Control of Mesenchymal Stromal Cell Senescence by Tryptophan Metabolites


1
Institute of Cellular and System Medicine, National Health Research Institutes, 35 Keyan Road, Zhunan Town, Miaoli County 35053, Taiwan
2
Institute of Biotechnology, College of Life Science, National Tsing-Hua University, Hsinchu 30071, Taiwan
Int. J. Mol. Sci. 2021, 22(2), 697; https://doi.org/10.3390/ijms22020697
Submission received: 4 December 2020
/
Revised: 3 January 2021
/
Accepted: 5 January 2021
/
Published: 12 January 2021
(This article belongs to the Special Issue Molecular Research of Aging Stress Response)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Cellular senescence contributes to aging and age-related disorders. High glucose (HG) induces mesenchymal stromal/stem cell (MSC) senescence, which hampers cell expansion and impairs MSC function. Intracellular HG triggers metabolic shift from aerobic glycolysis to oxidative phosphorylation, resulting in reactive oxygen species (ROS) overproduction. It causes mitochondrial dysfunction and morphological changes. Tryptophan metabolites such as 5-methoxytryptophan (5-MTP) and melatonin attenuate HG-induced MSC senescence by protecting mitochondrial integrity and function and reducing ROS generation. They upregulate the expression of antioxidant enzymes. Both metabolites inhibit stress-induced MSC senescence by blocking p38 MAPK signaling pathway, NF-κB, and p300 histone acetyltransferase activity. Furthermore, melatonin upregulates SIRT-1, which reduces NF-κB activity by de-acetylation of NF-κB subunits. Melatonin and 5-MTP are a new class of metabolites protecting MSCs against replicative and stress-induced cellular senescence. They provide new strategies to improve the efficiency of MSC-based therapy for diverse human diseases.
1. Introduction
Cellular senescence is a hallmark of aging [1]. Accumulation of senescent cells promotes aging and triggers age-related disorders [2]. Cellular senescence was originally observed in cultured fibroblasts following limited replications [3]. It was subsequently noted as a response to DNA damage, telomere attrition, mitochondrial dysfunction, and oncogenic, hyperglycemic, and oxidative stresses [4,5,6]. Cellular senescence plays an important role in parturition and embryo development [7,8]. It may influence the fate of tumorigenesis through senescence-associated secretory phenotype (SASP). It was reported that acute senescent cells induce immortalized prostate cells to undergo senescence via SASP but have no effect on metastatic prostate cancer cells [9]. Replicative, developmental, and stress-induced premature cell senescence share common cellular phenotypic changes including an increased expression of p16 and p21, cell cycle and proliferation arrest, senescence-associated (SA) heterochromatin foci, SA-β galactosidase (β gal), and SASP, as well as cellular morphological changes [10]. Phenotypic changes of senescent cells are mediated by multiple signaling pathways leading to complex transcriptional reprogramming [11,12].
Hyperglycemia due to type 2 diabetes (T2D) and pre-diabetic metabolic syndrome and obesity has emerged as a key extracellular stress signal to induce cellular senescence as well as cell death [13]. As T2D is increasing with aging and contributes to age-related chronic diseases [13], hyperglycemia has become a leading age stress factor. Hyperglycemia induces cellular senescence through metabolism shift, reactive oxygen species (ROS) generation, mitochondrial dysfunction, and aberrant gene expressions.
Replicative and stress (hyperglycemia and oxidative stress)-induced mesenchymal stromal cell senescence has been extensively investigated as it is critical for MSC-based cell therapy. Mesenchymal stromal cells (MSCs) are isolated and characterized according to a set of criteria [14,15]. Current isolation procedures generate heterogeneous nonclonal stromal cell populations with different multipotent and differentiation potentials [16]. MSCs possess immunosuppressive and anti-inflammatory properties [17,18]. As MSCs can be obtained and cultured with ease, they are popular sources for cell-based therapy of a variety of human diseases. More than 700 clinical trials have been registered [16]. However, MSC-based cell therapy faces challenging problems. Replicative senescence of cultured MSCs limits the cell expansion and its availability for cell therapy. Moreover, stress-induced premature senescence in vitro and in vivo reduces the efficacy of transplanted MSCs in tissue regeneration and treatment of autoimmune and inflammatory diseases. New strategies are actively being employed to develop new drugs to combat cellular senescence. The candidate drugs are either senolytic, which kill and remove senescent cells, or senomorphic, which modify senescent cell phenotypes to attenuate their tissue-damaging effects [19,20,21]. Senomorphic agents comprise a wide range of compounds with different targets aiming at reducing SASP and senescent markers without causing cell apoptosis [20]. Recent studies indicate that tryptophan metabolites produced via the tryptophan hydroxylase (TPH) pathway defend against replicative and hyperglycemia or oxidative stress-induced cell senescence. 5-methoxytryptophan (5-MTP) was reported to rescue bone marrow mesenchymal stromal cells (BM-MSCs) from high glucose (HG)-induced senescence [22], while melatonin protects MSC from replicative and stress-induced senescence [23]. Melatonin and 5-MTP represent a new class of senomorphic compounds which may be useful in protecting MSC against senescence and age-related diseases. This review will comment on the anti-senescence actions of 5-MTP and melatonin with a focus on 5-MTP biosynthesis, its defense of HG-induced MSC senescence, and mechanisms of actions.
2. Hyperglycemia Induces Cellular Senescence
High blood glucose levels (hyperglycemia) contribute to diabetic microvascular, renal, retinal and neural complications by multiple mechanisms including mitochondrial dysfunction and ROS generation [24,25]. Results from in vivo and in vitro experiments have shown that hyperglycemia induces cellular damage, apoptosis, and necrosis through ROS generation and mitochondrial dysfunction [24,25]. In addition, hyperglycemia was reported to induce renal tubular cell and retinal endothelial cell senescence in streptozotocin-induced diabetic mice [26,27] and HG in cultured media was reported to induce senescence of diverse cell types including MSCs [28,29,30]. Senescent cells cause further tissue damage through secretion of pro-inflammatory cytokines and proteolytic enzymes [1,31].
2.1. HG-Induced Cellular Senescence Is Attributed to Mitochondrial Dysfunction and ROS Generation
The exact mechanisms by which HG induces senescence are not entirely clear. Mitochondrial dysfunction and ROS generation are considered to be major players. HG induces mitochondrial ROS generation by enhancing mitochondrial metabolism via tricarboxylic acid (TCA) cycle and oxidative phosphorylation [24]. ROS generation is closely related to mitochondrial morphological changes. It was reported that HG-treated rat liver cells undergo mitochondrial fission, which was required for ROS generation [32]. ROS, in turn, cause mitochondrial fission [33], creating a vicious cycle (Figure 1). It was also reported that HG increases ROS through activation of NADPH oxidase [34,35], but its relevance to cell senescence is unclear and remains to be investigated. ROS overproduction is considered to be a major cause of cell damage and lethality. However, at sublethal concentrations, H2O2 induces cellular senescence as a way of protecting cells from ROS-induced death [36,37]. ROS represent a common mediator via which diverse stress signals induce cellular senescence. For example, Ras overexpression in fibroblasts induce cellular senescence by elevation of ROS generation [37].
ROS induces cellular senescence by oxidative damage to DNA, leading to telomere attrition and altered expression of p53, p16, and p21 [38,39]. However, increased ROS generation cannot explain all the phenotypic manifestations. Mitochondrial structural changes and functional defects contribute significantly to stress-induced senescence [40]. Changes in mitochondrial dynamics [41,42], metabolism, and signaling molecules such as AMPK [43,44,45] are considered to mediate senescence independent of ROS.
2.2. HG Induces MSC Senescence
MSCs in bone marrow reside in hypoxic microenvironment. They depend on glycolysis as energy source and thus express relatively low levels of oxidative phosphorylation (OXPHOS) proteins [46]. MSCs cultured in nutrient-rich normoxic conditions shifts the metabolism to OXPHOS [47]. MSC metabolism is altered during osteoblastic vs. chondrogenic differentiation. Osteoblastic differentiation requires OXPHOS, while chondrogenic differentiation uses aerobic glycolysis in energy generation [48]. When MSCs are incubated with HG medium, excessive intracellular glucose shifts metabolism from aerobic glycolysis to TCA cycle and OXPHOS. Consequently, a high level of ROS is leaked from the electron transport chain which induces MSC senescence [49,50]. Effects of HG on MSC mitochondrial biogenesis and metabolism have not been described. However, it is likely HG-induced mitochondrial structural and metabolic changes contribute to MSC senescence. MSC senescence puts a limit to MSC expansion, which hampers its use in cell therapy. Furthermore, it impairs MSC function which reduces its support for hematopoiesis and immunosuppressive properties [51].
