Insight into Cisplatin-Resistance Signaling of W1 Ovarian Cancer Cells Emerges mTOR and HSP27 as Targets for Sensitization Strategies

 and
and 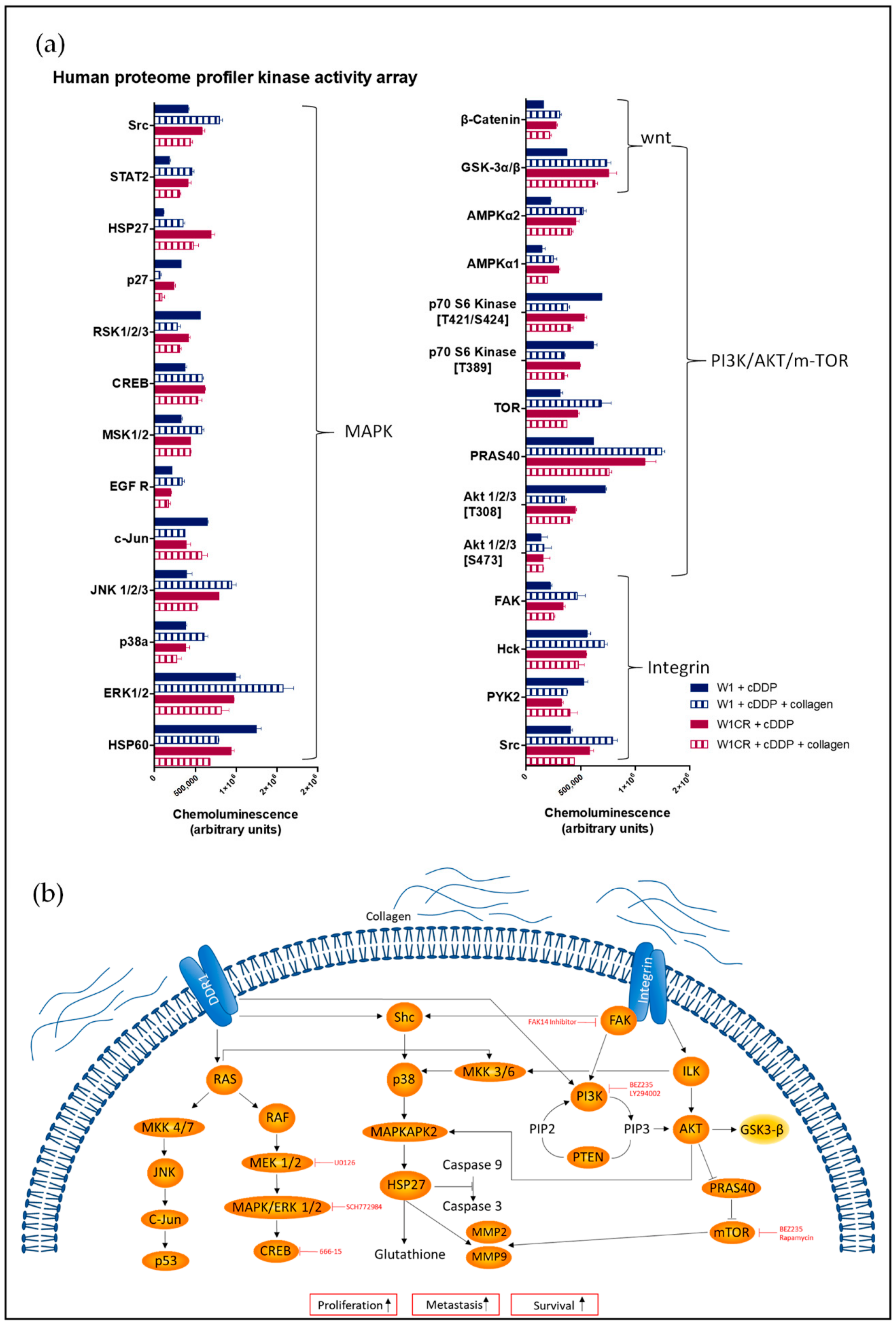{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Impact of COL1 Binding on Cellular Signaling Pathways in Cisplatin-Treated W1 and W1CR Ovarian Cancer Cells
2.2. The Role of the FAK/PI3K/AKT Pathway for COL1-Induced Cisplatin Resistance Formation in W1 and W1CR Cells
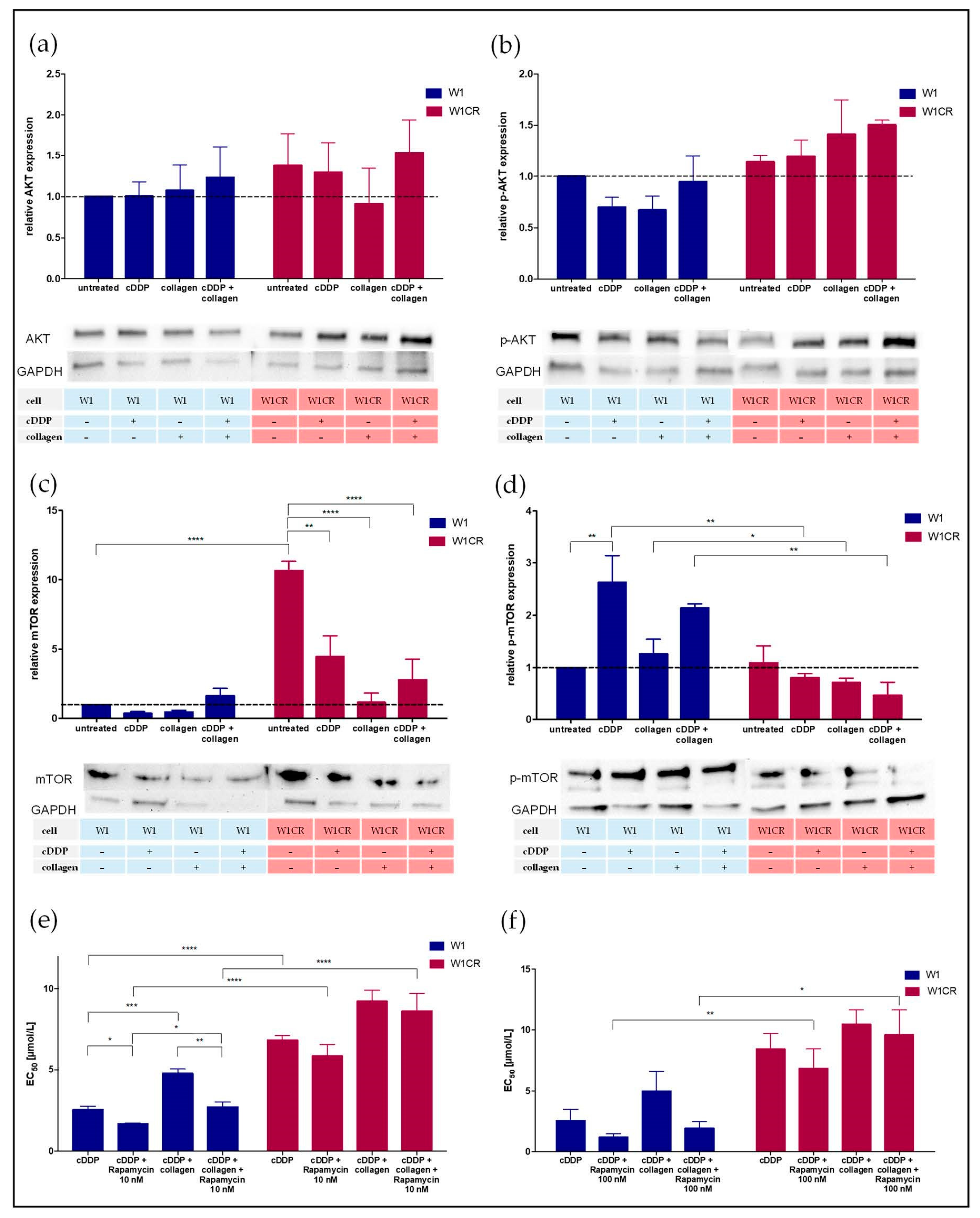
2.3. Insight into AKT/mTOR Axis Emerges mTOR as a Key Target for W1 Cell Sensitization
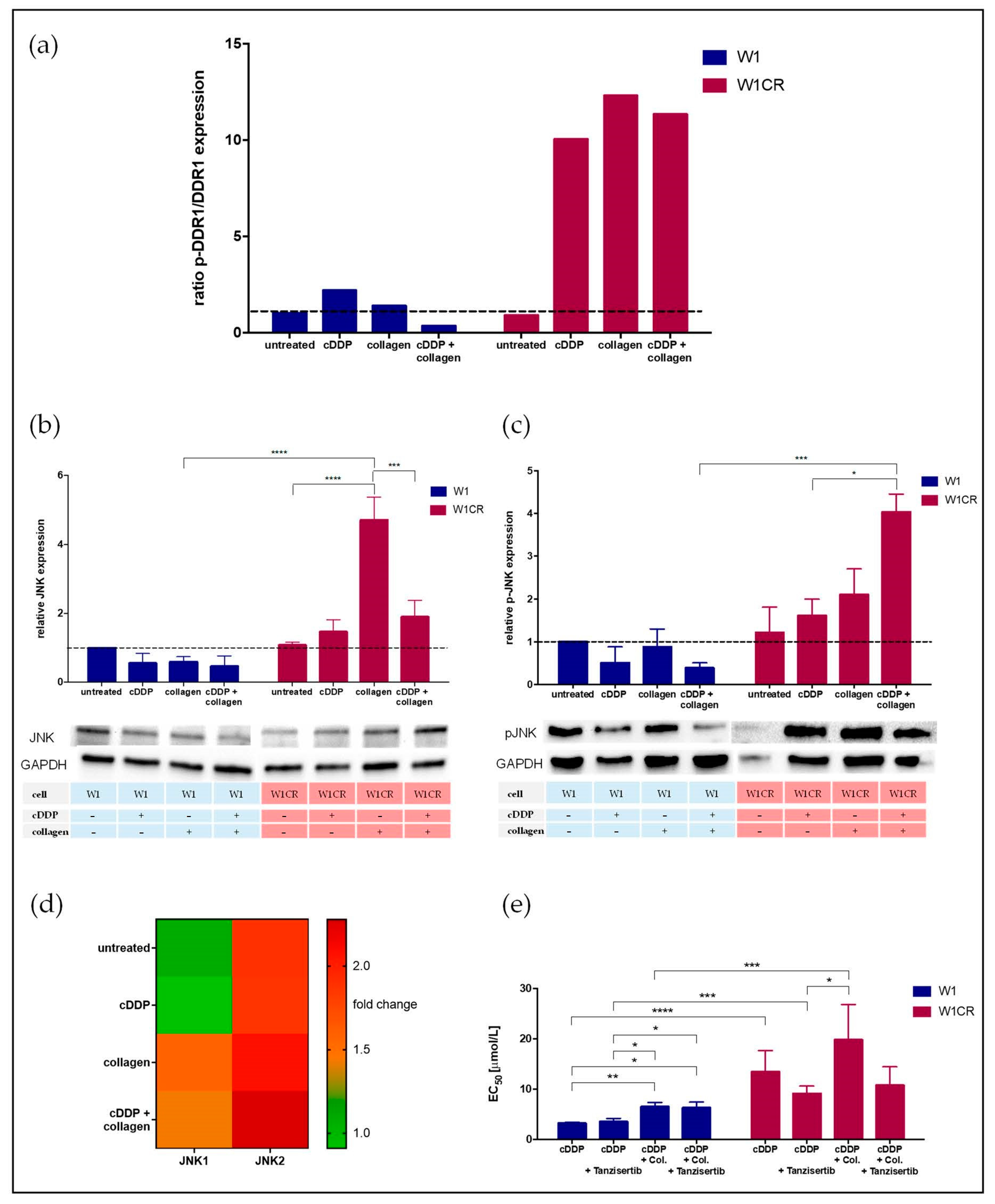
2.4. DDR1 Is a COL1 Sensor in W1CR Cells
2.5. JNK Appears as Crucial Downstream Component of COL1/DDR1 Resistance Axis in W1CR Cells
