
Gut Microbiota–Host Interactions in Inborn Errors of Immunity

 ,
,  , ,
, ,
Abstract
:1. Introduction
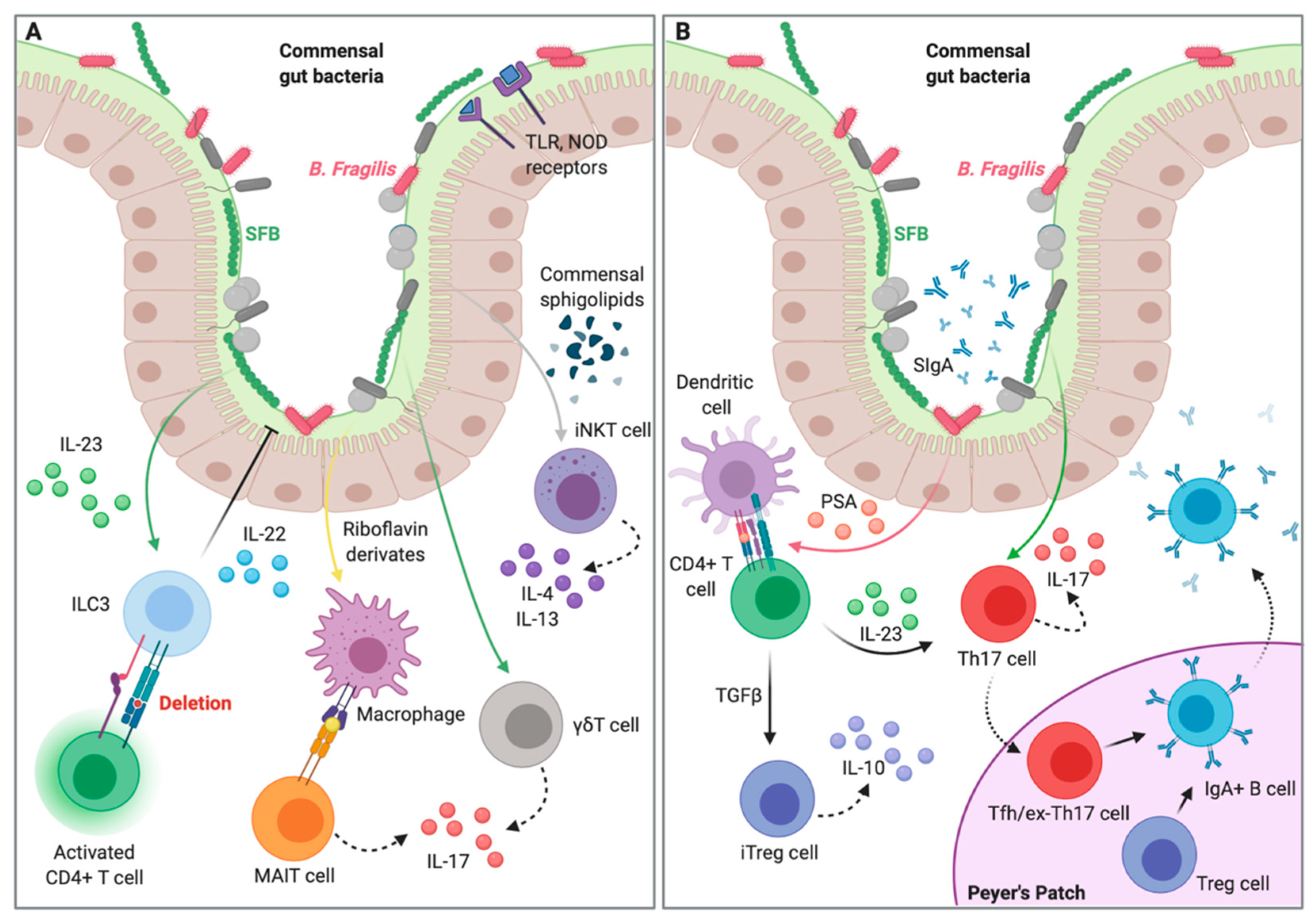
2. Overview on Molecular Mechanisms of Gut Microbiota–Host Interactions Leading to Intestinal Homeostasis
2.1. Epithelial Interaction and Compartmentalization of Gut Microbiota
2.2. Gut Microbiota–Innate Immunity Homeostatic Interplay
2.3. Gut Microbiota–Adaptive Immunity Homeostatic Interplay
3. Gut Microbiota–Host Interactions in Animal Models of Inborn Errors of Immunity
4. Gut Microbiota–Host Interactions in Human Inborn Errors of Immunity
4.1. Immunodeficiencies Affecting Cellular and Humoral Immunity
4.2. Predominantly Antibody Deficiencies
4.3. Studies in Other Human Inborn Errors of Immunity
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Notarangelo, L.D.; Baccheta, R.; Casanova, J.L.; Su, H.C. Human inborn errors of immunity: An expanding universe. Sci. Immunol. 2020, 5, eabb1662. [Google Scholar] [CrossRef]
- Tangye, S.G.; Al-Herz, W.; Bousfiha, A.; Chatila, T.; Cunningham-Rundles, C.; Etzioni, A.; Franco, J.L.; Holland, S.M.; Klein, C.; Morio, T.; et al. Human Inborn Errors of Immunity: 2019 Update on the Classification from the International Union of Immunological Societies Expert Committee. J. Clin. Immunol. 2020, 40, 24–64. [Google Scholar] [CrossRef] [Green Version]
- Hartono, S.; Ippoliti, M.R.; Mastroianni, M.; Torres, R.; Rider, N.L. Gastrointestinal Disorders Associated with Primary Immunodeficiency Diseases. Clin. Rev. Allergy Immunol. 2019, 57, 145–165. [Google Scholar] [CrossRef] [PubMed]
- Hooper, L.V.; Littman, D.R.; MacPherson, A.J. Interactions between the Microbiota and the Immune System. Science 2012, 336, 1268–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef] [PubMed]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Schumm, L.P.; Sharma, Y.; Anderson, C.A.; et al. Host–microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhlig, H.H. Monogenic diseases associated with intestinal inflammation: Implications for the understanding of inflammatory bowel disease. Gut 2013, 62, 1795–1805. [Google Scholar] [CrossRef] [PubMed]
- Pellicciotta, M.; Rigoni, R.; Falcone, E.L.; Holland, S.M.; Villa, A.; Cassani, B. The microbiome and immunodeficiencies: Lessons from rare diseases. J. Autoimmun. 2019, 98, 132–148. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Dittel, B.N. Interrelatedness between dysbiosis in the gut microbiota due to immunodeficiency and disease penetrance of colitis. Immunology 2015, 146, 359–368. [Google Scholar] [CrossRef] [Green Version]
- Arrieta, M.-C.; Stiemsma, L.T.; Amenyogbe, N.; Brown, E.M.; Finlay, B. The Intestinal Microbiome in Early Life: Health and Disease. Front. Immunol. 2014, 5, 427. [Google Scholar] [CrossRef] [Green Version]
- McGuckin, M.A.; Lindén, S.K.; Sutton, P.; Florin, T.H. Mucin dynamics and enteric pathogens. Nat. Rev. Microbiol. 2011, 9, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Bevins, C.L.; Salzman, N.H. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat. Rev. Microbiol. 2011, 9, 356–368. [Google Scholar] [CrossRef] [PubMed]
- Hooper, L.V.; MacPherson, A.J. Immune adaptations that maintain homeostasis with the intestinal microbiota. Nat. Rev. Immunol. 2010, 10, 159–169. [Google Scholar] [CrossRef]
- Cash, H.L.; Whitham, C.V.; Behrendt, C.L.; Hooper, L.V. Symbiotic Bacteria Direct Expression of an Intestinal Bactericidal Lectin. Science 2006, 313, 1126–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakoff-Nahoum, S.; Paglino, J.; Eslami-Varzaneh, F.; Edberg, S.; Medzhitov, R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 2004, 118, 229–241. [Google Scholar] [CrossRef] [Green Version]
- Elinav, E.; Strowig, T.; Kau, A.L.; Henao-Mejia, J.; Thaiss, C.A.; Booth, C.J.; Peaper, D.R.; Bertin, J.; Eisenbarth, S.C.; Gordon, J.I.; et al. NLRP6 Inflammasome Regulates Colonic Microbial Ecology and Risk for Colitis. Cell 2011, 145, 745–757. [Google Scholar] [CrossRef] [Green Version]
- Macia, L.; Tan, J.; Vieira, A.T.; Leach, K.; Stanley, D.; Luong, S.; Maruya, M.; Ian McKenzie, C.; Hijikata, A.; Wong, C.; et al. Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat. Commun. 2015, 6, 6734. [Google Scholar] [CrossRef] [Green Version]
- Vaishnava, S.; Yamamoto, M.; Severson, K.M.; Ruhn, K.A.; Yu, X.; Koren, O.; Ley, R.; Wakeland, E.K.; Hooper, L.V. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science 2011, 334, 255–258. [Google Scholar] [CrossRef] [Green Version]
- Chang, P.V.; Hao, L.; Offermanns, S.; Medzhitov, R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc. Natl. Acad. Sci. USA 2014, 111, 2247–2252. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.; Gurav, A.; Sivaprakasam, S.; Brady, E.; Padia, R.; Shi, H.; Thangaraju, M.; Prasad, P.D.; Manicassamy, S.; Munn, D.H.; et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity 2014, 40, 128–139. [Google Scholar] [CrossRef] [Green Version]
- Schulthess, J.; Pandey, S.; Capitani, M.; Rue-Albrecht, K.C.; Arnold, I.; Franchini, F.; Chomka, A.; Ilott, N.E.; Johnston, D.G.; Pires, E.; et al. The Short Chain Fatty Acid Butyrate Imprints an Antimicrobial Program in Macrophages. Immununity 2019, 50, 432–445. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.; Yuan, Y.; Yu, H.; Dai, X.; Wang, S.; Sun, Z.; Wang, F.; Fei, H.; Lin, Q.; Jiang, H.; et al. The gut microbial metabolite trimethylamine N-oxide aggravates GVHD by inducing M1 macrophage polarization in mice. Blood 2020. [Google Scholar] [CrossRef] [PubMed]
- Wojno, E.D.; Artis, D. Emerging concepts and future challenges in innate lymphoid cell biology. J. Exp. Med. 2016, 213, 2229–2248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bostick, J.W.; Wang, Y.; Shen, Z.; Ge, Y.; Brown, J.; Chen, Z.-M.E.; Mohamadzadeh, M.; Fox, J.G.; Zhou, L. Dichotomous regulation of group 3 innate lymphoid cells by nongastric Helicobacter species. Proc. Natl. Acad. Sci. USA 2019, 116, 24760–24769. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Liang, Y.; Zhang, Y.; Lasorella, A.; Kee, B.L.; Fu, Y.-X.; Yong, L. Innate Lymphoid Cells Control Early Colonization Resistance against Intestinal Pathogens through ID2-Dependent Regulation of the Microbiota. Immunity 2015, 42, 731–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atarashi, K.; Tanoue, T.; Ando, M.; Kamada, N.; Nagano, Y.; Narushima, S.; Suda, W.; Imaoka, A.; Setoyama, H.; Nagamori, T.; et al. Th17 Cell Induction by Adhesion of Microbes to Intestinal Epithelial Cells. Cell 2015, 163, 367–380. [Google Scholar] [CrossRef] [Green Version]