3. 5-MTP Rescues MSCs from HG-Induced Senescence
3.1. 5-MTP Biosynthesis and Its Perturbation by Environmental Stresses
5-MTP was originally identified as a cytoprotective molecule named cytoguardin [52]. It is produced in and released from human fibroblasts [52]. Its biosynthesis in fibroblasts is catalyzed by two enzymes: tryptophan hydroxylase (TPH), which converts L-tryptophan to 5-hydroxytryptophan (5-HTP) and hydroxyindole O-methyltransferase (HIOMT), which converts 5-HTP to 5-MTP [53] (Figure 2A). Of the two isoforms of TPH identified and characterized in human cells [54,55], TPH-1 is selectively expressed in human fibroblasts, and silencing of TPH-1 with siRNA results in diminished release of 5-MTP into the cultured medium [53]. Thus, TPH-1 is the functional isoform catalyzing 5-MTP synthesis. HIOMT was previously identified and characterized as the terminal enzyme in catalyzing melatonin (N-acetyl-5-methoxytryptamine) synthesis. As it catalyzes the conversion of N-acetylserotonin to melatonin in pineal cells (Figure 2B), it is commonly called N-acetylserotonin O-methyltransferase (ASMT). It is encoded by a single gene with three mRNA isoforms due to alternative splicing [56,57]. The full-length isoform which contains LINE 1 repeat sequences in exon 6 codes for a 373 aa protein [57]. The isoform that codes for a 345 aa protein has exon 6 spliced, while the isoform coding for a 298 aa protein loses exons 6 and 7 to splicing. The nomenclature for HIOMT isoforms is different between NCBI and Uniprot database. To avoid confusion, the isoforms are thus named HIOMT373, 345 and 298, respectively [58]. Bovine and macaque express only a single transcript which aligns with human HIOMT345. As only HIOMT345 from pineal tissues is catalytically active in melatonin synthesis, HIOMT345 is considered to be a wild-type ASMT [59]. By contrast, HIOMT345 is not involved in 5-MTP production. Fibroblasts express only HIOMT298 isoform which was shown to be active in catalyzing 5-MTP synthesis [58]. This is surprising because HIOMT298 is a truncated isoform and structural analysis suggests that it lacks binding site for S-adenosylmethionine, a co-factor required for melatonin synthesis [59]. It is unclear how this truncated isoform catalyzes conversion of 5-HTP to 5-MTP.
It was subsequently reported that 5-MTP production is not limited to fibroblasts. Vascular endothelial cells (ECs) and smooth muscle cells (SMCs) as well as bronchial and renal epithelial cells produce 5-MTP [60]. Human umbilical vein ECs express TPH-1 and HIOMT298. Immunofluorescent studies show that 5-MTP is detected in cytoplasm with an endoplasmic reticulum (ER) pattern, and biochemical studies suggest that 5-MTP is secreted via Golgi vesicular transport [60]. BM-MSCs, like fibroblasts and ECs, express TPH-1 and HIOMT298 and release 5-MTP into the conditioned medium.
Lipopolysaccharide (LPS) and pro-inflammatory cytokines inhibit 5-MTP production by suppressing TPH-1 expression in ECs [60,61]. Addition of 5-MTP alleviates LPS and cytokine-induced vascular permeability suggesting that 5-MTP plays an important role in protecting endothelial barrier function [60,61]. LPS and pro-inflammatory cytokines exert their actions via ROS [62,63]. BM-MSCs cultured in medium containing HG release a lower amount of 5-MTP into the medium than control. Incubation of BM-MSCs with sublethal concentrations of H2O2 results in reduction of 5-MTP in the medium. Reduction of 5-MTP may be due to TPH-1 suppression by ROS.
3.2. 5-MTP Rescues BM-MSC from HG-Induced Cellular Senescence by Suppressing ROS Generation
Addition of 5-MTP to BM-MSC cultured in HG medium prevents growth arrest, blocks p16 and p21 elevation, attenuates SA-β gal positive cells and reduces interleukin-6 (IL-6) [22]. Furthermore, 5-MTP preserves BM-MSC morphology. 5-MTP controls HG-induced MSC senescence by suppressing ROS accumulation [22]. HG enhances mitochondrial ROS generation through electron transport chain [24]. Excessive ROS causes cell damage and death. However, at sublethal concentrations, ROS induces cellular senescence. Pretreatment of BM-MSC with 5-MTP results in reduction of ROS [22]. ROS comprise several species of oxidants and oxygen radicals among which H2O2 is a key mediator of cellular senescence. H2O2 at sublethal concentrations induces characteristic BM-MSC senescence [22]. 5-MTP pretreatment alleviates H2O2-induced cellular senescence [22].
3.3. 5-MTP Upregulates MnSOD and Catalase via FOXO3a
ROS levels in mitochondria are controlled by several antioxidant enzymes including MnSOD (SOD-2) and catalase [64]. Hyperglycemia was reported to alter MnSOD and catalase activities in a cell-dependent manner: it downregulates MnSOD and catalase in human umbilical vein ECs but not in microvascular EC [65]. HG does not alter MnSOD or catalase activity in BM-MSC [22]. However, 5-MTP upregulates MnSOD and catalase in HG-treated BM-MSCs. MnSOD and catalase are mitochondrial antioxidant enzymes that have immediate access to ROS generated in mitochondrial matrix, converting superoxide to water and oxygen. Upregulation of both enzymes by 5-MTP increases ROS scavenging and thereby reduces the damaging effect of ROS.
FOXO3a is a pleotropic transcriptional activator that mediates expression of diverse genes including antioxidant genes [66,67]. 5-MTP upregulates FOXO3a expression, and silencing of FOXO3a in HG-treated BM-MSC abrogates 5-MTP-induced rise of MnSOD and catalase [22]. Importantly, FOXO3a silencing eliminates the protective effect of 5-MTP on HG-induced senescence [22]. 5-MTP protects HG-induced cellular senescence by upregulating FOXO3a-mediated MnSOD and catalase expression.
3.4. 5-MTP Restores BM-MSC Osteogenic Differentiation via Controlling ROS Levels
Excessive ROS generation was reported to induce defective MSC differentiation: it impairs osteogenic and promotes adipogenic differentiation [68]. The adverse effects of ROS on MSC differentiation play an important role in age-related skeletal disorders and obesity. It is worth noting that physiological osteogenic differentiation is accompanied by metabolism shift from aerobic glycolysis to oxidative phosphorylation [46]. Although ROS is expected to be elevated, it is not because MnSOD and catalase activities are upregulated [46]. Thus, the ROS level is controlled by a well-regulated redox balance during osteogenic differentiation. Oxidative stress disrupts the balance and tilts it toward ROS overproduction, which impairs osteogenic differentiation. 5-MTP restores the redox balance and thereby protects MSC osteogenic differentiation [22]. It is less clear whether excessive ROS influence MSC chondrocyte differentiation [69]. However, it was reported that chondrocytes from patients with osteoarthritis (OA) exhibit mitochondrial defects and senescent changes [70]. Oxidative stress-induced chondrocyte senescence contributes to joint dysfunction in OA [70]. As 5-MTP and melatonin reduce ROS accumulation, they may exert an effect on protecting chondrocyte function [22,71].
4. Melatonin Protects Against Replicative and Stress-Induced Cellular Senescence
Melatonin is produced primarily in pineal and retinal cells. Its synthesis from L-tryptophan shares with serotonin biosynthesis common enzymes, i.e., TPH-2 which converts L-tryptophan to 5-HTP and aromatic amino acid decarboxylase (AADC) which catalyzes decarboxylation of 5-HTP to form 5-hydroxytryptamine (5-HT, serotonin) (Figure 2B). 5-HT is N-acetylated by arylalkylamine N-acetyltransferase (AANAT) to form N-acetyl-5HT (N-acetylserotonin). AANAT expression is regulated by circadian rhysm and its expression in dark accounts for burst melatonin synthesis. The final step of melatonin synthesis is catalyzed by HIOMT (ASMT). All three isoforms are detected in pineal cells but only isoform 345 is catalytically active in melatonin synthesis [59]. As HIOMT298 is expressed in pineal cells, it is possible that they may produce 5-MTP. Melatonin plays a physiological role in regulating circadian rhythm and sleep [72]. In addition, a large number of reports suggest that melatonin possess anti-inflammatory actions by suppressing the expression of pro-inflammatory genes such as cyclooxygenase-2 [73]. It protects against stress-induced cell and tissue damage [74]. Melatonin inhibits cancer growth through melatonin-receptor-mediated signaling transduction [75,76]. Recent studies reveal that melatonin also possesses anti-senescence activities.
4.1. Melatonin Attenuates MSC Replicative Senescence by Restoring Mitochondrial Function and Reducing ROS
Cultured MSCs undergo progressive replicative senescence accompanied by mitochondrial dynamic changes, excessive ROS generation, and decreased mitochondrial membrane potential [23]. Melatonin pretreatment rescues MSC from replicative senescence by restoring mitochondrial dynamics, reducing ROS generation and maintaining membrane potential through upregulating heat shock protein 1L (HSPA1L) [18]. HSPA1L, a member of HSP70 family [77], functions as a chaperone protein facilitating protein folding and stabilizing prion protein Prpc [23]. Prpc regulates mitochondrial integrity and function through binding to HSPA1L. Melatonin-induced HSPA1L upregulation results in recruitment of Prpc to mitochondria to maintain mitochondrial integrity. HSPA1L expression is suppressed in replicative senescent MSCs with a lower level of Prpc recruitment [23]. Melatonin pretreatment restores HSPA1L expression and Prpc recruitment, thereby alleviating senescent changes and improving MSC functions. Melatonin-treated MSCs confer more effective revascularization when transplanted to a hindlimb ischemic murine model [23].
4.2. Melatonin Controls HG- and Oxidant-Induced Cellular Senescence by Upregulating Antioxidant Enzymes
Pancreatic β-cells incubated in HG medium undergo premature senescence which is associated with reduced expression of antioxidant enzymes and increased ROS [78]. HG-treated cells exhibit impaired insulin secretion. Melatonin attenuates HG-induced senescence and improves insulin secretory activity by restoring expression of antioxidant enzymes and suppressing ROS generation [78]. Melatonin was reported to protect MSC from oxidant-stress-induced senescence by scavenging ROS generation [79]. Melatonin is also effective in antagonizing the action of a uremic toxin, p-cresol, on MSC senescence [80]. Melatonin blocks HG- and oxidant-induced senescence in diverse cell types by scavenging ROS generation through upregulation of MnSOD and catalase activities [78,80] in a manner analogous to 5-MTP.