2.6. MAPK Pathway Did Not Provide Further Targets for Sensitizing Intrinsic Resistance in W1CR Cells
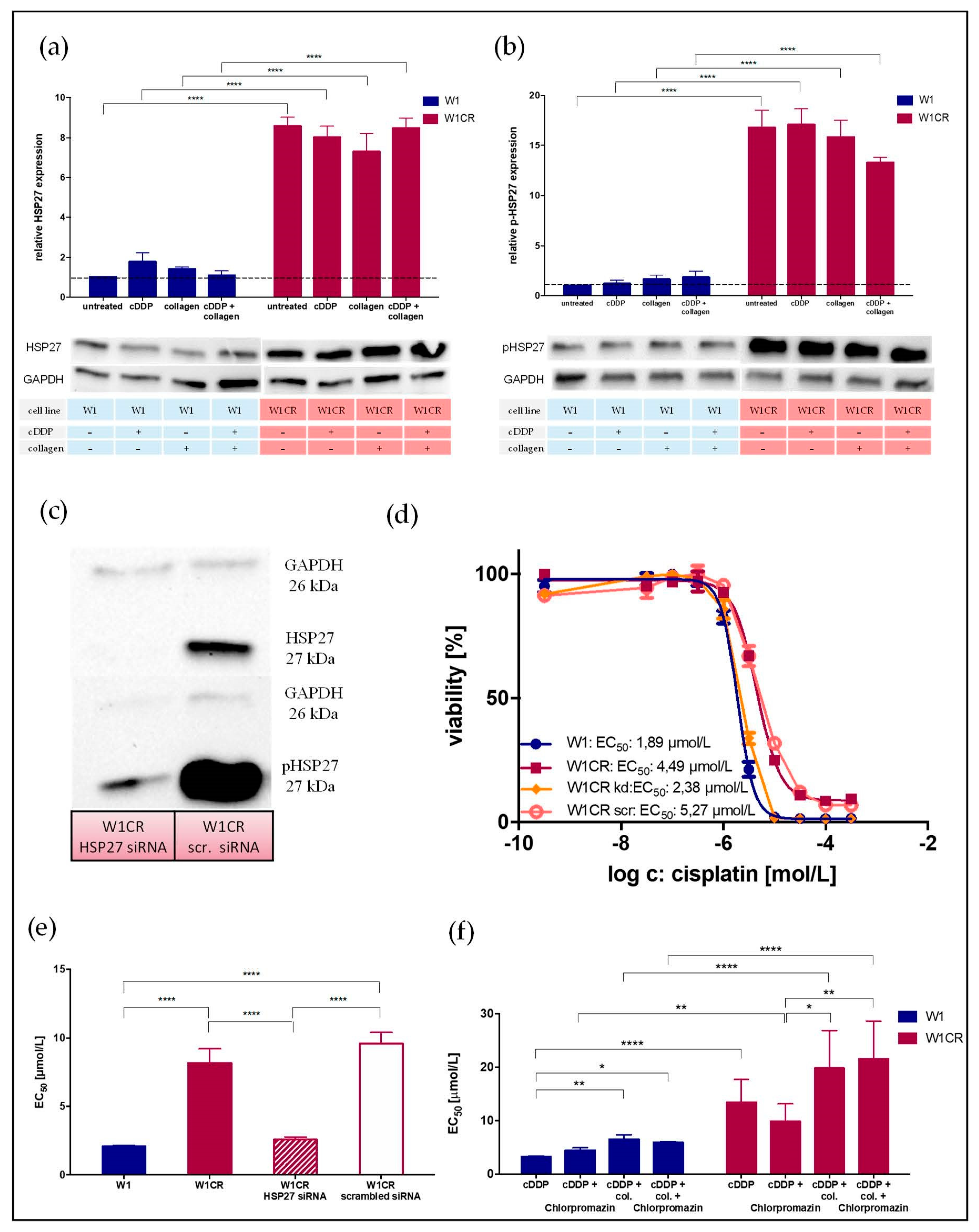
2.7. HSP27 Appears as Central Key for W1CR Resistance
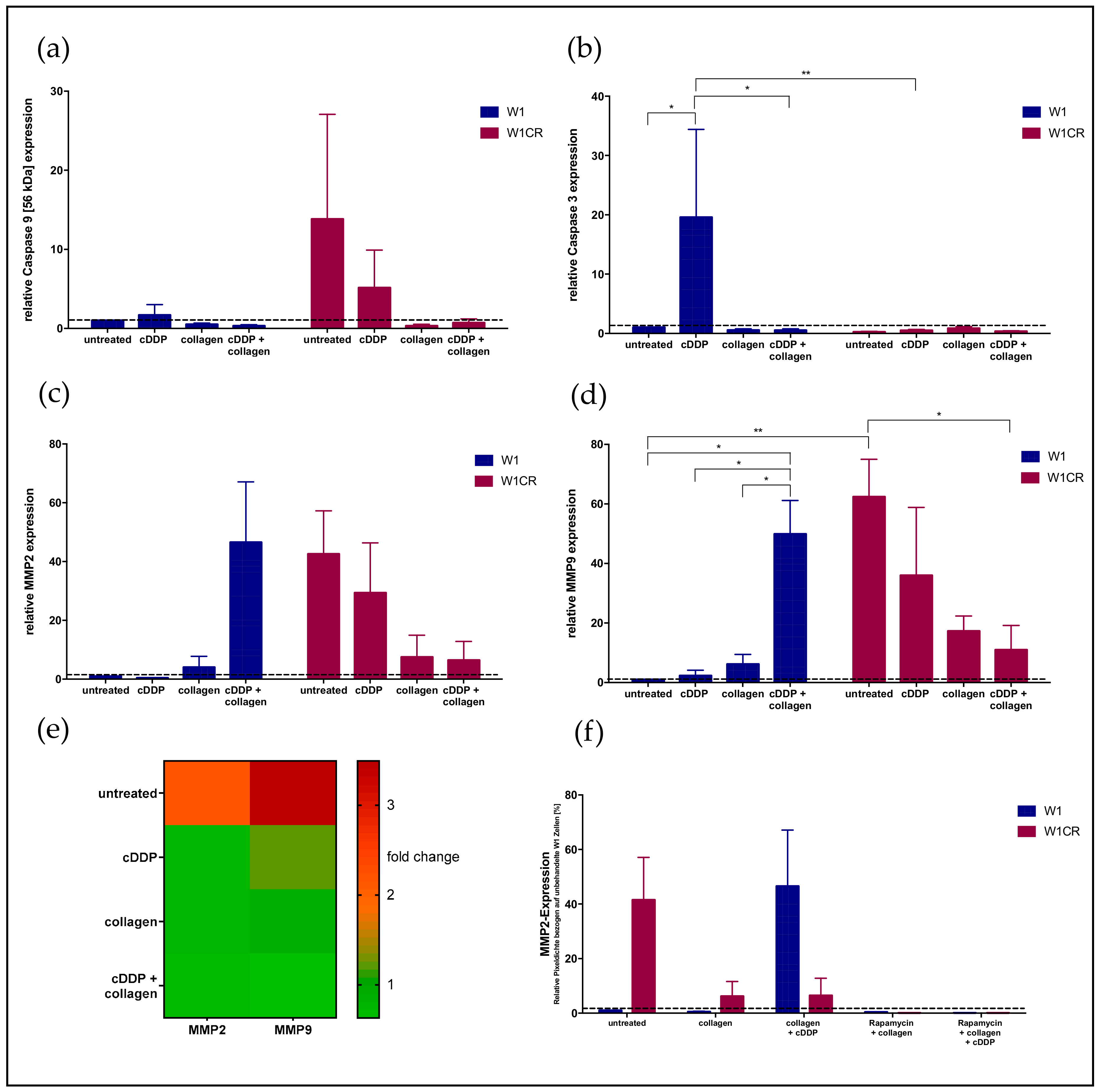
2.8. Downstream Effects on Caspases and MMPs
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. β1-Integrin Knockdown of W1 and W1CR Cells
4.3. Small-Interfering (siRNA) RNA Transfection of W1CR Cells to Knockdown HSP27
4.4. Cytotoxicity Assay
4.5. Western Blot
4.6. Proteome ProfilerTM Array
4.7. Microarray
4.8. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AKT | protein kinase B |
| CAM-DR | cell adhesion mediated drug resistance |
| cDDP | cisplatin |
| COL1 | collagen type 1 |
| DDR1/2 | discoidin domain receptor 1/2 |