- Sano, T.; Huang, W.; Hall, J.A.; Yang, Y.; Chen, A.; Gavzy, S.J.; Lee, J.-Y.; Ziel, J.W.; Miraldi, E.R.; Domingos, A.I.; et al. An IL-23R/IL-22 Circuit Regulates Epithelial Serum Amyloid A to Promote Local Effector Th17 Responses. Cell 2015, 163, 381–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hepworth, M.R.; Monticelli, L.A.; Fung, T.C.; Ziegler, C.G.K.; Grunberg, S.; Sinha, R.; Mantegazza, A.R.; Ma, H.-L.; Crawford, A.; Angelosanto, J.M.; et al. Innate lymphoid cells regulate CD4+ T-cell responses to intestinal commensal bacteria. Nature 2013, 498, 113–117. [Google Scholar] [CrossRef] [Green Version]
- Hepworth, M.R.; Fung, T.C.; Masur, S.H.; Kelsen, J.R.; McConnell, F.M.; Dubrot, J.; Withers, D.R.; Hugues, S.; Farrar, M.A.; Reith, W.; et al. Immune tolerance. Group 3 innate lymphoid cells mediate intestinal selection of commensal bacteria-specific CD4+ T cells. Science 2015, 348, 1031–1035. [Google Scholar] [CrossRef] [Green Version]
- Kabelitz, D.; Dchanet-Merville, J. Editorial: “Recent Advances in Gamma/Delta T Cell Biology: New Ligands, New Functions, and New Translational Perspectives”. Front. Immunol. 2015, 6, 371. [Google Scholar] [CrossRef] [Green Version]
- Duan, J.; Chung, H.; Troy, E.; Kasper, D.L. Microbial colonization drives expansion of IL-1 receptor 1-expressing and IL-17-producing gamma/delta T cells. Cell Host Microbe 2010, 7, 140–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paget, C.; Chow, M.T.; Gherardin, N.A.; Beavis, P.A.; Uldrich, A.P.; Duret, H.; Hassane, M.; Souza-Fonseca-Guimaraes, F.; Mogilenko, D.A.; Staumont-Salle, D.; et al. CD3bright signals on gd T cells identify IL-17A-producing Vg6Vd1+ T cells. Immunol. Cell Biol. 2015, 93, 198–212. [Google Scholar]
- Treiner, E.; Duban, L.; Bahram, S.; Radosavljevic, M.; Wanner, V.; Tilloy, F.; Affaticati, P.; Gilfillan, S.; Lantz, O. Selection of evolutionarily conserved mucosal-associated invariant T cells by MR1. Nature 2003, 422, 164–169. [Google Scholar] [CrossRef]
- Kjer-Nielsen, L.; Patel, O.; Corbett, A.J.; Le Nours, J.; Meehan, B.; Liu, L.; Bhati, M.; Chen, Z.; Kostenko, L.; Reantragoon, R.; et al. MR1 presents microbial vitamin B metabolites to MAIT cells. Nature 2012, 491, 717–723. [Google Scholar] [CrossRef] [PubMed]
- Le Bourhis, L.; Dusseaux, M.; Bohineust, A.; Bessoles, S.; Martin, E.; Premel, V.; Coré, M.; Sleurs, D.; Serriari, N.-E.; Treiner, E.; et al. MAIT Cells Detect and Efficiently Lyse Bacterially-Infected Epithelial Cells. PLOS Pathog. 2013, 9, e1003681. [Google Scholar] [CrossRef] [Green Version]
- Kurioka, A.; Ussher, J.E.; Cosgrove, C.; Clough, C.H.; Fergusson, J.R.; Smith, K.G.C.; Kang, Y.-H.; Walker, L.J.; Hansen, T.H.; Willberg, C.B.; et al. MAIT cells are licensed through granzyme exchange to kill bacterially sensitized targets. Mucosal Immunol. 2015, 8, 429–440. [Google Scholar] [CrossRef] [Green Version]
- An, D.; Oh, S.F.; Olszak, T.; Neves, J.F.; Avci, F.Y.; Erturk-Hasdemir, D.; Lu, X.; Zeissig, S.; Blumberg, R.S.; Kasper, D.L. Sphingolipids from a Symbiotic Microbe Regulate Homeostasis of Host Intestinal Natural Killer T Cells. Cell 2014, 156, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Olszak, T.; An, D.; Zeissig, S.; Vera, M.P.; Richter, J.; Franke, A.; Glickman, J.N.; Siebert, R.; Baron, R.M.; Kasper, D.L.; et al. Microbial Exposure During Early Life Has Persistent Effects on Natural Killer T Cell Function. Science 2012, 336, 489–493. [Google Scholar] [CrossRef] [Green Version]
- Peterson, D.A.; McNulty, N.P.; Guruge, J.L.; Gordon, J.I. IgA response to symbiotic bacteria as a mediator of gut homeostasis. Cell Host Microbe 2007, 2, 328–339. [Google Scholar] [CrossRef] [Green Version]
- Pietrzak, B.; Tomela, K.; Olejnik-Schmidt, A.; Mackiewicz, A.; Schmidt, M. Secretory IgA in Intestinal Mucosal Secretions as an Adaptive Barrier against Microbial Cells. Int. J. Mol. Sci. 2020, 21, 9254. [Google Scholar] [CrossRef]
- Kawamoto, S.; Maruya, M.; Kato, L.M.; Suda, W.; Atarashi, K.; Doi, Y.; Tsutsui, Y.; Qin, H.; Honda, K.; Okada, T.; et al. Foxp3(+) T cells regulate immunoglobulin a selection and facilitate diversification of bacterial species responsible for immune homeostasis. Immunity 2014, 41, 152–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutherland, D.B.; Suzuki, K.; Fagarasan, S. Fostering of advanced mutualism with gut microbiota by Immunoglobulin A. Immunol. Rev. 2016, 270, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Mucida, D.; Kutchukhidze, N.; Erazo, A.; Russo, M.; Lafaille, J.J.; De Lafaille, M.A.C. Oral tolerance in the absence of naturally occurring Tregs. J. Clin. Investig. 2005, 115, 1923–1933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lathrop, S.K.; Bloom, S.M.; Rao, S.M.; Nutsch, K.; Lio, C.-W.; Santacruz, N.; Peterson, D.A.; Stappenbeck, T.S.; Hsieh, C.-S. Peripheral education of the immune system by colonic commensal microbiota. Nature 2011, 478, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Macpherson, A.J.; Uhr, T. Induction of protective IgA by intestinal dendritic cells carrying commensal bacteria. Science 2004, 303, 1662–1665. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.; Maruya, M.; Kawamoto, S.; Sitnik, K.; Kitamura, H.; Agace, W.W.; Fagarasan, S. The sensing of environmental stimuli by follicular dendritic cells promotes immunoglobulin A generation in the gut. Immunity 2010, 33, 71–83. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Cho, B.; Suzuki, K.; Xu, Y.; Green, J.A.; An, J.; Cyster, J.G. Follicular dendritic cells help establish follicle identity and promote B cell retention in germinal centers. J. Exp. Med. 2011, 208, 2497–2510. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, I.I.; Atarashi, K.; Manel, N.; Brodie, E.L.; Shima, T.; Karaoz, U.; Wei, D.; Goldfarb, K.C.; Santee, C.A.; Lynch, S.V.; et al. Induction of Intestinal Th17 Cells by Segmented Filamentous Bacteria. Cell 2009, 139, 485–498. [Google Scholar] [CrossRef] [Green Version]
