Interestingly, HO-1 expression has been reported to decrease upon hypoxia while HO-2 expression was preserved in macrovascular human umbilical vein EC (HUVEC) [53]. However, HO-1 expression is inducible in microvascular peritubular capillaries’ EC under different conditions that may provoke hypoxia (heme, Angiotensin II, etc.). Physiological shear stress is a major determinant of EC survival, as shown in vivo in human carotid atherosclerotic plaques [54] and rabbit carotids with reduced blood flow [55]: it also induces the expression of anti-inflammatory factors (in vitro in HUVEC and in vivo in rabbit aorta [56]) and vasodilatory ones (in vitro and in vivo in mice [57]). Interestingly, HO-1 expression is induced by shear stress, in vitro in macrovascular (aorta) and microvascular (HMEC) human EC [58], and this expression was reported in rats to be directly related to the level of shear stress: higher expression at high flow levels (in arteries), but requiring other factors for HO-1 to be expressed at low flow levels (in microvessels) [59]. This phenomenon has so far only been reported in intestinal vessels. However, if these results are verified in other organs, it may explain decreased HO-1 induction in capillaries such as glomerular EC under stress [27]. EC are highly exposed to cytotoxic hemolysis-derived products and endothelial responses to these insults reflect the heterogeneity of EC. Microvascular EC, and particularly glomerular EC, are less prone to up-regulate HO-1 compared with macrovascular EC, both in vitro and in a mouse model of hemolysis [27]. This renders them particularly susceptible to injury through differences in their complement C3 regulation and heme degradation as compared with macrovascular EC [27]. Little is known about the HO-1 induction capacity of the peritubular capillaries’ EC.
Renal macrovascular EC remain poorly studied with respect to HO-1 expression and function. The most widely used model in the study of macrovascular EC is the human umbilical vein EC (HUVEC). Exposure to heme in HUVEC is responsible for rapid signal transduction, mobilization of the endothelial Weibel Palade bodies, and NF-kB activation, thus conferring them a pro-inflammatory and pro-thrombotic phenotype [60,61,62]. However, longer exposure allows them to develop adaptation mechanisms and become highly resistant to oxidative-stress-mediated injuries and lipid peroxidation products. These include the induction of ferritin and HO-1 [63], though HO-1 induction in glomerular EC in vitro and in mouse models of hemolysis is much weaker [27,64]. HO-1 induction in macrovascular EC notably modulated complement activation [27,65], thrombomodulin expression in hemolysis conditions [27] and in the context of septic kidney injury [66], and expression of adhesion molecules associated with EC activation [67].
Taken together, these data indicate that the kidney endothelium is strongly influenced by toxins and stressors as a result of its filtration function. Such stressors seem to trigger strong adaptation mechanisms in macrovascular EC which allow them to resist insults to a certain level, mainly by the upregulation of HO-1 and its associated genes. While there is a marked lack of data in the literature, available studies suggest that, in contrast to macrovascular EC, this HO-1 upregulation is less pronounced in the glomerular microvascular endothelium, making it particularly sensitive to environmental stress.
3.2. Podocytes
Podocytes are hyper-specialized cells that maintain the glomerular filtration barrier through the synthesis of glomerular basement membrane (GBM) components and the formation of the slit diaphragm by attaching their foot processes to the glomerular endothelial cells [68] (Figure 1). Their strategic localization, enabling their filtration function, makes them particularly sensitive to mechanical, oxidative and immunological stress. Podocytes have unique adaptive responses, including the upregulation of vimentin and desmin [69], or antioxidant proteins such as Sirtuin1 and metallothionein. Sirtuin upregulates Nrf2, thus enhancing HO-1 expression. [70,71] Interestingly, sirtuin-1 expression (together with Nrf2 and HO-1) has been reported as being increased by exposure to advanced glycation end products [71], and HO-1 was shown to be specifically enhanced in podocytes in diabetes patients [72]. Although less studied, the expression of HO-1 has also been described under hemolytic conditions [27,73], with heme inducing HO-1 expression in a Nrf2-dependent manner in podocytes. Interestingly, Nrf2-deficient mice spontaneously developed proteinuria with foot process effacement, decreased synaptopodin and nephrin expression, and podocyte apoptosis [73].
Though the literature on HO-1 in podocytes is not abundant, what evidence there is suggests a protective physiological role for the Nrf2/HO-1 axis by preserving pedicels and inhibiting the death and detachment of podocytes. In pathological contexts, HO-1 is overexpressed as a defense mechanism but could also be a byproduct of exhausted resistance mechanisms resulting from overwhelming stress, in which context it is insufficient to prevent podocyte injury.
3.3. Mesangial Cells
Mesangial cells, together with the mesangial matrix they produce, form the mesangium, a support tissue for the glomerulus floculus. Among mesangial cells are immune cells, monocyte/macrophage-like (5 to 15%) and contractile cells, which are similar to smooth muscle cells (85 to 95%) and provide structural support and contraction [74,75]. Heme promotes the proliferation of smooth muscle cells [76] and its receptor TLR4 is expressed on mesangial cells [77], but the role of HO-1 in these cells is poorly studied. Nitric oxide (NO) has been shown to induce HO-1 in mesangial cells [78], HO-1-deficient patients have a mild mesangial proliferation [17,18,43], and HO-1 has been shown to modulate mesangial cell proliferation via p21 upregulation [79]. Further work is needed to determine whether the large body of knowledge accumulated for macrophages is applicable for the macrophage-like mesangial cells.
3.4. Tubular Cells
If HO-1 expression was described in tubules some time ago, it is only recently that its regulation has been described as being segment specific; HO-1 also plays distinct roles in different segments of the tubule in order to maintain renal functions [80].
3.4.1. Proximal Tubule
The proximal tubules are the cells in the kidney which show the highest capacity for the overexpression of HO-1. Indeed, proximal tubular epithelial cells have been shown to be especially sensitive to oxidative stress in vitro [81]. They rely strongly upon HO-1 for protection from, and adaptation to, stressors, as evidenced by the fact that tubular injury is the cardinal pathologic feature in human HO-1 deficiency [82]. The main function of the renal proximal tubule is uptake of water, ions, amino acids and filtered proteins. In the absence of glomerular damage, filtered proteins are mostly of a size below 68kDa. Internalization of proteins, including heme-associated ones such as myoglobin or hemoglobin, but also hemopexin, occurs via the megalin and tubulin receptors [83]. This reabsorption function requires a large amount of energy (ATP) provided by mitochondrial beta-oxidation [84]. However, severe ATP depletion results in mitochondrial injury, causing further depletion of energy stores and formation of reactive oxygen species [85]. This high metabolic demand produces a state of relative hypoxia in kidney medulla, and the proximal tubules are particularly sensitivity to ischemia/reperfusion and heme excess [86,87]. The sensitivity to heme is also explained by the constitutive expression of TLR4 (a heme receptor) by the proximal tubules [61], and TLR4 expression is increased in renal ischemia–reperfusion injury and septic injuries [77].
While HO-1 is expressed only weakly in the kidney under normal conditions, it is strongly inducible in proximal tubules under different forms of stress [88], proteinuria among them [80]. The interplay among uptake of heme-containing proteins, osmotic pressure driving heme influx, TLR4 expression and increased synthesis of heme in proximal tubules may together explain this specific, strong upregulation of HO-1. It has been suggested that HO-1 expression in tubular epithelial cells depends more upon basolateral exposure to heme (from the peritubular capillaries under hemolytic conditions) than intraluminal heme content. However, under conditions of hypoxia and of altered polarity of renal EC, HO-1 upregulation has been reported to depend more upon apical exposure to heme [89]. In tubular epithelial cells, HO-1 upregulation by heme is dependent on Nrf2 stabilization [90]. HO-1 has a dual role in tubular EC, being simultaneously an anti-oxidative and a regulator of cell death. Indeed, HO-1 is responsible for the upregulation of p21, a cyclin-dependent kinase inhibitor responsible for stopping cell cycle progression, which reduces apoptosis in tubular EC [91,92]. Furthermore, HO-1 expression decreases ferroptosis in proximal tubular cells, a non-apoptotic regulated cell death associated with accumulation of reactive oxygen species (ROS) derived from lipid peroxidation [93]. Finally, HO-1 has been shown to inhibit autophagy in proximal tubular cells [94].
Altogether, proximal tubular cells are massively exposed to HO-1 inducers in pathological contexts (filtration of hemoglobin, myoglobin, hemopexin-heme complexes; ischemia, filtration of toxic products or drugs, etc.) and rely upon HO-1 for their protection, but HO-1 is also a marker of tubular stress and injury when their capacity for adaptation is overwhelmed.
3.4.2. Distal Tubule
Distal tubular EC along the medulla, notably in the thick, ascending limb of the loop of Henle (mTAL) and the distal convoluted tubule (DCT), are involved in the concentration and dilution of urine and the maintenance of homeostatic salt and solute levels by expressing different channels specific to their localization and cell subtype. During water reabsorption, the concentration of toxins (notably heme) becomes much higher in the distal nephron segments. Because of the low partial pressure of oxygen along the peritubular capillaries, these distal tubular EC are particularly vulnerable to hypoxia and rely more on anaerobic, glycolytic ATP production than proximal cells. Distal tubules have a greater ability to survive and adapt to hypoxic stress [95], being less sensitive to cell death, especially after ischemic injury [96].
Distal tubules, like proximal tubules, only weakly express HO-1 at a basal level; upon stimulation, however, its induction by heme has been called into question [88]. (Interestingly, HO-2 may be enhanced by heme in distal tubules [97].) However, HO-1’s role in distal tubules remains to be fully elucidated as its expression has also been associated with improved renal function after ischemia/reperfusion in cadaveric donor transplantation [98], as well as in CKD [99]. This may be related to the regeneration function of distal tubular epithelial cells, but further studies are needed to better describe the relationship between HO-1 and distal tubular epithelial cells’ functions.
To conclude, while data regarding distal tubules are lacking, some evidence suggests that enhanced HO-1 expression could confer protection to distal tubules’ EC.
4. HO-1 in Kidney Diseases
HO-1 and its induction in various kidney pathologies and physiological conditions aroused interest twenty years ago, particularly in the context of massive free heme release, such as that associated with hemolytic diseases and rhabdomyolysis [100]. However, HO-1 has exhibited a protective role in many other pathologies that are not classically considered as heme-mediated nephropathies. This encompasses ischemia/reperfusion renal injuries and tubular nephropathies (which may be toxic), septic and obstructive nephropathies and, moreover, glomerular nephropathies. This classification of disease represents a spectrum of different contributions from HO-1 stimulating stressors (Figure 3). Indeed, ischemia followed by reperfusion can be part of the mechanism of renal injuries in hemolytic pathologies [101] and rhabdomyolysis [102]. On the other hand, local heme release was recently reported in experimental ischemia-reperfusion [103], and may be a contributing factor in ischemia/reperfusion-related pathologies such as renal transplantation, though without it being the main damaging factor [104]. Furthermore, high plasma levels of free heme have been measured in some pathologies not classically considered as being heme-related, such as sepsis [105]. Having said this, we will consider the two extreme parts of the spectrum to illustrate the protective role of HO-1 in both the presence and absence of intrarenal heme overload.
4.1. Diseases Associated with a Massive Release of Free Heme
Two broad categories of pathology are associated with massive releases of heme, namely, hemolytic diseases and rhabdomyolysis. In hemolytic diseases, heme is released from hemoglobin after rupture of the red blood cells. The oxidation of Hb and liberation of heme can occur in the circulation or in the tubules, after filtration of the Hb dimers in the glomeruli. During rhabdomyolysis, myoglobin from injured skeletal muscle fibers enters the circulation, but heme is not released there due to its higher affinity for this protein compared to Hb. Instead, myoglobin is filtered by glomeruli and heme is released in the interstitium, especially after intake of this protein by the proximal tubules. In both cases, HO-1 is massively upregulated in the kidney. Using several examples, we will illustrate and analyze the role of HO-1 in diseases associated with a massive release of heme.
4.1.1. Hemolytic Diseases
Hemolysis is a pathological condition encompassing various etiologies which can occur either intra- or extravascularly. Intravascular hemolysis is the destruction of red blood cells (RBCs) in the circulation, which may be a complication of extravascular hemolysis, or else occur mechanically such as in thrombotic microangiopathy, malignant hypertension or from mechanical heart valve implants [28]. Extravascular hemolysis is the elimination of the RBCs in the spleen, liver or bone marrow by phagocytes. It is mainly linked to defects which may be intrinsic (including genetic diseases such as sickle cell anemia or acquired diseases such as paroxysmal nocturnal hemoglobinuria (PNH)) or extrinsic (such as autoimmune hemolytic anemia, hepatic cirrhosis). In both cases, lysed RBCs release hemoglobin, the auto-oxidization of which liberates free heme which possesses pro-oxidative and pro-inflammatory properties. Haptoglobin and hemopexin in the circulation scavenge hemoglobin and heme, respectively, and drive their elimination. However, these protective mechanisms are rapidly exhausted and cannot prevent hemoglobin and heme causing harm during a massive hemolytic crisis. Indeed, free heme levels have been shown to be elevated in the plasma of sickle cell disease (SCD) patients (4–50 µmol) [106,107,108] and a mouse model of SCD (75–120 μM) [109]. The pro-inflammatory effects of the remaining free heme and free hemoglobin may provoke organ damage, including of the kidney [110]. Notably, hemoglobin cast nephropathy has been reported in kidney biopsies of patients with hemolysis with various etiologies, such as PNH, autoimmune hemolytic anemia, hemoglobinopathies and transfusion accidents [111]. Regarding free heme that escapes hemopexin scavenging, only HO can degrade it and this detoxifying step is critical during hemolysis, as shown during hemoglobin infusion in HO-1-deficient mice which exhibited acute renal failure and marked mortality [112]. However, before describing the involvement of HO-1 in specific hemolytic pathologies, we will first examine the main results from a validated and widely used mouse model of intravascular hemolysis, namely, phenylhydrazine (PHZ) treatment. Thereafter, we will focus on the main hemolysis etiologies associated with kidney injuries and variations in HO-1 expression, namely: sickle cell anemia, hemolytic uremic syndrome, malaria and PNH.
PHZ-Induced Hemolysis Model
The most-used in vivo model for intravascular hemolysis employs an injection of the hemolytic drug PHZ. It induces massive RBC destruction via membrane lipid peroxidation [113] and destabilization of hemoglobin. Proposed as a treatment in the mid-20th century for patients with polycythemia (without much success), it has been used in over 200 papers to study hemolysis in experimental animals. PHZ-treated mice exhibit an increased tubular expression of HO-1 [114,115], which can be prevented by administration of Zinc Protoporphyrin (ZnPP) [116]. This upregulation is thought to be Nrf2 dependent since Nrf2-deficient mice exhibit a more severe, hemolysis-induced AKI phenotype, enhanced tubular injury markers (KIM-1 and NGAL) and cell death, as well as lower HO-1 and ferritin [117]. However, other studies suggest that despite elevated HO-1 expression, renal alterations in this model are at least partly independent of heme, since injection of free heme could not reproduce them and administration of the heme scavenger hemopexin could not prevent them in the PHZ model [115]. If complement overactivation in PHZ-induced hemolysis is involved in tissue damage [118], HO-1 expression seems to be independent of it, despite the kidney injury phenotype, since expression of HO-1 was comparable in wild type (WT) and C3−/−, PHZ-treated [118] and factor H (FH)-pretreated mice [119]. Therefore, HO-1 expression appears to be dependent on hemolytic status rather than complement-mediated tubular injury [119], and hemopexin prevented complement activation in the PHZ model without affecting HO-1 expression [120]. Thus HO-1 in the PHZ model protects the kidney from heme-dependent renal alterations, probably without influencing complement activation or other heme-independent injuries.
Sickle Cell Anemia
Sickle cell anemia (SCA) is a monogenic disease, characterized by a mutated Hb gene (HbS), which can polymerize and thus deform erythrocytes, reducing their lifespan [101]. Local hypoxia in a restricted area, for example, during a vaso-occlusive crisis, increases their polymerization capacity and instability and induces massive, intravascular hemolysis which can alter renal structure and function [121,122]. Both SCA and its heterozygous state (sickle cell trait, with an attenuated phenotype), are associated with an increased risk of CKD [123]. In addition to sickle cell susceptibility to hemolysis, HbS is unstable and prone to auto-oxidation and release of its cofactor heme. Kidney injuries in SCA are described as the result of systemic endothelial vasculopathy leading to impaired renal hemodynamics and accompanied by hyper glomerular filtration and hyperfunction of the proximal tubules. These features are associated with glomerular injuries such as Focal Segmental Glomerulosclerosis (FSGS) and interstitial chronic inflammation (secondary to medullary hypoperfusion), both of which are responsible for CKD [121]. HO-1 expression is increased in renal tubules, interstitial cells and in the vasculature of biopsies from SCA patients compared with normal kidneys [124]. However, HO-1 is not upregulated in mesangial cells, despite frequent mesangial cell proliferation in SCA [125], suggesting it could be mediated directly by heme as discussed above [76]. Interestingly, long GT tandem repeats, responsible for decreased inducibility of HO-1 expression [126], were independently associated with the occurrence of AKI in a monocentric SCA patients cohort [127], raising the possibility of a protective effect of HO-1 expression in patients; HO-1 expression is also induced in kidneys of various SCA mice models [124,128,129]. This murine expression seems to be relatively organ specific: compared with the control mice (Townes-AA), kidney and liver HO-1 expression was 10-fold greater in Townes SS mice (a validated SCD model), but only 3- to 4-fold greater in the heart and spleen [128]. In addition, in a global transcriptional analysis of SCA patients, HO-1 and its by-products were upregulated in circulating blood mononuclear cells together with genes involved in heme metabolism, cell-cycle regulation, antioxidant and stress responses, inflammation and angiogenesis [130], which suggests that circulating mononuclear cells could participate in compensating for sickle cell vascular injury. SCD patients displayed an increased population of circulating monocytes expressing high levels of HO-1 with anti-inflammatory properties (37% vs. 7% in controls), and could be involved in removing hemolysis-damaged endothelial cells, thus confirming the key role of circulant cells expressing HO-1 [131]. Interestingly, CO (one of the by-products of heme degradation by HO-1) has been suggested as being involved in hyperperfusion and hyperfiltration (both observed in the kidney of SCA patients [121,132,133]), and may thus counterbalance the vasoconstrictive effects of NO depletion by hemoglobin scavenging [108], as well as vasa recta rarefaction in medulla [134].
Microparticles (MPs—enriched in heme in SCA) are also involved in kidney pathology by inducing endothelial injury and facilitating acute, vaso-occlusive events in transgenic SAD mice [129]; they have also been shown to upregulate HO-1 expression in human EC [135]. Finally, recent studies have identified a key role for complement overactivation in the tissue damage seen in SCD [115,136,137], but as discussed in relation to the PHZ model, HO-1 expression seems to be independent of complement activation. Another model of mouse SCD reportedly showed decreased vascular congestion and down-regulation of injury-related genes in comparison with untreated mice (the SCD model had β-globin replaced by transgenes for human βS and βS-antilles globins on a C57Bl6 background, and was treated chronically with SnPP, a short-term HO-1 inhibitor which also induces HO-1 expression over the longer term [138]). This reflects the lack of specificity of SnPP, which has already been suggested for use against another protoporphyrin [139,140]. In conclusion, there is evidence for both a local and systemic (through circulating cells) protective effect of HO-1 on both kidney hemodynamics and levels of pro-oxidant stress in SCA-associated kidney injuries.
Thus, HO-1 has been implicated in SCA with respect to the preservation of local kidney hemodynamics and the protection of endothelial and tubular cells against oxidative stress.
Hemolytic Uremic Syndrome
Hemolytic uremic syndrome (HUS) is a rare, kidney-predominant, thrombotic microangiopathy (TMA) related to a dysregulation of the complement alternative pathway (AP—“atypical” or aHUS) and/or the presence of a Shiga-like toxin, secreted by a pathogen (“typical” or STEC-HUS) [141]. The formation of fibrin-platelet clots in the glomerular microvessels, promoted by endothelial pro-inflammatory activation, leads to intrarenal mechanical hemolysis. Genetic or acquired complement dysregulation in aHUS, or expression of Shiga-like toxin by a pathogen in STEC-HUS, nevertheless, does not fully explain the occurrence of these pathologies as its penetrance is incomplete in both cases and interest in heme as an additional factor in endothelial damage has increased [60]. Tubular HO-1 staining by immunohistochemistry was reported in kidney biopsies from two aHUS patients, at a proportional level to the degree of hemolysis; no glomerular endothelial HO-1 staining was observed, while positive staining was detected on podocytes [27]. Until recently in typical HUS, despite the hemolysis in this pathology suggesting involvement of heme and HO-1, data have been scarce. Only one article suggested that Shiga-like toxin (Stx), secreted by E. Coli, could inhibit HO-1 expression in renal epithelial cells [142]. However, a recent article reported significantly elevated levels of plasma heme in STEC-HUS patients compared with controls, inversely correlated with low plasma hemopexin levels but significantly associated with elevated plasma levels of HO-1 [64]. Moreover, microvascular glomerular EC incubated with heme upregulated HO-1 expression independent of the presence of STx.
The capacity of glomerular EC to upregulate HO-1 is weaker than other EC types [27]; however, the decreased inducibility of HO-1 in glomerular EC could explain their specific susceptibility in HUS.
Malaria
Infection with the parasite Plasmodium is responsible for extensive hemolysis with hemoglobin and free heme release. Acute kidney injury is a clinical hallmark of disease severity and malaria is also associated with collapsing glomerulopathy [143]. Labile heme concentrations are increased in the plasma of patients and in mouse models; interestingly, labile heme levels in the latter increased more strongly in urine compared to plasma [144]. HO-1 expression was shown to be protective in a cerebral malaria mice model of P. berghei ANKA via CO generation which, because it binds to hemoglobin, prevents its oxidation and the generation of free heme [145]. Malaria was associated with HO-1 upregulation in proximal tubular epithelial cells in the same model, as well as in a Plasmodium chabaudi chabaudi mice model [144,146]. In the latter model, HO-1−/− mice had extensive proximal tubular necrosis with hemoglobin casts and the beneficial effects of HO-1 expression for survival were dependent on its specific proximal tubular location (as opposed to hepatic, endothelial or cerebral expression) and was Nrf2-dependant [144].
HO-1 upregulation in tubular epithelial cells thus appears to be reno-protective in the context of malaria.
Paroxysmal Nocturne Hemoglobinuria
Paroxysmal nocturne hemoglobinuria (PNH) is an acquired clonal defect of phosphatidylinositol glycan anchor synthesis, responsible for enhanced RBC susceptibility to hemolysis [147]. PNH has been associated not only with AKI [148,149,150,151,152] but also with CKD (risk increased >6 fold) [153]. Interestingly, complement inhibition by Eculizumab improves renal function in all stages of CKD (within 6 months). The mechanism proposed for such improvement was the restoration of NO availability and subsequent changes in renal vascular tone. Increased expression of HO-1 has been reported in the cortex and outer medulla tubular EC and macrophages (notable in hemosiderin rich areas) [154,155]; in vitro, enhanced expression of HO-1 was observed when renal tubular epithelial cells were exposed to PNH patients’ urine [155]. However, the role of this enhanced HO-1 expression in PNH patients’ kidneys requires further study, notably, for example, in correlation with clinical data regarding kidney function and the effects of complement inhibition.
4.1.2. Rhabdomyolysis
Rhabdomyolysis is a massive skeletal muscle injury of diverse etiology, notably trauma (“crush syndrome”), muscle hypoxia, genetic defects, medication or drug abuse [102]. This destruction is responsible for excessive release into the circulation of the intracellular content of muscle fibers, including electrolytes, myoglobin, and other sarcoplasmic proteins (creatine kinase, aldolase, etc.). Myoglobin and its oxygen carrier, heme, are particularly involved in the most life-threatening complication of rhabdomyolysis, acute kidney injury (RIAKI), responsible for a significant morbi-mortality in intensive care units [156]. The mechanism for kidney injuries is classically described as an association of renal vasoconstriction [157], proximal tubular aggression (by oxidative stress, lipid peroxidation [158,159] and macrophage activation [160,161]) and intra-tubular precipitation of myoglobin. Recently, complement has also been reported to play a contributory role—in a partly heme-dependent manner—in RIAKI [30,162].