| ERK | extracellular signal regulated kinase |
| FAK | focal adhesion kinase |
| HSPs | heat shock proteins |
| ILK | integrin-linked kinase |
| ITGB1 | integrin β1 |
| JNK | c-Jun-N-terminal kinase |
| kd | knockdown |
| MMP | matrix metalloprotease |
| mTOR | mechanistic target of rapamycin |
| PI3K | phosphoinositide-3-kinase |
| RTK | receptor tyrosine kinases |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [Green Version]
- Reid, B.M.; Permuth, J.B.; Sellers, T.A. Epidemiology of ovarian cancer: A review. Cancer Biol. Med. 2017, 14, 9–32. [Google Scholar] [CrossRef] [Green Version]
- Wagner, U.; Reuß, A. S3-Leitlinie „Diagnostik, Therapie und Nachsorge maligner Ovarialtumoren“: Leitlinienprogramm Onkologie, Deutsche Krebsgesellschaft, Deutsche Krebshilfe, AWMF: Langversion 3.0, 2019, AWMF-Registernummer: 032/035OL. Forum 2019, 34, 413–415. [Google Scholar] [CrossRef]
- Buttmann-Schweiger, N.; Kraywinkel, K. Epidemiologie von Eierstockkrebs in Deutschland. Onkologe 2019, 25, 92–98. [Google Scholar] [CrossRef]
- Januchowski, R.; Wojtowicz, K.; Sujka-Kordowska, P.; Andrzejewska, M.; Zabel, M. MDR Gene Expression Analysis of Six Drug-Resistant Ovarian Cancer Cell Lines. BioMed Res. Int. 2013, 2013, 241763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherman-Baust, C.A.; Weeraratna, A.T.; Rangel, L.B.A.; Pizer, E.S.; Cho, K.R.; Schwartz, D.R.; Shock, T.; Morin, P.J. Remodeling of the extracellular matrix through overexpression of collagen VI contributes to cisplatin resistance in ovarian cancer cells. Cancer Cell 2003, 3, 377–386. [Google Scholar] [CrossRef] [Green Version]
- Su, C.; Su, B.; Tang, L.; Zhao, Y.; Zhou, C. Effects of Collagen IV on Cisplatin-Induced Apoptosis of Non-Small Cell Lung Cancer Cells. Cancer Investig. 2007, 25, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Wantoch von Rekowski, K.; König, P.; Henze, S.; Schlesinger, M.; Zawierucha, P.; Januchowski, R.; Bendas, G. The Impact of Integrin-Mediated Matrix Adhesion on Cisplatin Resistance of W1 Ovarian Cancer Cells. Biomolecules 2019, 9, 788. [Google Scholar] [CrossRef] [Green Version]
- Ma, B.; Rm, L. Adhesion Dependent Signalling in the Tumour Microenvironment: The Future of Drug Targetting. Available online: https://pubmed.ncbi.nlm.nih.gov/16918414/ (accessed on 28 May 2020).