- Omenetti, S.; Bussi, C.; Metidji, A.; Iseppon, A.; Lee, S.; Tolaini, M.; Li, Y.; Kelly, G.; Chakravarty, P.; Shoaie, S.; et al. The Intestine Harbors Functionally Distinct Homeostatic Tissue-Resident and Inflammatory Th17 Cells. Immunity 2019, 51, 77–89. [Google Scholar] [CrossRef] [Green Version]
- Bachem, A.; Makhlouf, C.; Binger, K.J.; De Souza, D.P.; Tull, D.; Hochheiser, K.; Whitney, P.G.; Fernandez-Ruiz, D.; Dähling, S.; Kastenmüller, W.; et al. Microbiota-Derived Short-Chain Fatty Acids Promote the Memory Potential of Antigen-Activated CD8+ T Cells. Immunity 2019, 51, 285–297. [Google Scholar] [CrossRef]
- Yin, Y.; Wang, Y.; Zhu, L.; Liu, W.; Liao, N.; Jiang, M.; Zhu, B.; Yu, H.D.; Xiang, C.; Wang, X. Comparative analysis of the distribution of segmented filamentous bacteria in humans, mice and chickens. ISME J. 2013, 7, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Hall, J.A.; Kroehling, L.; Wu, L.; Najar, T.; Nguyen, H.H.; Lin, W.Y.; Yeung, S.T.; Silva, H.M.; Li, D.; et al. Serum Amyloid A Proteins Induce Pathogenic Th17 Cells and Promote Inflammatory Disease. Cell 2020, 180, 79–91.e16. [Google Scholar] [CrossRef] [PubMed]
- Derebe, M.G.; Zlatkov, C.M.; Gattu, S.; Ruhn, K.A.; Vaishnava, S.; Diehl, G.E.; Macmillan, J.B.; Williams, N.S.; Hooper, L.V. Serum amyloid A is a retinol binding protein that transports retinol during bacterial infection. eLife 2014, 3, e03206. [Google Scholar] [CrossRef] [PubMed]
- Crotty, S. T Follicular Helper Cell Differentiation, Function, and Roles in Disease. Immunity 2014, 41, 529–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamoto, S.; Tran, T.H.; Maruya, M.; Suzuki, K.; Doi, Y.; Tsutsui, Y.; Kato, L.M.; Fagarasan, S. The Inhibitory Receptor PD-1 Regulates IgA Selection and Bacterial Composition in the Gut. Science 2012, 336, 485–489. [Google Scholar] [CrossRef] [PubMed]
- Teng, F.; Klinger, C.N.; Felix, K.M.; Bradley, C.P.; Wu, E.; Tran, N.L.; Umesaki, Y.; Wu, H.-J. Gut Microbiota Drive Autoimmune Arthritis by Promoting Differentiation and Migration of Peyer’s Patch T Follicular Helper Cells. Immunity 2016, 44, 875–888. [Google Scholar] [CrossRef] [Green Version]
- Kubinak, J.L.; Petersen, C.; Stephens, W.Z.; Soto, R.; Bake, E.; O’Connell, R.M.; Round, J.L. MyD88 signaling in T cells directs IgA-mediated control of the microbiota to promote health. Cell Host Microbe 2015, 17, 153–163. [Google Scholar] [CrossRef] [Green Version]
- Rosshart, S.P.; Herz, J.; Vassallo, B.G.; Hunter, A.; Wall, M.K.; Badger, J.H.; McCulloch, J.A.; Anastasakis, D.G.; Sarshad, A.A.; Leonardi, I.; et al. Laboratory mice born to wild mice have natural microbiota and model human immune responses. Science 2019, 365, eaaw4361. [Google Scholar] [CrossRef]
- Wagar, L.E.; DiFazio, R.M.; Davis, M.M. Advanced model systems and tools for basic and translational human immunology. Genome Med. 2018, 10, 73. [Google Scholar] [CrossRef]
- Zheng, S.; Zhao, T.; Yuan, S.; Yang, L.; Ding, J.; Cui, L.; Xu, M. Immunodeficiency Promotes Adaptive Alterations of Host Gut Microbiome: An Observational Metagenomic Study in Mice. Front. Microbiol. 2019, 10, 2415. [Google Scholar] [CrossRef]
- Berbers, R.-M.; Nierkens, S.; Van Laar, J.M.; Bogaert, D.; Leavis, H.L. Microbial Dysbiosis in Common Variable Immune Deficiencies: Evidence, Causes, and Consequences. Trends Immunol. 2017, 38, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Fagarasan, S.; Muramatsu, M.; Suzuki, K.; Nagaoka, H.; Hiai, H.; Honjo, T. Critical Roles of Activation-Induced Cytidine Deaminase in the Homeostasis of Gut Flora. Science 2002, 298, 1424–1427. [Google Scholar] [CrossRef] [PubMed]
- Rigoni, R.; Fontana, E.; Guglielmetti, S.; Fosso, B.; D’Erchia, A.M.; Maina, V.; Taverniti, V.; Castiello, M.C.; Mantero, S.; Pacchiana, G.; et al. Intestinal microbiota sustains inflammation and autoimmunity induced by hypomorphic RAG defects. J. Exp. Med. 2016, 213, 355–375. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Liu, Y.; Hoang, T.K.; Tian, X.; Taylor, C.M.; Luo, M.; Tran, D.Q.; Tatevian, N.; Rhoads, J.M. Antibiotic-modulated microbiome suppresses lethal inflammation and prolongs lifespan in Treg-deficient mice. Microbiome 2019, 7, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madsen, K.L. Inflammatory bowel disease: Lessons from the IL-10 gene-deficient mouse. Clin. Investig. Med. 2001, 24, 250–257. [Google Scholar]
- Delmonte, O.M.; Villa, A.; Notarangelo, L.D. Immune dysregulation in patients with RAG deficiency and other forms of combined immune deficiency. Blood 2020, 135, 610–619. [Google Scholar] [CrossRef]
- Villa, A.; Notarangelo, L.D. RAGgene defects at the verge of immunodeficiency and immune dysregulation. Immunol. Rev. 2019, 287, 73–90. [Google Scholar] [CrossRef]
- Notarangelo, L.D.; Kim, M.-S.; Walter, J.E.; Lee, Y.N. Human RAG mutations: Biochemistry and clinical implications. Nat. Rev. Immunol. 2016, 16, 234–246. [Google Scholar] [CrossRef]
- Chatila, T.A.; Blaeser, F.; Ho, N.; Lederman, H.M.; Voulgaropoulos, C.; Helms, C.; Bowcock, A.M. JM2, encoding a fork head–related protein, is mutated in X-linked autoimmunity–allergic disregulation syndrome. J. Clin. Investig. 2000, 106, R75–R81. [Google Scholar] [CrossRef]
- Bennett, C.L.; Christie, J.; Ramsdell, F.; Brunkow, M.E.; Ferguson, P.J.; Whitesell, L.; Kelly, T.E.; Saulsbury, F.T.; Chance, P.F.; Ochs, H.D. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat. Genet. 2001, 27, 20–21. [Google Scholar] [CrossRef] [PubMed]