Induction of HO-1 expression was discovered nearly 30 years ago in a rat model of glycerol-induced rhabdomyolysis [34]. In this model, a single infusion of hemoglobin prior to glycerol challenge enhanced HO-1 expression sufficiently to prevent kidney failure and mortality was drastically reduced (from 100% to 14%). On the contrary, competitive inhibition of HO-1 appears to increase AKI: in the same glycerol mice model, HO-1 −/− mice had fulminant, irreversible renal failure and 100% mortality [112]. HO-1 expression increased to a maximum 48h after glycerol injection and then decreased, whereupon a concomitant decrease in protective anti-inflammatory effects was observed [163]. HO-1 expression in kidney in response to glycerol challenge is particularly pronounced in comparison with other models of AKI (such as ureteral obstruction, cisplatin, ischemic/reperfusion injury), as plasmatic concentration and intrarenal HO-1 protein and mRNA reached maximal levels in this model [164,165]. Upregulation of HO-1 expression by pyruvate (a hydrogen peroxide scavenger) prior to glycerol injection was associated with renal protection against RIAKI [166], but also increased renal glucose concentrations and IL-10 mRNAs, while reducing monocyte chemoattractant protein 1(MCP-1) and tumor necrosis factor α (TNF-a) [167]. This suggests an immune-modulating role of HO-1 in RIAKI. However, the cytoprotective role for HO-1 in the kidney is insufficient for its protection against RIAKI; for example, despite significantly higher expression of HO-1, mice lacking heavy-chain ferritin had a more severe phenotype after glycerol injection [168]. HO-1 could thus play a passive, bystander role since HO-1 expression has been shown to be upregulated simultaneously with ferritin [34]. On the other hand, HO-1 is not alone in affording cytoprotection in glycerol-induced RIAKI, as N-acetylcystein treatment (a scavenger of free radicals) also showed beneficial effects [169].
In addition, preventive HO-1 induction, together with the induction of other anti-inflammatory and cytoprotective stress proteins such as ferritin, haptoglobin, hemopexin, alpha 1 antitrypsin and IL-10, has been suggested to procure protective effects, notably in a glycerol-induced RIAKI model. This has often been called a “preconditioning treatment” in the literature. For example, infusion of nitrated myoglobin or SnPP played a synergistic role in HO-1 induction, thus preserving renal function [170]. The anticholinergic agent Penehyclidine hydrochloride (PHC) [171], but also curcumin (a polyphenol with antioxidant, anti-apoptotic and anti-inflammatory properties) [172,173], have both been shown to exert a preventive effect on glycerol-induced AKI while also decreasing cell apoptosis in renal tissue, enhancing Nrf2 activation and HO-1 induction.
Interestingly, the heme receptor TLR4 may not be implicated in the effects observed, since TLR4−/− mice were not protected against glycerol-induced RIAKI, and the TLR4 inhibitor TAK-242 failed to preserve kidney function despite preserving blood flow [174]. Furthermore, HO-1 expression in tubular cells in vitro was not inhibited by TAK-242; this suggests that other heme receptors are involved in HO-1 induction. Receptor for advanced glycation end-products (RAGE) has also been confirmed as a receptor for heme [175] and is expressed in tubular cells [176]—it will be interesting to explore its role in RIAKI. Expression of HO-1 was reported to be independent of complement system activation in RIAKI, since HO-1 was upregulated equally in both wild type and C3−/− mice injected with glycerol, despite reduced kidney lesions in C3−/− mice [30].
Finally, HO-1 has a protective role mainly by improving renal microcirculation, reducing oxidative stress and probably also by regulating the immune response induced in RIAKI. This protective role is, however, insufficient by itself to completely protect the kidney from RIAKI lesions.
4.2. Diseases without Massive Cell-Free Heme Release
This category encompasses several pathologies affecting the kidney which are not classically associated with massive heme release, and, in accordance with the aim of this review, we will present the numerous pathologies in which HO-1’s role has elicited research interest. Our aim is to comprehensively examine how HO-1 expression is involved with the physiopathology of diseases with little in common except kidney damage rather than to be exhaustive. Indeed, HO-1 has been involved in toxicity (gentamycin, cisplatin, calcineurin inhibitor, radiocontrast product, mercuric chloride), septic, mechanic (obstruction), hemodynamic (hypertension, ischemia-reperfusion), metabolic (diabetes), genetic (Polycystic Kidney Disease (PKD)) and renal-specific (membranous nephropathies, anti GBM) and systemic (Lupus nephropathy) immunologic pathologies (Table 1). Moreover, these diseases affect podocytes, mesangial, tubular epithelial, endothelial and interstitial cells in a variety of ways, thus confirming the pleiotropic effects of HO-1 in both physiology and disease. Finally, HO-1 has been involved in the acute phase of kidney injury, but also in the chronic phases of renal damage. We chose to focus on deciphering HO-1’s roles in pathologies that are the subject of a substantial body of literature or the object of clinical trial(s) regarding HO-1 (with the exception of preeclampsia and PKD, for which data remains scarce). The protective functions of HO-1 in other pathologies, which from the literature could be equally important as those which are currently the subject of clinical trials, will be summarized in Table 1 and future therapeutic prospects regarding these will be discussed in the next section.
4.2.1. Ischemia/Reperfusion
Ischemia/reperfusion (I/R) injury is a result of an inflammatory process, triggered by transient reduction or interruption of blood flow in an organ and resulting in oxygen and nutrient deprivation, followed by a reperfusion. ATP depletion is responsible for cell injury and, if severe enough, cell death by necrosis or apoptosis [85]. Among relevant etiologies are embolic or thrombotic events, circulatory “shock” and hypotension, cardiac and vascular surgery and transplantation [289]
I/R can be induced in the kidney by unilateral or bilateral renal arterial clamping, which have been extensively studied in both mouse and rat models. In such models, HO-1 expression was notably increased in tubules [228]. HO-1 −/− mice exhibited severe renal dysfunction and IL-6-mediated mortality following ischemia, suggesting a protective role for its upregulation in I/R [229]. Pharmacological inhibition of HO-1 by tin mesoporphyrin (SnMP) was also responsible for renal failure [228]. On the contrary, tin protorporhyrin (SnPP, which has an immediate HO-1 inhibition effect, but induces HO-1 after a few hours) prevented injury when injected at a high concentration 24 h before I/R [230]. In addition, HO-1’s effects upon increased resistance to apoptosis in human tubular cells may be involved in protection against tubular injury [91]. However, tubular cells are not the only cell type to be involved, as ischemic injury in HO-1−/− mice also resulted in a significant increase in activated renal macrophages and neutrophils and a simultaneous decrease in intrarenal, dendritic resident cells [231]. Furthermore, upregulation of HO-1 has been shown to protect kidneys from ischemic renal failure [232], and mice with myeloid-restricted deletion of HO-1 exhibited significant renal damage, pro-inflammatory responses and oxidative stress 24 h after reperfusion, but also an impaired tubular repair 7 days after I/R [233]. Thus, HO-1’s upregulation also seems to have a role in renal protection through myeloid circulating cells, notably macrophages, in addition to tubular cells.
HO-1 inducers have generated significant interest in animal models of I/R and many of them have shown protective effects. Among them, hemin exhibited conflicting effects, probably owing to the timing and concentration of administration, namely, protective at low doses and before ischemic injury but deleterious in other cases [230,234]. HO-1 upregulation by Cobalt-Chloride (Co-Cl2) was associated with decreased basal NO concentration, and even inhibited the ischemic increase in NO while preserving post-ischemic glomerular filtration rate (GFR) and blood flow in the outer medullar. These effects where deleted when HO-1 was inhibited pharmacologically by SnMP [235]. Interestingly, statins’ anti-inflammatory effect on I/R injuries could be mediated by upregulation of HO-1 in circulating monocytes/macrophages [236]. Some HO-1 inducers with beneficial effects on I/R (adiponectin [290], bardoloxone methyl [205]) were shown to be Nrf2-independent, acting instead through a peroxisome-proliferator-activated-receptor-α (PPARα) which recognizes a specific region of the HO-1 promoter. Other studies suggested that NGAL (a tissue-specific marker of injury) and the hypoxia inducible factor (HIF) 1 alpha could also play a role in HO-1 upregulation in I/R [237,238].
Prevention of I/R injuries is a serious challenge during cardiac or vascular surgery. In patients, longer GT repeats in the HMOX1 gene promoter (leading to reduced HO-1 inducibility) is associated with an increased risk of AKI after cardiac surgery [38], hinting at a link with HO-1 expression. Indeed, ischemic preconditioning (by vascular clamping) showed promising protective effects in animal models of I/R and the potential implication of HO-1 in the observed effects was suggested [239,240], since a priori augmentation of the expression of HO-1 induces resistance to apoptosis in human tubular cells [91]. However, HO-1 expression was not increased in such models [241,242] and the results of such procedures on AKI incidence in human cardiac surgery were also contradictory [243,244].
Transplantation is, by definition, an I/R situation whose severity varies depending on the kind of the donor (living, brain dead or circulatory dead donor), the graft characteristics (extended criteria or not), but also on cold and warm ischemia times which can lead to delayed graft function or primary non-function. Lower expression of HO-1, VEGF165, and Bcl-2 has been evidenced in kidneys from cadaveric donors in comparison with living donors [98], but no difference was observed between extended criteria donors compared with normal donors [245]. In a rat model of transplantation, induction of HO-1 by hemin [246], by hyperthermic preconditioning [247,248] or by administration of cobalt protoporhyrin (CoPP) [248] or Fenoldopam [249] in the donor were all associated with preserved kidney graft function and prevention of post-reperfusion apoptosis. Duration of warm ischemia time is associated with delayed graft function and has a profound impact on I/R injuries. Indeed, long (45 min) but not short (15 min) warm ischemia induced inflammation, local tissue injury, higher cell-free heme concentrations and increases in HO-1 but also C5aR, IL-6 and TNF-α expression [103]. If cold ischemia time is also associated with delayed graft function, HO-1 expression during this period is probably a bystander effect of the preceding warm ischemia time, though its role has been poorly studied [250]. Interestingly, HO-1 also plays a determining role in transplantation, besides this major protective role in I/R, by preventing Rapamycine toxicity [221], acute rejection (affecting the recipient cellular response and by protecting the graft itself) [251,252] and improving long-term graft survival [256]. Genetic polymorphisms in the HO-1 promoter have been suggested to be associated with better kidney function in the receiver, but with conflicting results [39,253,254,255], and HO-1 has generated interest in xenotransplantation [257,258].
To conclude, HO-1 upregulation during I/R in tubular cells has an essential protective effect, notably by modulating local hemodynamics through NO synthesis regulation, but HO-1 expression by myeloid cells is also key in preserving kidneys from pro-inflammatory effects of recruited circulating immune cells. Many therapeutic molecules have been tested in animals, providing new insights into HO-1 regulation and promising results that could improve clinical practice, in particular for interventions that are (at least partly) scheduled, such as cardiovascular surgery or transplantation.
4.2.2. Sepsis
Sepsis-associated AKI (S-AKI) is a frequent complication in critically ill patients and is associated with morbidity and mortality [291]. Sepsis has a complex and unique pathophysiology, incompletely understood, that distinguishes it from other AKI phenotypes. If S-AKI was previously considered to be merely the natural evolution from decreased renal perfusion to tubular epithelial cell death (or acute tubular necrosis), the broader modern interpretation includes microvascular dysfunction, inflammation, and metabolic reprogramming [291,292]. Resistance and tolerance have been suggested as two different mechanisms that preserve kidney function and sustain wound healing during sepsis [293]. Lipopolysaccharide (LPS) has been known to induce HO-1 expression for almost 50 years [294]. It has been implied that HO-1 confers specific kidney resistance against LPS because HO-1−/− mice exhibited a greater reduction in GFR and renal blood flow, increased renal cytokine expression, and increased activation of NF-κB after LPS injection [279]. In a model of polymicrobial sepsis (cecal ligation and puncture), HO-1-deficient mice displayed a higher mortality rate [280], but also increased Blood Urea Nitrogen (BUN) levels and tubular epithelial necrosis [105]. Levels of plasmatic, cell-free heme were elevated after high-grade infection while hemopexin treatment reduced mortality and improved kidney function (notably improved BUN levels), thereby suggesting a heme-dependent mechanism being responsible for the observed results [105]. Enhanced HO-1 expression, and in particular its capacity for CO formation in macrophages, was reported as essential for pathogen clearance and as contributing to renal protection against AKI [281].
Taken together, HO-1 expression, which is enhanced by endotoxins and probably by elevated heme levels during sepsis, is critical in renal resistance to pathogen-induced tubular epithelial necrosis but also more generally for the clearance of pathogens at the whole-body level.
4.2.3. Hypertensive Nephropathy
Hypertension is the first or second primary cause of progressive CKD (depending on geographic location) and is one of the most important and frequent causes of renal complications. During hypertension, the role of Angiotensin II (Ang II) is central, besides its hemodynamic role, and has been associated in animal models with podocyte loss [295], enhancement of matrix production by mesangial cells [296] and glomerulosclerosis, but also with changes in transporter expression in the tubule [297] and interstitial fibrosis [298], as well as oxidative stress and inflammation [299].
In animals, HO-1−/− mice reportedly had the same blood pressure as controls suggesting HO-1 is not involved in the maintenance of blood pressure under physiological conditions [261]; nevertheless, unilateral kidney clamping in HO-1−/− animals induced more severe hypertension and cardiac hypertrophy than in heterozygous or HO-1+/+ mice, with extensive ischemic injury at the corticomedullary junction [261]. Infusion of Angiotensin II (Ang II) increased HO-1 mRNA levels and its expression in adventitial and endothelial cells in Ang II–induced, hypertensive rat aortas, but losartan or hydralazine treatments prevented both Ang II induction of hypertension and upregulation of HO-1 expression [262]. Overexpression of HO-1, and subsequent increased generation of CO, was shown to reduce the effects of Ang II on blood pressure [263]. HO-1 −/− mice also had increased blood pressure compared with HO +/+ after Ang II infusion, and this was associated with endothelial inflammation in vivo, and with aortic infiltration of pro-inflammatory monocytes and neutrophils [264]. The effect of Ang II on HO-1 have been suggested to be mediated by the Angiotensin Receptor 1 (AT1) [265].
Heme administration, which increases renal HO-1 expression, decreased blood pressure in spontaneously hypertensive rats, both when coupled with administration of arginate [266] or on its own [267]. The effects of heme on blood pressure were counterbalanced by administration of the HO-1 inhibitor ZnPP [267].
The medullar expression of HO-1 has been iteratively explored: indeed, medullary interstitial infusion of the HO-1 inhibitor zinc deuteroporphyrin 2,4-bis glycol (ZnDPBG – by an intramedullary dialysis probe) decreased medullar blood flow and suggested an important role for HO-1 in its maintenance [25]. On the other hand, medullary infusion of CoPP, a potent HO-1 inductor, preserved uni-nephrectomized mice from Ang II–dependent hypertension [268]. Several models of HO-1-specific overexpression in TALH were developed and have indicated a specific role for HO-1 regarding the protection of TALH cells from Ang II (notably decreased prostaglandin E2 (PGE2) levels and Ang II-induced DNA damage) [269] and in decreasing blood pressure, but also in reducing medullary Na-K-Cl cotransporter 2 (NKCC2) transporter expression and sensibility to furosemide [270], thereby modulating natriuresis as previously suggested [271].
In conclusion, if HO-1 is not essential to preserve arterial blood pressure in physiological conditions, it nevertheless has a critical role in reducing blood pressure under pathological conditions, notably under increases in Ang II. Furthermore, medullar and, notably, TALH expression of HO-1 seems to influence many factors implicated in chronic vascular nephropathy, such as decreased blood flow and natriuresis, thus making it highly eligible as a target for future therapeutics.
4.2.4. Diabetic Nephropathy
Diabetic nephropathy (DN), which is a major microangiopathic complication of diabetes, is described as the result of chronic hyperglycemia and accumulation of the resultant advanced glycated products [300], podocyte loss [301], enhanced oxidative stress [302] and impaired microcirculation [303], orchestrated notably by innate immunity [304] leading to fibrosis, matrix deposition, and progressive renal injury [305].
Glomerular HO-1 expression, which is weak in the majority of renal diseases, has unexpectedly been reported as increased in different animal models of diabetes such as streptozotocin (STZ)-induced diabetes mellitus [72,187,188], and in a genetic model of diabetic db/db mice [189]. Even partial genetic deficiency in HO-1 is sufficient to sensitize mice to the development of diabetic glomerular microvascular lesions [190]. In vitro, high-glucose concentrations increased HO-1 expression in podocytes, and also their apoptosis. HO-1 inhibition (by ZnPP or HO-1 siRNA) increases the number of apoptotic podocytes both in vitro and in vivo, again suggesting a protective role for HO-1 [72]. The effects of HO-1 induction by CoPP on apoptosis and glomerular injury were confirmed in a model of STZ-treated, spontaneously hypertensive rats (SHR), who exhibited decreased NF-κB-induced inflammation and oxidative stress [191].
The protective role of HO-1 in DN affects not only podocytes but also mesangial cells, via the influence of Nrf2 [192]. Mouse mesangial cells overexpressing Nrf2 and exposed to high glucose showed upregulated expression of HO-1, as well as decreased ROS production and cell proliferation. These observations were confirmed by experiments with Nrf2-knockout mice in which the opposite was reported.
Enhanced HO-1 expression also plays a role in renal hemodynamics, as shown in STZ-treated rats receiving CoPP, through upregulation of extracellular superoxide dismutase (SOD) and endothelial Nitric Oxid Synthase (NOS) leading to endothelial relaxation and decreased ROS production [193]. Furthermore, HO-1 inhibition by SnMP increased renal vascular resistance and altered GFR and renal blood flow, and this effect was reversed by co-administration of an SOD mimetic or a CO-releasing molecule [194].
Finally, HO-1 also plays a role in insulin sensitivity: its enhancement may reduce adipose tissue volume and cause remodeling of adipose tissue in a diabetic rat model of obesity-induced insulin resistance [195]. In type 2 diabetes patients with poor glycemic control, T(-413)A SNP in the HO-1 promoter was significantly associated with albuminuria development [196], but larger studies are required to confirm these results.
Among HO-1 inducers, hemin has been closely examined in co-administration with STZ and was associated with drastically increased HO-1 expression, especially in tubules, which interestingly was associated with improved renal function, decreased inducible NOS (iNOS), blood glucose levels, micro-albuminuria, glomerulosclerosis and fibrosis at 60 days [197]. These observations further suggest a crosstalk between glomeruli and tubules regarding HO-1. Hemin has been proposed to act through selective enhancement of an anti-inflammatory macrophage-M2 phenotype and IL-10, following a study inhibiting pro-inflammatory macrophage-M1 phenotype and suppressing the extracellular matrix/profibrotic factors responsible for renal lesions and interstitial macrophage infiltration in STZ ND rats [198]. Hemin could also exert protective functions on endothelium through synthesis of bilirubin, as proposed in the db/db genetic mouse model of diabetes where hemin and bilirubin were reportedly enhanced, similar to the endothelium-mediated relaxation of aortas [189]. Several other HO-1 inducers, namely, sinapic acid [199], artemisin [200] and puerarin [201], reportedly reduced kidney injuries in STZ-treated rodents, offering up the possibility of new therapeutic approaches.
As mentioned above, HO-1 is not the only HO implicated in the protection against diabetic nephropathy, and HO-2 deficiency has been shown to enhance STZ-induced renal dysfunction and injury, while HO-1 upregulation prevented these negative effects of DN [306].
To conclude, HO-1 plays a multifaceted and unexpected role in DN by limiting apoptosis of podocytes and mesangial proliferation, modulating renal hemodynamics and bolstering insulin sensitivity, but it may also influence glomerulo-tubular crosstalk in a heme-dependent manner.
4.2.5. Chronic Kidney Disease
While HO-1 has pleiotropic roles in specific nephropathies, these do not add up to the totality of known kidney pathologies. Indeed, each of the pathologies described previously in this section, as well as numerous others not mentioned here, can lead to progressive and permanent death of renal tissue and the replacement of functional nephrons with interstitial fibrosis characteristic of chronic kidney disease (CKD). Unilateral ureteral obstruction (UUO) is the gold standard rodent model for studying rapid, progressive interstitial fibrosis [272]. Early HO-1 expression has been observed in the peri-glomerular and peritubular interstitium [273]. Interestingly, preventive enhancement of HO-1 expression by hemin 48h before UUO attenuated fibrosis in rats by down-regulating inflammatory and pro-fibrotic genes, and decreasing pro-apoptotic pathways (caspase-3 activation), proteinuria and renal dysfunction [274,275]; ZnPP (an HO-1 inhibitor) prevented these protective effects. Confirming this protective role, HO-1 −/− mice with UUO exhibited increased fibrosis, tubular TGF-ß1 expression and inflammation, and had enhanced epithelial to mesenchymal transition with increased infiltration of macrophages [276], as well as increased expression of M2 markers and heavy-chain ferritin H [277]. HO-1 overexpression in transgenic mice was reported to significantly reduce renal interstitial fibrosis, inhibit the loss of peritubular capillaries, suppress activation and proliferation of myofibroblasts, limit the tubule-interstitial infiltration of macrophages and to regulate the secretion of inflammatory cytokines in UUO mice [278]. Taken together, these studies indicate a pleiotropic role for HO-1 in accelerated, obstruction-induced fibrosis, limiting macrophage infiltration while conferring upon them a regulatory phenotype, promoting microcirculation maintenance and inhibiting pro-fibrotic processes. It is, therefore, unsurprising that there is increasing interest in HO-1-targeted therapeutics to treat accelerated fibrosis.
Among the key maintenance factors important during CKD are the metabolic wastes and toxins, which the remaining nephrons are eventually unable to eliminate. Among them, indoxyl sulfate, a uremic toxin, has been shown to downregulate renal expression of Nrf2 through activation of NF-κB, thus down-regulating HO-1 and NQO1 and increasing production of ROS [307]. Indeed, ineffective HO-1 upregulation has been described in a mouse model of progressive post-ischemic inflammation [308], while a decreased Nrf2 expression (HO-1 transcription factor) was also reported in another mouse model of CKD [309,310] following upregulation of the Nrf2 repressor Keap1. This inefficient upregulation of HO-1 was also reported in patients, with significant suppression of plasmatic HO-1 upregulation after administration of low concentrations of SnPP in CKD patients versus healthy volunteers [311].
Interestingly, among patients with coronary artery disease, a greater number of GT dinucleotide repeats in the HMOX1 gene promoter, reducing its expression, has been associated with an increased risk of CKD [312]. Thus, the failure of anti-inflammatory enzymes such as HO-1 to be upregulated may provoke oxidative stress and inflammation, which in turn could contribute to this self-sustaining, injury-promoting state [308]. Despite impaired oxygen supply in CKD, HIF is down-regulated and has been reported as being involved in HO-1 downregulation: HIF induction in a mouse model of CKD restored HO-1 expression, together with VEGF and angiogenesis [313]. Finally, HO-1 in the proximal tubule may reduce albumin-stimulated production of cytokines such as MCP-1 [314]. Thus, repression of HO-1 expression by either Nrf2 or HIF seems to participate in CKD progression by unfavorably influencing angiogenesis and proteinuria.
To conclude, deficiencies in HO-1 upregulation during CKD may favor fibrosis, notably by decreasing angiogenesis and establishing and maintaining a pro-inflammatory state particularly with respect to the macrophage phenotype.
5. Modulation of the HO-1 Axis: Past, Current and Future Strategies
Evidence of the health impact of HO-1 and its involvement in kidney diseases is accumulating and has motivated many researchers to investigate it as a therapeutic target. While this has led to some treatment suggestions, no drugs focusing on the HO system have yet been translated to clinic practice. The complexity of the disease processes studied, and sometimes conflicting results from animal models, contribute to difficulties in its clinical application. In this section, we will briefly describe recent and ongoing human clinical trials, before discussing some issues where HO-1 may be a promising therapeutic avenue.