5. Melatonin and 5-MTP Target p38 MAPK
p38 MAPK occupies a central position in transducing signals from environmental insults [81]. It mediates inflammation by activating pro-inflammatory activators [81]. Furthermore, it mediates stress-induced cellular senescence through interaction with ROS [82,83]. ROS alters p38 MAPK activities by oxidative modification of kinases in the p38 MAPK signaling cascade [84]. ROS may sustain p38 MAPK activation by inhibiting MAPK phosphatases (MKP) [85,86].
5-MTP was reported to be an arsenal against LPS- induced systemic inflammation by blocking p38 MAPK activation [60]. It protects vascular barrier function and endothelial integrity by inhibiting p38-mediated damage [61,87,88]. 5-MTP effectively prevents vascular intimal hyperplasia and vascular smooth muscle cell migration by blocking p38 MAPK activity [89]. Thus, p38 MAPK inactivation is a key mechanism by which 5-MTP exerts its biological actions. It is highly likely that 5-MTP attenuates HG-induced MSC senescence by suppressing p38 MAPK signaling pathway. Melatonin controls inflammation and tumorigenesis also by targeting p38 MAPK. Melatonin was reported to inhibit breast cancer cell invasion and control glial cell-mediated inflammation by blocking p38 MAPK activation [90,91]. Furthermore, it protects BM-MSC from oxidant-induced senescence and differentiation defects through inhibiting p38 MAPK [92].
5-MTP and melatonin protect stress-induced cell damage and senescence by targeting ROS and p38 MAPK-signaling pathway suggesting that they share a common mechanism. Melatonin is known to exert its actions by interacting with membrane receptors. It is unknown whether 5-MTP acts via a specific receptor. There is suggestive evidence that macrophage plasma membrane expresses 5-MTP receptors [60]. It remains unclear whether 5-MTP receptors are related to melatonin receptors and whether the receptor-mediated signaling pathway cross-talks with the p38 MAPK signaling cascade.
6. Melatonin and 5-MTP Control Cellular Senescence through Inhibition of NF-κB
Cellular senescence is accompanied by transcriptome rearrangement and altered gene expressions [11,12]. Several transactivators are activated to promote SASP and senescence. Genome-wide search and proteomic analysis identify NF-κB as a master regulator of cellular senescence and SASP [93,94]. ROS and redox transitions are the key drivers of NF-κB activation [95,96,97]. H2O2 activates IκB kinase (IKK) via which it phosphorylates and degrades IκB and the consequent liberation of p65/p50 NF-κB [95]. NF-κB enters nucleus where it is phosphorylated and binds to specific binding motifs of a large repertoire of pro-inflammatory and pro-senescent genes and promotes their expression. C/EBPβ was reported to regulate stress-induced senescence and SASP via ROS and p38 MAPK [98]. NF-κB and C/EBPβ act in concert to promote senescence, and mediate age-related chronic inflammation and tissue damages through SASP. Melatonin and 5-MTP block NF-κB and C/EBPβ activation and thereby attenuate stress-induced senescence and SASP-mediated chronic inflammation.
Melatonin and 5-MTP Modify Histone and NF-κB Acetylation
Gene transcription is regulated by epigenetic modification of histones and transactivators. Acetylation of histones by transcriptional co-activators such as p300 histone acetyltransferase (HAT) enhances promoter activities by altering chromatin structure [99,100]. Furthermore, p300 HAT acetylates myriade transactivators to strengthen their binding [99,100]. Histone and transactivator acetylation are controlled by histone deacetylases and a dynamic balance between p300 HAT and deacetylases maintains a normal state of gene expression. NF-κB binding and transcriptional activity are enhanced by p300 HAT [101,102]. HG increases expression of p300 [103]. 5-MTP inhibits p300 HAT activation and reduces NF-κB-mediated expression of cyclooxygenase-2 (COX-2) and pro-inflammatory cytokines [60,104]. 5-MTP probably controls stress-induced MSC senescence by blocking p300 HAT activation thereby reducing NF-κB acetylation and NF-κB mediated transcription of pro-senescent genes.
Melatonin protects cells and tissues from inflammatory damage by inhibiting NF-κB activation and NF-κB-mediated expression of COX-2 and cytokines [105,106,107]. Furthermore, its control of H2O2-induced cellular senescence is mediated by Sirt-1 dependent deacetylation of p65 submit of NF-κB, thereby reducing NF-κB activity [108]. Sirt-1 is a NAD-dependent deacetylase, which performs diverse biological functions including regulation of cellular senescence [109]. Sirt-1 is suppressed in senescent cells [110] and its supplement alters senescent phenotype and reduces secretion of pro-inflammatory cytokines through histone de-acetylation [111]. H2O2 at sublethal concentrations suppresses Sirt 1 expression accompanied by p38 MAPK activation. Melatonin restores Sirt 1 levels while inhibiting p38 MAPK suggesting that Sirt 1 is regulated via p38 MAPK and melatonin upregulates Sirt 1 by controlling p38 MAPK.
Transcription of pro-senescent and pro-inflammatory genes is regulated by a delicate balance between p300 HAT and Sirt 1 deacetylase. Oxidative and metabolic stresses tilt the balance to histone and NF-κB acetylation through p300 HAT activation and Sirt 1 downregulation (Figure 3). Melatonin restores the balance and maintains a normal level of histone and NF-κB acetylation. Given that 5-MTP inhibits p300 HAT, 5-MTP acts on histone and NF-κB acetylation in a manner similar to melatonin. This regulatory mechanism is of particular importance to SASP as it contributes significantly to age-related chronic inflammatory disorders.
7. Conclusions
Melatonin and 5-MTP are structurally related metabolites derived from L-tryptophan via the TPH pathway (Figure 2C). They control replicative and/or HG- and oxidant-induced MSC senescence by scavenging ROS. Replicative and oxidative stress-induced cellular senescence is characterized by an early event of mitochondrial structural changes and metabolic shift, resulting in excessive ROS generation. Melatonin and 5-MTP reduce ROS by upregulating the expression of mitochondrial MnSOD and catalase. Melatonin protects mitochondrial integrity and function by upregulating a p70 heat shock family protein, HSPA1L, which recruits Prion protein, Prpc, to maintain mitochondrial homeostasis and reduce ROS generation.
Proteomic analysis and genome-wide search identified NF-κB (p65/p50) as a master promoter of cellular senescence. Stress signals activate IKK, which phosphorylates IκB, resulting in IκB degradation and p65/p50 activation. NF-κB activity is post-translationally enhanced by phosphorylation and epigenetic modification. The p300 HAT acetylates NF-κB subunits and augments its binding activity, while Sirt 1 deacetylates NF-κB and reduce its activity. Pro-inflammatory mediators and HG increase p300 HAT activity, thereby enhancing NF-κB-mediated senescence. Stress-induced senescence is accompanied by downregulation of Sirt 1, enforcing NF-κB acetylation and activation. 5-MTP inhibits p300 HAT activity, thereby reducing NF-κB binding and transcriptional activity, while melatonin upregulates Sirt 1 and the consequent deacetylation of p65 subunit. Epigenetic modification of NF-κB and histone is a common transcriptional mechanism by which tryptophan metabolites combat cellular senescence.
Several triggering events of cellular senescence are mediated via p38 MAPK. The p38 MAPK mediates ROS-induced cellular senescence and NF-κB activation. Melatonin and 5-MTP target p38 MAPK for their anti-senescent and anti-inflammatory actions. It is unclear how these two metabolites block p38 MAPK activation. Melatonin exerts its actions by interaction with membrane receptors and receptor-mediated signaling transduction. It is likely that melatonin inhibits p38 MAPK and transcriptional mechanism through cross-talk between its receptor-mediated signaling and the p38 MAPK activation cascade. Preliminary data suggest that 5-MTP acts via interaction with a membrane receptor. However, the receptor has not been isolated and characterized and it’s signaling pathway remains to be investigated.
Melatonin-treated MSCs in culture improve revascularization when transplanted to a hindlimb ischemia mouse model through attenuation of cellular senescence. Melatonin and 5-MTP are potentially useful for improving MSC expansion and restoring MSC function in MSC-based cell therapy of diverse age-related disorders.
Funding
This research was funded by Excellent Team Research Program of Ministry of Science and Technology, Taiwan ((102-106)-2321-B-039-001; 107-2321-B-400-011; 108-2321-B-400-008).
Institutional Review Board Statement
Non-applicable.
Informed Consent Statement
Non-applicable.
Data Availability Statement
Non-applicable.
Conflicts of Interest
The author declares no conflict of interest.