- Brunkow, M.E.; Jeffery, E.W.; Hjerrild, K.A.; Paeper, B.; Clark, L.B.; Yasayko, S.-A.; Wilkinson, J.E.; Galas, D.; Ziegler, S.F.; Ramsdell, F. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat. Genet. 2001, 27, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.C.; Tonkonogy, S.L.; Albright, C.A.; Tsang, J.; Balish, E.J.; Braun, J.; Huycke, M.M.; Sartor, R.B. Variable phenotypes of enterocolitis in interleukin 10-deficient mice monoassociated with two different commensal bacteria. Gastroenterololgy 2005, 128, 891–906. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, J.; O’Mahony, L.; O’Callaghan, L.; Sheil, B.; Vaughan, E.E.; Fitzsimons, N.; FitzGibbon, J.; O’Sullivan, G.C.; Kiely, B.; Collins, J.K.; et al. Double blind, placebo controlled trial of two probiotic strains in interleukin 10 knockout mice and mechanistic link with cytokine balance. Gut 2003, 52, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Burich, A.; Hershberg, R.; Waggie, K.; Zeng, W.; Brabb, T.; Westrich, G.; Viney, J.L.; Maggio-Price, L. Helicobacter-induced inflammatory bowel disease in IL-10- and Tcell-deficient mice, Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G764–G778. [Google Scholar]
- Balish, E.; Warner, T. Enterococcus faecalis Induces Inflammatory Bowel Disease in Interleukin-10 Knockout Mice. Am. J. Pathol. 2002, 160, 2253–2257. [Google Scholar] [CrossRef] [Green Version]
- Schultz, M.; Veltkamp, C.; Dieleman, L.A.; Grenther, W.B.; Wyrick, P.B.; Tonkonogy, S.L.; Sartor, R.B. Lactobacillus plantarum 299V in the treatment and prevention of spontaneous colitis in interleukin-10-deficient mice. Inflamm. Bowel Dis. 2002, 8, 71–80. [Google Scholar] [CrossRef]
- Falcone, E.L.; Abusleme, L.; Swamydas, M.; Lionakis, M.S.; Ding, L.; Hsu, A.P.; Zelazny, A.M.; Moutsopoulos, N.M.; Kuhns, D.B.; Deming, C.; et al. Colitis susceptibility in p47(phox−/−) mice is mediated by the microbiome. Microbiome 2016, 4, 13. [Google Scholar] [CrossRef] [Green Version]
- Fischer, A.; Notarangelo, L.D.; Neven, B.; Cavazzana, M.; Puck, J.M. Severe combined immunodeficiencies and related disorders. Nat. Rev. Dis. Prim. 2015, 1, 15061. [Google Scholar] [CrossRef] [PubMed]
- Picard, C.; Fusaro, M.; Kashef, S.; Ziegler, J.B.; Su, H.C.; Lenardo, M.J. Chapter 8—Combined immune deficiencies (CIDs). In Stiehm’s Immune Deficiencies, 2nd ed.; Kathleen, E., Sullivan, E., Stiehm, R., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 207–268. ISBN 9780128167687. [Google Scholar] [CrossRef]
- Al-Herz, W.; Chou, J.; Delmonte, O.M.; Massaad, M.J.; Bainter, W.; Castagnoli, R.; Klein, C.; Bryceson, Y.T.; Geha, R.S.; Notarangelo, L.D. Comprehensive Genetic Results for Primary Immunodeficiency Disorders in a Highly Consanguineous Population. Front. Immunol. 2019, 9, 3146. [Google Scholar] [CrossRef] [Green Version]
- Castagnoli, R.; Delmonte, O.M.; Calzoni, E.; Notarangelo, L.D. Hematopoietic Stem Cell Transplantation in Primary Immunodeficiency Diseases: Current Status and Future Perspectives. Front. Pediatr. 2019, 7, 295. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, G.; Thrasher, A.J.; Aiuti, A. Gene therapy using haematopoietic stem and progenitor cells. Nat. Rev. Genet. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kohn, D.B.; Hershfield, M.S.; Puck, J.M.; Aiuti, A.; Blincoe, A.; Gaspar, H.B.; Notarangelo, L.D.; Grunebaum, E. Consensus approach for the management of severe combined immune deficiency caused by adenosine deaminase deficiency. J. Allergy Clin. Immunol. 2019, 143, 852–863. [Google Scholar] [CrossRef] [PubMed]
- King, J.R.; Hammarström, L. Newborn Screening for Primary Immunodeficiency Diseases: History, Current and Future Practice. J. Clin. Immunol. 2018, 38, 56–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delmonte, O.M.; Castagnoli, R.; Calzoni, E.; Notarangelo, L.D. Inborn Errors of Immunity With Immune Dysregulation: From Bench to Bedside. Front. Pediatr. 2019, 7, 353. [Google Scholar] [CrossRef] [Green Version]
- Delmonte, O.M.; Rowe, J.H.; Dobbs, A.K.; Palterer, B.; Castagnoli, R.; Notarangelo, L.D. Complete Absence of CD3γ Protein Expression Is Responsible for Combined Immunodeficiency with Autoimmunity Rather than SCID. J. Clin. Immunol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Daley, S.R.; Koay, H.-F.; Dobbs, K.; Bosticardo, M.; Wirasinha, R.C.; Pala, F.; Castagnoli, R.; Rowe, J.H.; De Bruin, L.M.O.; Keles, S.; et al. Cysteine and hydrophobic residues in CDR3 serve as distinct T-cell self-reactivity indices. J. Allergy Clin. Immunol. 2019, 144, 333–336. [Google Scholar] [CrossRef] [Green Version]
- Lane, J.; Stewart, C.J.; Cummings, S.P.; Gennery, A.R. Gut microbiome variations during hematopoietic stem cell transplant in severe combined immunodeficiency. J. Allergy Clin. Immunol. 2015, 135, 1654–1656. [Google Scholar] [CrossRef] [PubMed]
- Lane, J.; Stewart, C.J.; Cummings, S.P.; Gennery, A.R. Functional changes in gut microbiota during hematopoietic stem cell transplantation for severe combined immunodeficiency. J. Allergy Clin. Immunol. 2016, 138, 622–625.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zama, D.; Biagi, E.; Masetti, R.; Gasperini, P.; Prete, A.; Candela, M.; Brigidi, P.; Pession, A. Gut microbiota and hematopoietic stem cell transplantation: Where do we stand? Bone Marrow Transplant. 2016, 52, 7–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Sun, H.; Cao, W.; Han, L.; Song, Y.; Wan, D.; Jiang, Z. Applications of gut microbiota in patients with hematopoietic stem-cell transplantation. Exp. Hematol. Oncol. 2020, 9, 35. [Google Scholar] [CrossRef]
- Fredricks, D.N. The gut microbiota and graft-versus-host disease. J. Clin. Investig. 2019, 129, 1808–1817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biagi, E.; Zama, D.; Rampelli, S.; Turroni, S.; Brigidi, P.; Consolandi, C.; Severgnini, M.; Picotti, E.; Gasperini, P.; Merli, P.; et al. Early gut microbiota signature of aGvHD in children given allogeneic hematopoietic cell transplantation for hematological disorders. BMC Med. Genom. 2019, 12, 49. [Google Scholar] [CrossRef] [PubMed]
- Ilett, E.E.; Jørgensen, M.; Noguera-Julian, M.; Nørgaard, J.C.; Daugaard, G.; Helleberg, M.; Paredes, R.; Murray, D.D.; Lundgren, J.; MacPherson, C.; et al. Associations of the gut microbiome and clinical factors with acute GVHD in allogeneic HSCT recipients. Blood Adv. 2020, 4, 5797–5809. [Google Scholar] [CrossRef] [PubMed]