- Schmidmaier, R.; Baumann, P. ANTI-ADHESION Evolves to a Promising Therapeutic Concept in Oncology. Available online: https://pubmed.ncbi.nlm.nih.gov/18393855/ (accessed on 28 May 2020).
- Shen, B.; Delaney, M.K.; Du, X. Inside-out, outside-in, and inside-outside-in: G protein signaling in integrin-mediated cell adhesion, spreading, and retraction. Curr. Opin. Cell Biol. 2012, 24, 600–606. [Google Scholar] [CrossRef] [Green Version]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasahara, T.; Koguchi, E.; Funakoshi, M.; Aizu-Yokota, E.; Sonoda, Y. Antiapoptotic Action of Focal Adhesion Kinase (FAK) Against Ionizing Radiation. Antioxid. Redox Signal. 2002, 4, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.; Czarnecki, A.A.; Dean, M.; Modi, D.A.; Lantvit, D.D.; Hardy, L.; Baligod, S.; Davis, D.A.; Wei, J.-J.; Burdette, J.E. PTEN loss in the fallopian tube induces hyperplasia and ovarian tumor formation. Oncogene 2018, 37, 1976–1990. [Google Scholar] [CrossRef] [PubMed]
- Milella, M.; Falcone, I.; Conciatori, F.; Cesta Incani, U.; Del Curatolo, A.; Inzerilli, N.; Nuzzo, C.M.A.; Vaccaro, V.; Vari, S.; Cognetti, F.; et al. PTEN: Multiple Functions in Human Malignant Tumors. Front Oncol. 2015, 5, 24. [Google Scholar] [CrossRef] [Green Version]
- Ediriweera, M.K.; Tennekoon, K.H.; Samarakoon, S.R. Role of the PI3K/AKT/mTOR signaling pathway in ovarian cancer: Biological and therapeutic significance. Semin. Cancer Biol. 2019, 59, 147–160. [Google Scholar] [CrossRef]
- Deng, J.; Bai, X.; Feng, X.; Ni, J.; Beretov, J.; Graham, P.; Li, Y. Inhibition of PI3K/Akt/mTOR signaling pathway alleviates ovarian cancer chemoresistance through reversing epithelial-mesenchymal transition and decreasing cancer stem cell marker expression. BMC Cancer 2019, 19, 618. [Google Scholar] [CrossRef]
- Dancey, J.E. Therapeutic targets: MTOR and related pathways. Cancer Biol. Ther. 2006, 5, 1065–1073. [Google Scholar] [CrossRef] [Green Version]
- Madhunapantula, S.V.; Sharma, A.; Robertson, G.P. PRAS40 Deregulates Apoptosis in Malignant Melanoma. Cancer Res. 2007, 67, 3626–3636. [Google Scholar] [CrossRef] [Green Version]
- Shipitsin, M.; Small, C.; Giladi, E.; Siddiqui, S.; Choudhury, S.; Hussain, S.; Huang, Y.E.; Chang, H.; Rimm, D.L.; Berman, D.M.; et al. Automated quantitative multiplex immunofluorescence in situ imaging identifies phospho-S6 and phospho-PRAS40 as predictive protein biomarkers for prostate cancer lethality. Proteome Sci. 2014, 12, 40. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Harris, T.E.; Roth, R.A.; Lawrence, J.C. PRAS40 Regulates mTORC1 Kinase Activity by Functioning as a Direct Inhibitor of Substrate Binding. J. Biol. Chem. 2007, 282, 20036–20044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyriakis, J.M.; Avruch, J. Mammalian MAPK Signal Transduction Pathways Activated by Stress and Inflammation: A 10-Year Update. Physiol. Rev. 2012, 92, 689–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, W.; Brakebusch, C.; Fässler, R.; Alves, F.; Ruggiero, F.; Pawson, T. Discoidin Domain Receptor 1 Is Activated Independently of β1 Integrin. J. Biol. Chem. 2000, 275, 5779–5784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reel, B.; Korkmaz, C.G.; Arun, M.Z.; Yildirim, G.; Ogut, D.; Kaymak, A.; Micili, S.C.; Ergur, B.U. The Regulation of Matrix Metalloproteinase Expression and the Role of Discoidin Domain Receptor 1/2 Signalling in Zoledronate-treated PC3 Cells. J. Cancer 2015, 6, 1020–1029. [Google Scholar] [CrossRef] [Green Version]