Abbreviations
HG, High glucose; MSC, mesenchymal stromal/stem cell; ROS, reactive oxygen species; 5-MTP, 5-methoxytryptophan; SA, senescence-associated; SASP, senescence-associated secretory phenotype; β gal, β galactosidase;; T2D, Type 2 diabetes; MSCs, mesenchymal stromal cells; TCA, tricarboxylic acid; OXPHOS, oxidative phosphorylation; TPH, tryptophan hydroxylase; 5-HTP, 5-hydroxytryptophan; ECs, endothelial cells; SMCs, smooth muscle cells; ER, endoplasmic reticulum; LPS, lipopolysaccharide; IL-6, interleukin-6; OA, osteoarthritis; 5-HT, 5-hydroxytryptamine; AANAT, arylalkylamine N-acetyltransferase; HSPA1L, heat shock protein 1L; MKP, MAPK phosphatases; IKK, IκB kinase; HAT, histone acetyltransferase; COX-2, cyclooxygenase-2
References
- McHugh, D.; Gil, J. Senescence and aging: Causes, consequences, and therapeutic avenues. J. Cell Biol. 2018, 217, 65–77. [Google Scholar] [CrossRef]
- Van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [Green Version]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Rodier, F.; Campisi, J. Four faces of cellular senescence. J. Cell Biol. 2011, 192, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salama, R.; Sadaie, M.; Hoare, M.; Narita, M. Cellular senescence and its effector programs. Genes Dev. 2014, 28, 99–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behnia, F.; Taylor, B.D.; Woodson, M.; Kacerovsky, M.; Hawkins, H.; Fortunato, S.J.; Saade, G.R.; Menon, R. Chorioamniotic membrane senescence: A signal for parturition? Am. J. Obstet. Gynecol. 2015, 213, 359.e1–359.e16. [Google Scholar] [CrossRef] [PubMed]
- Storer, M.; Mas, A.; Robert-Moreno, A.; Pecoraro, M.; Ortells, M.C.; Di Giacomo, V.; Yosef, R.; Pilpel, N.; Krizhanovsky, V.; Sharpe, J.; et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 2013, 155, 1119–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alessio, N.; Aprile, D.; Squillaro, T.; Di Bernardo, G.; Finicelli, M.; Melone, M.A.; Peluso, G.; Galderisi, U. The senescence-associated secretory phenotype (SASP) from mesenchymal stromal cells impairs growth of immortalized prostate cells but has no effect on metastatic prostatic cancer cells. Aging 2019, 11, 5817–5828. [Google Scholar] [CrossRef]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Alfego, D.; Rodeck, U.; Kriete, A. Global mapping of transcription factor motifs in human aging. PLoS ONE 2018, 13, e0190457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanigan, F.; Geraghty, J.G.; Bracken, A.P. Transcriptional regulation of cellular senescence. Oncogene 2011, 30, 2901–2911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, A.K.; Tchkonia, T.; LeBrasseur, N.K.; Chini, E.N.; Xu, M.; Kirkland, J.L. Cellular Senescence in Type 2 Diabetes: A Therapeutic Opportunity. Diabetes 2015, 64, 2289–2298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horwitz, E.M.; Le Blanc, K.; Dominici, M.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Deans, R.J.; Krause, D.S.; Keating, A.; International Society for Cellular Therapy. Clarification of the nomenclature for MSC: The International Society for Cellular Therapy position statement. Cytotherapy 2005, 7, 393–395. [Google Scholar] [CrossRef] [PubMed]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Squillaro, T.; Peluso, G.; Galderisi, U. Clinical Trials with Mesenchymal Stem Cells: An Update. Cell Transpl. 2016, 25, 829–848. [Google Scholar] [CrossRef] [Green Version]
- Uccelli, A.; Moretta, L.; Pistoia, V. Mesenchymal stem cells in health and disease. Nat. Rev. Immunol. 2008, 8, 726–736. [Google Scholar] [CrossRef]
- English, K. Mechanisms of mesenchymal stromal cell immunomodulation. Immunol. Cell Biol. 2013, 91, 19–26. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef]
- Childs, B.G.; Gluscevic, M.; Baker, D.J.; Laberge, R.M.; Marquess, D.; Dananberg, J.; van Deursen, J.M. Senescent cells: An emerging target for diseases of ageing. Nat. Rev. Drug Discov. 2017, 16, 718–735. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Ding, Y.; Liu, Z.; Liang, X. Senescence in Mesenchymal Stem Cells: Functional Alterations, Molecular Mechanisms, and Rejuvenation Strategies. Front. Cell Dev. Biol. 2020, 8, 258. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.C.; Hsu, M.F.; Shih, C.Y.; Wu, K.K. 5-methoxytryptophan protects MSCs from stress induced premature senescence by upregulating FoxO3a and mTOR. Sci. Rep. 2017, 7, 11133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Yoon, Y.M.; Song, K.H.; Noh, H.; Lee, S.H. Melatonin suppresses senescence-derived mitochondrial dysfunction in mesenchymal stem cells via the HSPA1L-mitophagy pathway. Aging Cell 2020, 19, e13111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef]
- Brownlee, M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef] [Green Version]
- Kitada, K.; Nakano, D.; Ohsaki, H.; Hitomi, H.; Minamino, T.; Yatabe, J.; Felder, R.A.; Mori, H.; Masaki, T.; Kobori, H.; et al. Hyperglycemia causes cellular senescence via a SGLT2- and p21-dependent pathway in proximal tubules in the early stage of diabetic nephropathy. J. Diabetes Complicat. 2014, 28, 604–611. [Google Scholar] [CrossRef] [Green Version]
- Shosha, E.; Xu, Z.; Narayanan, S.P.; Lemtalsi, T.; Fouda, A.Y.; Rojas, M.; Xing, J.; Fulton, D.; Caldwell, R.W.; Caldwell, R.B. Mechanisms of Diabetes-Induced Endothelial Cell Senescence: Role of Arginase 1. Int. J. Mol. Sci. 2018, 19, 1215. [Google Scholar] [CrossRef] [Green Version]
- Blazer, S.; Khankin, E.; Segev, Y.; Ofir, R.; Yalon-Hacohen, M.; Kra-Oz, Z.; Gottfried, Y.; Larisch, S.; Skorecki, K.L. High glucose-induced replicative senescence: Point of no return and effect of telomerase. Biochem. Biophys. Res. Commun. 2002, 296, 93–101. [Google Scholar] [CrossRef]
- Zhong, W.; Zou, G.; Gu, J.; Zhang, J. L-arginine attenuates high glucose-accelerated senescence in human umbilical vein endothelial cells. Diabetes Res. Clin. Pract. 2010, 89, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Maeda, M.; Hayashi, T.; Mizuno, N.; Hattori, Y.; Kuzuya, M. Intermittent high glucose implements stress-induced senescence in human vascular endothelial cells: Role of superoxide production by NADPH oxidase. PLoS ONE 2015, 10, e0123169. [Google Scholar] [CrossRef]