- Yoshifuji, K.; Inamoto, K.; Kiridoshi, Y.; Takeshita, K.; Sasajima, S.; Shiraishi, Y.; Yamashita, Y.; Nisaka, Y.; Ogura, Y.; Takeuchi, R.; et al. Prebiotics protect against acute graft-versus-host disease and preserve the gut microbiota in stem cell transplantation. Blood Adv. 2020, 4, 4607–4617. [Google Scholar] [CrossRef] [PubMed]
- Vossen, J.M.; Guiot, H.F.L.; Lankester, A.C.; Vossen, A.C.T.M.; Bredius, R.G.M.; Wolterbeek, R.; Bakker, H.D.J.; Heidt, P.J. Complete Suppression of the Gut Microbiome Prevents Acute Graft-Versus-Host Disease following Allogeneic Bone Marrow Transplantation. PLoS ONE 2014, 9, e105706. [Google Scholar] [CrossRef] [PubMed]
- Henig, I.; Yehudai-Ofir, D.; Zuckerman, T. The clinical role of the gut microbiome and fecal microbiota transplantation in allogeneic stem cell transplantation. Haematologica 2020. [Google Scholar] [CrossRef] [PubMed]
- Merli, P.; Putignani, L.; Ruggeri, A.; Del Chierico, F.; Gargiullo, L.; Galaverna, F.; Gaspari, S.; Pagliara, D.; Russo, A.; Pane, S.; et al. Decolonization of multi-drug resistant bacteria by fecal microbiota transplantation in five pediatric patients before allogeneic hematopoietic stem cell transplantation: Gut microbiota profiling, infectious and clinical outcomes. Haematologica 2020, 105, 244210. [Google Scholar] [CrossRef]
- Clarke, E.L.; Connell, A.J.; Six, E.; Kadry, N.A.; Abbas, A.A.; Hwang, Y.; Everett, J.K.; Hofstaedter, C.E.; Marsh, R.; Armant, M.; et al. T cell dynamics and response of the microbiota after gene therapy to treat X-linked severe combined immunodeficiency. Genome Med. 2018, 10, 70. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Li, Y.Y.; Tang, X.; Zhao, X. Faecal microbial dysbiosis in children with Wiskott-Aldrich syndrome. Scand. J. Immunol. 2019, 91, e12805. [Google Scholar] [CrossRef] [PubMed]
- Candotti, F. Clinical Manifestations and Pathophysiological Mechanisms of the Wiskott-Aldrich Syndrome. J. Clin. Immunol. 2018, 38, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Blundell, M.P.; Worth, A.; Bouma, G.; Thrasher, A.J. The Wiskott-Aldrich Syndrome: The Actin Cytoskeleton and Immune Cell Function. Dis. Markers 2010, 29, 157–175. [Google Scholar] [CrossRef] [PubMed]
- Ohya, T.; Yanagimachi, M.; Iwasawa, K.; Umetsu, S.; Sogo, T.; Inui, A.; Fujisawa, T.; Ito, S. Childhood-onset inflammatory bowel diseases associated with mutation of Wiskott-Aldrich syndrome protein gene. World J. Gastroenterol. 2017, 23, 8544–8552. [Google Scholar] [CrossRef] [PubMed]
- Conte, M.P.; Schippa, S.; Zamboni, I.; Penta, M.; Chiarini, F.; Seganti, L.; Osborn, J.F.; Falconieri, P.; Borrelli, O.; Cucchiara, S. Gut-associated bacterial microbiota in paediatric patients with inflammatory bowel disease. Gut 2006, 55, 1760–1768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haberman, Y.; Tickle, T.L.; Dexheimer, P.J.; Kim, M.; Tang, D.; Karns, R.; Baldassano, R.N.; Noe, J.D.; Rosh, J.; Markowitz, J.; et al. Pediatric Crohn disease patients exhibit specific ileal transcriptome and microbiome signature. J. Clin. Invest. 2015, 124, 3617–3633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lepage, P.; Häsler, R.; Spehlmann, M.E.; Rehman, A.; Zvirbliene, A.; Begun, A.; Ott, S.; Kupcinskas, L.; Dore, J.; Raedler, A.; et al. Twin study indicates loss of interaction between microbiota and mucosa of patients with ulcerative colitis. Gastroenterology 2011, 141, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Man, S.M.; Kaakoush, N.O.; Mitchell, H.M. The role of bacteria and pattern-recognition receptors in Crohn’s disease. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 152–168. [Google Scholar] [CrossRef]
- Manichanh, C.; Rigottier-Gois, L.; Bonnaud, E.; Gloux, K.; Pelletier, E.; Frangeul, L.; Jarrin, C.; Chardon, P.; Marteau, P.; Roca, J.; et al. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut 2006, 55, 205–212. [Google Scholar] [CrossRef] [Green Version]
- Maukonen, J.; Kolho, K.L.; Paasela, M.; Honkanen, J.; Klemetti, P.; Vaarala, O.; Saarela, M. Altered fecal microbiota in paediatric inflammatory bowel disease. J. Crohns Colitis 2015, 9, 1088–1095. [Google Scholar] [CrossRef] [Green Version]
- Michail, S.; Durbin, M.; Turner, D.; Griffiths, A.M.; Mack, D.R.; Hyams, J.; Leleiko, N.; Kenche, H.; Stolfi, A.; Wine, E. Alterations in the gut microbiome of children with severe ulcerative colitis. Inflamm. Bowel Dis. 2012, 18, 1799–1808. [Google Scholar] [CrossRef]
- Wang, F.; Kaplan, J.L.; Gold, B.D.; Bhasin, M.; Ward, N.L.; Kellermayer, R.; Kirschner, B.S.; Heyman, M.B.; Dowd, S.E.; Cox, S.B.; et al. Detecting microbial dysbiosis associated with Pediatric Crohn’s disease despite the high variability of the gut microbiota. Cell Rep. 2016, 14, 945–955. [Google Scholar] [CrossRef] [Green Version]
- Cunningham-Rundles, C. How I treat common variable immune deficiency. Blood 2010, 116, 7–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ameratunga, R.; Ebrewerton, M.; Eslade, C.; Ejordan, A.; Egillis, D.; Esteele, R.; Ekoopmans, W.; Ewoon, S.-T. Comparison of Diagnostic Criteria for Common Variable Immunodeficiency Disorder. Front. Immunol. 2014, 5, 415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salzer, U.; Warnatz, K.; Peter, H.-H. Common variable immunodeficiency: An update. Arthritis Res. Ther. 2012, 14, 223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jolles, S. The Variable in Common Variable Immunodeficiency: A Disease of Complex Phenotypes. J. Allergy Clin. Immunol. Pr. 2013, 1, 545–556. [Google Scholar] [CrossRef] [PubMed]
- Chapel, H.; Lucas, M.; Lee, M.; Bjorkander, J.; Webster, D.; Grimbacher, B.; Fieschi, C.; Thon, V.; Abedi, M.R.; Hammarstrom, L. Common variable immunodeficiency disorders: Division into distinct clinical phenotypes. Blood 2008, 112, 277–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, H.-E.; Cunningham-Rundles, C. Non-infectious Complications of Common Variable Immunodeficiency: Updated Clinical Spectrum, Sequelae, and Insights to Pathogenesis. Front. Immunol. 2020, 11, 149. [Google Scholar] [CrossRef] [Green Version]