5.1. Past and Current Clinical Trials in Humans
In the context of an acute exposure that exceeds the regulation systems, free heme is noxious. However, heme is also one of the main inducers of HO-1. Therefore, HO-1 upregulation by controlled administration of low doses of heme (or another HO-1 inducer) could result in a favorable biological response via the production of antioxidant and cytoprotective metabolites. The administration of heme as an HO-1 inducer has been considered as a therapeutic approach and, as described above, pre-treatment with low doses of heme in different animal models of kidney disease has been associated with a decrease in inflammation, oxidative stress and tissue damage [315,316,317]. In humans, both heme and heme arginate (a therapeutic agent for porphyria) induced HO-1 expression in healthy volunteers [318,319], while heme arginate improved experimental ischemia-reperfusion lesions in healthy individuals [320]. These data raised hopes for the therapeutic application of HO-1 induction using heme. However, hemin (the oxidized form of heme) treatment is not without adverse effects: after intravenous administration of hemin in healthy volunteers, thrombophlebitis occurred in 45% of cases (4/9 patients) [321]. Thrombotic complications have also been described in patients with intermittent acute porphyria treated with hemin infusion. Few studies of efficacy exist and their results are inconclusive. In a study of patients with metabolic syndrome, heme arginate did not improve endothelial function or insulin sensitivity, but significantly reduced the vasodilatory response to nitroglycerin [322]. These negative results contrast starkly with the preclinical data, possibly due to the short duration of treatment and limited induction of HO-1, as well as the interference of significantly elevated plasma heme levels [322]. Future studies should focus not only on accurately determining an effective dose and its timing, but also in fully describing the specific conditions under which the normalization of plasma heme levels is accomplished. Other molecules, such as statins [323,324] and 5-aminosalicylic acid [325], lead to positive regulation of HO-1. A clinical trial in a SCD population to evaluate the effect of atorvastatin on endothelial function and inflammatory and oxidative stress markers, including HO-1 activity, is currently under way (Table 2).
The paucity of clinical trials targeting HO-1 in humans is noteworthy and Table 2 details the trials currently referenced on the ClinicalTrials.gov website. A search using the keywords “heme-oxygenase-1” AND “kidney” identified only 15 clinical trials, three of which had not had their status updated in more than two years, despite their completion date having passed, and were therefore not included in Table 2.
5.2. Therapeutic Challenges
The apparent imbalance between the scale of research interest in HO-1 induction—both in vitro and in vivo—and the relatively few ongoing clinical trials is remarkable. Indeed, many gray areas remain regarding the consequences of HO-1 induction, hampering the transition from in vivo experiments to clinical trials. The long-term effects of HO-1 induction in tissues and cells are poorly understood and could have detrimental effects regarding, for example, tumorigenesis, efficacy of treatments for cancer or, more widely, susceptibility to infection.
Indeed, in contrast with all the protective effects described in kidney pathologies, HO-1 pro-survival properties favor cancer progression [326]. Indeed, enhanced HO-1 expression has been correlated with tumor growth, aggressiveness, metastatic and angiogenic potential, resistance to therapy, tumor escape and poor prognosis [327]. Furthermore, HO-1 inhibition showed benefits in a number of cancer models [328]. Even if HO-1’s role in cancer is probably dual (both protective and detrimental), chronic induction of HO-1 could play a role in tumor progression by aiding tumors to escape the host immune system, which remains to be properly studied.
HO-1 induction could also influence the efficacy of cancer treatments, which should be monitored in potential future studies. For example, HO-1 induction has been thoroughly studied to prevent cisplatin toxicity [8,23,94,202,203,204], which still frequently leads to acute kidney injury through glomerular, vascular and tubular damage [329]. Different molecules which increase HO-1 expression have been studied to prevent cisplatin-induced apoptosis and inflammation in the kidneys of mouse or rat models: microRNA-140-5p [206], JQ1 [207], farrerol [208], zinc oxide nanoparticles [209], human growth factor [210], bardoxolone methyl [205] and capsaicin (an active component of chili peppers) [211] have all been studied in this regard. Unfortunately, only a few of these articles reported upon the effect of these molecules on the proapoptotic, antitumoral action of cisplatin on tumoral cells [207], which is the major stumbling block for their use in offsetting iatrogenic renal damage in cancer patients.
Finally, immunoregulatory effects of HO-1 in circulating and resident monocytes and macrophages could influence sensitivity to infection. Indeed, all the pathologies described here, whether mediated by heme or not, are associated with an increased incidence of infections: in transplantation or autoimmune diseases, it is partly a result of immunosuppression; in chronic, heme-associated pathologies such as PNH (including before treatment by Eculizumab [330]), SCD, etc., recurrent infections are a hallmark of disease. HO-1 is thought to be essential for preventing tissue damage during infection; however, this role is dual—notably according to the pathogen type, as described for intracellular pathogens [331]—and its tolerogenic properties may reduce pathogen clearance [332]. Exploring to what extent the immunoregulatory functions of HO-1 could be immunosuppressive (perhaps only moderately so), in particular during long-term HO-1 induction, should be assessed in future studies to guarantee the safety of such applications.
Thus, while in vitro and in vivo data show promising results in several kidney pathologies, more data on the potential bystander effects of long-term HO-1 induction are required to improve our understanding of how HO-1 could be targeted, and to enlarge the spectrum of clinical trials regarding HO-1 induction to other pathologies. The research interest over the past decade in other strategies, such as the administration of heme degradation products, reflects the need for researchers to bypass the duality of heme and HO-1 effects. Increasing levels of CO, through administration of CO-releasing molecules, has notably showed promising results in AKI and in improving transplantation success [333,334,335].
6. Conclusions
HO-1 has essential functions in preserving the kidney against injury, not only in heme-mediated pathologies, but also in a wide range of diseases which differentially affect glomeruli, vessels and tubules in an acute or chronic manner. While major benefits of its induction are expected, the wealth of HO-1 literature in vitro and in vivo contrasts with the small number of clinical trials under way to date. This suggests a need for data regarding the side effects of HO-1 induction, but also for more detailed studies aimed at developing therapeutic tools for specific induction of HO-1 in different structures/cell types.
Author Contributions
A.G., L.T.R. and M.F. were all involved in planning and writing this review. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants from Francophone Society of Nephrology, Dialysis and Transplantation—ASTELLAS 2020 to M.F. and Fondation pour la Recherche Médicale to A.G.
Acknowledgments
The authors are grateful to Mike Howsam for English proofreading.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Jager, K.J.; Kovesdy, C.; Langham, R.; Rosenberg, M.; Jha, V.; Zoccali, C. A Single Number for Advocacy and Communication—Worldwide More than 850 Million Individuals Have Kidney Diseases. Kidney Int. 2019, 96, 1048–1050. [Google Scholar] [CrossRef]
- Foreman, K.J.; Marquez, N.; Dolgert, A.; Fukutaki, K.; Fullman, N.; McGaughey, M.; Pletcher, M.A.; Smith, A.E.; Tang, K.; Yuan, C.-W.; et al. Forecasting Life Expectancy, Years of Life Lost, and All-Cause and Cause-Specific Mortality for 250 Causes of Death: Reference and Alternative Scenarios for 2016–40 for 195 Countries and Territories. Lancet 2018, 392, 2052–2090. [Google Scholar] [CrossRef] [Green Version]
- Luyckx, V.A.; Al-Aly, Z.; Bello, A.K.; Bellorin-Font, E.; Carlini, R.G.; Fabian, J.; Garcia-Garcia, G.; Iyengar, A.; Sekkarie, M.; van Biesen, W.; et al. Sustainable Development Goals Relevant to Kidney Health: An Update on Progress. Nat. Rev. Nephrol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Cassis, P.; Zoja, C.; Perico, L.; Remuzzi, G. A Preclinical Overview of Emerging Therapeutic Targets for Glomerular Diseases. Expert Opin. Ther. Targets 2019, 23, 593–606. [Google Scholar] [CrossRef]
- Ruiz-Ortega, M.; Rayego-Mateos, S.; Lamas, S.; Ortiz, A.; Rodrigues-Diez, R.R. Targeting the Progression of Chronic Kidney Disease. Nat. Rev. Nephrol. 2020, 16, 269–288. [Google Scholar] [CrossRef] [PubMed]
- Tenhunen, R.; Marver, H.S.; Schmid, R. Microsomal Heme Oxygenase Charactarization of the Enzyme. J. Biol. Chem. 1969, 244, 6388–6394. [Google Scholar] [CrossRef]
- Maines, M.D.; Gibbs, P.E.M. 30 Some Years of Heme Oxygenase: From a “Molecular Wrecking Ball” to a “Mesmerizing” Trigger of Cellular Events. Biochem. Biophys. Res. Commun 2005, 338, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Bolisetty, S.; Zarjou, A.; Agarwal, A. Heme Oxygenase 1 as a Therapeutic Target in Acute Kidney Injury. Am. J. Kidney Dis. 2017, 69, 531–545. [Google Scholar] [CrossRef] [Green Version]
- Lever, J.M.; Boddu, R.; George, J.F.; Agarwal, A. Heme Oxygenase-1 in Kidney Health and Disease. Antioxid. Redox Signal. 2016, 25, 165–183. [Google Scholar] [CrossRef] [Green Version]
- Drummond, G.S.; Baum, J.; Greenberg, M.; Lewis, D.; Abraham, N.G. HO-1 Overexpression and Underexpression: Clinical Implications. Arch. Biochem. Biophys. 2019, 673, 108073. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, Y.; Kondo, K.; Momiyama, Y. The Protective Role of Heme Oxygenase-1 in Atherosclerotic Diseases. Int. J. Mol. Sci. 2019, 20, 628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Layton, A.T. Recent Advances in Renal Hypoxia: Insights from Bench Experiments and Computer Simulations. Am. J. Physiol. Ren. Physiol. 2016, 311, F162–F165. [Google Scholar] [CrossRef] [Green Version]
- Ware, L.B.; Johnson, A.C.M.; Zager, R.A. Renal Cortical Albumin Gene Induction and Urinary Albumin Excretion in Response to Acute Kidney Injury. Am. J. Physiol. Ren. Physiol. 2011, 300, F628–F638. [Google Scholar] [CrossRef] [Green Version]
- Zager, R.A.; Vijayan, A.; Johnson, A.C.M. Proximal Tubule Haptoglobin Gene Activation Is an Integral Component of the Acute Kidney Injury “Stress Response.”. Am. J. Physiol. Ren. Physiol. 2012, 303, F139–F148. [Google Scholar] [CrossRef] [Green Version]
- Gazzin, S.; Vitek, L.; Watchko, J.; Shapiro, S.M.; Tiribelli, C. A Novel Perspective on the Biology of Bilirubin in Health and Disease. Trends Mol. Med. 2016, 22, 758–768. [Google Scholar] [CrossRef] [PubMed]
- Nath, K.A.; Garovic, V.D.; Grande, J.P.; Croatt, A.J.; Ackerman, A.W.; Farrugia, G.; Katusic, Z.S.; Belcher, J.D.; Vercellotti, G.M. Heme Oxygenase-2 Protects against Ischemic Acute Kidney Injury: Influence of Age and Sex. Am. J. Physiol. Ren. Physiol. 2019, 317, F695–F704. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, A.; Oda, Y.; Yachie, A.; Koizumi, S.; Nakanishi, I. Heme Oxygenase-1 Deficiency: The First Autopsy Case. Hum. Pathol. 2002, 33, 125–130. [Google Scholar] [CrossRef]
- Radhakrishnan, N.; Yadav, S.P.; Sachdeva, A.; Pruthi, P.K.; Sawhney, S.; Piplani, T.; Wada, T.; Yachie, A. Human Heme Oxygenase-1 Deficiency Presenting with Hemolysis, Nephritis, and Asplenia. J. Pediatr. Hematol. Oncol. 2011, 33, 74–78. [Google Scholar] [CrossRef]
- Poss, K.D.; Tonegawa, S. Reduced Stress Defense in Heme Oxygenase 1-Deficient Cells. Proc. Natl. Acad. Sci. USA 1997, 94, 10925–10930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atsaves, V.; Detsika, M.G.; Poulaki, E.; Gakiopoulou, H.; Lianos, E.A. Phenotypic Characterization of a Novel HO-1 Depletion Model in the Rat. Transgenic Res. 2017, 26, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Kovtunovych, G.; Eckhaus, M.A.; Ghosh, M.C.; Ollivierre-Wilson, H.; Rouault, T.A. Dysfunction of the Heme Recycling System in Heme Oxygenase 1–Deficient Mice: Effects on Macrophage Viability and Tissue Iron Distribution. Blood 2010, 116, 6054–6062. [Google Scholar] [CrossRef] [Green Version]
- Nath, M.; Agarwal, A. New Insights into the Role of Heme Oxygenase-1 in Acute Kidney Injury. Kidney Res. Clin. Pr. 2020. [Google Scholar] [CrossRef]
- Kim, J.; Zarjou, A.; Traylor, A.M.; Bolisetty, S.; Jaimes, E.A.; Hull, T.D.; George, J.F.; Mikhail, F.M.; Agarwal, A. In Vivo Regulation of the Heme Oxygenase-1 Gene in Humanized Transgenic Mice. Kidney Int. 2012, 82, 278–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morimoto, K.; Ohta, K.; Yachie, A.; Yang, Y.; Shimizu, M.; Goto, C.; Toma, T.; Kasahara, Y.; Yokoyama, H.; Miyata, T.; et al. Cytoprotective Role of Heme Oxygenase (HO)-1 in Human Kidney with Various Renal Diseases. Kidney Int. 2001, 60, 1858–1866. [Google Scholar] [CrossRef] [Green Version]
- Ai-Ping, Z.; Billington, H.; Su, N.; Cowley, A.W. Expression and Actions of Heme Oxygenase in the Renal Medulla of Rats. Hypertension 2000, 35, 342–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosley, K.; Wembridge, D.E.; Cattell, V.; Cook, H.T. Heme Oxygenase Is Induced in Nephrotoxic Nephritis and Hemin, a Stimulator of Heme Oxygenase Synthesis, Ameliorates Disease. Kidney Int. 1998, 53, 672–678. [Google Scholar] [CrossRef] [Green Version]
- May, O.; Merle, N.S.; Grunenwald, A.; Gnemmi, V.; Leon, J.; Payet, C.; Robe-Rybkine, T.; Paule, R.; Delguste, F.; Satchell, S.C.; et al. Heme Drives Susceptibility of Glomerular Endothelium to Complement Overactivation Due to Inefficient Upregulation of Heme Oxygenase-1. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Frimat, M.; Boudhabhay, I.; Roumenina, L.T. Hemolysis Derived Products Toxicity and Endothelium: Model of the Second Hit. Toxins 2019, 11, 660. [Google Scholar] [CrossRef] [Green Version]
- Alvarado, G.; Tóth, A.; Csősz, É.; Kalló, G.; Dankó, K.; Csernátony, Z.; Smith, A.; Gram, M.; Akerström, B.; Édes, I.; et al. Heme-Induced Oxidation of Cysteine Groups of Myofilament Proteins Leads to Contractile Dysfunction of Permeabilized Human Skeletal Muscle Fibres. Int. J. Mol. Sci. 2020, 21, 8172. [Google Scholar] [CrossRef] [PubMed]
- Boudhabhay, I.; Poillerat, V.; Grunenwald, A.; Torset, C.; Leon, J.; Daugan, M.V.; Lucibello, F.; El Karoui, K.; Ydee, A.; Chauvet, S.; et al. Complement Activation Is a Crucial Driver of Acute Kidney Injury in Rhabdomyolysis. Kidney Int. 2020. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Ferris, C.D.; Jaffrey, S.R.; Sawa, A.; Takahashi, M.; Brady, S.D.; Barrow, R.K.; Tysoe, S.A.; Wolosker, H.; Barañano, D.E.; Doré, S.; et al. Haem Oxygenase-1 Prevents Cell Death by Regulating Cellular Iron. Nat. Cell Biol. 1999, 1, 152–157. [Google Scholar] [CrossRef]
- Vítek, L. Bilirubin as a Signaling Molecule. Med. Res. Rev. 2020, 40, 1335–1351. [Google Scholar] [CrossRef]
- Nath, K.A.; Balla, G.; Vercellotti, G.M.; Balla, J.; Jacob, H.S.; Levitt, M.D.; Rosenberg, M.E. Induction of Heme Oxygenase Is a Rapid, Protective Response in Rhabdomyolysis in the Rat. J. Clin. Investig. 1992, 90, 267–270. [Google Scholar] [CrossRef] [Green Version]
- Nath, K.A. Heme Oxygenase-1 and Acute Kidney Injury. Curr. Opin. Nephrol. Hypertens. 2014, 23, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Kaneda, H.; Ohno, M.; Taguchi, J.; Togo, M.; Hashimoto, H.; Ogasawara, K.; Aizawa, T.; Ishizaka, N.; Nagai, R. Heme Oxygenase-1 Gene Promoter Polymorphism Is Associated with Coronary Artery Disease in Japanese Patients with Coronary Risk Factors. Arter. Thromb. Vasc. Biol. 2002, 22, 1680–1685. [Google Scholar] [CrossRef] [Green Version]
- Katana, E.; Skoura, L.; Giakoustidis, D.; Takoudas, D.; Malisiovas, N.; Daniilidis, M. Association between the Heme Oxygenase-1 Promoter Polymorphism and Renal Transplantation Outcome in Greece. Transpl. Proc. 2010, 42, 2479–2485. [Google Scholar] [CrossRef] [PubMed]
- Leaf, D.E.; Body, S.C.; Muehlschlegel, J.D.; McMahon, G.M.; Lichtner, P.; Collard, C.D.; Shernan, S.K.; Fox, A.A.; Waikar, S.S. Length Polymorphisms in Heme Oxygenase-1 and AKI after Cardiac Surgery. J. Am. Soc. Nephrol. 2016, 27, 3291–3297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courtney, A.E.; McNamee, P.T.; Middleton, D.; Heggarty, S.; Patterson, C.C.; Maxwell, A.P. Association of Functional Heme Oxygenase-1 Gene Promoter Polymorphism with Renal Transplantation Outcomes. Am. J. Transpl. 2007, 7, 908–913. [Google Scholar] [CrossRef]
- Teissier, T.; Quersin, V.; Gnemmi, V.; Daroux, M.; Howsam, M.; Delguste, F.; Lemoine, C.; Fradin, C.; Schmidt, A.-M.; Cauffiez, C.; et al. Knockout of Receptor for Advanced Glycation End-Products Attenuates Age-Related Renal Lesions. Aging Cell 2019, 18, e12850. [Google Scholar] [CrossRef] [PubMed]
- Ferenbach, D.A.; Nkejabega, N.C.J.; McKay, J.; Choudhary, A.K.; Vernon, M.A.; Beesley, M.F.; Clay, S.; Conway, B.C.; Marson, L.P.; Kluth, D.C.; et al. The Induction of Macrophage Hemeoxygenase-1 Is Protective during Acute Kidney Injury in Aging Mice. Kidney Int. 2011, 79, 966–976. [Google Scholar] [CrossRef] [Green Version]
- Roumenina, L.T.; Rayes, J.; Frimat, M.; Fremeaux-Bacchi, V. Endothelial Cells: Source, Barrier, and Target of Defensive Mediators. Immunol. Rev. 2016, 274, 307–329. [Google Scholar] [CrossRef]
- Yachie, A.; Niida, Y.; Wada, T.; Igarashi, N.; Kaneda, H.; Toma, T.; Ohta, K.; Kasahara, Y.; Koizumi, S. Oxidative Stress Causes Enhanced Endothelial Cell Injury in Human Heme Oxygenase-1 Deficiency. J. Clin. Investig. 1999, 103, 129–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loboda, A.; Jazwa, A.; Grochot-Przeczek, A.; Rutkowski, A.J.; Cisowski, J.; Agarwal, A.; Jozkowicz, A.; Dulak, J. Heme Oxygenase-1 and the Vascular Bed: From Molecular Mechanisms to Therapeutic Opportunities. Antioxid. Redox Signal. 2008, 10, 1767–1812. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-M.; Pae, H.-O.; Park, J.E.; Lee, Y.C.; Woo, J.M.; Kim, N.-H.; Choi, Y.K.; Lee, B.-S.; Kim, S.R.; Chung, H.-T. Heme Oxygenase in the Regulation of Vascular Biology: From Molecular Mechanisms to Therapeutic Opportunities. Antioxid. Redox Signal. 2010, 14, 137–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jourde-Chiche, N.; Fakhouri, F.; Dou, L.; Bellien, J.; Burtey, S.; Frimat, M.; Jarrot, P.-A.; Kaplanski, G.; Le Quintrec, M.; Pernin, V.; et al. Endothelium Structure and Function in Kidney Health and Disease. Nat. Rev. Nephrol. 2019, 15, 87–108. [Google Scholar] [CrossRef]
- Rostgaard, J.; Qvortrup, K. Electron Microscopic Demonstrations of Filamentous Molecular Sieve Plugs in Capillary Fenestrae. Microvasc. Res. 1997, 53, 1–13. [Google Scholar] [CrossRef]
- Satchell, S. The Role of the Glomerular Endothelium in Albumin Handling. Nat. Rev. Nephrol. 2013, 9, 717–725. [Google Scholar] [CrossRef]