- Roberts, M.E.; Magowan, L.; Hall, I.P.; Johnson, S.R. Discoidin domain receptor 1 regulates bronchial epithelial repair and matrix metalloproteinase production. Eur. Resp. J. 2011, 37, 1482–1493. [Google Scholar] [CrossRef] [Green Version]
- Baltes, F.; Caspers, J.; Henze, S.; Schlesinger, M.; Bendas, G. Targeting Discoidin Receptor 1 (DDR1) Signaling and Its Crosstalk with β1-integrin emerges as a Key Factor for Breast Cancer Chemosensitization upon Collagen Type 1 Binding. Int. J. Mol. Sci. 2020, 21, 4956. [Google Scholar] [CrossRef]
- Kim, E.K.; Choi, E.-J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2010, 1802, 396–405. [Google Scholar] [CrossRef] [Green Version]
- Cargnello, M.; Roux, P.P. Activation and Function of the MAPKs and Their Substrates, the MAPK-Activated Protein Kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [Green Version]
- Martini, E.; Schneider, E.; Neufert, C.; Neurath, M.F.; Becker, C. Survivin is a guardian of the intestinal stem cell niche and its expression is regulated by TGF-β. Cell Cycle 2016, 15, 2875–2881. [Google Scholar] [CrossRef] [Green Version]
- Seino, M.; Okada, M.; Sakaki, H.; Takeda, H.; Watarai, H.; Suzuki, S.; Seino, S.; Kuramoto, K.; Ohta, T.; Nagase, S.; et al. Time-staggered inhibition of JNK effectively sensitizes chemoresistant ovarian cancer cells to cisplatin and paclitaxel. Oncol. Rep. 2015, 35, 593–601. [Google Scholar] [CrossRef]
- Šuštić, T.; van Wageningen, S.; Bosdriesz, E.; Reid, R.J.D.; Dittmar, J.; Lieftink, C.; Beijersbergen, R.L.; Wessels, L.F.A.; Rothstein, R.; Bernards, R. A role for the unfolded protein response stress sensor ERN1 in regulating the response to MEK inhibitors in KRAS mutant colon cancers. Genome Med. 2018, 10, 1–3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galmiche, A.; Sauzay, C.; Chevet, E.; Pluquet, O. Role of the unfolded protein response in tumor cell characteristics and cancer outcome. Curr. Opin. Oncol. 2017, 29, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Ojha, R.; Amaravadi, R.K. Targeting the unfolded protein response in cancer. Pharmacol. Res. 2017, 120, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Mehlen, P.; Arrigo, A.-P. The serum-induced phosphorylation of mammalian hsp27 correlates with changes in its intracellular localization and levels of oligomerization. Eur. J. Biochem. 1994, 221, 327–334. [Google Scholar] [CrossRef]
- Welch, W.J. Mammalian stress response: Cell physiology, structure/function of stress proteins, and implications for medicine and disease. Physiol. Rev. 1992, 72, 1063–1081. [Google Scholar] [CrossRef]
- Sarto, C.; Binz, P.A.; Mocarelli, P. Heat Shock Proteins in Human Cancer. Available online: https://pubmed.ncbi.nlm.nih.gov/10786894/ (accessed on 28 May 2020).
- Rocchi, P.; So, A.; Kojima, S.; Signaevsky, M.; Beraldi, E.; Fazli, L.; Hurtado-coll, A.; Yamanaka, K.; Gleave, M. Heat Shock Protein 27 Increases after Androgen Ablation and Plays a Cytoprotective Role in Hormone-Refractory Prostate Cancer. Cancer Res. 2004, 64, 6595–6602. [Google Scholar] [CrossRef] [Green Version]
- Shiota, M.; Bishop, J.L.; Nip, K.M.; Zardan, A.; Takeuchi, A.; Cordonnier, T.; Beraldi, E.; Bazov, J.; Fazli, L.; Chi, K.; et al. Hsp27 Regulates Epithelial Mesenchymal Transition, Metastasis, and Circulating Tumor Cells in Prostate Cancer. Cancer Res. 2013, 73, 3109–3119. [Google Scholar] [CrossRef] [Green Version]
- Hansen, R.K.; Parra, I.; Lemieux, P.; Oesterreich, S.; Hilsenbeck, S.G.; Fuqua, S.A. Hsp27 Overexpression Inhibits Doxorubicin-Induced Apoptosis in Human Breast Cancer Cells. Available online: https://pubmed.ncbi.nlm.nih.gov/10573111/ (accessed on 28 May 2020).