- Berbers, R.-M.; Drylewicz, J.; Ellerbroek, P.M.; Van Montfrans, J.M.; Dalm, V.A.S.H.; Van Hagen, P.M.; Keller, B.; Warnatz, K.; Van De Ven, A.; Van Laar, J.M.; et al. Targeted Proteomics Reveals Inflammatory Pathways that Classify Immune Dysregulation in Common Variable Immunodeficiency. J. Clin. Immunol. 2020. [Google Scholar] [CrossRef] [PubMed]
- De Valles-Ibáñez, G.; Esteve-Solé, A.; Piquer, M.; González-Navarro, E.A.; Hernandez-Rodriguez, J.; Laayouni, H.; González-Roca, E.; Plaza-Martin, A.M.; Deyà-Martínez, Á.; Martín-Nalda, A.; et al. Evaluating the Genetics of Common Variable Immunodeficiency: Monogenetic Model and Beyond. Front. Immunol. 2018, 9, 636. [Google Scholar] [CrossRef]
- Maffucci, P.; Filion, C.A.; Boisson, B.; Itan, Y.; Shang, L.; Casanova, J.-L.; Cunningham-Rundles, C. Genetic Diagnosis Using Whole Exome Sequencing in Common Variable Immunodeficiency. Front. Immunol. 2016, 7, 220. [Google Scholar] [CrossRef]
- Quinti, I.; Soresina, A.; Spadaro, G.; Martino, S.; Donnanno, S.; Agostini, C.; Claudio, P.; Franco, D.; Pesce, A.M.; Borghese, F.; et al. Long-Term Follow-Up and Outcome of a Large Cohort of Patients with Common Variable Immunodeficiency. J. Clin. Immunol. 2007, 27, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Smereka, P.; Harpaz, N.; Cunningham-Rundles, C.; Mayer, L. Characterization of immunologic defects in patients with common variable immunodeficiency (CVID) with intestinal disease. Inflamm. Bowel Dis. 2011, 17, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Dhalla, F.; Da Silva, S.P.; Lucas, M.; Travis, S.; Chapel, H. Review of gastric cancer risk factors in patients with common variable immunodeficiency disorders, resulting in a proposal for a surveillance programme. Clin. Exp. Immunol. 2011, 165, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, S.F.; Fevang, B.; Aukrust, P. Autoimmunity and Inflammation in CVID: A Possible Crosstalk between Immune Activation, Gut Microbiota, and Epigenetic Modifications. J. Clin. Immunol. 2019, 39, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Fiedorová, K.; Radvanský, M.; Bosák, J.; Grombiříková, H.; Němcová, E.; Králíčková, P.; Černochová, M.; Kotásková, I.; Lexa, M.; Litzman, J.; et al. Bacterial but Not Fungal Gut Microbiota Alterations Are Associated With Common Variable Immunodeficiency (CVID) Phenotype. Front. Immunol. 2019, 10, 1914. [Google Scholar] [CrossRef] [Green Version]
- Jørgensen, S.F.; Trøseid, M.; Kummen, M.; Anmarkrud, J.A.; Michelsen, A.E.; Osnes, L.T.; Holm, K.; Høivik, M.L.; Rashidi, A.; Dahl, C.P.; et al. Altered gut microbiota profile in common variable immunodeficiency associates with levels of lipopolysaccharide and markers of systemic immune activation. Mucosal Immunol. 2016, 9, 1455–1465. [Google Scholar] [CrossRef] [PubMed]
- Shulzhenko, N.; Dong, X.; Vyshenska, D.; Greer, R.L.; Gurung, M.; Vasquez-Perez, S.; Peremyslova, E.; Sosnovtsev, S.; Quezado, M.; Yao, M.; et al. CVID enteropathy is characterized by exceeding low mucosal IgA levels and interferon-driven inflammation possibly related to the presence of a pathobiont. Clin. Immunol. 2018, 197, 139–153. [Google Scholar] [CrossRef]
- Berbers, R.-M.; Franken, I.A.; Leavis, H.L. Immunoglobulin A and microbiota in primary immunodeficiency diseases. Curr. Opin. Allergy Clin. Immunol. 2019, 19, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Fadlallah, J.; El Kafsi, H.; Sterlin, D.; Juste, C.; Parizot, C.; Dorgham, K.; Autaa, G.; Gouas, D.; Almeida, M.; Lepage, P.; et al. Microbial ecology perturbation in human IgA deficiency. Sci. Transl. Med. 2018, 10, eaan1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sterlin, D.; Fieschi, C.; Malphettes, M.; Larsen, M.; Gorochov, G.; Fadlallah, J. Immune/microbial interface perturbation in human IgA deficiency. Gut Microbes 2019, 10, 429–433. [Google Scholar] [CrossRef]
- Fadlallah, J.; Sterlin, D.; Fieschi, C.; Parizot, C.; Dorgham, K.; El Kafsi, H.; Autaa, G.; Ghillani-Dalbin, P.; Juste, C.; Lepage, P.; et al. Synergistic convergence of microbiota-specific systemic IgG and secretory IgA. J. Allergy Clin. Immunol. 2019, 143, 1575–1585.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacPherson, M.E.; Hov, J.R.; Ueland, T.; Dahl, T.B.; Kummen, M.; Otterdal, K.; Holm, K.; Berge, R.K.; Mollnes, T.E.; Trøseid, M.; et al. Gut Microbiota-Dependent Trimethylamine N-Oxide Associates With Inflammation in Common Variable Immunodeficiency. Front. Immunol. 2020, 11, 574500. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, S.F.; MacPherson, M.E.; Bjørnetrø, T.; Holm, K.; Kummen, M.; Rashidi, A.; Michelsen, A.E.; Lekva, T.; Halvorsen, B.; Trøseid, M.; et al. Rifaximin alters gut microbiota profile, but does not affect systemic inflammation—A randomized controlled trial in common variable immunodeficiency. Sci. Rep. 2019, 9, 167. [Google Scholar] [CrossRef]
- Sokol, H.; Mahlaoui, N.; Aguilar, C.; Bach, P.; Join-Lambert, O.; Garraffo, A.; Seksik, P.; Danion, F.; Jegou, S.; Straube, M.; et al. Intestinal dysbiosis in inflammatory bowel disease associated with primary immunodeficiency. J. Allergy Clin. Immunol. 2019, 143, 775–778.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segal, B.H.; Leto, T.L.; Gallin, J.I.; Malech, H.L.; Holland, S.M. Genetic, Biochemical, and Clinical Features of Chronic Granulomatous Disease. Medicine 2000, 79, 170–200. [Google Scholar] [CrossRef] [PubMed]
- Holland, S.M. Chronic granulomatous disease. Hematol. Oncol. Clin. North Am. 2013, 27, 89–99. [Google Scholar] [CrossRef] [Green Version]