- Liu, X.-M.; Peyton, K.J.; Durante, W. Ammonia Promotes Endothelial Cell Survival via the Heme Oxygenase-1-Mediated Release of Carbon Monoxide. Free Radic. Biol. Med. 2017, 102, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Hong, S.W.; Kim, M.-O.; Kim, H.-S.; Jang, J.E.; Leem, J.; Park, I.-S.; Lee, K.-U.; Koh, E.H. S-Adenosyl Methionine Prevents Endothelial Dysfunction by Inducing Heme Oxygenase-1 in Vascular Endothelial Cells. Mol. Cells 2013, 36, 376–384. [Google Scholar] [CrossRef] [Green Version]
- Ali, F.; Hamdulay, S.S.; Kinderlerer, A.R.; Boyle, J.J.; Lidington, E.A.; Yamaguchi, T.; Soares, M.P.; Haskard, D.O.; Randi, A.M.; Mason, J.C. Statin-Mediated Cytoprotection of Human Vascular Endothelial Cells: A Role for Kruppel-like Factor 2-Dependent Induction of Heme Oxygenase-1. J. Thromb. Haemost. 2007, 5, 2537–2546. [Google Scholar] [CrossRef]
- Ungvari, Z.; Orosz, Z.; Rivera, A.; Labinskyy, N.; Xiangmin, Z.; Olson, S.; Podlutsky, A.; Csiszar, A. Resveratrol Increases Vascular Oxidative Stress Resistance. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H2417–H2424. [Google Scholar] [CrossRef]
- He, J.Z.; Ho, J.J.D.; Gingerich, S.; Courtman, D.W.; Marsden, P.A.; Ward, M.E. Enhanced Translation of Heme Oxygenase-2 Preserves Human Endothelial Cell Viability during Hypoxia. J. Biol. Chem. 2010, 285, 9452–9461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tricot, O.; Mallat, Z.; Heymes, C.; Belmin, J.; Lesèche, G.; Tedgui, A. Relation between Endothelial Cell Apoptosis and Blood Flow Direction in Human Atherosclerotic Plaques. Circulation 2000, 101, 2450–2453. [Google Scholar] [CrossRef] [Green Version]
- Aesim, C.; Lylieth, M.; Koopmans, D.; Langille, B. Lowell Effects of Changes in Blood Flow Rate on Cell Death and Cell Proliferation in Carotid Arteries of Immature Rabbits. Circ. Res. 1997, 81, 328–337. [Google Scholar] [CrossRef]
- Yamawaki, H.; Pan, S.; Lee, R.T.; Berk, B.C. Fluid Shear Stress Inhibits Vascular Inflammation by Decreasing Thioredoxin-Interacting Protein in Endothelial Cells. J. Clin. Investig. 2005, 115, 733–738. [Google Scholar] [CrossRef] [Green Version]
- Iring, A.; Jin, Y.-J.; Albarrán-Juárez, J.; Siragusa, M.; Wang, S.; Dancs, P.T.; Nakayama, A.; Tonack, S.; Chen, M.; Künne, C.; et al. Shear Stress-Induced Endothelial Adrenomedullin Signaling Regulates Vascular Tone and Blood Pressure. J. Clin. Investig. 2019, 129, 2775–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.-L.; Varner, S.E.; Rao, A.S.; Grey, J.Y.; Thomas, S.; Cook, C.K.; Wasserman, M.A.; Medford, R.M.; Jaiswal, A.K.; Kunsch, C. Laminar Flow Induction of Antioxidant Response Element-Mediated Genes in Endothelial Cells. A Novel Anti-Inflammatory Mechanism. J. Biol. Chem. 2003, 278, 703–711. [Google Scholar] [CrossRef] [Green Version]
- Freidja, M.L.; Toutain, B.; Caillon, A.; Desquiret, V.; Lambert, D.; Loufrani, L.; Procaccio, V.; Henrion, D. Heme Oxygenase 1 Is Differentially Involved in Blood Flow-Dependent Arterial Remodeling: Role of Inflammation, Oxidative Stress, and Nitric Oxide. Hypertension 2011, 58, 225–231. [Google Scholar] [CrossRef] [Green Version]
- Frimat, M.; Tabarin, F.; Dimitrov, J.D.; Poitou, C.; Halbwachs-Mecarelli, L.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement Activation by Heme as a Secondary Hit for Atypical Hemolytic Uremic Syndrome. Blood 2013, 122, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Belcher, J.D.; Chen, C.; Nguyen, J.; Milbauer, L.; Abdulla, F.; Alayash, A.I.; Smith, A.; Nath, K.A.; Hebbel, R.P.; Vercellotti, G.M. Heme Triggers TLR4 Signaling Leading to Endothelial Cell Activation and Vaso-Occlusion in Murine Sickle Cell Disease. Blood 2014, 123, 377–390. [Google Scholar] [CrossRef] [Green Version]
- Merle, N.S.; Paule, R.; Leon, J.; Daugan, M.; Robe-Rybkine, T.; Poillerat, V.; Torset, C.; Frémeaux-Bacchi, V.; Dimitrov, J.D.; Roumenina, L.T. P-Selectin Drives Complement Attack on Endothelium during Intravascular Hemolysis in TLR-4/Heme-Dependent Manner. Proc. Natl. Acad. Sci. USA 2019, 116, 6280–6285. [Google Scholar] [CrossRef] [Green Version]
- Balla, G.; Jacob, H.S.; Balla, J.; Rosenberg, M.; Nath, K.; Apple, F.; Eaton, J.W.; Vercellotti, G.M. Ferritin: A Cytoprotective Antioxidant Strategem of Endothelium. J. Biol. Chem. 1992, 267, 18148–18153. [Google Scholar] [CrossRef]
- Wijnsma, K.L.; Veissi, S.T.; de Wijs, S.; van der Velden, T.; Wagener, F.A.D.T.G.; van de Kar, N.C.A.J.; van den Heuvel, L.P. Heme as Possible Contributing Factor in the Evolvement of Shiga Toxin Escherichia Coli Induced Hemolytic Uremic Syndrome. Front. Immunol. 2020. [Google Scholar] [CrossRef]
- Kinderlerer, A.R.; Gregoire, I.P.; Hamdulay, S.S.; Ali, F.; Steinberg, R.; Silva, G.; Ali, N.; Wang, B.; Haskard, D.O.; Soares, M.P.; et al. Heme Oxygenase-1 Expression Enhances Vascular Endothelial Resistance to Complement-Mediated Injury through Induction of Decay-Accelerating Factor: A Role for Increased Bilirubin and Ferritin. Blood 2009, 113, 1598–1607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, K.; Nan, C.; Fei, D.; Meng, X.; Liu, W.; Zhang, W.; Jiang, L.; Zhao, M.; Pan, S.; Zhao, M. Heme Oxygenase 1 Modulates Thrombomodulin and Endothelial Protein C Receptor Levels to Attenuate Septic Kidney Injury. Shock 2013, 40, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Soares, M.P.; Seldon, M.P.; Gregoire, I.P.; Vassilevskaia, T.; Berberat, P.O.; Yu, J.; Tsui, T.-Y.; Bach, F.H. Heme Oxygenase-1 Modulates the Expression of Adhesion Molecules Associated with Endothelial Cell Activation. J. Immunol. 2004, 172, 3553–3563. [Google Scholar] [CrossRef] [Green Version]
- Nagata, M. Podocyte Injury and Its Consequences. Kidney Int. 2016, 89, 1221–1230. [Google Scholar] [CrossRef] [PubMed]
- Shimojo, H. Adaptation and Distortion of Podocytes in Rat Remnant Kidney. Pathol. Int. 1998, 48, 368–383. [Google Scholar] [CrossRef]
- Matsui, S.; Sasaki, T.; Kohno, D.; Yaku, K.; Inutsuka, A.; Yokota-Hashimoto, H.; Kikuchi, O.; Suga, T.; Kobayashi, M.; Yamanaka, A.; et al. Neuronal SIRT1 Regulates Macronutrient-Based Diet Selection through FGF21 and Oxytocin Signalling in Mice. Nat. Commun. 2018, 9, 4604. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.-J.; Wu, W.-J.; Zhou, Q.; Jie, J.-P.; Chen, X.; Wang, F.; Gong, X.-H. Advanced Glycation End-Products Induce Oxidative Stress through the Sirt1/Nrf2 Axis by Interacting with the Receptor of AGEs under Diabetic Conditions. J. Cell Biochem. 2018. [Google Scholar] [CrossRef]
- Lee, S.C.; Han, S.H.; Li, J.J.; Lee, S.H.; Jung, D.-S.; Kwak, S.-J.; Kim, S.H.; Kim, D.K.; Yoo, T.-H.; Kim, J.H.; et al. Induction of Heme Oxygenase-1 Protects against Podocyte Apoptosis under Diabetic Conditions. Kidney Int. 2009, 76, 838–848. [Google Scholar] [CrossRef] [Green Version]
- Rubio-Navarro, A.; Sanchez-Niño, M.D.; Guerrero-Hue, M.; García-Caballero, C.; Gutiérrez, E.; Yuste, C.; Sevillano, Á.; Praga, M.; Egea, J.; Román, E.; et al. Podocytes Are New Cellular Targets of Haemoglobin-Mediated Renal Damage. J. Pathol. 2018, 244, 296–310. [Google Scholar] [CrossRef]
- Dean, D.F.; Molitoris, B.A. Chapter 7—The Physiology of the Glomerulus. In Critical Care Nephrology, 3rd ed.; Ronco, C., Bellomo, R., Kellum, J.A., Ricci, Z., Eds.; Elsevier: Philadelphia, PA, USA, 2019; pp. 35–42. ISBN 978-0-323-44942-7. [Google Scholar]
- Ghayur, M.N.; Krepinsky, J.C.; Janssen, L.J. Contractility of the renal glomerulus and mesangial cells: Lingering doubts and strategies for the future. Med. Hypotheses Res. 2008, 4, 1–9. [Google Scholar]
- Moraes, J.A.; Barcellos-de-Souza, P.; Rodrigues, G.; Nascimento-Silva, V.; Silva, S.V.; Assreuy, J.; Arruda, M.A.; Barja-Fidalgo, C. Heme Modulates Smooth Muscle Cell Proliferation and Migration via NADPH Oxidase: A Counter-Regulatory Role for Heme Oxygenase System. Atherosclerosis 2012, 224, 394–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Chen, G.; Wyburn, K.R.; Yin, J.; Bertolino, P.; Eris, J.M.; Alexander, S.I.; Sharland, A.F.; Chadban, S.J. TLR4 Activation Mediates Kidney Ischemia/Reperfusion Injury. J. Clin. Investig. 2007, 117, 2847–2859. [Google Scholar] [CrossRef] [PubMed]
- Datta, P.K.; Lianos, E.A. Nitric Oxide Induces Heme Oxygenase-1 Gene Expression in Mesangial Cells. Kidney Int. 1999, 55, 1734–1739. [Google Scholar] [CrossRef] [Green Version]
- Kumar, D.; Bhaskaran, M.; Alagappan, L.; Tori, D.; Yadav, I.; Konkimalla, S.; Magoon, S.; Singhal, P.C. Heme Oxygenase-1 Modulates Mesangial Cell Proliferation by P21 Waf1 Upregulation. Ren. Fail. 2010, 32, 254–258. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, M.; Ohta, K.; Yang, Y.; Nakai, A.; Toma, T.; Saikawa, Y.; Kasahara, Y.; Yachie, A.; Yokoyama, H.; Seki, H.; et al. Glomerular Proteinuria Induces Heme Oxygenase-1 Gene Expression within Renal Epithelial Cells. Pediatr. Res. 2005, 58, 666–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Ohta, K.; Shimizu, M.; Morimoto, K.; Goto, C.; Nakai, A.; Toma, T.; Kasahara, Y.; Yachie, A.; Seki, H.; et al. Selective Protection of Renal Tubular Epithelial Cells by Heme Oxygenase (HO)-1 during Stress-Induced Injury. Kidney Int. 2003, 64, 1302–1309. [Google Scholar] [CrossRef] [Green Version]
- Ohta, K.; Yachie, A.; Fujimoto, K.; Kaneda, H.; Wada, T.; Toma, T.; Seno, A.; Kasahara, Y.; Yokoyama, H.; Seki, H.; et al. Tubular Injury as a Cardinal Pathologic Feature in Human Heme Oxygenase-1 Deficiency. Am. J. Kidney Dis. 2000, 35, 863–870. [Google Scholar] [CrossRef]
- Nielsen, R.; Christensen, E.I.; Birn, H. Megalin and Cubilin in Proximal Tubule Protein Reabsorption: From Experimental Models to Human Disease. Kidney Int. 2016, 89, 58–67. [Google Scholar] [CrossRef] [Green Version]
- Uchida, S.; Endou, H. Substrate Specificity to Maintain Cellular ATP along the Mouse Nephron. Am. J. Physiol. 1988, 255, F977–F983. [Google Scholar] [CrossRef] [PubMed]
- Sharfuddin, A.A.; Molitoris, B.A. Pathophysiology of Ischemic Acute Kidney Injury. Nat. Rev. Nephrol. 2011, 7, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Nath, K.A.; Grande, J.P.; Croatt, A.J.; Likely, S.; Hebbel, R.P.; Enright, H. Intracellular Targets in Heme Protein-Induced Renal Injury. Kidney Int. 1998, 53, 100–111. [Google Scholar] [CrossRef] [Green Version]
- Tracz, M.J.; Alam, J.; Nath, K.A. Physiology and Pathophysiology of Heme: Implications for Kidney Disease. J. Am. Soc. Nephrol. 2007, 18, 414–420. [Google Scholar] [CrossRef]
- Da Silva, J.-L.; Zand, B.A.; Yang, L.M.; Sabaawy, H.E.; Lianos, E.; Abraham, N.G. Heme Oxygenase Isoform-Specific Expression and Distribution in the Rat Kidney. Kidney Int. 2001, 59, 1448–1457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basireddy, M.; Lindsay, J.T.; Agarwal, A.; Balkovetz, D.F. Epithelial Cell Polarity and Hypoxia Influence Heme Oxygenase-1 Expression by Heme in Renal Epithelial Cells. Am. J. Physiol. Ren. Physiol. 2006, 291, F790–F795. [Google Scholar] [CrossRef] [PubMed]
- Alam, J.; Killeen, E.; Gong, P.; Naquin, R.; Hu, B.; Stewart, D.; Ingelfinger, J.R.; Nath, K.A. Heme Activates the Heme Oxygenase-1 Gene in Renal Epithelial Cells by Stabilizing Nrf2. Am. J. Physiol. Ren. Physiol. 2003, 284, F743–F752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inguaggiato, P.; Gonzalez-Michaca, L.; Croatt, A.J.; Haggard, J.J.; Alam, J.; Nath, K.A. Cellular Overexpression of Heme Oxygenase-1 up-Regulates P21 and Confers Resistance to Apoptosis. Kidney Int. 2001, 60, 2181–2191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Michaca, L.; Farrugia, G.; Croatt, A.J.; Alam, J.; Nath, K.A. Heme: A Determinant of Life and Death in Renal Tubular Epithelial Cells. Am. J. Physiol. Ren. Physiol. 2004, 286, F370–F377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adedoyin, O.; Boddu, R.; Traylor, A.; Lever, J.M.; Bolisetty, S.; George, J.F.; Agarwal, A. Heme Oxygenase-1 Mitigates Ferroptosis in Renal Proximal Tubule Cells. Am. J. Physiol. Ren. Physiol. 2018, 314, F702–F714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolisetty, S.; Traylor, A.M.; Kim, J.; Joseph, R.; Ricart, K.; Landar, A.; Agarwal, A. Heme Oxygenase-1 Inhibits Renal Tubular Macroautophagy in Acute Kidney Injury. J. Am. Soc. Nephrol. 2010, 21, 1702–1712. [Google Scholar] [CrossRef] [Green Version]
- Fine, L.G.; Norman, J.T. The Breathing Kidney. JASN 2002, 13, 1974–1976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gobe, G.C.; Johnson, D.W. Distal Tubular Epithelial Cells of the Kidney: Potential Support for Proximal Tubular Cell Survival after Renal Injury. Int. J. Biochem. Cell Biol. 2007, 39, 1551–1561. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Ma, N.; Yang, M.; Semba, R. Expression and Distribution of Heme Oxygenase-2 MRNA and Protein in Rat Kidney. J. Histochem. Cytochem. 1998, 46, 249–256. [Google Scholar] [CrossRef] [Green Version]
- Lemos, F.B.C. Differential Expression of Heme Oxygenase-1 and Vascular Endothelial Growth Factor in Cadaveric and Living Donor Kidneys after Ischemia-Reperfusion. J. Am. Soc. Nephrol. 2003, 14, 3278–3287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Raaij, S.; van Swelm, R.; Bouman, K.; Cliteur, M.; van den Heuvel, M.C.; Pertijs, J.; Patel, D.; Bass, P.; van Goor, H.; Unwin, R.; et al. Tubular Iron Deposition and Iron Handling Proteins in Human Healthy Kidney and Chronic Kidney Disease. Sci. Rep. 2018, 8, 9353. [Google Scholar] [CrossRef]
- Nath, K.A.; Croatt, A.J.; Haggard, J.J.; Grande, J.P. Renal Response to Repetitive Exposure to Heme Proteins: Chronic Injury Induced by an Acute Insult. Kidney Int. 2000, 57, 2423–2433. [Google Scholar] [CrossRef] [Green Version]
- Kato, G.J.; Piel, F.B.; Reid, C.D.; Gaston, M.H.; Ohene-Frempong, K.; Krishnamurti, L.; Smith, W.R.; Panepinto, J.A.; Weatherall, D.J.; Costa, F.F.; et al. Sickle Cell Disease. Nat. Rev. Dis. Primers 2018, 4, 18010. [Google Scholar] [CrossRef] [Green Version]
- Bosch, X.; Poch, E.; Grau, J.M. Rhabdomyolysis and Acute Kidney Injury. N. Engl. J. Med. 2009, 361, 62–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Vijayan, V.; Jang, M.-S.; Thorenz, A.; Greite, R.; Rong, S.; Chen, R.; Shushakova, N.; Tudorache, I.; Derlin, K.; et al. Labile Heme Aggravates Renal Inflammation and Complement Activation After Ischemia Reperfusion Injury. Front. Immunol. 2019, 10, 2975. [Google Scholar] [CrossRef]
- Maines, M.D.; Mayer, R.D.; Ewing, J.F.; McCoubrey, W.K. Induction of Kidney Heme Oxygenase-1 (HSP32) MRNA and Protein by Ischemia/Reperfusion: Possible Role of Heme as Both Promotor of Tissue Damage and Regulator of HSP32. J. Pharm. Exp. Ther. 1993, 264, 457–462. [Google Scholar]
- Larsen, R.; Gozzelino, R.; Jeney, V.; Tokaji, L.; Bozza, F.A.; Japiassú, A.M.; Bonaparte, D.; Cavalcante, M.M.; Chora, A.; Ferreira, A.; et al. A Central Role for Free Heme in the Pathogenesis of Severe Sepsis. Sci. Transl. Med. 2010, 2, 51ra71. [Google Scholar] [CrossRef] [Green Version]
- Muller-Eberhard, U.; Javid, J.; Liem, H.H.; Hanstein, A.; Hanna, M. Plasma Concentrations of Hemopexin, Haptoglobin and Heme in Patients with Various Hemolytic Diseases. Blood 1968, 32, 811–815. [Google Scholar] [CrossRef]
- Naumann, H.N.; Diggs, L.W.; Barreras, L.; Williams, B.J. Plasma Hemoglobin and Hemoglobin Fractions in Sickle Cell Crisis. Am. J. Clin. Pathol. 1971, 56, 137–147. [Google Scholar] [CrossRef]
- Reiter, C.D.; Wang, X.; Tanus-Santos, J.E.; Hogg, N.; Cannon, R.O.; Schechter, A.N.; Gladwin, M.T. Cell-Free Hemoglobin Limits Nitric Oxide Bioavailability in Sickle-Cell Disease. Nat. Med. 2002, 8, 1383–1389. [Google Scholar] [CrossRef]
- Vinchi Francesca; De Franceschi Lucia; Ghigo Alessandra; Townes Tim; Cimino James; Silengo Lorenzo; Hirsch Emilio; Altruda Fiorella; Tolosano Emanuela Hemopexin Therapy Improves Cardiovascular Function by Preventing Heme-Induced Endothelial Toxicity in Mouse Models of Hemolytic Diseases. Circulation 2013, 127, 1317–1329. [CrossRef] [Green Version]
- Van Avondt, K.; Nur, E.; Zeerleder, S. Mechanisms of Haemolysis-Induced Kidney Injury. Nat. Rev. Nephrol. 2019, 15, 671–692. [Google Scholar] [CrossRef] [PubMed]
- Dvanajscak, Z.; Walker, P.D.; Cossey, L.N.; Messias, N.C.; Boils, C.L.; Kuperman, M.B.; Larsen, C.P. Hemolysis-Associated Hemoglobin Cast Nephropathy Results from a Range of Clinicopathologic Disorders. Kidney Int. 2019, 96, 1400–1407. [Google Scholar] [CrossRef] [Green Version]
- Nath, K.A.; Haggard, J.J.; Croatt, A.J.; Grande, J.P.; Poss, K.D.; Alam, J. The Indispensability of Heme Oxygenase-1 in Protecting against Acute Heme Protein-Induced Toxicity in Vivo. Am. J. Pathol. 2000, 156, 1527–1535. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, B.D.; Rozen, M.G.; Kunis, R.L. Role of Red Cell Membrane Lipid Peroxidation in Hemolysis Due to Phenylhydrazine. Biochem. Pharmacol. 1980, 29, 1355–1359. [Google Scholar] [CrossRef]
- Lim, Y.K.; Jenner, A.; Ali, A.B.; Wang, Y.; Stephen, I.; Hsu, H.; Chong, S.M.; Baumman, H.; Halliwell, B.; Lim, S.-K. Haptoglobin Reduces Renal Oxidative DNA and Tissue Damage during Phenylhydrazine-Induced Hemolysis. Kidney Int. 2000, 58, 1033–1044. [Google Scholar] [CrossRef] [Green Version]
- Merle, N.S.; Grunenwald, A.; Figueres, M.-L.; Chauvet, S.; Daugan, M.; Knockaert, S.; Robe-Rybkine, T.; Noe, R.; May, O.; Frimat, M.; et al. Characterization of Renal Injury and Inflammation in an Experimental Model of Intravascular Hemolysis. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Maines, M.D.; Veltman, J.C. Phenylhydrazine-Mediated Induction of Haem Oxygenase Activity in Rat Liver and Kidney and Development of Hyperbilirubinaemia. Inhibition by Zinc-Protoporphyrin. Biochem. J. 1984, 217, 409–417. [Google Scholar] [CrossRef] [Green Version]
- Rubio-Navarro, A.; Vázquez-Carballo, C.; Guerrero-Hue, M.; García-Caballero, C.; Herencia, C.; Gutiérrez, E.; Yuste, C.; Sevillano, Á.; Praga, M.; Egea, J.; et al. Nrf2 Plays a Protective Role Against Intravascular Hemolysis-Mediated Acute Kidney Injury. Front. Pharmacol. 2019, 10, 740. [Google Scholar] [CrossRef] [Green Version]
- Merle, N.S.; Grunenwald, A.; Rajaratnam, H.; Gnemmi, V.; Frimat, M.; Figueres, M.-L.; Knockaert, S.; Bouzekri, S.; Charue, D.; Noe, R.; et al. Intravascular Hemolysis Activates Complement via Cell-Free Heme and Heme-Loaded Microvesicles. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Merle, N.S.; Leon, J.; Poillerat, V.; Grunenwald, A.; Boudhabhay, I.; Knockaert, S.; Robe-Rybkine, T.; Torset, C.; Pickering, M.C.; Chauvet, S.; et al. Circulating FH Protects Kidneys From Tubular Injury During Systemic Hemolysis. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Poillerat, V.; Gentinetta, T.; Leon, J.; Wassmer, A.; Edler, M.; Torset, C.; Luo, D.; Tuffin, G.; Roumenina, L.T. Hemopexin as an Inhibitor of Hemolysis-Induced Complement Activation. Front. Immunol. 2020, 11, 1684. [Google Scholar] [CrossRef]
- Nath, K.A.; Hebbel, R.P. Sickle Cell Disease: Renal Manifestations and Mechanisms. Nat. Rev. Nephrol. 2015, 11, 161–171. [Google Scholar] [CrossRef] [Green Version]
- Scheinman, J.I. Sickle Cell Disease and the Kidney. Nat. Clin. Pract. Nephrol. 2009, 5, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Olaniran, K.O.; Allegretti, A.S.; Zhao, S.H.; Achebe, M.M.; Eneanya, N.D.; Thadhani, R.I.; Nigwekar, S.U.; Kalim, S. Kidney Function Decline among Black Patients with Sickle Cell Trait and Sickle Cell Disease: An Observational Cohort Study. J. Am. Soc. Nephrol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Nath, K.A.; Grande, J.P.; Haggard, J.J.; Croatt, A.J.; Katusic, Z.S.; Solovey, A.; Hebbel, R.P. Oxidative Stress and Induction of Heme Oxygenase-1 in the Kidney in Sickle Cell Disease. Am. J. Pathol. 2001, 158, 893–903. [Google Scholar] [CrossRef] [Green Version]