- Palmer, A.K.; Gustafson, B.; Kirkland, J.L.; Smith, U. Cellular senescence: At the nexus between ageing and diabetes. Diabetologia 2019, 62, 1835–1841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, T.; Robotham, J.L.; Yoon, Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2006, 103, 2653–2658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abuarab, N.; Munsey, T.S.; Jiang, L.H.; Li, J.; Sivaprasadarao, A. High glucose-induced ROS activates TRPM2 to trigger lysosomal membrane permeabilization and Zn 2+-mediated mitochondrial fission. Sci. Signal. 2017, 10, eaal4161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoguchi, T.; Li, P.; Umeda, F.; Yu, H.Y.; Kakimoto, M.; Imamura, M.; Aoki, T.; Etoh, T.; Hashimoto, T.; Naruse, M.; et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C--dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 2000, 49, 1939–1945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balteau, M.; Tajeddine, N.; de Meester, C.; Ginion, A.; Des Rosiers, C.; Brady, N.R.; Sommereyns, C.; Horman, S.; Vanoverschelde, J.L.; Gailly, P.; et al. NADPH oxidase activation by hyperglycaemia in cardiomyocytes is independent of glucose metabolism but requires SGLT1. Cardiovasc. Res. 2011, 9, 237–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.M.; Bartholomew, J.C.; Campisi, J.; Acosta, M.; Reagan, J.D.; Ames, B.N. Molecular analysis of H2O2-induced senescent-like growth arrest in normal human fibroblasts: p53 and Rb control G1 arrest but not cell replication. Biochem. J. 1998, 332, 43–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.C.; Fenster, B.E.; Ito, H.; Takeda, K.; Bae, N.S.; Hirai, T.; Yu, Z.X.; Ferrans, V.J.; Howard, B.H.; Finkel, T. Ras proteins induce senescence by altering the intracellular levels of reactive oxygen species. J. Biol. Chem. 1999, 274, 7936–7940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macip, S.; Igarashi, M.; Berggren, P.; Yu, J.; Lee, S.W.; Aaronson, S.A. Influence of induced reactive oxygen species in p53-mediated cell fate decisions. Mol. Cell Biol. 2003, 23, 8576–8585. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Fischer, A.; Reagan, J.D.; Yan, L.J.; Ames, B.N. Oxidative DNA damage and senescence of human diploid fibroblast cells. Proc. Natl. Acad. Sci. USA 1995, 92, 4337–4341. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, D.V.; Wiley, C.D.; Velarde, M.C. Mitochondrial effectors of cellular senescence: Beyond the free radical theory of aging. Aging Cell 2015, 14, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Chomyn, A.; Chan, D.C. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J. Biol. Chem. 2005, 280, 26185–26192. [Google Scholar] [CrossRef] [Green Version]
- Yoon, Y.S.; Yoon, D.S.; Lim, I.K.; Yoon, S.H.; Chung, H.Y.; Rojo, M.; Malka, F.; Jou, M.J.; Martinou, J.C.; Yoon, G. Formation of elongated giant mitochondria in DFO-induced cellular senescence: Involvement of enhanced fusion process through modulation of Fis1. J. Cell Physiol. 2006, 209, 468–480. [Google Scholar] [CrossRef]
- Stöckl, P.; Hütter, E.; Zwerschke, W.; Jansen-Dürr, P. Sustained inhibition of oxidative phosphorylation impairs cell proliferation and induces premature senescence in human fibroblasts. Exp. Gerontol. 2006, 41, 674–682. [Google Scholar] [CrossRef]
- Borradaile, N.M.; Pickering, J.G. Nicotinamide phosphoribosyltransferase imparts human endothelial cells with extended replicative lifespan and enhanced angiogenic capacity in a high glucose environment. Aging Cell 2009, 8, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.; van der Veer, E.; Akawi, O.; Pickering, J.G. SIRT1 markedly extends replicative lifespan if the NAD+ salvage pathway is enhanced. FEBS Lett. 2009, 583, 3081–3085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.T.; Shih, Y.R.; Kuo, T.K.; Lee, O.K.; Wei, Y.H. Coordinated changes of mitochondrial biogenesis and antioxidant enzymes during osteogenic differentiation of human mesenchymal stem cells. Stem Cells 2008, 26, 960–968. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Suda, T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat. Rev. Mol. Cell Biol. 2014, 15, 243–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pattappa, G.; Heywood, H.K.; de Bruijn, J.D.; Lee, D.A. The metabolism of human mesenchymal stem cells during proliferation and differentiation. J. Cell Physiol. 2011, 226, 2562–2570. [Google Scholar] [CrossRef]
- Stolzing, A.; Coleman, N.; Scutt, A. Glucose-induced replicative senescence in mesenchymal stem cells. Rejuvenation Res. 2006, 9, 31–35. [Google Scholar] [CrossRef]
- Zhang, D.; Lu, H.; Chen, Z.; Wang, Y.; Lin, J.; Xu, S.; Zhang, C.; Wang, B.; Yuan, Z.; Feng, X.; et al. High glucose induces the aging of mesenchymal stem cells via Akt/mTOR signaling. Mol. Med. Rep. 2017, 16, 1685–1690. [Google Scholar] [CrossRef] [Green Version]
- Ferraro, F.; Lymperi, S.; Méndez-Ferrer, S.; Saez, B.; Spencer, J.A.; Yeap, B.Y.; Masselli, E.; Graiani, G.; Prezioso, L.; Rizzini, E.L.; et al. Diabetes impairs hematopoietic stem cell mobilization by altering niche function. Sci. Transl. Med. 2011, 3, 104ra101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, W.G.; Saunders, M.; Gilroy, D.; He, X.Z.; Yeh, H.; Zhu, Y.; Shtivelband, M.I.; Ruan, K.H.; Wu, K.K. Purification and characterization of a cyclooxygenase-2 and angiogenesis suppressing factor produced by human fibroblasts. FASEB J. 2002, 16, 1286–1288. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.H.; Kuo, C.C.; Yan, J.L.; Chen, H.L.; Lin, W.C.; Wang, K.H.; Tsai, K.K.; Guvén, H.; Flaberg, E.; Szekely, L.; et al. Control of cyclooxygenase-2 expression and tumorigenesis by endogenous 5-methoxytryptophan. Proc. Nat. Acad. Sci. USA 2012, 109, 13231–13236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovenberg, W.; Jequier, E.; Sjoerdsma, A. Tryptophan hydroxylation: Measurement in pineal gland, brainstem, and carcinoid tumor. Science 1967, 155, 217–219. [Google Scholar] [CrossRef] [PubMed]
- Walther, D.J.; Peter, J.U.; Bashammakh, S.; Hörtnagl, H.; Voits, M.; Fink, H.; Bader, M. Synthesis of serotonin by a second tryptophan hydroxylase isoform. Science 2003, 299, 76. [Google Scholar] [CrossRef]
- Rodriguez, I.R.; Mazuruk, K.; Schoen, T.J.; Chader, G.J. Structural analysis of the human hydroxyindole-O-methyltransferase gene. Presence of two distinct promoters. J. Biol. Chem. 1994, 269, 31969–31977. [Google Scholar] [CrossRef] [PubMed]
- Donohue, S.J.; Roseboom, P.H.; Illnerova, H.; Weller, J.L.; Klein, D.C. Human hydroxyindole-O-methyltransferase: Presence of LINE-1 fragment in a cDNA clone and pineal mRNA. DNA Cell Biol. 1993, 12, 715–727. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.L.; Yuan, C.Y.; Cheng, H.H.; Chang, T.C.; Huang, S.K.; Kuo, C.C.; Wu, K.K. Restoration of hydroxyindole O-methyltransferase levels in human cancer cells induces a tryptophan-metabolic switch and attenuates cancer progression. J. Biol. Chem. 2018, 293, 11131–11142. [Google Scholar] [CrossRef] [Green Version]
- Botros, H.G.; Legrand, P.; Pagan, C.; Bondet, V.; Weber, P.; Ben-Abdallah, M.; Lemière, N.; Huguet, G.; Bellalou, J.; Maronde, E.; et al. Crystal structure and functional mapping of human ASMT, the last enzyme of the melatonin synthesis pathway. J. Pineal Res. 2013, 54, 46–57. [Google Scholar] [CrossRef]