- Winkelstein, J.A.; Marino, M.C.; Johnston, R.B.; Boyle, J.; Curnutte, J.; Gallin, J.I.; Malech, H.L.; Holland, S.M.; Ochs, H.; Quie, P.; et al. Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine 2000, 79, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Notarangelo, L.D. The long road to optimal management for chronic granulomatous disease. J. Allergy Clin. Immunol. 2013, 132, 1164–1165. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Abbasakoor, F.; Vaizey, C.J. Gastrointestinal manifestations of chronic granulomatous disease. Colorectal Dis. 2006, 8, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Marks, D.J.; Miyagi, K.; Rahman, F.Z.; Novelli, M.; Bloom, S.L.; Segal, A.W. Inflammatory Bowel Disease in CGD Reproduces the Clinicopathological Features of Crohn’s Disease. Am. J. Gastroenterol. 2009, 104, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Alimchandani, M.; Lai, J.-P.; Aung, P.P.; Khangura, S.; Kamal, N.; Gallin, J.I.; Holland, S.M.; Malech, H.L.; Heller, T.; Miettinen, M.; et al. Gastrointestinal Histopathology in Chronic Granulomatous Disease. Am. J. Surg. Pathol. 2013, 37, 1365–1372. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.E.; De Ravin, S.S.; Uzel, G.; Landers, C.; Targan, S.; Malech, H.L.; Holland, S.M.; Cao, W.; Harpaz, N.; Mayer, L.; et al. High levels of Crohn’s disease-associated anti-microbial antibodies are present and independent of colitis in chronic granulomatous disease. Clin. Immunol. 2011, 138, 14–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khangura, S.K.; Kamal, N.; Ho, N.; Quezado, M.; Zhao, X.; Marciano, B.; Simpson, J.; Zerbe, C.S.; Uzel, G.; Yao, M.D.; et al. Gastrointestinal Features of Chronic Granulomatous Disease Found During Endoscopy. Clin. Gastroenterol. Hepatol. 2016, 14, 395–402.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magnani, A.; Brosselin, P.; Beauté, J.; De Vergnes, N.; Mouy, R.; Debré, M.; Suarez, F.; Hermine, O.; Lortholary, O.; Blanche, S.; et al. Inflammatory manifestations in a single-center cohort of patients with chronic granulomatous disease. J. Allergy Clin. Immunol. 2014, 134, 655–662.e8. [Google Scholar] [CrossRef] [PubMed]
- Van De Geer, A.; Nieto-Patlán, A.; Kuhns, D.B.; Tool, A.T.; Arias, A.A.; Bouaziz, M.; De Boer, M.; Franco, J.; Gazendam, R.P.; Van Hamme, J.L.; et al. Inherited p40phox deficiency differs from classic chronic granulomatous disease. J. Clin. Investig. 2018, 128, 3957–3975. [Google Scholar] [CrossRef] [Green Version]
- Marsh, R.A.; Leiding, J.W.; Logan, B.R.; Griffith, L.M.; Arnold, D.E.; Haddad, E.; Falcone, E.L.; Yin, Z.; Patel, K.; Arbuckle, E.; et al. Chronic Granulomatous Disease-Associated IBD Resolves and Does Not Adversely Impact Survival Following Allogeneic HCT. J. Clin. Immunol. 2019, 39, 653–667. [Google Scholar] [CrossRef]
- Aguilar, C.; Latour, S. X-linked Inhibitor of Apoptosis Protein Deficiency: More than an X-linked Lymphoproliferative Syndrome. J. Clin. Immunol. 2015, 35, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Latour, S.; Aguilar, C. XIAP deficiency syndrome in humans. Semin. Cell Dev. Biol. 2015, 39, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Canna, S.W.; Marsh, R.A. Pediatric hemophagocytic lymphohistiocytosis. Blood 2020, 135, 1332–1343. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, O.H.; Lacasse, E.C. How genetic testing can lead to targeted management of XIAP deficiency–related inflammatory bowel disease. Genet. Med. 2017, 19, 133–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.; Giliani, S.; Lanzi, G.; Mias, G.I.; Lonardi, S.; Dobbs, K.; Manis, J.; Im, H.; Gallagher, J.E.; Phanstiel, D.H.; et al. Whole-exome sequencing identifies tetratricopeptide repeat domain 7A (TTC7A) mutations for combined immunodeficiency with intestinal atresias. J. Allergy Clin. Immunol. 2013, 132, 656–664.e17. [Google Scholar] [CrossRef] [Green Version]
- Samuels, M.E.; Majewski, J.; Alirezaie, N.; Fernandez, I.; Casals, F.; Patey, N.; Decaluwe, H.; Gosselin, I.; Haddad, E.; Hodgkinson, A.; et al. Exome sequencing identifies mutations in the geneTTC7Ain French-Canadian cases with hereditary multiple intestinal atresia. J. Med Genet. 2013, 50, 324–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, L.A.; Gottrand, F.; Turck, D.; Manouvrier-Hanu, S.; Mazingue, F.; Morisot, C.; Le Deist, F.; Ricour, C.; Nihoul-Feketé, C.; Debeugny, P.; et al. Severe combined immunodeficiency syndrome associated with autosomal recessive familial multiple gastrointestinal atresias: Study of a family. Am. J. Med Genet. 1990, 37, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Notarangelo, L.D. Multiple intestinal atresia with combined immune deficiency. Curr. Opin. Pediatr. 2014, 26, 690–696. [Google Scholar] [CrossRef] [PubMed]
- Lien, R.; Lin, Y.-F.; Lai, M.-W.; Weng, H.-Y.; Wu, R.; Jaing, T.-H.; Huang, J.-L.; Tsai, S.-F.; Lee, W.-I. Novel Mutations of the Tetratricopeptide Repeat Domain 7A Gene and Phenotype/Genotype Comparison. Front. Immunol. 2017, 8, 1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kummen, M.; Holm, K.; Anmarkrud, J.A.; Nygård, S.; Vesterhus, M.; Høivik, M.L.; Trøseid, M.; Marschall, H.-U.; Schrumpf, E.; Moum, B.; et al. The gut microbial profile in patients with primary sclerosing cholangitis is distinct from patients with ulcerative colitis without biliary disease and healthy controls. Gut 2017, 66, 611–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokol, H.; Leducq, V.; Aschard, H.; Pham, H.-P.; Jegou, S.; Landman, C.; Cohen, D.; Liguori, G.; Bourrier, A.; Nion-Larmurier, I.; et al. Fungal microbiota dysbiosis in IBD. Gut 2017, 66, 1039–1048. [Google Scholar] [CrossRef] [Green Version]
- Xue, A.-J.; Miao, S.-J.; Sun, H.; Qiu, X.-X.; Wang, S.-N.; Wang, L.; Ye, Z.-Q.; Zheng, C.-F.; Huang, Z.-H.; Wang, Y.-H.; et al. Intestinal dysbiosis in pediatric Crohn’s disease patients with IL10RA mutations. World J. Gastroenterol. 2020, 26, 3098–3109. [Google Scholar] [CrossRef]
- Glocker, E.; Frede, N.; Perro, M.; Sebire, N.; Elawad, M.; Shah, N.; Grimbacher, B. Infant colitis—it’s in the genes. Lancet 2010, 376, 1272. [Google Scholar] [CrossRef] [PubMed]