- Falk, R.J.; Scheinman, J.; Phillips, G.; Orringer, E.; Johnson, A.; Jennette, J.C. Prevalence and Pathologic Features of Sickle Cell Nephropathy and Response to Inhibition of Angiotensin-Converting Enzyme. N. Engl. J. Med. 1992, 326, 910–915. [Google Scholar] [CrossRef]
- Yamada, N.; Yamaya, M.; Okinaga, S.; Nakayama, K.; Sekizawa, K.; Shibahara, S.; Sasaki, H. Microsatellite Polymorphism in the Heme Oxygenase-1 Gene Promoter Is Associated with Susceptibility to Emphysema. Am. J. Hum. Genet. 2000, 66, 187–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saraf, S.L.; Viner, M.; Rischall, A.; Raslan, R.; Shah, B.N.; Zhang, X.; Han, J.; Gowhari, M.; Jain, S.; Molokie, R.E.; et al. HMOX1 and Acute Kidney Injury in Sickle Cell Anemia. Blood 2018, 132, 1621–1625. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Tan, F.; Yu, T.; Li, Y.; Adisa, O.; Mosunjac, M.; Ofori-Acquah, S.F. Global Gene Expression Profiling of Endothelium Exposed to Heme Reveals an Organ-Specific Induction of Cytoprotective Enzymes in Sickle Cell Disease. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camus, S.M.; De Moraes, J.A.; Bonnin, P.; Abbyad, P.; Le Jeune, S.; Lionnet, F.; Loufrani, L.; Grimaud, L.; Lambry, J.-C.; Charue, D.; et al. Circulating Cell Membrane Microparticles Transfer Heme to Endothelial Cells and Trigger Vaso-Occlusions in Sickle Cell Disease. Blood 2015. [Google Scholar] [CrossRef] [Green Version]
- Jison, M.L.; Munson, P.J.; Barb, J.J.; Suffredini, A.F.; Talwar, S.; Logun, C.; Raghavachari, N.; Beigel, J.H.; Shelhamer, J.H.; Danner, R.L.; et al. Blood Mononuclear Cell Gene Expression Profiles Characterize the Oxidant, Hemolytic, and Inflammatory Stress of Sickle Cell Disease. Blood 2004, 104, 270–280. [Google Scholar] [CrossRef]
- Liu, Y.; Jing, F.; Yi, W.; Mendelson, A.; Shi, P.; Walsh, R.; Friedman, D.F.; Minniti, C.; Manwani, D.; Chou, S.T.; et al. HO-1hi Patrolling Monocytes Protect against Vaso-Occlusion in Sickle Cell Disease. Blood 2018, 131, 1600–1610. [Google Scholar] [CrossRef]
- Haymann, J.-P.; Stankovic, K.; Levy, P.; Avellino, V.; Tharaux, P.-L.; Letavernier, E.; Grateau, G.; Baud, L.; Girot, R.; Lionnet, F. Glomerular Hyperfiltration in Adult Sickle Cell Anemia: A Frequent Hemolysis Associated Feature. Clin. J. Am. Soc. Nephrol. 2010, 5, 756–761. [Google Scholar] [CrossRef]
- Nath, K.A.; Katusic, Z.S. Vasculature and Kidney Complications in Sickle Cell Disease. J. Am. Soc. Nephrol. 2012, 23, 781–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Eps, L.W.S.; Pinedo-Veels, C.; De Vries, G.H.; De Koning, J. NATURE OF CONCENTRATING DEFECT IN SICKLE-CELL NEPHROPATHY: Microradioangiographic Studies. Lancet 1970, 295, 450–452. [Google Scholar] [CrossRef]
- Jana, S.; Strader, M.B.; Meng, F.; Hicks, W.; Kassa, T.; Tarandovskiy, I.; De Paoli, S.; Simak, J.; Heaven, M.R.; Belcher, J.D.; et al. Hemoglobin Oxidation-Dependent Reactions Promote Interactions with Band 3 and Oxidative Changes in Sickle Cell-Derived Microparticles. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Lombardi, E.; Matte, A.; Risitano, A.M.; Ricklin, D.; Lambris, J.D.; Zanet, D.D.; Jokiranta, S.T.; Martinelli, N.; Scambi, C.; Salvagno, G.; et al. Factor H Interferes with the Adhesion of Sickle Red Cells to Vascular Endothelium: A Novel Disease-Modulating Molecule. Haematologica 2019, 104, 919–928. [Google Scholar] [CrossRef] [Green Version]
- Vercellotti, G.M.; Dalmasso, A.P.; Schaid, T.R.; Nguyen, J.; Chen, C.; Ericson, M.E.; Abdulla, F.; Killeen, T.; Lindorfer, M.A.; Taylor, R.P.; et al. Critical Role of C5a in Sickle Cell Disease. Am. J. Hematol. 2019, 94, 327–337. [Google Scholar] [CrossRef]
- Juncos, J.P.; Grande, J.P.; Murali, N.; Croatt, A.J.; Juncos, L.A.; Hebbel, R.P.; Katusic, Z.S.; Nath, K.A. Anomalous Renal Effects of Tin Protoporphyrin in a Murine Model of Sickle Cell Disease. Am. J. Pathol. 2006, 169, 21–31. [Google Scholar] [CrossRef] [Green Version]
- Loboda, A.; Jazwa, A.; Wegiel, B.; Jozkowicz, A.; Dulak, J. Heme Oxygenase-1-Dependent and -Independent Regulation of Angiogenic Genes Expression: Effect of Cobalt Protoporphyrin and Cobalt Chloride on VEGF and IL-8 Synthesis in Human Microvascular Endothelial Cells. Cell Mol. Biol. (Noisy-Le-Grand) 2005, 51, 347–355. [Google Scholar]
- Wang, S.; Avery, J.E.; Hannafon, B.N.; Lind, S.E.; Ding, W.-Q. Zinc Protoporphyrin Suppresses Cancer Cell Viability through a Heme Oxygenase-1-Independent Mechanism: The Involvement of the Wnt/β-Catenin Signaling Pathway. Biochem. Pharm. 2013, 85, 1611–1618. [Google Scholar] [CrossRef] [PubMed]
- Fakhouri, F.; Zuber, J.; Frémeaux-Bacchi, V.; Loirat, C. Haemolytic Uraemic Syndrome. Lancet 2017, 390, 681–696. [Google Scholar] [CrossRef]
- Bitzan, M.; Bickford, B.B.; Foster, G.H. Verotoxin (Shiga Toxin) Sensitizes Renal Epithelial Cells to Increased Heme Toxicity: Possible Implications for the Hemolytic Uremic Syndrome. J. Am. Soc. Nephrol. 2004, 15, 2334–2343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amoura, A.; Moktefi, A.; Halfon, M.; Karras, A.; Rafat, C.; Gibier, J.-B.; Gleeson, P.J.; Servais, A.; Argy, N.; Maillé, P.; et al. Malaria, Collapsing Glomerulopathy, and Focal and Segmental Glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 2020, 15, 964–972. [Google Scholar] [CrossRef]
- Ramos, S.; Carlos, A.R.; Sundaram, B.; Jeney, V.; Ribeiro, A.; Gozzelino, R.; Bank, C.; Gjini, E.; Braza, F.; Martins, R.; et al. Renal Control of Disease Tolerance to Malaria. Proc. Natl. Acad. Sci. USA 2019, 116, 5681–5686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pamplona, A.; Ferreira, A.; Balla, J.; Jeney, V.; Balla, G.; Epiphanio, S.; Chora, Â.; Rodrigues, C.D.; Gregoire, I.P.; Cunha-Rodrigues, M.; et al. Heme Oxygenase-1 and Carbon Monoxide Suppress the Pathogenesis of Experimental Cerebral Malaria. Nat. Med. 2007, 13, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Amodu, A.S.; Pitts, S.; Patrickson, J.; Hibbert, J.M.; Battle, M.; Ofori-Acquah, S.F.; Stiles, J.K. Heme Mediated STAT3 Activation in Severe Malaria. PLoS ONE 2012, 7, e34280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brodsky, R.A. Paroxysmal Nocturnal Hemoglobinuria. Blood 2014, 124, 2804–2811. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.A.; Butler, S.A.; Braren, V.; Hartmann, R.C.; Jenkins, D.E. The Kidneys in Paroxysmal Nocturnal Hemoglobinuria. Blood 1981, 57, 83–89. [Google Scholar] [CrossRef] [Green Version]
- Chow, K.M.; Lai, F.M.; Wang, A.Y.; Chan, Y.L.; Tang, N.L.; Li, P.K. Reversible Renal Failure in Paroxysmal Nocturnal Hemoglobinuria. Am. J. Kidney Dis. 2001, 37, E17. [Google Scholar] [CrossRef]
- Nguyen, J.S.; Marinopoulos, S.S.; Ashar, B.H.; Flynn, J.A. More Than Meets the Eye. N. Engl. J. Med. 2006, 355, 1048–1052. [Google Scholar] [CrossRef]
- Tsai, C.-W.; Wu, V.-C.; Lin, W.-C.; Huang, J.-W.; Wu, M.-S. Acute Renal Failure in a Patient with Paroxysmal Nocturnal Hemoglobinuria. Kidney Int. 2007, 71, 1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandran, A.V.; Nair, R.; Chandran, P.K.G. The Case | Acute Renal Injury in a Young Burmese Immigrant. Paroxysmal Nocturnal Hemoglobinuria. Kidney Int. 2014, 85, 719–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hillmen, P.; Elebute, M.; Kelly, R.; Urbano-Ispizua, A.; Hill, A.; Rother, R.P.; Khursigara, G.; Fu, C.-L.; Omine, M.; Browne, P.; et al. Long-Term Effect of the Complement Inhibitor Eculizumab on Kidney Function in Patients with Paroxysmal Nocturnal Hemoglobinuria. Am. J. Hematol. 2010, 85, 553–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballarín, J.; Arce, Y.; Torra Balcells, R.; Diaz Encarnación, M.; Manzarbeitia, F.; Ortiz, A.; Egido, J.; Moreno, J.A. Acute Renal Failure Associated to Paroxysmal Nocturnal Haemoglobinuria Leads to Intratubular Haemosiderin Accumulation and CD163 Expression. Nephrol. Dial. Transpl. 2011, 26, 3408–3411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nath, K.A.; Vercellotti, G.M.; Grande, J.P.; Miyoshi, H.; Paya, C.V.; Manivel, J.C.; Haggard, J.J.; Croatt, A.J.; Payne, W.D.; Alam, J. Heme Protein-Induced Chronic Renal Inflammation: Suppressive Effect of Induced Heme Oxygenase-11. Kidney Int. 2001, 59, 106–117. [Google Scholar] [CrossRef] [Green Version]
- Candela, N.; Silva, S.; Georges, B.; Cartery, C.; Robert, T.; Moussi-Frances, J.; Rondeau, E.; Rebibou, J.-M.; Lavayssiere, L.; Belliere, J.; et al. Short- and Long-Term Renal Outcomes Following Severe Rhabdomyolysis: A French Multicenter Retrospective Study of 387 Patients. Ann. Intensive Care 2020, 10. [Google Scholar] [CrossRef] [Green Version]
- Kurtz, T.W.; Maletz, R.M.; Hsu, C.H. Renal Cortical Blood Flow in Glycerol-Induced Acute Renal Failure in the Rat. Circ. Res. 1976, 38, 30–35. [Google Scholar] [CrossRef] [Green Version]
- Zager, R.A. Mitochondrial Free Radical Production Induces Lipid Peroxidation during Myohemoglobinuria. Available online: https://pubmed.ncbi.nlm.nih.gov/8648915/ (accessed on 14 October 2020).
- Boutaud, O.; Roberts, L.J. Mechanism-Based Therapeutic Approaches to Rhabdomyolysis-Induced Renal Failure. Free Radic. Biol. Med. 2011, 51, 1062–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belliere, J.; Casemayou, A.; Ducasse, L.; Zakaroff-Girard, A.; Martins, F.; Iacovoni, J.S.; Guilbeau-Frugier, C.; Buffin-Meyer, B.; Pipy, B.; Chauveau, D.; et al. Specific Macrophage Subtypes Influence the Progression of Rhabdomyolysis-Induced Kidney Injury. J. Am. Soc. Nephrol. 2015, 26, 1363–1377. [Google Scholar] [CrossRef] [Green Version]
- Okubo, K.; Kurosawa, M.; Kamiya, M.; Urano, Y.; Suzuki, A.; Yamamoto, K.; Hase, K.; Homma, K.; Sasaki, J.; Miyauchi, H.; et al. Macrophage Extracellular Trap Formation Promoted by Platelet Activation Is a Key Mediator of Rhabdomyolysis-Induced Acute Kidney Injury. Nat. Med. 2018, 24, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zhao, W.; Zhang, L.; Yang, X.; Wang, L.; Chen, Y.; Wang, J.; Zhang, C.; Wu, G. The Role of Complement Activation in Rhabdomyolysis-Induced Acute Kidney Injury. PLoS ONE 2018, 13. [Google Scholar] [CrossRef] [Green Version]
- Zager, R.A.; Johnson, A.C.M. Progressive Histone Alterations and Proinflammatory Gene Activation: Consequences of Heme Protein/Iron-Mediated Proximal Tubule Injury. Am. J. Physiol. Ren. Physiol. 2010, 298, F827–F837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zager, R.A.; Johnson, A.C.M.; Becker, K. Plasma and Urinary Heme Oxygenase-1 in AKI. J. Am. Soc. Nephrol. 2012, 23, 1048–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, A.C.M.; Zager, R.A. Mechanisms Underlying Increased TIMP2 and IGFBP7 Urinary Excretion in Experimental AKI. J. Am. Soc. Nephrol. 2018, 29, 2157–2167. [Google Scholar] [CrossRef]
- Salahudeen, A.K.; Clark, E.C.; Nath, K.A. Hydrogen Peroxide-Induced Renal Injury. A Protective Role for Pyruvate in Vitro and in Vivo. J. Clin. Investig. 1991, 88, 1886–1893. [Google Scholar] [CrossRef]
- Zager, R.A.; Johnson, A.C.M.; Becker, K. Renal Cortical Pyruvate Depletion during AKI. J. Am. Soc. Nephrol. 2014, 25, 998–1012. [Google Scholar] [CrossRef] [Green Version]
- Zarjou, A.; Bolisetty, S.; Joseph, R.; Traylor, A.; Apostolov, E.O.; Arosio, P.; Balla, J.; Verlander, J.; Darshan, D.; Kuhn, L.C.; et al. Proximal Tubule H-Ferritin Mediates Iron Trafficking in Acute Kidney Injury. J. Clin. Investig. 2013, 123, 4423–4434. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Lee, S.S.; Jung, M.H.; Yeo, H.D.; Kim, H.-J.; Yang, J.I.; Roh, G.S.; Chang, S.-H.; Park, D.J. N-Acetylcysteine Attenuates Glycerol-Induced Acute Kidney Injury by Regulating MAPKs and Bcl-2 Family Proteins. Nephrol. Dial. Transpl. 2010, 25, 1435–1443. [Google Scholar] [CrossRef] [Green Version]
- Zager, R.A. Marked Protection against Acute Renal and Hepatic Injury after Nitrited Myoglobin + Tin Protoporphyrin Administration. Transl. Res. 2015, 166, 485–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.; Huang, X.; Zhang, L.; Yang, X.; Wang, L.; Chen, Y.; Wang, J.; Wu, G. Penehyclidine Hydrochloride Pretreatment Ameliorates Rhabdomyolysis-Induced AKI by Activating the Nrf2/HO-1 Pathway and Alleviating [Corrected] Endoplasmic Reticulum Stress in Rats. The. PLoS ONE 2016, 11, e0151158. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Pan, X.; Fu, H.; Zheng, Y.; Dai, Y.; Yin, Y.; Chen, Q.; Hao, Q.; Bao, D.; Hou, D. Effect of Curcumin on Glycerol-Induced Acute Kidney Injury in Rats. Sci. Rep. 2017, 7, 10114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerrero-Hue, M.; García-Caballero, C.; Palomino-Antolín, A.; Rubio-Navarro, A.; Vázquez-Carballo, C.; Herencia, C.; Martín-Sanchez, D.; Farré-Alins, V.; Egea, J.; Cannata, P.; et al. Curcumin Reduces Renal Damage Associated with Rhabdomyolysis by Decreasing Ferroptosis-Mediated Cell Death. FASEB J. 2019, 33, 8961–8975. [Google Scholar] [CrossRef]
- Nath, K.A.; Belcher, J.D.; Nath, M.C.; Grande, J.P.; Croatt, A.J.; Ackerman, A.W.; Katusic, Z.S.; Vercellotti, G.M. Role of TLR4 Signaling in the Nephrotoxicity of Heme and Heme Proteins. Am. J. Physiol. Ren. Physiol. 2018, 314, F906–F914. [Google Scholar] [CrossRef]
- May, O.; Yatime, L.; Merle, N.S.; Delguste, F.; Howsam, M.; Daugan, M.V.; Paul-Constant, C.; Billamboz, M.; Ghinet, A.; Lancel, S.; et al. The Receptor for Advanced Glycation End Products Is a Sensor for Cell-Free Heme. FEBS J. 2020. [Google Scholar] [CrossRef]
- Liu, J.; Huang, K.; Cai, G.-Y.; Chen, X.-M.; Yang, J.-R.; Lin, L.-R.; Yang, J.; Huo, B.-G.; Zhan, J.; He, Y.-N. Receptor for Advanced Glycation End-Products Promotes Premature Senescence of Proximal Tubular Epithelial Cells via Activation of Endoplasmic Reticulum Stress-Dependent P21 Signaling. Cell Signal. 2014, 26, 110–121. [Google Scholar] [CrossRef]
- Takeda, Y.; Takeno, M.; Iwasaki, M.; Kobayashi, H.; Kirino, Y.; Ueda, A.; Nagahama, K.; Aoki, I.; Ishigatsubo, Y. Chemical Induction of HO-1 Suppresses Lupus Nephritis by Reducing Local INOS Expression and Synthesis of Anti-DsDNA Antibody. Clin. Exp. Immunol. 2004, 138, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Yoh, K.; Itoh, K.; Enomoto, A.; Hirayama, A.; Yamaguchi, N.; Kobayashi, M.; Morito, N.; Koyama, A.; Yamamoto, M.; Takahashi, S. Nrf2-Deficient Female Mice Develop Lupus-like Autoimmune Nephritis. Kidney Int 2001, 60, 1343–1353. [Google Scholar] [CrossRef] [Green Version]
- Cuitino, L.; Obreque, J.; Gajardo-Meneses, P.; Villarroel, A.; Crisóstomo, N.; San Francisco, I.F.; Valenzuela, R.A.; Méndez, G.P.; Llanos, C. Heme-Oxygenase-1 Is Decreased in Circulating Monocytes and Is Associated With Impaired Phagocytosis and ROS Production in Lupus Nephritis. Front. Immunol. 2019, 10, 2868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebihara, S.; Tajima, H.; Ono, M. Nuclear Factor Erythroid 2-Related Factor 2 Is a Critical Target for the Treatment of Glucocorticoid-Resistant Lupus Nephritis. Arthritis Res. Ther. 2016, 18, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishimoto, D.; Kirino, Y.; Tamura, M.; Takeno, M.; Kunishita, Y.; Takase-Minegishi, K.; Nakano, H.; Kato, I.; Nagahama, K.; Yoshimi, R.; et al. Dysregulated Heme Oxygenase-1low M2-like Macrophages Augment Lupus Nephritis via Bach1 Induced by Type I Interferons. Arthritis Res. Ther. 2018, 20, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, P.K.; Koukouritaki, S.B.; Hopp, K.A.; Lianos, E.A. Heme Oxygenase-1 Induction Attenuates Inducible Nitric Oxide Synthase Expression and Proteinuria in Glomerulonephritis. J. Am. Soc. Nephrol. 1999, 10, 2540–2550. [Google Scholar]
- Datta, P.K.; Gross, E.J.; Lianos, E.A. Interactions between Inducible Nitric Oxide Synthase and Heme Oxygenase-1 in Glomerulonephritis. Kidney Int. 2002, 61, 847–850. [Google Scholar] [CrossRef] [Green Version]
- Datta, P.K.; Dhupar, S.; Lianos, E.A. Regulatory Effects of Inducible Nitric Oxide Synthase on Cyclooxygenase-2 and Heme Oxygenase-1 Expression in Experimental Glomerulonephritis. Nephrol. Dial. Transpl. 2006, 21, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-C.; Lu, K.-C.; Chen, J.-S.; Hsieh, H.-Y.; Lin, S.-H.; Chu, P.; Wang, J.-Y.; Sytwu, H.-K.; Lin, Y.-F. HO-1 Induction Ameliorates Experimental Murine Membranous Nephropathy: Anti-Oxidative, Anti-Apoptotic and Immunomodulatory Effects. Nephrol. Dial. Transpl. 2008, 23, 3082–3090. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-C.; Huang, Y.-S.; Chen, J.-S.; Huang, C.-F.; Su, S.-L.; Lu, K.-C.; Lin, Y.-F.; Chu, P.; Lin, S.-H.; Sytwu, H.-K. Resveratrol Ameliorates Renal Damage, Increases Expression of Heme Oxygenase-1, and Has Anti-Complement, Anti-Oxidative, and Anti-Apoptotic Effects in a Murine Model of Membranous Nephropathy. PLoS ONE 2015, 10, e0125726. [Google Scholar] [CrossRef]
- Agarwal, A.; Nick, H.S. Renal Response to Tissue Injury: Lessons from Heme Oxygenase-1 Gene Ablation and Expression. JASN 2000, 11, 965–973. [Google Scholar] [PubMed]
- Hayashi, K.; Haneda, M.; Koya, D.; Maeda, S.; Isshiki, K.; Kikkawa, R. Enhancement of Glomerular Heme Oxygenase-1 Expression in Diabetic Rats. Diabetes Res. Clin. Pract. 2001, 52, 85–96. [Google Scholar] [CrossRef]
- Liu, J.; Wang, L.; Tian, X.Y.; Liu, L.; Wong, W.T.; Zhang, Y.; Han, Q.-B.; Ho, H.-M.; Wang, N.; Wong, S.L.; et al. Unconjugated Bilirubin Mediates Heme Oxygenase-1–Induced Vascular Benefits in Diabetic Mice. Diabetes 2015, 64, 1564–1575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenoir, O.; Gaillard, F.; Lazareth, H.; Robin, B.; Tharaux, P.-L. Hmox1 Deficiency Sensitizes Mice to Peroxynitrite Formation and Diabetic Glomerular Microvascular Injuries. J. Diabetes Res. 2017, 2017, 9603924. [Google Scholar] [CrossRef] [Green Version]
- Elmarakby, A.A.; Faulkner, J.; Baban, B.; Saleh, M.A.; Sullivan, J.C. Induction of Hemeoxygenase-1 Reduces Glomerular Injury and Apoptosis in Diabetic Spontaneously Hypertensive Rats. Am. J. Physiol. Ren. Physiol. 2012, 302, F791–F800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Wang, F.; Zhang, L.; Cao, Y.; Liu, W.; Hao, J.; Liu, Q.; Duan, H. Modulation of Nrf2 Expression Alters High Glucose-Induced Oxidative Stress and Antioxidant Gene Expression in Mouse Mesangial Cells. Cell Signal. 2011, 23, 1625–1632. [Google Scholar] [CrossRef]
- Turkseven, S.; Kruger, A.; Mingone, C.J.; Kaminski, P.; Inaba, M.; Rodella, L.F.; Ikehara, S.; Wolin, M.S.; Abraham, N.G. Antioxidant Mechanism of Heme Oxygenase-1 Involves an Increase in Superoxide Dismutase and Catalase in Experimental Diabetes. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H701–H707. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, F.; Lopez, B.; Perez, C.; Fenoy, F.J.; Hernandez, I.; Stec, D.E.; Li Volti, G.; Salom, M.G. Chronic Tempol Treatment Attenuates the Renal Hemodynamic Effects Induced by a Heme Oxygenase Inhibitor in Streptozotocin Diabetic Rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R1540–R1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolai, A.; Li, M.; Kim, D.H.; Peterson, S.J.; Vanella, L.; Positano, V.; Gastaldelli, A.; Rezzani, R.; Rodella, L.F.; Drummond, G.; et al. Heme Oxygenase-1 Induction Remodels Adipose Tissue and Improves Insulin Sensitivity in Obesity-Induced Diabetic Rats. Hypertension 2009, 53, 508–515. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.Y.; Lee, Y.-H.; Kim, S.H.; Jung, K.S.; Chung, K.S.; Kwon, O.; Kim, B.S.; Nam, C.M.; Park, C.S.; Lee, B.-W.; et al. Association Between Heme Oxygenase-1 Promoter Polymorphisms and the Development of Albuminuria in Type 2 Diabetes: A Case-Control Study. Medicine 2015, 94, e1825. [Google Scholar] [CrossRef]