- Wang, Y.F.; Hsu, Y.J.; Wu, H.F.; Lee, G.L.; Yang, Y.S.; Wu, J.Y.; Yet, S.F.; Wu, K.K.; Kuo, C.C. Endothelium-derived 5-methoxytryptophan is a circulating anti-inflammatory molecule that blocks systemic inflammation. Circ. Res. 2016, 119, 222–236. [Google Scholar] [CrossRef] [Green Version]
- Chu, L.Y.; Wang, Y.F.; Cheng, H.H.; Kuo, C.C.; Wu, K.K. Endothelium-derived 5-methoxytryptophan protects endothelial barrier function by blocking p38 MAPK activation. PLoS ONE 2016, 11, e0152166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, T.C.; Hsu, M.F.; Wu, K.K. High glucose induces bone marrow-derived mesenchymal stem cell senescence by upregulating autophagy. PLoS ONE 2015, 10, e0126537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxid. Med. Cell Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivitz, W.I.; Yorek, M.A. Mitochondrial dysfunction in diabetes: From molecular mechanisms to functional significance and therapeutic opportunities. Antioxid. Redox Signal. 2010, 12, 537–577. [Google Scholar] [CrossRef] [Green Version]
- Patel, H.; Chen, J.; Das, K.C.; Kavdia, M. Hyperglycemia induces differential change in oxidative stress at gene expression and functional levels in HUVEC and HMVEC. Cardiovasc. Diabetol. 2013, 12, 142. [Google Scholar] [CrossRef] [Green Version]
- Eijkelenboom, A.; Burgering, B.M. FOXOs: Signaling integrators for homeostasis maintenance. Nat. Rev. Mol. Cell Biol. 2013, 14, 83–97. [Google Scholar] [CrossRef]
- Marinkovic, D.; Zhang, X.; Yalcin, S.; Luciano, J.P.; Brugnara, C.; Huber, T.; Ghaffari, S. Foxo3 is required for the regulation of oxidative stress in erythropoiesis. J. Clin. Investig. 2007, 117, 2133–2144. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Marsboom, G.; Toth, P.T.; Rehman, J. Mitochondrial respiration regulates adipogenic differentiation of human mesenchymal stem cells. PLoS ONE 2013, 8, e77077. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Gao, Z.; Chen, Y.; Guan, M.X. The role of mitochondria in osteogenic, adipogenic and chondrogenic differentiation of mesenchymal stem cells. Protein Cell 2017, 8, 439–445. [Google Scholar] [CrossRef] [Green Version]
- Martin, J.A.; Buckwalter, J.A. Aging, articular cartilage chondrocyte senescence and osteoarthritis. Biogerontology 2002, 3, 257–264. [Google Scholar] [CrossRef]
- Luchetti, F.; Canonico, B.; Bartolini, D.; Arcangeletti, M.; Ciffolilli, S.; Murdolo, G.; Piroddi, M.; Papa, S.; Reiter, R.J.; Galli, F. Melatonin regulates mesenchymal stem cell differentiation: A review. J. Pineal Res. 2014, 56, 382–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardeland, R.; Pandi-Perumal, S.R.; Cardinali, D.P. Melatonin. Int. J. Biochem. Cell Biol. 2006, 38, 313–316. [Google Scholar] [CrossRef] [PubMed]
- García, J.J.; López-Pingarrón, L.; Almeida-Souza, P.; Tres, A.; Escudero, P.; García-Gil, F.A.; Tan, D.X.; Reiter, R.J.; Ramírez, J.M.; Bernal-Pérez, M. Protective effects of melatonin in reducing oxidative stress and in preserving the fluidity of biological membranes: A review. J. Pineal Res. 2014, 56, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Mayo, J.C.; Tan, D.X.; Sainz, R.M.; Alatorre-Jimenez, M.; Qin, L. Melatonin as an antioxidant: Under promises but over delivers. J. Pineal Res. 2016, 61, 253–278. [Google Scholar] [CrossRef]
- Blask, D.E.; Sauer, L.A.; Dauchy, R.T.; Holowachuk, E.W.; Ruhoff, M.S.; Kopff, H.S. Melatonin inhibition of cancer growth in vivo involves suppression of tumor fatty acid metabolism via melatonin receptor-mediated signal transduction events. Cancer Res. 1999, 59, 4693–4701. [Google Scholar]
- Reiter, R.J.; Rosales-Corral, S.A.; Tan, D.X.; Acuna-Castroviejo, D.; Qin, L.; Yang, S.F.; Xu, K. Melatonin, a Full Service Anti-Cancer Agent: Inhibition of Initiation, Progression and Metastasis. Int. J. Mol. Sci. 2017, 18, 843. [Google Scholar] [CrossRef]
- Mayer, M.P.; Bukau, B. Hsp70 chaperones: Cellular functions and molecular mechanism. Cell Mol. Life Sci. 2005, 62, 670–684. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.H.; Jung, H.S.; Kwon, M.J.; Jang, J.E.; Kim, T.N.; Lee, S.H.; Kim, M.K.; Park, J.H. Melatonin protects INS-1 pancreatic β-cells from apoptosis and senescence induced by glucotoxicity and glucolipotoxicity. Islets 2020, 12, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Yang, L.; Li, Y.; Yan, G.; Feng, C.; Liu, T.; Gong, R.; Yuan, Y.; Wang, N.; Idiiatullina, E.; et al. Melatonin protects bone marrow mesenchymal stem cells against iron overload-induced aberrant differentiation and senescence. J. Pineal Res. 2017, 63. [Google Scholar] [CrossRef]
- Yun, S.P.; Han, Y.S.; Lee, J.H.; Kim, S.M.; Lee, S.H. Melatonin Rescues Mesenchymal Stem Cells from Senescence Induced by the Uremic Toxin p-Cresol via Inhibiting mTOR-Dependent Autophagy. Biomol. Ther. 2018, 26, 389–398. [Google Scholar] [CrossRef]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signaling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borodkina, A.; Shatrova, A.; Abushik, P.; Nikolsky, N.; Burova, E. Interaction between ROS dependent DNA damage, mitochondria and p38 MAPK underlies senescence of human adult stem cells. Aging 2014, 6, 481–495. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Li, N.; Xiang, R.; Sun, P. Emerging roles of the p38 MAPK and PI3K/AKT/mTOR pathways in oncogene-induced senescence. Trends Biochem. Sci. 2014, 39, 268–276. [Google Scholar] [CrossRef] [Green Version]
- Son, Y.; Cheong, Y.K.; Kim, N.H.; Chung, H.T.; Kang, D.G.; Pae, H.O. Mitogen-Activated Protein Kinases and Reactive Oxygen Species: How Can ROS Activate MAPK Pathways? J. Signal Transduct. 2011, 2011, 792639. [Google Scholar] [CrossRef] [PubMed]
- Hou, N.; Torii, S.; Saito, N.; Hosaka, M.; Takeuchi, T. Reactive oxygen species-mediated pancreatic beta-cell death is regulated by interactions between stress-activated protein kinases, p38 and c-Jun N-terminal kinase, and mitogen-activated protein kinase phosphatases. Endocrinology 2008, 149, 1654–1665. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.Y.; Liu, Y.; Wu, G.S. The role of mitogen-activated protein kinase phosphatase-1 in oxidative damage-induced cell death. Cancer Res. 2006, 66, 4888–4894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.H.; Ho, Y.C.; Ho, H.H.; Liang, L.Y.; Jiang, W.C.; Lee, G.L.; Lee, J.K.; Hsu, Y.J.; Kuo, C.C.; Wu, K.K.; et al. Tryptophan metabolite 5-methoxytryptophan ameliorates arterial denudation-induced intimal hyperplasia via opposing effects on vascular endothelial and smooth muscle cells. Aging 2019, 11, 8604–8622. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.K.; Kuo, C.C.; Yet, S.F.; Lee, C.M.; Liou, J.Y. 5-methoxytryptophan: An arsenal against vascular injury and inflammation. J. Biomed. Sci. 2020, 27, 79. [Google Scholar] [CrossRef]
- Ho, Y.C.; Wu, M.L.; Su, C.H.; Chen, C.H.; Ho, H.H.; Lee, G.L.; Lin, W.S.; Lin, W.Y.; Hsu, Y.J.; Kuo, C.C.; et al. A Novel Protective Function of 5-Methoxytryptophan in Vascular Injury. Sci. Rep. 2016, 6, 25374. [Google Scholar] [CrossRef]