- Kotlarz, D.; Beier, R.; Murugan, D.; Diestelhorst, J.; Jensen, O.; Boztug, K.; Pfeifer, D.; Kreipe, H.; Pfister, E.; Baumann, U.; et al. Loss of Interleukin-10 Signaling and Infantile Inflammatory Bowel Disease: Implications for Diagnosis and Therapy. Gastroenterology 2012, 143, 347–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gevers, D.; Kugathasan, S.; Denson, L.A.; Vázquez-Baeza, Y.; Van Treuren, W.; Ren, B.; Schwager, E.; Knights, D.; Song, S.J.; Yassour, M.; et al. The Treatment-Naive Microbiome in New-Onset Crohn’s Disease. Cell Host Microbe 2014, 15, 382–392. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Shen, N.; Luo, L.; Deng, Z.; Chen, J.; Tao, Y.; Mo, X.; Cao, Q. Fecal microbiota transplantation before hematopoietic stem cell transplantation in a pediatric case of chronic diarrhea with a FOXP3 mutation. Pediatr. Neonatol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Cepika, A.-M.; Sato, Y.; Liu, J.M.-H.; Uyeda, M.J.; Baccheta, R.; Roncarolo, M.-G. Tregopathies: Monogenic diseases resulting in regulatory T-cell deficiency. J. Allergy Clin. Immunol. 2018, 142, 1679–1695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gambineri, E.; Mannurita, S.C.; Hagin, D.; Vignoli, M.; Anover-Sombke, S.; DeBoer, S.; Segundo, G.R.S.; Allenspach, E.J.; Favre, C.; Ochs, H.D.; et al. Clinical, Immunological, and Molecular Heterogeneity of 173 Patients With the Phenotype of Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked (IPEX) Syndrome. Front. Immunol. 2018, 9, 2411. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Motal, U.M.; Al-Shaibi, A.; Elawad, M.; Lo, B. Zero tolerance! A perspective on monogenic disorders with defective regulatory T cells and IBD-like disease. Immunol. Rev. 2019, 287, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Patron, R.L.; Hartmann, C.A.; Allen, S.; Griesbach, C.L.; Kosiorek, H.E.; DiBaise, J.K.; Orenstein, R. Vancomycin Taper and Risk of Failure of Fecal Microbiota Transplantation in Patients With Recurrent Clostridium difficile Infection. Clin. Infect. Dis. 2017, 65, 1214–1217. [Google Scholar] [CrossRef] [PubMed]
- Sacco, K.; Pongdee, T.; Binnicker, M.J.; Espy, M.; Pardi, D.; Khanna, S.; Joshi, A. Presence of immune deficiency increases the risk of hospitalization in patients with norovirus infection. Diagn. Microbiol. Infect. Dis. 2018, 90, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Kao, D.; Mehta, S.R.; Martin, T.; Dimitry, J.; Keshteli, A.H.; Cook, G.K.; Phelps, E.; Sipe, B.W.; Xu, H.; et al. Predictors of Early Failure After Fecal Microbiota Transplantation for the Therapy of Clostridium Difficile Infection: A Multicenter Study. Am. J. Gastroenterol. 2016, 111, 1024–1031. [Google Scholar] [CrossRef] [PubMed]
- Loke, P.; Heine, R.G.; McWilliam, V.; Cameron, D.J.S.; Tang, M.L.K.; Allen, K.J. Fecal microbial transplantation in a pediatric case of recurrentClostridium difficileinfection and specific antibody deficiency. Pediatr. Allergy Immunol. 2016, 27, 872–874. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Disease | Genetic Defect | Inheritance | Main Findings | Reference |
|---|---|---|---|---|
| Immunodeficiencies Affecting Cellular and Humoral Immunity (Including CID with Associated or Syndromic Features) | ||||
| SCID | IL2RG (X-SCID) RAG1 | XL AR | Gut microbiota and fecal metabolite composition can be differentiated into pre- and post-HSCT groups. Gut microbiota of X-SCID patients changes to more resemble those of healthy children after successful gene therapy. | [88,89] [99] |
| Wiskott-Aldrich syndrome | WAS | XL | Reduced fecal microbial community richness and diversity in WAS patients compared to age-matched healthy controls. Among WAS children, those with IBD and those who failed to express WASP, presented with more severe microbial dysbiosis. | [100] |
| Predominantly antibody deficiencies | ||||
| CVID | Multiple genetic defects | Variable | In CVID patients with immune dysregulation, reduced microbial alpha diversity correlates with increased levels of LPS, soluble CD14 and CD25, and reduced IgA serum levels. IgG from healthy subjects targets the microbiota of CVID patients much less effectively than the microbiota of healthy subjects. Elevated concentrations of the gut microbiota-dependent metabolite TMAO is associated with systemic inflammation and increased gut microbial abundance of Gammaproteobacteria in CVID patients A single broad-spectrum antibiotic (rifaximin) does not modify microbial translocation, immune cell activation, and immune dysregulation | [126] [131] [132] [133] |
| sIgAD | Unknown | Unknown | Adequate IgM and/or IgG induction in sIgAD may protect from endotoxemia, while this compensatory response is lacking in CVID. | [129,130] |
| Other IEI | ||||
| CGD XIAP deficiency TTC7A deficiency | CYBB, CYBA, CYBC1, NCF1, NCF2, NCF4 XIAP TTC7A | XL AR XL AR | Gut microbiota of patients with different genetic defects has distinct alterations; moreover, patients with the same gene defect who differ for the presence or absence of GI involvement display different microbial communities. | [134] |
| IL-10 receptor deficiency | IL10RA | AR | The degree of gut dysbiosis (calculated based on the relative abundance of five taxa at the order level: Lactobacillales, Micrococcales, Veillonellaceae, Clostridiales, and Selenomonadales) appears to be directly associated to disease severity. | [158] |
| IPEX | FOXP3 | XL | First report on the effect of FMT before HSCT in a child with IPEX | [162] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castagnoli, R.; Pala, F.; Bosticardo, M.; Licari, A.; Delmonte, O.M.; Villa, A.; Marseglia, G.L.; Notarangelo, L.D. Gut Microbiota–Host Interactions in Inborn Errors of Immunity. Int. J. Mol. Sci. 2021, 22, 1416. https://doi.org/10.3390/ijms22031416
Castagnoli R, Pala F, Bosticardo M, Licari A, Delmonte OM, Villa A, Marseglia GL, Notarangelo LD. Gut Microbiota–Host Interactions in Inborn Errors of Immunity. International Journal of Molecular Sciences. 2021; 22(3):1416. https://doi.org/10.3390/ijms22031416
Chicago/Turabian StyleCastagnoli, Riccardo, Francesca Pala, Marita Bosticardo, Amelia Licari, Ottavia M. Delmonte, Anna Villa, Gian Luigi Marseglia, and Luigi Daniele Notarangelo. 2021. "Gut Microbiota–Host Interactions in Inborn Errors of Immunity" International Journal of Molecular Sciences 22, no. 3: 1416. https://doi.org/10.3390/ijms22031416