- Ptilovanciv, E.O.; Fernandes, G.S.; Teixeira, L.C.; Reis, L.A.; Pessoa, E.A.; Convento, M.B.; Simões, M.J.; Albertoni, G.A.; Schor, N.; Borges, F.T. Heme Oxygenase 1 Improves Glucoses Metabolism and Kidney Histological Alterations in Diabetic Rats. Diabetol. Metab. Syndr. 2013, 5, 3. [Google Scholar] [CrossRef] [Green Version]
- Ndisang, J.F.; Jadhav, A. Hemin Therapy Improves Kidney Function in Male Streptozotocin-Induced Diabetic Rats: Role of the Heme Oxygenase/Atrial Natriuretic Peptide/Adiponectin Axis. Endocrinology 2014, 155, 215–229. [Google Scholar] [CrossRef] [Green Version]
- Alaofi, A.L. Sinapic Acid Ameliorates the Progression of Streptozotocin (STZ)-Induced Diabetic Nephropathy in Rats via NRF2/HO-1 Mediated Pathways. Front. Pharmacol. 2020, 11, 1119. [Google Scholar] [CrossRef]
- Zhang, H.; Qi, S.; Song, Y.; Ling, C. Artemisinin Attenuates Early Renal Damage on Diabetic Nephropathy Rats through Suppressing TGF-Β1 Regulator and Activating the Nrf2 Signaling Pathway. Life Sci. 2020, 256, 117966. [Google Scholar] [CrossRef]
- Li, X.; Zhu, Q.; Zheng, R.; Yan, J.; Wei, M.; Fan, Y.; Deng, Y.; Zhong, Y. Puerarin Attenuates Diabetic Nephropathy by Promoting Autophagy in Podocytes. Front. Physiol. 2020, 11, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, A.; Balla, J.; Alam, J.; Croatt, A.J.; Nath, K.A. Induction of Heme Oxygenase in Toxic Renal Injury: A Protective Role in Cisplatin Nephrotoxicity in the Rat. Kidney Int. 1995, 48, 1298–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiraishi, F.; Curtis, L.M.; Truong, L.; Poss, K.; Visner, G.A.; Madsen, K.; Nick, H.S.; Agarwal, A. Heme Oxygenase-1 Gene Ablation or Expression Modulates Cisplatin-Induced Renal Tubular Apoptosis. Am. J. Physiol. Ren. Physiol. 2000, 278, F726–F736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Periyasamy-Thandavan, S.; Jiang, M.; Wei, Q.; Smith, R.; Yin, X.-M.; Dong, Z. Autophagy Is Cytoprotective during Cisplatin Injury of Renal Proximal Tubular Cells. Kidney Int. 2008, 74, 631–640. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.Q.; Wang, Y.; Senitko, M.; Meyer, C.; Wigley, W.C.; Ferguson, D.A.; Grossman, E.; Chen, J.; Zhou, X.J.; Hartono, J.; et al. Bardoxolone Methyl (BARD) Ameliorates Ischemic AKI and Increases Expression of Protective Genes Nrf2, PPARγ, and HO-1. Am. J. Physiol. Ren. Physiol. 2011, 300, F1180–F1192. [Google Scholar] [CrossRef] [Green Version]
- Liao, W.; Fu, Z.; Zou, Y.; Wen, D.; Ma, H.; Zhou, F.; Chen, Y.; Zhang, M.; Zhang, W. MicroRNA-140-5p Attenuated Oxidative Stress in Cisplatin Induced Acute Kidney Injury by Activating Nrf2/ARE Pathway through a Keap1-Independent Mechanism. Exp. Cell Res. 2017, 360, 292–302. [Google Scholar] [CrossRef]
- Sun, L.; Liu, J.; Yuan, Y.; Zhang, X.; Dong, Z. Protective Effect of the BET Protein Inhibitor JQ1 in Cisplatin-Induced Nephrotoxicity. Am. J. Physiol. Ren. Physiol. 2018, 315, F469–F478. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Wei, W.; Fan, X.; Ci, X. Farrerol Attenuates Cisplatin-Induced Nephrotoxicity by Inhibiting the Reactive Oxygen Species-Mediated Oxidation, Inflammation, and Apoptotic Signaling Pathways. Front. Physiol. 2019, 10, 1419. [Google Scholar] [CrossRef] [PubMed]
- Barakat, L.A.A.; Barakat, N.; Zakaria, M.M.; Khirallah, S.M. Protective Role of Zinc Oxide Nanoparticles in Kidney Injury Induced by Cisplatin in Rats. Life Sci. 2020, 262, 118503. [Google Scholar] [CrossRef]
- Mahran, Y.F. New Insights into the Protection of Growth Hormone in Cisplatin-Induced Nephrotoxicity: The Impact of IGF-1 on the Keap1-Nrf2/HO-1 Signaling. Life Sci. 2020, 253, 117581. [Google Scholar] [CrossRef]
- Jung, S.-H.; Kim, H.-J.; Oh, G.-S.; Shen, A.; Lee, S.; Choe, S.-K.; Park, R.; So, H.-S. Capsaicin Ameliorates Cisplatin-Induced Renal Injury through Induction of Heme Oxygenase-1. Mol. Cells 2014, 37, 234–240. [Google Scholar] [CrossRef] [Green Version]
- Bolisetty, S.; Traylor, A.; Joseph, R.; Zarjou, A.; Agarwal, A. Proximal Tubule-Targeted Heme Oxygenase-1 in Cisplatin-Induced Acute Kidney Injury. Am. J. Physiol. Ren. Physiol. 2016, 310, F385–F394. [Google Scholar] [CrossRef] [Green Version]
- Sue, Y.-M.; Cheng, C.-F.; Chang, C.-C.; Chou, Y.; Chen, C.-H.; Juan, S.-H. Antioxidation and Anti-Inflammation by Haem Oxygenase-1 Contribute to Protection by Tetramethylpyrazine against Gentamicin-Induced Apoptosis in Murine Renal Tubular Cells. Nephrol. Dial. Transpl. 2009, 24, 769–777. [Google Scholar] [CrossRef] [Green Version]
- Jaikumkao, K.; Pongchaidecha, A.; Chattipakorn, N.; Chatsudthipong, V.; Promsan, S.; Arjinajarn, P.; Lungkaphin, A. Atorvastatin Improves Renal Organic Anion Transporter 3 and Renal Function in Gentamicin-Induced Nephrotoxicity in Rats. Exp. Physiol. 2016, 101, 743–753. [Google Scholar] [CrossRef] [Green Version]
- Polte, T.; Hemmerle, A.; Berndt, G.; Grosser, N.; Abate, A.; Schröder, H. Atrial Natriuretic Peptide Reduces Cyclosporin Toxicity in Renal Cells: Role of CGMP and Heme Oxygenase-1. Free Radic. Biol. Med. 2002, 32, 56–63. [Google Scholar] [CrossRef]
- Shin, D.; Park, H.-M.; Jung, K.-A.; Choi, H.-G.; Kim, J.-A.; Kim, D.-D.; Kim, S.G.; Kang, K.W.; Ku, S.K.; Kensler, T.W.; et al. The NRF2-Heme Oxygenase-1 System Modulates Cyclosporin A-Induced Epithelial-Mesenchymal Transition and Renal Fibrosis. Free Radic. Biol. Med. 2010, 48, 1051–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, Y.A.; Lim, J.H.; Kim, M.Y.; Kim, E.N.; Koh, E.S.; Shin, S.J.; Choi, B.S.; Park, C.W.; Chang, Y.S.; Chung, S. Delayed Treatment with Oleanolic Acid Attenuates Tubulointerstitial Fibrosis in Chronic Cyclosporine Nephropathy through Nrf2/HO-1 Signaling. J. Transl. Med. 2014, 12, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, Q.; Luo, Z.; Wu, C.; Lai, S.; Wei, H.; Li, T.; Wang, Q.; Yu, Y. Attenuation of Cyclosporine A Induced Nephrotoxicity by Schisandrin B through Suppression of Oxidative Stress, Apoptosis and Autophagy. Int. Immunopharmacol. 2017, 52, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Koh, E.S.; Kim, S.; Kim, M.; Hong, Y.A.; Shin, S.J.; Park, C.W.; Chang, Y.S.; Chung, S.; Kim, H.-S. D-Pinitol Alleviates Cyclosporine A-induced Renal Tubulointerstitial Fibrosis via Activating Sirt1 and Nrf2 Antioxidant Pathways. Int. J. Mol. Med. 2018, 41, 1826–1834. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhu, P.; Fujino, M.; Isaka, Y.; Ito, H.; Takahashi, K.; Nakajima, M.; Tanaka, T.; Zhuang, J.; Li, X.-K. 5-Aminolaevulinic Acid (ALA), Enhances Heme Oxygenase (HO)-1 Expression and Attenuates Tubulointerstitial Fibrosis and Renal Apoptosis in Chronic Cyclosporine Nephropathy. Biochem. Biophys. Res. Commun. 2019, 508, 583–589. [Google Scholar] [CrossRef]
- Gonçalves, G.M.; Cenedeze, M.A.; Feitoza, C.Q.; Wang, P.M.H.; Bertocchi, A.P.F.; Damião, M.J.; Pinheiro, H.S.; Antunes Teixeira, V.P.; dos Reis, M.A.; Pacheco-Silva, A.; et al. The Role of Heme Oxygenase 1 in Rapamycin-Induced Renal Dysfunction after Ischemia and Reperfusion Injury. Kidney Int. 2006, 70, 1742–1749. [Google Scholar] [CrossRef] [Green Version]
- Cicora, F.; Lausada, N.; Vasquez, D.N.; Cicora, P.; Guerrieri, D.; Gonzalez, P.; Zalazar, G.; Stringa, P.; Raimondi, C. Protective Effect of Immunosuppressive Treatment before Orthotopic Kidney Autotransplantation. Transpl. Immunol. 2011, 24, 107–112. [Google Scholar] [CrossRef]
- Feitoza, C.Q.; Gonçalves, G.M.; Bertocchi, A.P.F.; Wang, P.W.; Damião, M.J.; Cenedeze, M.A.; Teixeira, V.P.A.; Dos Reis, M.A.; Pacheco-Silva, A.; Câmara, N.O.S. A Role for HO-1 in Renal Function Impairment in Animals Subjected to Ischemic and Reperfusion Injury and Treated with Immunosuppressive Drugs. Transpl. Proc. 2007, 39, 424–426. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.I.; Olszanecki, R.; Yang, L.M.; Quan, S.; Li, M.; Omura, S.; Stec, D.E.; Abraham, N.G. Heme Oxygenase-1 Protects against Radiocontrast-Induced Acute Kidney Injury by Regulating Anti-Apoptotic Proteins. Kidney Int. 2007, 72, 945–953. [Google Scholar] [CrossRef] [Green Version]
- Nath, K.A.; Croatt, A.J.; Likely, S.; Behrens, T.W.; Warden, D. Renal Oxidant Injury and Oxidant Response Induced by Mercury. Kidney Int. 1996, 50, 1032–1043. [Google Scholar] [CrossRef] [Green Version]
- Horikawa, S.; Ito, K.; Ikeda, S.; Shibata, T.; Ishizuka, S.; Yano, T.; Hagiwara, K.; Ozasa, H.; Katsuyama, I. Induction of Heme Oxygenase-1 in Toxic Renal Injury: Mercuric Chloride-Induced Acute Renal Failure in Rat. Toxicol. Lett. 1998, 94, 57–64. [Google Scholar] [CrossRef]
- Yoneya, R.; Ozasa, H.; Nagashima, Y.; Koike, Y.; Teraoka, H.; Hagiwara, K.; Horikawa, S. Hemin Pretreatment Ameliorates Aspects of the Nephropathy Induced by Mercuric Chloride in the Rat. Toxicol. Lett. 2000, 116, 223–229. [Google Scholar] [CrossRef]
- Shimizu, H.; Takahashi, T.; Suzuki, T.; Yamasaki, A.; Fujiwara, T.; Odaka, Y.; Hirakawa, M.; Fujita, H.; Akagi, R. Protective Effect of Heme Oxygenase Induction in Ischemic Acute Renal Failure. Crit. Care Med. 2000, 28, 809–817. [Google Scholar] [CrossRef]
- Tracz, M.J.; Juncos, J.P.; Croatt, A.J.; Ackerman, A.W.; Grande, J.P.; Knutson, K.L.; Kane, G.C.; Terzic, A.; Griffin, M.D.; Nath, K.A. Deficiency of Heme Oxygenase-1 Impairs Renal Hemodynamics and Exaggerates Systemic Inflammatory Responses to Renal Ischemia. Kidney Int. 2007, 72, 1073–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaizu, T.; Tamaki, T.; Tanaka, M.; Uchida, Y.; Tsuchihashi, S.-I.; Kawamura, A.; Kakita, A. Preconditioning with Tin-Protoporphyrin IX Attenuates Ischemia/Reperfusion Injury in the Rat Kidney. Kidney Int. 2003, 63, 1393–1403. [Google Scholar] [CrossRef] [Green Version]
- Hull, T.D.; Kamal, A.I.; Boddu, R.; Bolisetty, S.; Guo, L.; Tisher, C.C.; Rangarajan, S.; Chen, B.; Curtis, L.M.; George, J.F.; et al. Heme Oxygenase-1 Regulates Myeloid Cell Trafficking in AKI. J. Am. Soc. Nephrol. 2015. [Google Scholar] [CrossRef] [Green Version]
- Ferenbach, D.A.; Ramdas, V.; Spencer, N.; Marson, L.; Anegon, I.; Hughes, J.; Kluth, D.C. Macrophages Expressing Heme Oxygenase-1 Improve Renal Function in Ischemia/Reperfusion Injury. Mol. Ther. 2010, 18, 1706–1713. [Google Scholar] [CrossRef]
- Rossi, M.; Thierry, A.; Delbauve, S.; Preyat, N.; Soares, M.P.; Roumeguère, T.; Leo, O.; Flamand, V.; Le Moine, A.; Hougardy, J.-M. Specific Expression of Heme Oxygenase-1 by Myeloid Cells Modulates Renal Ischemia-Reperfusion Injury. Sci. Rep. 2017, 7, 197. [Google Scholar] [CrossRef]
- Rossi, M.; Delbauve, S.; Wespes, E.; Roumeguère, T.; Leo, O.; Flamand, V.; Le Moine, A.; Hougardy, J.-M. Dual Effect of Hemin on Renal Ischemia-Reperfusion Injury. Biochem. Biophys. Res. Commun. 2018, 503, 2820–2825. [Google Scholar] [CrossRef]
- Salom, M.G.; Cerón, S.N.; Rodriguez, F.; Lopez, B.; Hernández, I.; Martínez, J.G.; Losa, A.M.; Fenoy, F.J. Heme Oxygenase-1 Induction Improves Ischemic Renal Failure: Role of Nitric Oxide and Peroxynitrite. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H3542–H3549. [Google Scholar] [CrossRef]
- Gueler, F.; Park, J.-K.; Rong, S.; Kirsch, T.; Lindschau, C.; Zheng, W.; Elger, M.; Fiebeler, A.; Fliser, D.; Luft, F.C.; et al. Statins Attenuate Ischemia-Reperfusion Injury by Inducing Heme Oxygenase-1 in Infiltrating Macrophages. Am. J. Pathol. 2007, 170, 1192–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, K.; Lee, H.T.; Rapoport, D.; Drexler, I.R.; Foster, K.; Yang, J.; Schmidt-Ott, K.M.; Chen, X.; Li, J.Y.; Weiss, S.; et al. Endocytic Delivery of Lipocalin-Siderophore-Iron Complex Rescues the Kidney from Ischemia-Reperfusion Injury. J. Clin. Investig. 2005, 115, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Conde, E.; Alegre, L.; Blanco-Sánchez, I.; Sáenz-Morales, D.; Aguado-Fraile, E.; Ponte, B.; Ramos, E.; Sáiz, A.; Jiménez, C.; Ordoñez, A.; et al. Hypoxia Inducible Factor 1-Alpha (HIF-1 Alpha) Is Induced during Reperfusion after Renal Ischemia and Is Critical for Proximal Tubule Cell Survival. PLoS ONE 2012, 7, e33258. [Google Scholar] [CrossRef]
- Zager, R.A.; Baltes, L.A.; Sharma, H.M.; Jurkowitz, M.S. Responses of the Ischemic Acute Renal Failure Kidney to Additional Ischemic Events. Kidney Int. 1984, 26, 689–700. [Google Scholar] [CrossRef] [Green Version]
- Wever, K.E.; Menting, T.P.; Rovers, M.; van der Vliet, J.A.; Rongen, G.A.; Masereeuw, R.; Ritskes-Hoitinga, M.; Hooijmans, C.R.; Warlé, M. Ischemic Preconditioning in the Animal Kidney, a Systematic Review and Meta-Analysis. PLoS ONE 2012, 7, e32296. [Google Scholar] [CrossRef] [Green Version]
- Joo, J.D.; Kim, M.; D’Agati, V.D.; Lee, H.T. Ischemic Preconditioning Provides Both Acute and Delayed Protection against Renal Ischemia and Reperfusion Injury in Mice. JASN 2006, 17, 3115–3123. [Google Scholar] [CrossRef]
- Soendergaard, P.; Krogstrup, N.V.; Secher, N.G.; Ravlo, K.; Keller, A.K.; Toennesen, E.; Bibby, B.M.; Moldrup, U.; Ostraat, E.O.; Pedersen, M.; et al. Improved GFR and Renal Plasma Perfusion Following Remote Ischaemic Conditioning in a Porcine Kidney Transplantation Model. Transpl. Int. 2012, 25, 1002–1012. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, X.; Chi, D.; Wang, S.; Wei, H.; Yu, H.; Li, Q.; Liu, B. Remote Ischemic Preconditioning for Prevention of Acute Kidney Injury in Patients Undergoing On-Pump Cardiac Surgery. Medicine 2016, 95. [Google Scholar] [CrossRef]
- Meersch, M.; Küllmar, M.; Pavenstädt, H.; Rossaint, J.; Kellum, J.A.; Martens, S.; Klausmeyer, P.; Schmidt, E.A.; Kerschke, L.; Zarbock, A. Effects of Different Doses of Remote Ischemic Preconditioning on Kidney Damage Among Patients Undergoing Cardiac Surgery: A Single-Center Mechanistic Randomized Controlled Trial. Crit. Care Med. 2020, 48, e690–e697. [Google Scholar] [CrossRef]
- Florim, G.M.S.; Caldas, H.C.; Gonçalves, N.N.; Bueno, G.O.B.E.; Baptista, M.A.S.F.; Fernandes-Charpiot, I.M.M.; Abbud-Filho, M. Activation of HMGB1–TLR4 Pathway and Inflammasome Contribute to Enhanced Inflammatory Response in Extended Criteria and Kidneys With KDPI ≥85%. Transplantation 2020, 104, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Hölzen, J.P.; August, C.; Bahde, R.; Minin, E.; Lang, D.; Heidenreich, S.; Dietl, K.-H.; Spiegel, H.-U. Influence of Heme Oxygenase-1 on Microcirculation after Kidney Transplantation. J. Surg. Res. 2008, 148, 126–135. [Google Scholar] [CrossRef]
- Redaelli, C.A.; Wagner, M.; Kulli, C.; Tian, Y.-H.; Schilling, M.K.; Wagner, A.C.C.; Mazzucchelli, L.; Kubulus, D. Hyperthermia-Induced HSP Expression Correlates with Improved Rat Renal Isograft Viability and Survival in Kidneys Harvested from Non-Heart-Beating Donors. Transpl. Int. 2001, 14, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.; Cadetg, P.; Ruf, R.; Mazzucchelli, L.; Ferrari, P.; Redaelli, C.A. Heme Oxygenase-1 Attenuates Ischemia/Reperfusion-Induced Apoptosis and Improves Survival in Rat Renal Allografts. Kidney Int. 2003, 63, 1564–1573. [Google Scholar] [CrossRef] [Green Version]
- Salahudeen, A.K.; Yang, M.; Huang, H.; Dore, S.; Stec, D.E. Fenoldopam-Preconditioning: Role of Heme Oxygenase-1 in Protecting Human Tubular Cells and Rodent Kidneys against Cold-Hypoxic Injury. Transplantation 2011, 91, 176–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corona, D.; Ekser, B.; Gioco, R.; Caruso, M.; Schipa, C.; Veroux, P.; Giaquinta, A.; Granata, A.; Veroux, M. Heme-Oxygenase and Kidney Transplantation: A Potential for Target Therapy? Biomolecules 2020, 10, 840. [Google Scholar] [CrossRef]
- Agarwal, A.; Kim, Y.; Matas, A.J.; Alam, J.; Nath, K.A. Gas-Generating Systems in Acute Renal Allograft Rejection in the Rat. Co-Induction of Heme Oxygenase and Nitric Oxide Synthase. Transplantation 1996, 61, 93–98. [Google Scholar] [CrossRef]
- Öllinger, R.; Pratschke, J. Role of Heme Oxygenase-1 in Transplantation: HO-1 in Transplantation. Transpl. Int. 2010, 23, 1071–1081. [Google Scholar] [CrossRef] [PubMed]
- Baan, C.; Peeters, A.; Lemos, F.; Uitterlinden, A.; Doxiadis, I.; Claas, F.; Ijzermans, J.; Roodnat, J.; Weimar, W. Fundamental Role for HO-1 in the Self-Protection of Renal Allografts. Am. J. Transplant. 2004, 4, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Exner, M.; Böhmig, G.A.; Schillinger, M.; Regele, H.; Watschinger, B.; Hörl, W.H.; Raith, M.; Mannhalter, C.; Wagner, O.F. Donor Heme Oxygenase-1 Genotype Is Associated with Renal Allograft Function. Transplantation 2004, 77, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Israni, A.K.; Li, N.; Cizman, B.B.; Snyder, J.; Abrams, J.; Joffe, M.; Rebbeck, T.; Feldman, H.I. Association of Donor Inflammation– and Apoptosis-Related Genotypes and Delayed Allograft Function after Kidney Transplantation. Am. J. Kidney Dis. 2008, 52, 331–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bédard, E.L.R.; Jiang, J.; Parry, N.; Wang, H.; Liu, W.; Garcia, B.; Kim, P.; Chakrabarti, S.; Buelow, R.; Zhong, R. Peritransplant Treatment with Cobalt Protoporphyrin Attenuates Chronic Renal Allograft Rejection. Transpl. Int. 2005, 18, 341–349. [Google Scholar] [CrossRef]
- Petersen, B.; Ramackers, W.; Lucas-Hahn, A.; Lemme, E.; Hassel, P.; Queisser, A.-L.; Herrmann, D.; Barg-Kues, B.; Carnwath, J.W.; Klose, J.; et al. Transgenic Expression of Human Heme Oxygenase-1 in Pigs Confers Resistance against Xenograft Rejection during Ex Vivo Perfusion of Porcine Kidneys. Xenotransplantation 2011, 18, 355–368. [Google Scholar] [CrossRef]
- Yeom, H.-J.; Koo, O.J.; Yang, J.; Cho, B.; Hwang, J.-I.; Park, S.J.; Hurh, S.; Kim, H.; Lee, E.M.; Ro, H.; et al. Generation and Characterization of Human Heme Oxygenase-1 Transgenic Pigs. PLoS ONE 2012, 7, e46646. [Google Scholar] [CrossRef]
- Hancock, W.W.; Buelow, R.; Sayegh, M.H.; Turka, L.A. Antibody-Induced Transplant Arteriosclerosis Is Prevented by Graft Expression of Anti-Oxidant and Anti-Apoptotic Genes. Nat. Med. 1998, 4, 1392–1396. [Google Scholar] [CrossRef]
- Yamashita, K.; Ollinger, R.; McDaid, J.; Sakahama, H.; Wang, H.; Tyagi, S.; Csizmadia, E.; Smith, N.R.; Soares, M.P.; Bach, F.H. Heme Oxygenase-1 Is Essential for and Promotes Tolerance to Transplanted Organs. FASEB J. 2006, 20, 776–778. [Google Scholar] [CrossRef]
- Wiesel, P.; Patel, A.P.; Carvajal, I.M.; Wang, Z.Y.; Pellacani, A.; Maemura, K.; DiFonzo, N.; Rennke, H.G.; Layne, M.D.; Yet, S.F.; et al. Exacerbation of Chronic Renovascular Hypertension and Acute Renal Failure in Heme Oxygenase-1-Deficient Mice. Circ. Res. 2001, 88, 1088–1094. [Google Scholar] [CrossRef] [Green Version]
- Ishizaka, N.; de León, H.; Laursen, J.B.; Fukui, T.; Wilcox, J.N.; De Keulenaer, G.; Griendling, K.K.; Alexander, R.W. Angiotensin II-Induced Hypertension Increases Heme Oxygenase-1 Expression in Rat Aorta. Circulation 1997, 96, 1923–1929. [Google Scholar] [CrossRef]
- Yang, L.; Quan, S.; Nasjletti, A.; Laniado-Schwartzman, M.; Abraham, N.G. Heme Oxygenase-1 Gene Expression Modulates Angiotensin II-Induced Increase in Blood Pressure. Hypertension 2004, 43, 1221–1226. [Google Scholar] [CrossRef] [Green Version]
- Wenzel, P.; Rossmann, H.; Müller, C.; Kossmann, S.; Oelze, M.; Schulz, A.; Arnold, N.; Simsek, C.; Lagrange, J.; Klemz, R.; et al. Heme Oxygenase-1 Suppresses a pro-Inflammatory Phenotype in Monocytes and Determines Endothelial Function and Arterial Hypertension in Mice and Humans. Eur. Heart J. 2015, 36, 3437–3446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konvalinka, A.; Zhou, J.; Dimitromanolakis, A.; Drabovich, A.P.; Fang, F.; Gurley, S.; Coffman, T.; John, R.; Zhang, S.-L.; Diamandis, E.P.; et al. Determination of an Angiotensin II-Regulated Proteome in Primary Human Kidney Cells by Stable Isotope Labeling of Amino Acids in Cell Culture (SILAC). J. Biol. Chem. 2013, 288, 24834–24847. [Google Scholar] [CrossRef] [Green Version]