- Mao, L.; Yuan, L.; Slakey, L.M.; Jones, F.E.; Burow, M.E.; Hill, S.M. Inhibition of breast cancer cell invasion by melatonin is mediated through regulation of the p38 mitogen-activated protein kinase signaling pathway. Breast Cancer Res. 2010, 12, R107. [Google Scholar] [CrossRef] [Green Version]
- Vilar, A.; de Lemos, L.; Patraca, I.; Martínez, N.; Folch, J.; Junyent, F.; Verdaguer, E.; Pallàs, M.; Auladell, C.; Camins, A. Melatonin suppresses nitric oxide production in glial cultures by pro-inflammatory cytokines through p38 MAPK inhibition. Free Radic. Res. 2014, 48, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Chen, X.; Liu, T.; Gong, Y.; Chen, S.; Pan, G.; Cui, W.; Luo, Z.P.; Pei, M.; Yang, H.; et al. Melatonin reverses H2O2-induced premature senescence in mesenchymal stem cells via the SIRT1-dependent pathway. J. Pineal Res. 2015, 59, 190–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, Y.; Scuoppo, C.; Wang, X.; Fang, X.; Balgley, B.; Bolden, J.E.; Premsrirut, P.; Luo, W.; Chicas, A.; Lee, C.S.; et al. Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes Dev. 2011, 25, 2125–2136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernard, D.; Gosselin, K.; Monte, D.; Vercamer, C.; Bouali, F.; Pourtier, A.; Vandenbunder, B.; Abbadie, C. Involvement of Rel/nuclear factor-kappaB transcription factors in keratinocyte senescence. Cancer Res. 2004, 64, 472–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takada, Y.; Mukhopadhyay, A.; Kundu, G.C.; Mahabeleshwar, G.H.; Singh, S.; Aggarwal, B.B. Hydrogen peroxide activates NF-kappa B through tyrosine phosphorylation of I kappa B alpha and serine phosphorylation of p65: Evidence for the involvement of I kappa B alpha kinase and Syk protein-tyrosine kinase. J. Biol. Chem. 2003, 278, 24233–24241. [Google Scholar] [CrossRef] [Green Version]
- Reynaert, N.L.; van der Vliet, A.; Guala, A.S.; McGovern, T.; Hristova, M.; Pantano, C.; Heintz, N.H.; Heim, J.; Ho, Y.S.; Matthews, D.E.; et al. Dynamic redox control of NF-kappaB through glutaredoxin-regulated S-glutathionylation of inhibitory kappaB kinase beta. Proc. Natl. Acad. Sci. USA 2006, 103, 13086–13091. [Google Scholar] [CrossRef] [Green Version]
- Schoonbroodt, S.; Ferreira, V.; Best-Belpomme, M.; Boelaert, J.R.; Legrand-Poels, S.; Korner, M.; Piette, J. Crucial role of the amino-terminal tyrosine residue 42 and the carboxyl-terminal PEST domain of I kappa B alpha in NF-kappa B activation by an oxidative stress. J. Immunol. 2000, 164, 4292–4300. [Google Scholar] [CrossRef]
- Salotti, J.; Johnson, P.F. Regulation of senescence and the SASP by the transcription factor C/EBPβ. Exp. Gerontol. 2019, 128, 110752. [Google Scholar] [CrossRef]
- Goodman, R.H.; Smolik, S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000, 14, 1553–1577, Review. [Google Scholar]
- Grunstein, M. Histone acetylation in chromatin structure and transcription. Nature 1997, 389, 349–352. [Google Scholar] [CrossRef]
- Deng, W.G.; Zhu, Y.; Wu, K.K. Up-regulation of p300 binding and p50 acetylation in tumor necrosis factor-alpha-induced cyclooxygenase-2 promoter activation. J. Biol. Chem. 2003, 278, 4770–4777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, W.G.; Zhu, Y.; Wu, K.K. Role of p300 and PCAF in regulating cyclooxygenase-2 promoter activation by inflammatory mediators. Blood 2004, 103, 2135–2142. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Feng, B.; George, B.; Chakrabarti, R.; Chen, M.; Chakrabarti, S. Transcriptional coactivator p300 regulates glucose-induced gene expression in endothelial cells. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E127–E137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, H.H.; Wang, K.H.; Chu, L.Y.; Chang, T.C.; Kuo, C.C.; Wu, K.K. Quiescent and proliferative fibroblasts exhibit differential p300 HAT activation through control of 5-methoxytryptophan production. PLoS ONE 2014, 9, e88507. [Google Scholar] [CrossRef] [Green Version]
- Favero, G.; Franceschetti, L.; Bonomini, F.; Rodella, L.F.; Rezzani, R. Melatonin as an Anti-Inflammatory Agent Modulating Inflammasome Activation. Int. J. Endocrinol. 2017, 2017, 1835195. [Google Scholar] [CrossRef] [Green Version]
- Deng, W.G.; Tang, S.T.; Tseng, H.P.; Wu, K.K. Melatonin suppresses macrophage cyclooxygenase-2 and inducible nitric oxide synthase expression by inhibiting p52 acetylation and binding. Blood 2006, 108, 518–524. [Google Scholar] [CrossRef]
- Wang, J.; Xiao, X.; Zhang, Y.; Shi, D.; Chen, W.; Fu, L.; Liu, L.; Xie, F.; Kang, T.; Huang, W.; et al. Simultaneous modulation of COX-2, p300, Akt, and Apaf-1 signaling by melatonin to inhibit proliferation and induce apoptosis in breast cancer cells. J. Pineal Res. 2012, 53, 77–90. [Google Scholar] [CrossRef]
- Nopparat, C.; Sinjanakhom, P.; Govitrapong, P. Melatonin reverses H2O2-induced senescence in SH-SY5Y cells by enhancing autophagy via sirtuin 1 deacetylation of the RelA/p65 subunit of NF-κB. J. Pineal Res. 2017, 63. [Google Scholar] [CrossRef]
- Rahman, S.; Islam, R. Mammalian Sirt1: Insights on its biological functions. Cell Commun. Signal 2011, 9, 11. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Wang, L.; Fozouni, P.; Evjen, G.; Chandra, V.; Jiang, J.; Lu, C.; Nicastri, M.; Bretz, C.; Winkler, J.D.; et al. SIRT1 is downregulated by autophagy in senescence and ageing. Nat. Cell Biol. 2020, 22, 1170–1179. [Google Scholar] [CrossRef]
- Hayakawa, T.; Iwai, M.; Aoki, S.; Takimoto, K.; Maruyama, M.; Maruyama, W.; Motoyama, N. SIRT1 suppresses the senescence-associated secretory phenotype through epigenetic gene regulation. PLoS ONE 2015, 10, e0116480. [Google Scholar] [CrossRef] [PubMed]
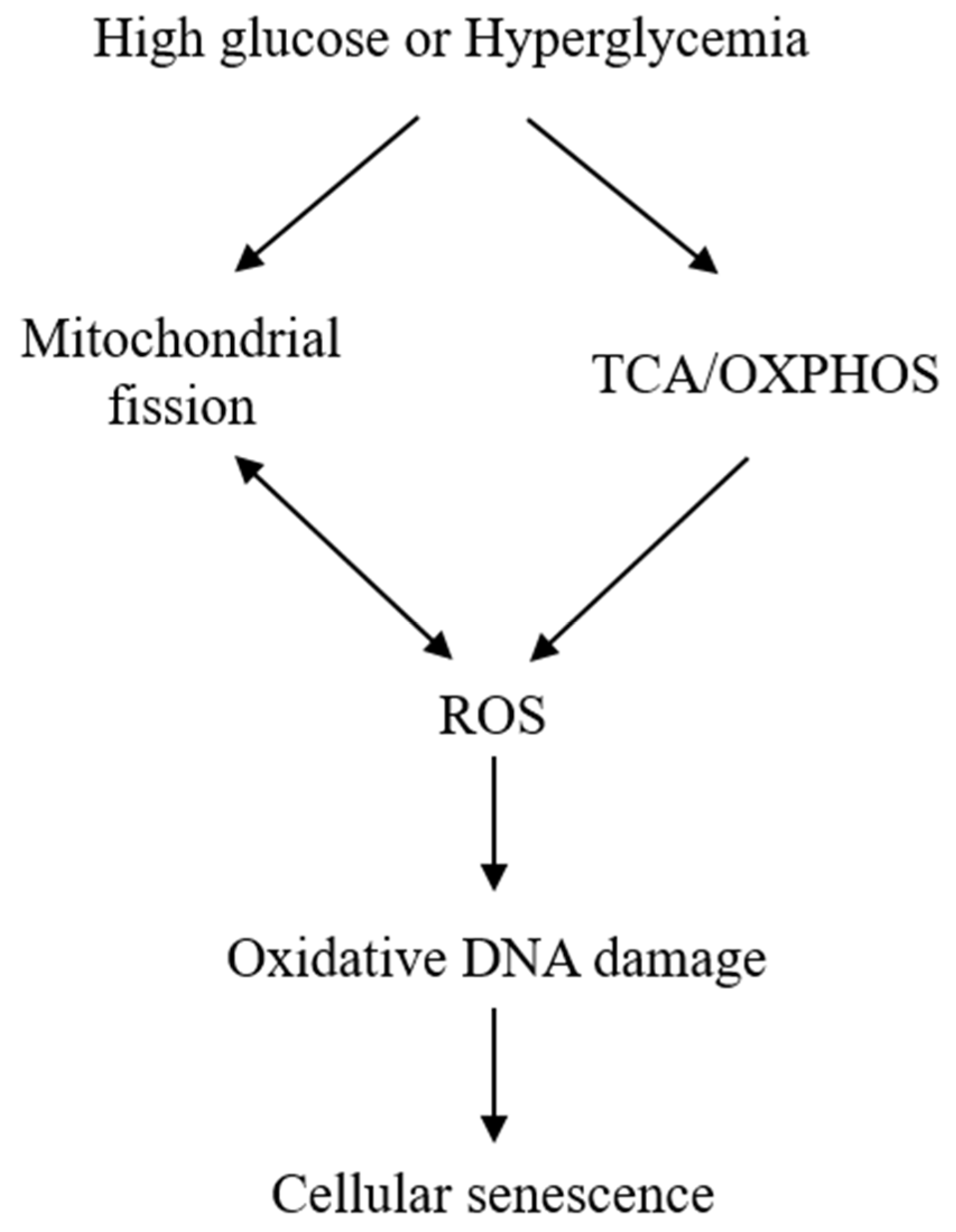
Figure 1.
Simplified scheme illustrating the mechanism by which high glucose (HG) induces cellular senescence. HG enhances tricarboxylic acid (TCA) cycle and oxidative phosphorylation (OXPHOS), which results in reactive oxygen species (ROS) generation. HG induces mitochondrial fission, which is accompanied by ROS accumulation. ROS, in turn, induces mitochondrial fission as indicated by a two-head arrow. Sublethal ROS (H2O2) induces oxidative DNA damage and senescent phenotypic changes.
Figure 1.
Simplified scheme illustrating the mechanism by which high glucose (HG) induces cellular senescence. HG enhances tricarboxylic acid (TCA) cycle and oxidative phosphorylation (OXPHOS), which results in reactive oxygen species (ROS) generation. HG induces mitochondrial fission, which is accompanied by ROS accumulation. ROS, in turn, induces mitochondrial fission as indicated by a two-head arrow. Sublethal ROS (H2O2) induces oxidative DNA damage and senescent phenotypic changes.

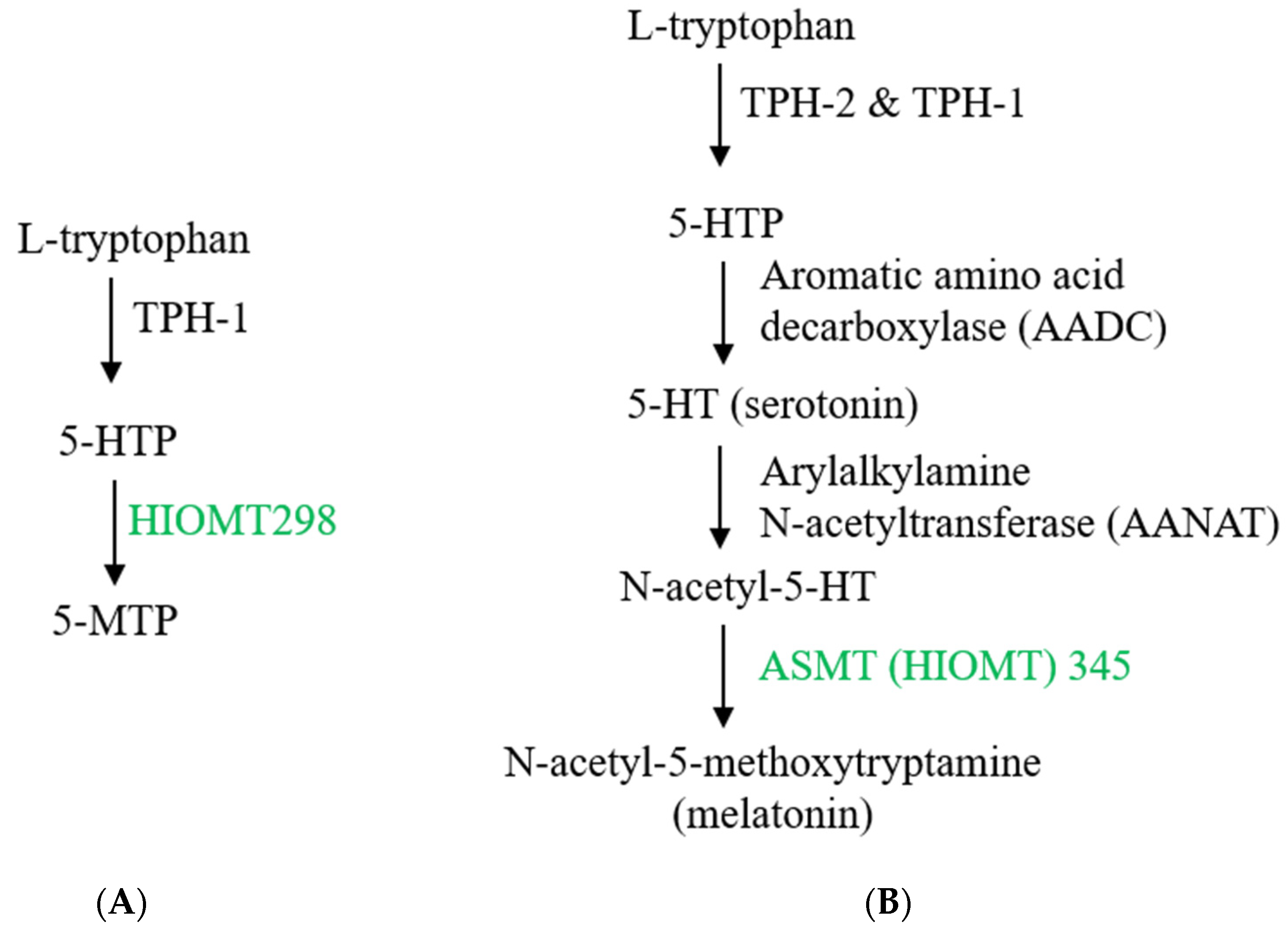
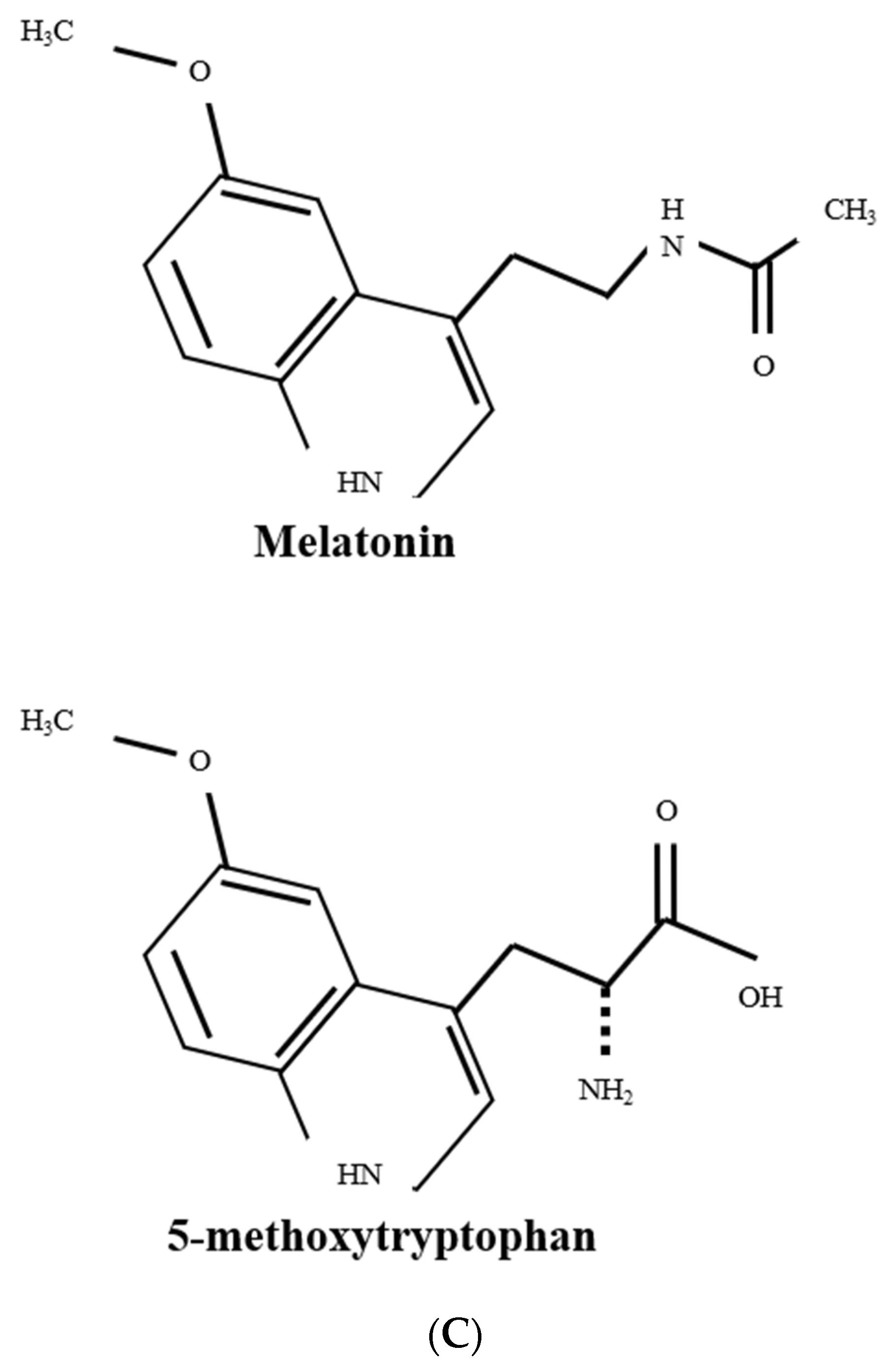
Figure 2.