- Levere, R.D.; Martasek, P.; Escalante, B.; Schwartzman, M.L.; Abraham, N.G. Effect of Heme Arginate Administration on Blood Pressure in Spontaneously Hypertensive Rats. J. Clin. Investig. 1990, 86, 213–219. [Google Scholar] [CrossRef] [Green Version]
- Martasek, P.; Schwartzman, M.L.; Goodman, A.I.; Solangi, K.B.; Levere, R.D.; Abraham, N.G. Hemin and L-Arginine Regulation of Blood Pressure in Spontaneous Hypertensive Rats. JASN 1991, 2, 1078–1084. [Google Scholar]
- Vera, T.; Kelsen, S.; Stec, D.E. Kidney-Specific Induction of Heme Oxygenase-1 Prevents Angiotensin II Hypertension. Hypertension 2008, 52, 660–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quan, S.; Yang, L.; Shnouda, S.; Schwartzman, M.L.; Nasjletti, A.; Goodman, A.I.; Abraham, N.G. Expression of Human Heme Oxygenase-1 in the Thick Ascending Limb Attenuates Angiotensin II-Mediated Increase in Oxidative Injury. Kidney Int. 2004, 65, 1628–1639. [Google Scholar] [CrossRef] [Green Version]
- Stec, D.E.; Drummond, H.A.; Gousette, M.U.; Storm, M.V.; Abraham, N.G.; Csongradi, E. Expression of Heme Oxygenase-1 in Thick Ascending Loop of Henle Attenuates Angiotensin II-Dependent Hypertension. J. Am. Soc. Nephrol. 2012, 23, 834–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Yi, F.; dos Santos, E.A.; Donley, D.K.; Li, P.-L. Role of Renal Medullary Heme Oxygenase in the Regulation of Pressure Natriuresis and Arterial Blood Pressure. Hypertension 2007, 49, 148–154. [Google Scholar] [CrossRef] [Green Version]
- Bascands, J.-L.; Schanstra, J.P. Obstructive Nephropathy: Insights from Genetically Engineered Animals. Kidney Int. 2005, 68, 925–937. [Google Scholar] [CrossRef] [Green Version]
- Kawada, N.; Moriyama, T.; Ando, A.; Fukunaga, M.; Miyata, T.; Kurokawa, K.; Imai, E.; Hori, M. Increased Oxidative Stress in Mouse Kidneys with Unilateral Ureteral Obstruction. Kidney Int. 1999, 56, 1004–1013. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Yang, J.I.; Jung, M.H.; Hwa, J.S.; Kang, K.R.; Kang, K.-R.; Park, D.J.; Roh, G.S.; Cho, G.J.; Choi, W.S.; et al. Heme Oxygenase-1 Protects Rat Kidney from Ureteral Obstruction via an Antiapoptotic Pathway. J. Am. Soc. Nephrol. 2006, 17, 1373–1381. [Google Scholar] [CrossRef] [Green Version]
- Correa-Costa, M.; Semedo, P.; Monteiro, A.P.F.S.; Silva, R.C.; Pereira, R.L.; Gonçalves, G.M.; Marques, G.D.M.; Cenedeze, M.A.; Faleiros, A.C.G.; Keller, A.C.; et al. Induction of Heme Oxygenase-1 Can Halt and Even Reverse Renal Tubule-Interstitial Fibrosis. PLS ONE 2010, 5, e14298. [Google Scholar] [CrossRef] [Green Version]
- Kie, J.-H.; Kapturczak, M.H.; Traylor, A.; Agarwal, A.; Hill-Kapturczak, N. Heme Oxygenase-1 Deficiency Promotes Epithelial-Mesenchymal Transition and Renal Fibrosis. J. Am. Soc. Nephrol. 2008, 19, 1681–1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolisetty, S.; Zarjou, A.; Hull, T.D.; Traylor, A.; Perianayagam, A.; Joseph, R.; Kamal, A.I.; Arosio, P.; Soares, M.P.; Jeney, V.; et al. Macrophage and Epithelial Cell H-Ferritin Expression Regulates Renal Inflammation. Kidney Int. 2015, 88, 95–108. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Wei, S.-Y.; Li, J.-S.; Zhang, Q.-F.; Wang, Y.-X.; Zhao, S.-L.; Yu, J.; Wang, C.; Qin, Y.; Wei, Q.-J.; et al. Overexpression of Heme Oxygenase-1 Prevents Renal Interstitial Inflammation and Fibrosis Induced by Unilateral Ureter Obstruction. PLoS ONE 2016, 11, e0147084. [Google Scholar] [CrossRef] [Green Version]
- Tracz, M.J.; Juncos, J.P.; Grande, J.P.; Croatt, A.J.; Ackerman, A.W.; Rajagopalan, G.; Knutson, K.L.; Badley, A.D.; Griffin, M.D.; Alam, J.; et al. Renal Hemodynamic, Inflammatory, and Apoptotic Responses to Lipopolysaccharide in HO-1−/− Mice. Am. J. Pathol. 2007, 170, 1820–1830. [Google Scholar] [CrossRef] [Green Version]
- Chung, S.W.; Liu, X.; Macias, A.A.; Baron, R.M.; Perrella, M.A. Heme Oxygenase-1–Derived Carbon Monoxide Enhances the Host Defense Response to Microbial Sepsis in Mice. J. Clin. Investig. 2008, 118, 239–247. [Google Scholar] [CrossRef] [Green Version]
- Wegiel, B.; Larsen, R.; Gallo, D.; Chin, B.Y.; Harris, C.; Mannam, P.; Kaczmarek, E.; Lee, P.J.; Zuckerbraun, B.S.; Flavell, R.; et al. Macrophages Sense and Kill Bacteria through Carbon Monoxide-Dependent Inflammasome Activation. J. Clin. Investig. 2014, 124, 4926–4940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cudmore, M.; Ahmad, S.; Al-Ani, B.; Fujisawa, T.; Coxall, H.; Chudasama, K.; Devey, L.R.; Wigmore, S.J.; Abbas, A.; Hewett, P.W.; et al. Negative Regulation of Soluble Flt-1 and Soluble Endoglin Release by Heme Oxygenase-1. Circulation 2007, 115, 1789–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Ani, B.; Hewett, P.W.; Cudmore, M.J.; Fujisawa, T.; Saifeddine, M.; Williams, H.; Ramma, W.; Sissaoui, S.; Jayaraman, P.-S.; Ohba, M.; et al. Activation of Proteinase-Activated Receptor 2 Stimulates Soluble Vascular Endothelial Growth Factor Receptor 1 Release via Epidermal Growth Factor Receptor Transactivation in Endothelial Cells. Hypertension 2010, 55, 689–697. [Google Scholar] [CrossRef] [Green Version]
- Craici, I.M.; Wagner, S.J.; Weissgerber, T.L.; Grande, J.P.; Garovic, V.D. Advances in the Pathophysiology of Pre-Eclampsia and Related Podocyte Injury. Kidney Int. 2014, 86, 275–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakrania, B.A.; Spradley, F.T.; Satchell, S.C.; Stec, D.E.; Rimoldi, J.M.; Gadepalli, R.S.V.; Granger, J.P. Heme Oxygenase-1 Is a Potent Inhibitor of Placental Ischemia-Mediated Endothelin-1 Production in Cultured Human Glomerular Endothelial Cells. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 314, R427–R432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maser, R.L.; Vassmer, D.; Magenheimer, B.S.; Calvet, J.P. Oxidant Stress and Reduced Antioxidant Enzyme Protection in Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2002, 13, 991–999. [Google Scholar] [PubMed]
- Zhou, J.; Ouyang, X.; Schoeb, T.R.; Bolisetty, S.; Cui, X.; Mrug, S.; Yoder, B.K.; Johnson, M.R.; Szalai, A.J.; Mrug, M. Kidney Injury Accelerates Cystogenesis via Pathways Modulated by Heme Oxygenase and Complement. JASN 2012, 23, 1161–1171. [Google Scholar] [CrossRef] [Green Version]
- Fujiki, T.; Ando, F.; Murakami, K.; Isobe, K.; Mori, T.; Susa, K.; Nomura, N.; Sohara, E.; Rai, T.; Uchida, S. Tolvaptan Activates the Nrf2/HO-1 Antioxidant Pathway through PERK Phosphorylation. Sci. Rep. 2019, 9, 9245. [Google Scholar] [CrossRef] [Green Version]
- Pefanis, A.; Ierino, F.L.; Murphy, J.M.; Cowan, P.J. Regulated Necrosis in Kidney Ischemia-Reperfusion Injury. Kidney Int. 2019, 96, 291–301. [Google Scholar] [CrossRef]
- Cheng, C.-F.; Lian, W.-S.; Chen, S.-H.; Lai, P.-F.; Li, H.-F.; Lan, Y.-F.; Cheng, W.T.-K.; Lin, H. Protective Effects of Adiponectin against Renal Ischemia-Reperfusion Injury via Prostacyclin-PPARα-Heme Oxygenase-1 Signaling Pathway. J. Cell Physiol. 2012, 227, 239–249. [Google Scholar] [CrossRef]
- Peerapornratana, S.; Manrique-Caballero, C.L.; Gómez, H.; Kellum, J.A. Acute Kidney Injury from Sepsis: Current Concepts, Epidemiology, Pathophysiology, Prevention and Treatment. Kidney Int. 2019, 96, 1083–1099. [Google Scholar] [CrossRef]
- Schrier, R.W.; Wang, W. Acute Renal Failure and Sepsis. N. Engl. J. Med. 2004, 351, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Gómez, H.; Kellum, J.A.; Ronco, C. Metabolic Reprogramming and Tolerance during Sepsis-Induced AKI. Nat. Rev. Nephrol. 2017, 13, 143–151. [Google Scholar] [CrossRef] [Green Version]
- Gemsa, D.; Woo, C.H.; Fudenberg, H.H.; Schmid, R. Stimulation of Heme Oxygenase in Macrophages and Liver by Endotoxin. J. Clin. Investig. 1974, 53, 647–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, A.; Wickman, L.T.; Venkatareddy, M.P.; Sato, Y.; Chowdhury, M.A.; Wang, S.Q.; Shedden, K.A.; Dysko, R.C.; Wiggins, J.E.; Wiggins, R.C. Angiotensin II-Dependent Persistent Podocyte Loss from Destabilized Glomeruli Causes Progression of End Stage Kidney Disease. Kidney Int. 2012, 81, 40–55. [Google Scholar] [CrossRef] [Green Version]
- Kagami, S.; Border, W.A.; Miller, D.E.; Noble, N.A. Angiotensin II Stimulates Extracellular Matrix Protein Synthesis through Induction of Transforming Growth Factor-Beta Expression in Rat Glomerular Mesangial Cells. J. Clin. Investig. 1994, 93, 2431–2437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bautista, R.; Manning, R.; Martinez, F.; del Carmen Avila-Casado, M.; Soto, V.; Medina, A.; Escalante, B. Angiotensin II-Dependent Increased Expression of Na+-Glucose Cotransporter in Hypertension. Am. J. Physiol. Ren. Physiol. 2004, 286, F127–F133. [Google Scholar] [CrossRef] [Green Version]
- Cuevas, C.A.; Gonzalez, A.A.; Inestrosa, N.C.; Vio, C.P.; Prieto, M.C. Angiotensin II Increases Fibronectin and Collagen I through the β-Catenin-Dependent Signaling in Mouse Collecting Duct Cells. Am. J. Physiol. Ren. Physiol. 2015, 308, F358–F365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, T.-D.; Yang, X.-P.; Liu, Y.-H.; Shesely, E.G.; Cavasin, M.A.; Kuziel, W.A.; Pagano, P.J.; Carretero, O.A. Role of Inflammation in the Development of Renal Damage and Dysfunction in Angiotensin II–Induced Hypertension. Hypertension 2008, 52, 256–263. [Google Scholar] [CrossRef]
- Daroux, M.; Prévost, G.; Maillard-Lefebvre, H.; Gaxatte, C.; D’Agati, V.D.; Schmidt, A.M.; Boulanger, E. Advanced Glycation End-Products: Implications for Diabetic and Non-Diabetic Nephropathies. Diabetes Metab. 2010, 36, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Pagtalunan, M.E.; Miller, P.L.; Jumping-Eagle, S.; Nelson, R.G.; Myers, B.D.; Rennke, H.G.; Coplon, N.S.; Sun, L.; Meyer, T.W. Podocyte Loss and Progressive Glomerular Injury in Type II Diabetes. J. Clin. Investig. 1997, 99, 342–348. [Google Scholar] [CrossRef] [Green Version]
- Forbes, J.M.; Coughlan, M.T.; Cooper, M.E. Oxidative Stress as a Major Culprit in Kidney Disease in Diabetes. Diabetes 2008, 57, 1446–1454. [Google Scholar] [CrossRef] [Green Version]
- Lindenmeyer, M.T.; Kretzler, M.; Boucherot, A.; Berra, S.; Yasuda, Y.; Henger, A.; Eichinger, F.; Gaiser, S.; Schmid, H.; Rastaldi, M.P.; et al. Interstitial Vascular Rarefaction and Reduced VEGF-A Expression in Human Diabetic Nephropathy. J. Am. Soc. Nephrol. 2007, 18, 1765–1776. [Google Scholar] [CrossRef] [Green Version]
- Wada, J.; Makino, H. Innate Immunity in Diabetes and Diabetic Nephropathy. Nat. Rev. Nephrol. 2016, 12, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Chow, F.; Ozols, E.; Nikolic-Paterson, D.J.; Atkins, R.C.; Tesch, G.H. Macrophages in Mouse Type 2 Diabetic Nephropathy: Correlation with Diabetic State and Progressive Renal Injury. Kidney Int. 2004, 65, 116–128. [Google Scholar] [CrossRef] [Green Version]
- Goodman, A.I.; Chander, P.N.; Rezzani, R.; Schwartzman, M.L.; Regan, R.F.; Rodella, L.; Turkseven, S.; Lianos, E.A.; Dennery, P.A.; Abraham, N.G. Heme Oxygenase-2 Deficiency Contributes to Diabetes-Mediated Increase in Superoxide Anion and Renal Dysfunction. J. Am. Soc. Nephrol. 2006, 17, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Bolati, D.; Shimizu, H.; Yisireyili, M.; Nishijima, F.; Niwa, T. Indoxyl Sulfate, a Uremic Toxin, Downregulates Renal Expression of Nrf2 through Activation of NF-ΚB. BMC Nephrol. 2013, 14, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zager, R.A.; Johnson, A.C.M.; Becker, K. Acute Unilateral Ischemic Renal Injury Induces Progressive Renal Inflammation, Lipid Accumulation, Histone Modification, and “End-Stage” Kidney Disease. Am. J. Physiol. Ren. Physiol. 2011, 301, F1334–F1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aminzadeh, M.A.; Nicholas, S.B.; Norris, K.C.; Vaziri, N.D. Role of Impaired Nrf2 Activation in the Pathogenesis of Oxidative Stress and Inflammation in Chronic Tubulo-Interstitial Nephropathy. Nephrol. Dial. Transpl. 2013, 28, 2038–2045. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Vaziri, N.D. Contribution of Impaired Nrf2-Keap1 Pathway to Oxidative Stress and Inflammation in Chronic Renal Failure. Am. J. Physiol. Ren. Physiol. 2010, 298, F662–F671. [Google Scholar] [CrossRef] [Green Version]
- Zager, R.A.; Johnson, A.C.M.; Guillem, A.; Keyser, J.; Singh, B. A Pharmacologic “Stress Test” for Assessing Select Antioxidant Defenses in Patients with CKD. CJASN 2020, 15, 633–642. [Google Scholar] [CrossRef]
- Chen, H.-H.; Lu, P.-J.; Chen, B.-R.; Hsiao, M.; Ho, W.-Y.; Tseng, C.-J. Heme Oxygenase-1 Ameliorates Kidney Ischemia-Reperfusion Injury in Mice through Extracellular Signal-Regulated Kinase 1/2-Enhanced Tubular Epithelium Proliferation. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 2195–2201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schellinger, I.N.; Cordasic, N.; Panesar, J.; Buchholz, B.; Jacobi, J.; Hartner, A.; Klanke, B.; Jakubiczka-Smorag, J.; Burzlaff, N.; Heinze, E.; et al. Hypoxia Inducible Factor Stabilization Improves Defective Ischemia-Induced Angiogenesis in a Rodent Model of Chronic Kidney Disease. Kidney Int. 2017, 91, 616–627. [Google Scholar] [CrossRef] [Green Version]
- Murali, N.S.; Ackerman, A.W.; Croatt, A.J.; Cheng, J.; Grande, J.P.; Sutor, S.L.; Bram, R.J.; Bren, G.D.; Badley, A.D.; Alam, J.; et al. Renal Upregulation of HO-1 Reduces Albumin-Driven MCP-1 Production: Implications for Chronic Kidney Disease. Am. J. Physiol. Ren. Physiol. 2007, 292, F837–F844. [Google Scholar] [CrossRef] [PubMed]
- Soares, M.P.; Lin, Y.; Anrather, J.; Csizmadia, E.; Takigami, K.; Sato, K.; Grey, S.T.; Colvin, R.B.; Choi, A.M.; Poss, K.D.; et al. Expression of Heme Oxygenase-1 Can Determine Cardiac Xenograft Survival. Nat. Med. 1998, 4, 1073–1077. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Jiang, L.; Kang, K.; Fei, D.; Meng, X.; Nan, C.; Pan, S.; Zhao, M.; Zhao, M. Hemin Inhibits NLRP3 Inflammasome Activation in Sepsis-Induced Acute Lung Injury, Involving Heme Oxygenase-1. Int. Immunopharmacol. 2014, 20, 24–32. [Google Scholar] [CrossRef]
- Chi, X.; Guo, N.; Yao, W.; Jin, Y.; Gao, W.; Cai, J.; Hei, Z. Induction of Heme Oxygenase-1 by Hemin Protects Lung against Orthotopic Autologous Liver Transplantation-Induced Acute Lung Injury in Rats. J. Transl. Med. 2016, 14. [Google Scholar] [CrossRef] [Green Version]
- Bharucha, A.E.; Kulkarni, A.; Choi, K.M.; Camilleri, M.; Lempke, M.; Brunn, G.J.; Gibbons, S.J.; Zinsmeister, A.R.; Farrugia, G. First-in-Human Study Demonstrating Pharmacological Activation of Heme Oxygenase-1 in Humans. Clin. Pharm. Ther. 2010, 87, 187–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doberer, D.; Haschemi, A.; Andreas, M.; Zapf, T.-C.; Clive, B.; Jeitler, M.; Heinzl, H.; Wagner, O.; Wolzt, M.; Bilban, M. Haem Arginate Infusion Stimulates Haem Oxygenase-1 Expression in Healthy Subjects. Br. J. Pharm. 2010, 161, 1751–1762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreas, M.; Schmid, A.I.; Doberer, D.; Schewzow, K.; Weisshaar, S.; Heinze, G.; Bilban, M.; Moser, E.; Wolzt, M. Heme Arginate Improves Reperfusion Patterns after Ischemia: A Randomized, Placebo-Controlled Trial in Healthy Male Subjects. J. Cardiovasc. Magn. Reson. 2012, 14, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simionatto, C.S.; Cabal, R.; Jones, R.L.; Galbraith, R.A. Thrombophlebitis and Disturbed Hemostasis Following Administration of Intravenous Hematin in Normal Volunteers. Am. J. Med. 1988, 85, 538–540. [Google Scholar] [CrossRef]
- Dekker, D.; Dorresteijn, M.J.; Peters, W.H.; Bilos, A.; Pennings, S.W.C.; Wagener, F.A.D.T.G.; Smits, P. Vascular and Metabolic Effects of the Haem Oxygenase-1 Inducer Haem Arginate in Subjects with the Metabolic Syndrome: A Translational Cross-over Study. Diabetes Vasc. Dis. Res. 2016, 13, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Grosser, N.; Hemmerle, A.; Berndt, G.; Erdmann, K.; Hinkelmann, U.; Schürger, S.; Wijayanti, N.; Immenschuh, S.; Schröder, H. The Antioxidant Defense Protein Heme Oxygenase 1 Is a Novel Target for Statins in Endothelial Cells. Free Radic. Biol. Med. 2004, 37, 2064–2071. [Google Scholar] [CrossRef]
- Lee, T.-S.; Chang, C.-C.; Zhu, Y.; Shyy, J.Y.-J. Simvastatin Induces Heme Oxygenase-1: A Novel Mechanism of Vessel Protection. Circulation 2004, 110, 1296–1302. [Google Scholar] [CrossRef]
- Horváth, K.; Varga, C.; Berkó, A.; Pósa, A.; László, F.; Whittle, B.J.R. The Involvement of Heme Oxygenase-1 Activity in the Therapeutic Actions of 5-Aminosalicylic Acid in Rat Colitis. Eur. J. Pharm. 2008, 581, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Chiang, S.-K.; Chen, S.-E.; Chang, L.-C. A Dual Role of Heme Oxygenase-1 in Cancer Cells. Int. J. Mol. Sci. 2019, 20, 39. [Google Scholar] [CrossRef] [Green Version]
- Nitti, M.; Piras, S.; Marinari, U.M.; Moretta, L.; Pronzato, M.A.; Furfaro, A.L. HO-1 Induction in Cancer Progression: A Matter of Cell Adaptation. Antioxidants 2017, 6, 29. [Google Scholar] [CrossRef]
- Loboda, A.; Jozkowicz, A.; Dulak, J. HO-1/CO System in Tumor Growth, Angiogenesis and Metabolism—Targeting HO-1 as an Anti-Tumor Therapy. Vasc. Pharmacol. 2015, 74, 11–22. [Google Scholar] [CrossRef]
- Pabla, N.; Dong, Z. Cisplatin Nephrotoxicity: Mechanisms and Renoprotective Strategies. Kidney Int. 2008, 73, 994–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craddock, P.R.; Fehr, J.; Jacob, H.S. Complement-Mediated Granulocyte Dysfunction in Paroxysmal Nocturnal Hemoglobinuria. Blood 1976, 47, 931–939. [Google Scholar] [CrossRef] [Green Version]
- Costa, D.L.; Amaral, E.P.; Andrade, B.B.; Sher, A. Modulation of Inflammation and Immune Responses by Heme Oxygenase-1: Implications for Infection with Intracellular Pathogens. Antioxidants 2020, 9, 1205. [Google Scholar] [CrossRef] [PubMed]
- Nithichanon, A.; Tussakhon, I.; Samer, W.; Kewcharoenwong, C.; Ato, M.; Bancroft, G.J.; Lertmemongkolchai, G. Immune Responses in Beta-Thalassaemia: Heme Oxygenase 1 Reduces Cytokine Production and Bactericidal Activity of Human Leucocytes. Sci. Rep. 2020, 10, 10297. [Google Scholar] [CrossRef]
- Caumartin, Y.; Stephen, J.; Deng, J.P.; Lian, D.; Lan, Z.; Liu, W.; Garcia, B.; Jevnikar, A.M.; Wang, H.; Cepinskas, G.; et al. Carbon Monoxide-Releasing Molecules Protect against Ischemia-Reperfusion Injury during Kidney Transplantation. Kidney Int. 2011, 79, 1080–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Huang, J.; Li, Y.; Chang, R.; Wu, H.; Lin, J.; Huang, Z. Exogenous Carbon Monoxide Decreases Sepsis-Induced Acute Kidney Injury and Inhibits NLRP3 Inflammasome Activation in Rats. Int. J. Mol. Sci. 2015, 16, 20595–20608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharjee, R.N.; Richard-Mohamed, M.; Sun, Q.; Haig, A.; Aboalsamh, G.; Barrett, P.; Mayer, R.; Alhasan, I.; Pineda-Solis, K.; Jiang, L.; et al. CORM-401 Reduces Ischemia Reperfusion Injury in an Ex Vivo Renal Porcine Model of the Donation After Circulatory Death. Transplantation 2018, 102, 1066–1074. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic view of kidney structures describing cortical and medullar compartments, components of glomerular basement membrane and tubular segments and structures. GBM: Glomerular basement membrane, TALH: Thick ascending loop of Henle (Created with BioRender.com).