Biosynthesis of 5-methoxytryptophan (5-MTP) and melatonin. (A) 5-MTP is produced in human fibroblasts, bone marrow mesenchymal stromal cells (BM-MSCs), vascular endothelial cells (ECs) and SMCs, bronchial and renal epithelial cells. Its synthesis is catalyzed by TPH-1 (tryptophan hydroxylase-1) followed by hydroxyindole O-methyltransferase (HIOMT, also known as N-acetylserotonin O-methyltransferase, ASMT). HIOMT298 denotes HIOMT isoform coding for 298 aa HIOMT. (B) For comparison, melatonin biosynthesis in pineal cells is shown, highlighting HIOMT345 (ASMT 345) as the functional isoform. (C) Structural comparison between 5-MTP and melatonin.
Figure 2.
Biosynthesis of 5-methoxytryptophan (5-MTP) and melatonin. (A) 5-MTP is produced in human fibroblasts, bone marrow mesenchymal stromal cells (BM-MSCs), vascular endothelial cells (ECs) and SMCs, bronchial and renal epithelial cells. Its synthesis is catalyzed by TPH-1 (tryptophan hydroxylase-1) followed by hydroxyindole O-methyltransferase (HIOMT, also known as N-acetylserotonin O-methyltransferase, ASMT). HIOMT298 denotes HIOMT isoform coding for 298 aa HIOMT. (B) For comparison, melatonin biosynthesis in pineal cells is shown, highlighting HIOMT345 (ASMT 345) as the functional isoform. (C) Structural comparison between 5-MTP and melatonin.


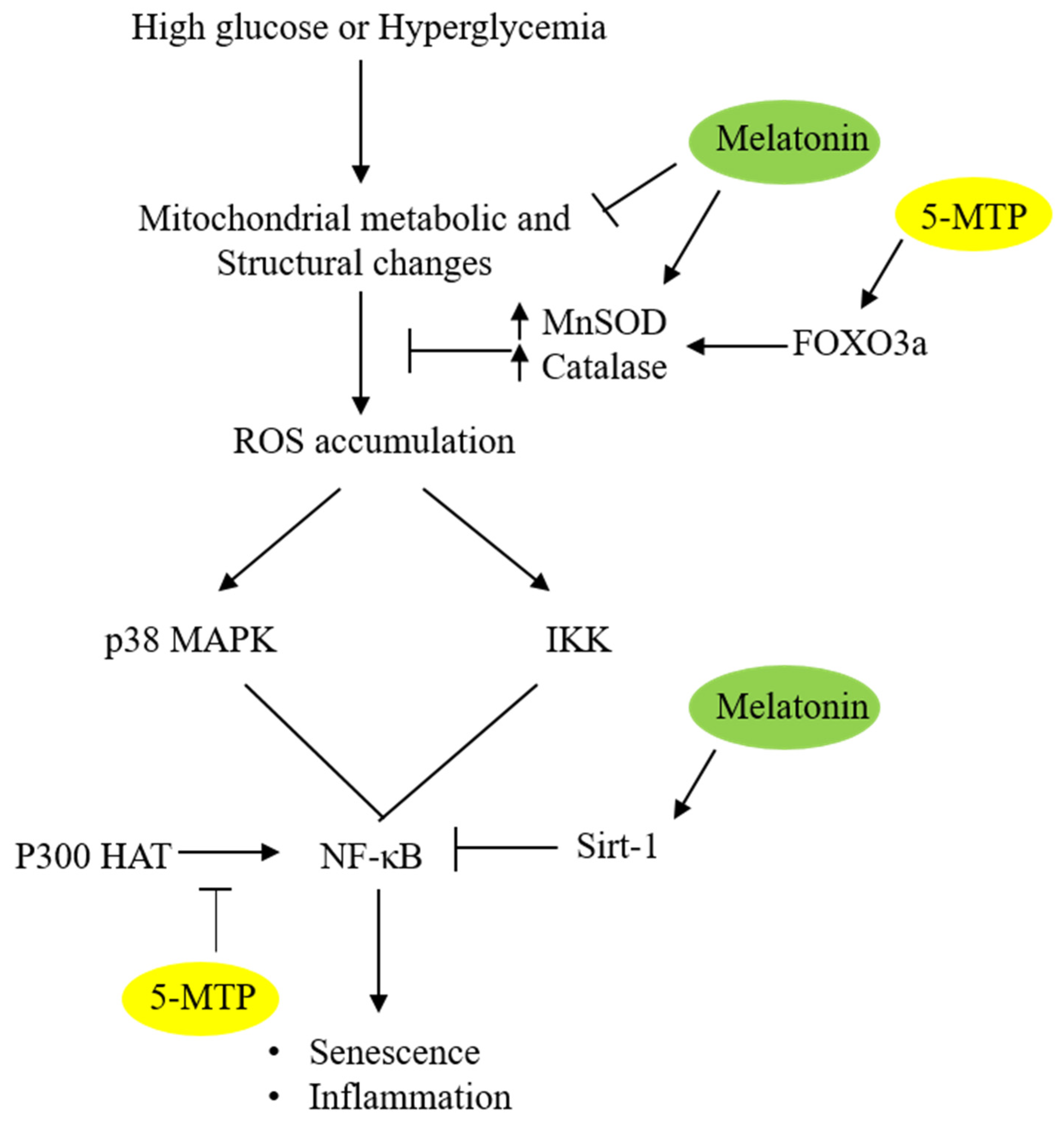
Figure 3.
Schematic illustration of the anti-senescent actions of 5-MTP and melatonin. 5-MTP upregulates MnSOD and catalase activities via FOXO3a, thereby reducing ROS accumulation. Melatonin reduces ROS by upregulating MnSOD and catalase possibly via maintaining mitochondrial integrity. 5-MTP inhibits p300 HAT thereby reducing NF-κB activity while melatonin upregulates Sirt 1 which de-acetylates p65 and suppresses NF-κB activity.
Figure 3.
Schematic illustration of the anti-senescent actions of 5-MTP and melatonin. 5-MTP upregulates MnSOD and catalase activities via FOXO3a, thereby reducing ROS accumulation. Melatonin reduces ROS by upregulating MnSOD and catalase possibly via maintaining mitochondrial integrity. 5-MTP inhibits p300 HAT thereby reducing NF-κB activity while melatonin upregulates Sirt 1 which de-acetylates p65 and suppresses NF-κB activity.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wu, K.K. Control of Mesenchymal Stromal Cell Senescence by Tryptophan Metabolites. Int. J. Mol. Sci. 2021, 22, 697. https://doi.org/10.3390/ijms22020697
AMA Style
Wu KK. Control of Mesenchymal Stromal Cell Senescence by Tryptophan Metabolites. International Journal of Molecular Sciences. 2021; 22(2):697. https://doi.org/10.3390/ijms22020697
Chicago/Turabian StyleWu, Kenneth K. 2021. "Control of Mesenchymal Stromal Cell Senescence by Tryptophan Metabolites" International Journal of Molecular Sciences 22, no. 2: 697. https://doi.org/10.3390/ijms22020697
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.