Figure 1.
Schematic view of kidney structures describing cortical and medullar compartments, components of glomerular basement membrane and tubular segments and structures. GBM: Glomerular basement membrane, TALH: Thick ascending loop of Henle (Created with BioRender.com).

Figure 2.
Renal expression of HO1 (adapted from [27]). HO-1 staining in mouse kidneys in IF (A) and IHC (B). A- HO-1 (red) staining, vWF (green) staining, and colocalization on frozen kidney (x15) sections of mice, injected with PBS (upper panel) or heme as HO1 inducer (lower panel), studied by IF (A); B- HO-1 staining appears in brown on frozen kidneys sections of mice treated with PBS or heme. (C) HO-1 expression in human kidneys: hemosiderin deposits in human kidney biopsies evaluated by Perls’ coloration and HO-1 (brown) staining were performed by IHC. A normal protocol, kidney allograft biopsy performed 3 months after transplant was used as a negative control. Patients 1 and 2 suffered chronic hemolysis associated with prosthetic heart valves, and patient 3 carried the C3 mutation complicated with atypical hemolytic uremic syndrome (arrows indicate glomeruli).
Figure 2.
Renal expression of HO1 (adapted from [27]). HO-1 staining in mouse kidneys in IF (A) and IHC (B). A- HO-1 (red) staining, vWF (green) staining, and colocalization on frozen kidney (x15) sections of mice, injected with PBS (upper panel) or heme as HO1 inducer (lower panel), studied by IF (A); B- HO-1 staining appears in brown on frozen kidneys sections of mice treated with PBS or heme. (C) HO-1 expression in human kidneys: hemosiderin deposits in human kidney biopsies evaluated by Perls’ coloration and HO-1 (brown) staining were performed by IHC. A normal protocol, kidney allograft biopsy performed 3 months after transplant was used as a negative control. Patients 1 and 2 suffered chronic hemolysis associated with prosthetic heart valves, and patient 3 carried the C3 mutation complicated with atypical hemolytic uremic syndrome (arrows indicate glomeruli).

Figure 3.
Schematic description of HO-1 regulation in renal pathologies. Plain arrows: proved link, Dotted arrows: suspected link, AKI: acute kidney injury, SCA: sickle cell anemia, HUS: hemolytic uremic syndrom, PNH: paroxysmal nocturnal haemoglobinuria (Created with BioRender.com).
Figure 3.
Schematic description of HO-1 regulation in renal pathologies. Plain arrows: proved link, Dotted arrows: suspected link, AKI: acute kidney injury, SCA: sickle cell anemia, HUS: hemolytic uremic syndrom, PNH: paroxysmal nocturnal haemoglobinuria (Created with BioRender.com).


{kind=link}
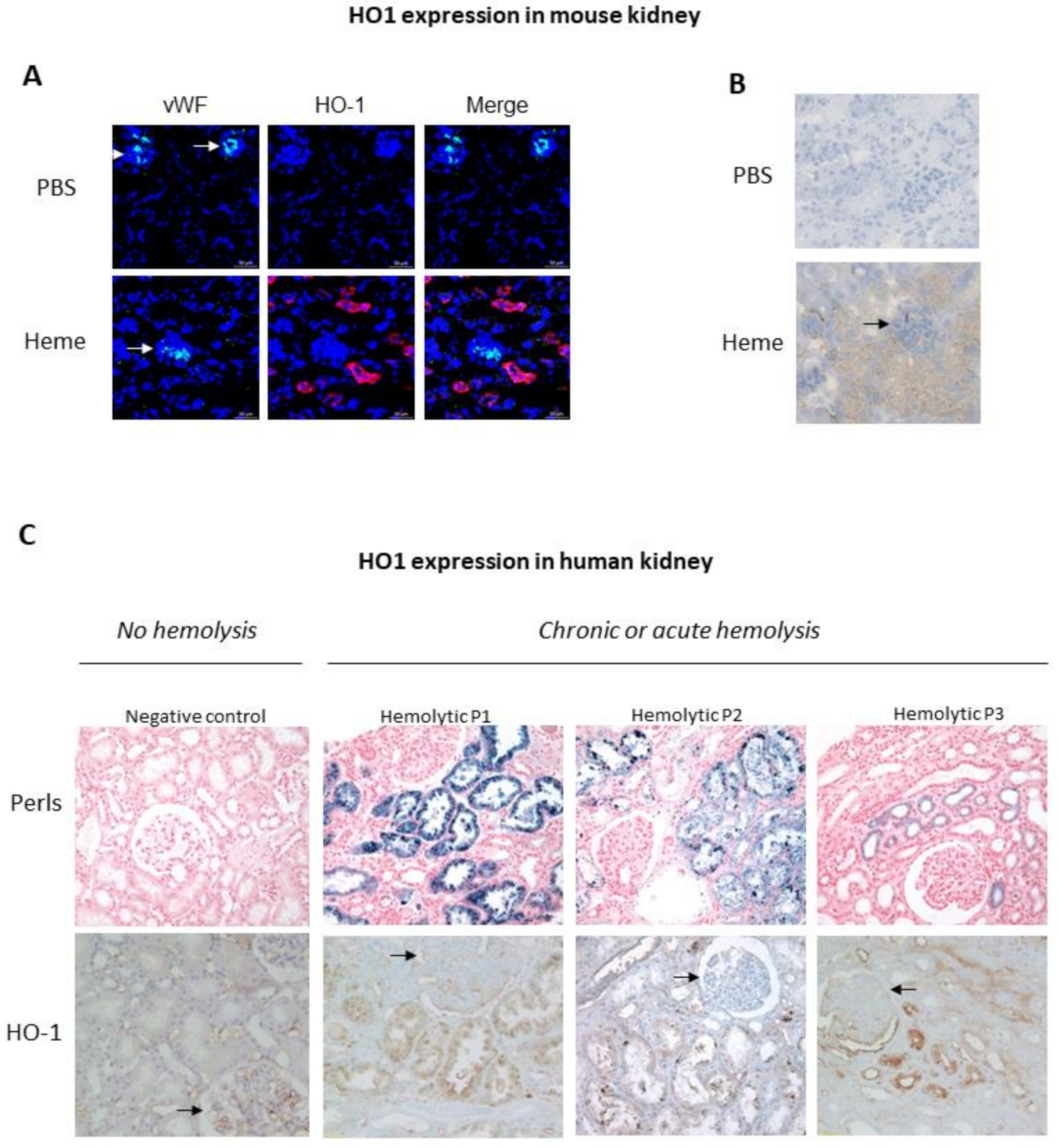{kind=link}
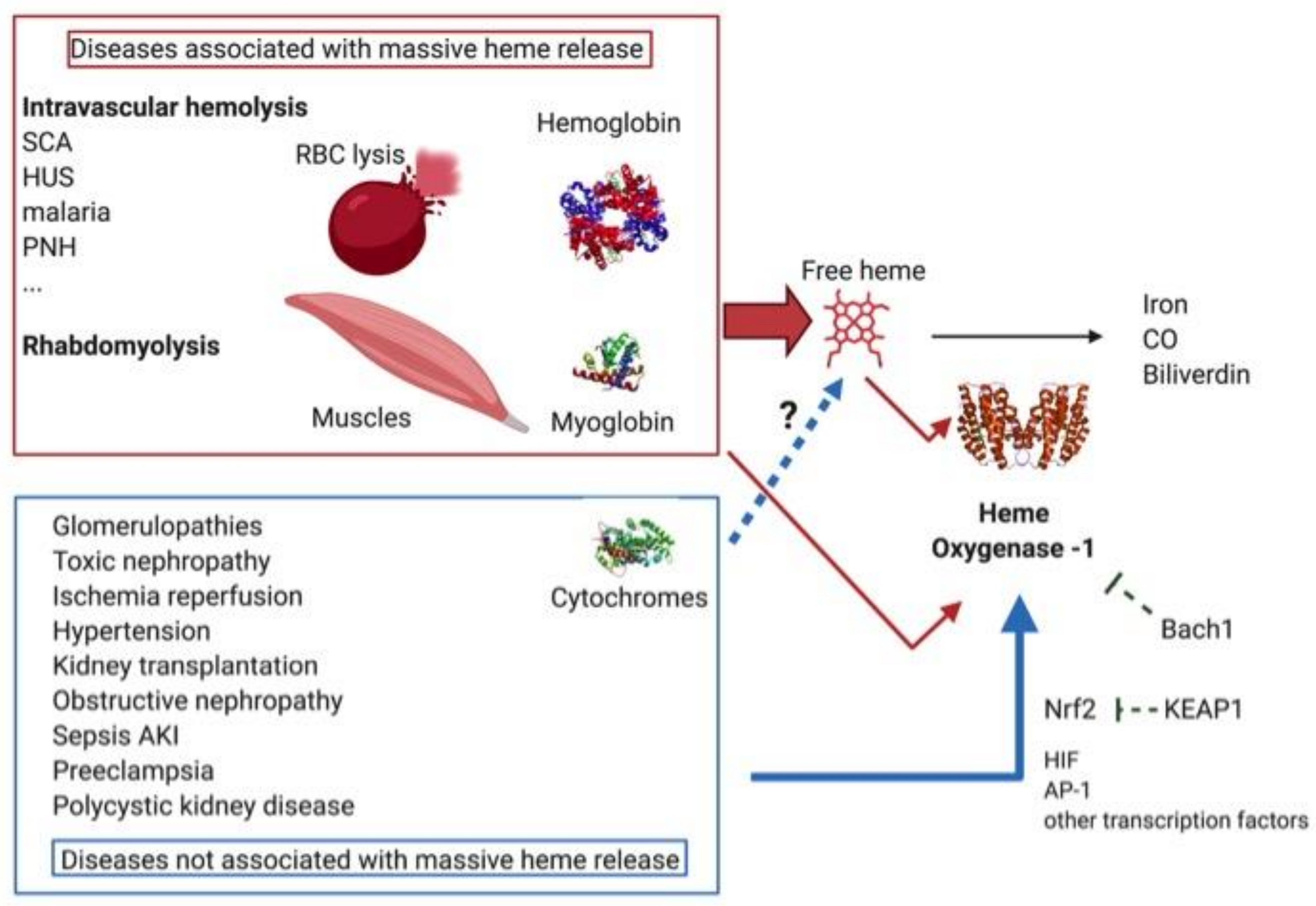{kind=link}
Table 1.
Main models and functions of HO-1 in kidney pathologies.
| Pathology | Number Pub. | Models | HO-1 Main Functions in the Pathology | References | ||
|---|---|---|---|---|---|---|
| Heme dependent | Hemolysis | Sickle cell disease | 14 | Mice (Townes SS) EC + MPs from SCA patients βS/βS-antilles mice + SnPP | Preserves local kidney hemodynamics Decreases exposure to oxidative stress | [123,124,125,126,127,128,129,130,132,137] |
| Hemolytic Uremic Anemia | 4 | Microvascular glomerular cells + heme | Weakly expressed in glomerular EC increasing its susceptibility | [26,64,141,142] | ||
| Malaria | 5 | HO-1 −/− mice + P. berghei ANKA Mice + P. berghei ANKA +/− FePP or CoPP or ZnPP HO-1 −/− mice + P. chabaudi chabaudi | Decreases proximal tubular necrosis | [143,144,145] | ||
| Paroxysmal nocturne hemoglobinuria | 4 | ns | unknown | [153,154] | ||
| Rhabdomyolysis | 24 | Mice +/− Hb or heme + glycerol HO-1 −/− mice + glycerol | Improves renal microcirculation Reduces oxidative stress Regulates immune response | [33,111,162,163,164,165,166,167] | ||
| Not heme dependent | Glomerular nephropathies | Lupus | 13 | MRL/lpr mice + hemin MRL/lpr bach1−/− mice Nrf2−/− mice Pristane-induced LN mice + Nrf2 activators Patients’ mph, Mo, PNN | Reduces proteinuria, Decreases proliferation and deposition in glomeruli Is weakly expressed in renal and circulating monocytes/mph | [177,178,179,180,181] |
| Anti-GBM | 6 | Anti-rat nephrotoxic globulin + hemin Rat +/− hemin + anti-GBM | Reduces proteinuria Induces switch to a M2 macrophage phenotype | [182,183,184] | ||
| Membranous nephropathy | 4 | Albumin induced membranous nephropathy + CoPP | Reduces proteinuria Improves glomerular lesions Decreases immune-complex deposition, oxidative stress, | [185,186] | ||
| Diabetes | 136 | STZ- induced DM mice/rat +/− HO-1 siRNA +/− hemin +/− ZnPP or CoPP or SnMP Genetic diabetic db/db mice Diabetic fat rat | Decreases albuminuria, Limit podocytes apoptosis, Decreases mesangial proliferation, Modulate renal hemodynamics, Preserves insulino-sensitivity Influences glomerulo-tubular crosstalk in a heme-dependant manner. | [72,187,188,189,190,191,192,193,194,195,196,197,198,199,200,201] | ||
| Toxic nephropathy | Cisplatin toxicity | 86 | Rat +/− SnPP + cisplatin Targeted HO-1 deletion in PT of mice (CreLox) + cisplatin HO−/− mice + cisplatin HO+/+ mice +/− hemin + cisplatin | Improves renal function Decreases tubular epithelial cells – notably proximal - necrosis Increases autophagy | [23,94,202,203,204,205,206,207,208,209,210,211,212] | |
| Gentamycin toxicity | 7 | Rat +/− SnPP + gentamycin HO-1 overexpressing or KD NRK-52E cells Mice overexpressing HO-1 in kidney (adenoviral construct) | Decreases apoptosis in tubular cells | [202,213,214] | ||
| Ciclosporin toxicity | 35 | LLC-K1 + CsA +/− SnPP NRK-52E + CsA Nrf2 −/− mice + CsA | Modulates EMT Decreases PT cells apoptosis and oxidative stress | [215,216,217,218,219,220] | ||
| Rapamycin toxicity | 13 | Rat transplantation+Rapamycin Mice with pedicle ligation + rapamycin | Preserves kidney dysfunction | [221,222,223] | ||
| Radiocontrast product | 3 | Rat +/− SnPP or CoPP + sodium iothalamate | Preserves kidney function+ decreases apoptosis | [224] | ||
| Mercuric chloride toxicity | 7 | Mice +/− hemin+/− HgCl2 | Improves renal function and Decreases distal tubule necrosis | [225,226,227] | ||
| Ischemia reperfusion | 183 | Unilateral/bilateral renal arteries clamping in - HO−/− mice - HO+/+ mice preventively treated by SnPP/hemin - mice overexpressing HO-1in BMDM (adenoviral construct) - myeloid-restricted deletion of HO-1 mice (Cre-Lox) HO-1 overexpressing LLC-PK1 cells | Improves renal function -in tubular cells, * modulating local hemodynamic through NO synthesis regulation * conferring resistance to apoptosis - in myeloid cells * preserve kidneys from their pro inflammatory effects | [91,103,205,221,228,229,230,231,232,233,234,235,236,237,238,239,240,241,242,243,244,245,246,247,248,249,250,251,252,253,254,255] | ||
| Kidney transplantation | 215 | Hemin or CoPP in rat donor transplant CoPP or ZnPP in rat or mouse transplant receivers | Prevents acute rejection Prevents IS toxicity In donor, preserves kidney graft function and prevents post reperfusion apoptosis | [39,221,251,252,253,254,256,257,258,259,260] | ||
| Hypertension | 146 | Unilateral kidney artery clamping in HO-1 −/− mice Spontaneously hypertensive rats +/− heme +/− ZnPP Ang II induced hypertension in HO-1 −/− mice HO-1-specific overexpression in TALH | Not essential to preserve arterial blood pressure in physiological conditions Reduce blood pressure under pathological conditions, (increase of Angiotensin II) In TALH: maintains blood flow and natriuresis | [25,261,262,263,264,265,266,267,268,269,270,271] | ||
| Obstructive nephropathy | 43 | Unilateral or bilateral obstruction * +/− preventive heme +/− ZnPP * in HO-1 −/− mice * in transgenic mice overexpressing HO-1 | Limits macrophages infiltrate Confers mph a regulatory phenotype, Promotes microcirculation maintenance Inhibits profibrotic processes | [272,273,274,275,276,277,278] | ||
| Sepsis-AKI | 46 | HO-1 −/− mice + LPS HO-1−/− mice + cecal ligation Myeloid-restricted deletion of HO-1 mice (Cre-Lox) + Enteroccocus faecalis/Escherichia coli | Preserves glomerular filtration rate Preserves renal blood flow, Decreases renal cytokine expression Decreases tubular necrosis Degrades heme In macrophages, contributes to pathogen clearance and decreased AKI | [105,279,280,281] | ||
| Preeclampsia and kidney | 4 | HUVEC +/− overexpressing HO-1 (adenoviral construct) + PAR2 activating peptides HUVEC + VEGF-E Reduced uterine perfusion pressure rats +/− CoPP | Promotes proper placental vascular development Decreases endothelial PAR-2 activation Reduces Edothelin-1 production (in GEC) Attenuates hypertension | [282,283,284,285] | ||
| Polycystic kidney disease | 6 | Cys1cpk/cpk mice + CoPP or SnPP | Decreases kidney weight Decreases kidney injury markers Decreases cystogenesis | [286,287,288] | ||
Number of articles referenced in Pubmed.ncbi.nlm.nih.gov, identified by the key words "heme-oxygenase-1" AND ""Kidney"", and the name of the pathology. AKI: acute kidney injury; Ang II: Angiotensin II; CoPP: cobalt protoporphyrin; EC: endothelial cells; EMT: epithelial–mesenchymal transition; FePP: iron protoporphyrin; GBM: glomerular basement membrane; GEC: glomerular endothelial cells; HUVEC: human umbilical vein EC; IS: immunosuppressors; Pub.: publication; LPS: lipopolysaccharide; Mo: monocytes; mph: macrophages; SnMP: tin mesoporphyrin; SnPP: tin protoporhyrin; ZnPP: zinc protoporphyrin. Asterisks (*) and hyphens (-) indicat detailed explanations of functions or models, the right above ones.
Table 2.
Clinical trials for HO-1 and kidney.
| Study Topic | Population | Study Design | Intervention/Treatment | Outcome Measures | ClinicalTrials.gov Identifier | Sponsor |
|---|---|---|---|---|---|---|
| SnPP in Healthy Volunteers and Stage 3–4 CKD patients | Healthy volunteers Stage 3–4 CKD | Interventional no-random. | A single dose of 9, 27 or 90 mg of SnPP (followed for 28 days) | Measuring Haptoglobin, Ferritin, Bilirubin, Hemopexin, IL-10, and HO-1, the P21 biomarker systemic levels | NCT04072861 | Renibus Therapeutics, Inc. |
| RBT-1 in Healthy Volunteers and Stage 3–4 CKD patients | Healthy volunteers Stage 3–4 CKD | Interventional no-random. | A single dose of 240 mg Iron Sucrose and 9, 27 or 90 mg of SnPP (followed for 28 days) | Change in biomarkers of cytoprotective activity: Haptoglobin, Ferritin, Bilirubin, Hemopexin, IL-10, HO-1, the P21 biomarker systemic levels | NCT03893799 | Renibus Therapeutics, Inc. |
| Effects of Resveratrol in CKD Patients | Stage 3 - 4 CKD | Interventional random. CO | 500mg of trans-resveratrol per day for 4 weeks vs. placebo | Antioxidants and anti-inflammatory biomarkers: Nrf2, GPx, HO-1; NFkB, IL-6, TNF-alfa | NCT02433925 | Universidade Federal Fluminense |
| Propolis in CKD Patients | CKD under conservative treatment | Interventional random. CO | 20 drops of standardized propolis daily for 2 months vs. non-intervention. | Change in cytokines plasma levels, Nrf2 and NF-kB, antioxidant enzymes (NQO1, HO-1), NLRP3 receptor, PPAR-γ. Change the profile of the intestinal microbiota | NCT04411758 | Universidade Federal Fluminense |
| Chocolate in CKD Patients | CKD in hemodialysis | Interventional no-random. | 40g dark chocolate per day for 2 months vs. no intervention | Change in antioxidants and anti-inflammatory biomarkers: Nrf2, GPx, HO-1, NFkB, IL6, TNFa. Uremic toxins. | NCT04600258 | Universidade Federal Fluminense |
| Effects of Curcumin Supplementation in HD patients | CKD in hemodialysis | Interventional random. CO, | 3 capsules per day with 500mg of curcumin and 5mg of piperine for 4 weeks vs. placebo. | Change in antioxidants and anti-inflammatory biomarkers: Nrf2, glutathioneperoxidase (GPx), HO-1, NFkB, IL6, TNFa | NCT03475017 | Universidade Federal Fluminense |
| Effects of Curcumin Supplementation in PD Patients | CKD in PD | Interventional random. CO | 3 capsules with 500mg of curcumin and piperine per day, for 12 weeks vs. placebo | Change in antioxidants and anti-inflammatory biomarkers: Nrf2, GPx, HO-1, NFkB, IL6, TNFa, PCR, IL-18, Inflammasome. | NCT04413266 | Universidade Federal Fluminense |
| Tacrolimus After rATG and Infliximab Induction IS | KTR | Interventional no-random. | rATG induction (2 × 1.5 mg/kg) in 1st KTR given perioperatively and on first postoperative day, then Infliximab mAb at day 2. | Composite endpoint of efficacy failure and renal function of the induction regimen at 12 months post transplantation. HO-1 polymorphisms | NCT04114188 | Charite University, Berlin, Germany |
| Induction of HO-1 to Reduce Ischemia Reperfusion Injury in KTR | KTR | Interventional Random. | Heme arginate (3mg/kg) or placebo prior to transplant and repeat again on day 2 post-transplant | Measure the level of HO-1 protein in isolated macrophages/monocytes in a blood sample taken at 24 h after drug infusion | NCT01430156 | University of Edinburgh |
| Characterization of the Nrf2 Response in Patients With ADPKD | ADPKD | Observational Prospective | No intervention | 8-oxodeoxyguanosine, F2-isoprostanes, HO-1, SOD, catalase, GPx, NAD(P)H dehydrogenase, glutathione, Nrf2.Total kidney volume determined by MRI. | NCT04344769 | Mayo Clinic |
| Effect of Atorvastatin on Endothelial Dysfunction and Albuminuria in SCD | SCD | Interventional random. CO | Atorvastatin 40 mg once daily for 6 weeks vs. placebo. | Change from Baseline to Week 6 in endothelial function, in plasma markers of endothelial activation, in HO activity. | NCT01732718 | University of North Carolina, Chapel Hill |
Clinical trials referenced in ClinicalTrial.gov, identified by the key words “heme-oxygenase-1” AND “Kidney” (OR “renal”). ADPKD: autosomal dominant polycystic kidney disease; CKD: chronic kidney disease; CO: crossover; GPx: glutathioneperoxidase; GSR: glutathione reductase; KTR: kidney transplant recipient; HD: hemodialysis; PD: peritoneal dialysis; PPAR-γ: peroxisome proliferator-activated receptor-γ; Random.: randomized; SnPP: stannous protoporphyrin; SOD: superoxide dismutase. Bold letters present emphasis and highligt.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Grunenwald, A.; Roumenina, L.T.; Frimat, M. Heme Oxygenase 1: A Defensive Mediator in Kidney Diseases. Int. J. Mol. Sci. 2021, 22, 2009. https://doi.org/10.3390/ijms22042009
AMA Style
Grunenwald A, Roumenina LT, Frimat M. Heme Oxygenase 1: A Defensive Mediator in Kidney Diseases. International Journal of Molecular Sciences. 2021; 22(4):2009. https://doi.org/10.3390/ijms22042009
Chicago/Turabian StyleGrunenwald, Anne, Lubka T. Roumenina, and Marie Frimat. 2021. "Heme Oxygenase 1: A Defensive Mediator in Kidney Diseases" International Journal of Molecular Sciences 22, no. 4: 2009. https://doi.org/10.3390/ijms22042009
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.