- Beurel, E.; Grieco, S.F.; Jope, R.S. Glycogen synthase kinase-3 (GSK3): Regulation, actions, and diseases. Pharmacol. Ther. 2015, 148, 114–131. [Google Scholar] [CrossRef] [Green Version]
- Gadiya, M.; Chakraborty, G. Signaling by discoidin domain receptor 1 in cancer metastasis. Cell Adh. Migr. 2018, 12, 315–323. [Google Scholar] [CrossRef]
- Santarpia, L.; Lippman, S.L.; El-Naggar, A.K. Targeting the Mitogen-Activated Protein Kinase RAS-RAF Signaling Pathway in Cancer Therapy. Exp. Opin. Ther. Targets 2012, 16, 103–119. [Google Scholar] [CrossRef] [Green Version]
- Vidyasagar, A.; Wilson, N.A.; Djamali, A. Heat shock protein 27 (HSP27): Biomarker of disease and therapeutic target. Fibrogen. Tissue Rep. 2012, 5, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Kang, D.; Sun, B.K.; Kim, J.-H.; Song, J.J. TRAIL/MEKK4/p38/HSP27/Akt survival network is biphasically modulated by the Src/CIN85/c-Cbl complex. Cell. Signal. 2013, 25, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Qi, S.; Xin, Y.; Qi, Z.; Xu, Y.; Diao, Y.; Lan, L.; Luo, L.; Yin, Z. HSP27 phosphorylation modulates TRAIL-induced activation of Src-Akt/ERK signaling through interaction with β-arrestin2. Cell. Signal. 2014, 26, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, J.C.; Donakonda, S.; Haupt, V.J.; Lennig, P.; Zhang, Y.; Schroeder, M. New HSP27 inhibitors efficiently suppress drug resistance development in cancer cells. Oncotarget 2016, 7, 68156–68169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fong, A.-C.W.T.; Thavasu, P.; Gagrica, S.; Swales, K.E.; Leach, M.O.; Cosulich, S.C.; Chung, Y.-L.; Banerji, U. Evaluation of the combination of the dual m-TORC1/2 inhibitor vistusertib (AZD2014) and paclitaxel in ovarian cancer models. Oncotarget 2017, 8, 113874–113884. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.J.; Heo, J.H.; Park, J.Y.; Jeong, J.Y.; Cho, H.J.; Park, K.S.; Kim, S.H.; Moon, Y.W.; Kim, J.S.; An, H.J. A novel PI3K/mTOR dual inhibitor, CMG002, overcomes the chemoresistance in ovarian cancer. Gynecol. Oncol. 2019, 153, 135–148. [Google Scholar] [CrossRef]
- Quan, J.; Yahata, T.; Adachi, S.; Yoshihara, K.; Tanaka, K. Identification of receptor tyrosine kinase, discoidin domain receptor 1 (DDR1), as a potential biomarker for serous ovarian cancer. Int. J. Mol. Sci. 2011, 12, 971–982. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Zhao, F.; Hui, L.; Li, X.; Zhang, D.; Lin, W.; Chen, Z.; Ning, Y. Suppressing miR-199a-3p by promoter methylation contributes to tumor aggressiveness and cisplatin resistance of ovarian cancer through promoting DDR1 expression. J. Ovar. Res. 2017, 10, 50. [Google Scholar] [CrossRef] [Green Version]
- Baltes, F.; Pfeifer, V.; Silbermann, K.; Caspers, J.; Wantoch von Rekowski, K.; Schlesinger, M.; Bendas, G. β1-Integrin binding to collagen type 1 transmits breast cancer cells into chemoresistance by activating ABC efflux transporters. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118663. [Google Scholar] [CrossRef]
- Braicu, C.; Buse, M.; Busuioc, C.; Drula, R.; Gulei, D.; Raduly, L.; Rusu, A.; Irimie, A.; Atanasov, A.G.; Slaby, O.; et al. A Comprehensive Review on MAPK: A Promising Therapeutic Target in Cancer. Cancers 2019, 11, 1618. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.V.; Sleiman, M.; Moriarty, T.; Herrick, W.G.; Peyton, S.R. Sorafenib resistance and JNK signaling in carcinoma during extracellular matrix stiffening. Biomaterials 2014, 35, 5749–5759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Rauch, J.; Kolch, W. Targeting MAPK Signaling in Cancer: Mechanisms of Drug Resistance and Sensitivity. Int. J. Mol. Sci. 2020, 21, 1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, T.F.; Zhang, Z.F.; Liu, L.; Yang, T.; Jiang, J.; Li, P. Small interfering RNA-mediated silencing of heat shock protein 27 (HSP27) Increases chemosensitivity to paclitaxel by increasing production of reactive oxygen species in human ovarian cancer cells (HO8910). J. Int. Med. Res. 2009, 37, 1375–1388. [Google Scholar] [CrossRef] [PubMed]
- Rajesh, Y.; Banerjee, A.; Pal, I.; Biswas, A.; Das, S.; Dey, K.K.; Kapoor, N.; Ghosh, A.K.; Mitra, P.; Mandal, M. Delineation of crosstalk between HSP27 and MMP-2/MMP-9: A synergistic therapeutic avenue for glioblastoma management. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 1196–1209. [Google Scholar] [CrossRef]
- Zhou, H.Y.; Wong, A.S.T. Activation of p70S6K induces expression of matrix metalloproteinase 9 associated with hepatocyte growth factor-mediated invasion in human ovarian cancer cells. Endocrinology 2006, 147, 2557–2566. [Google Scholar] [CrossRef] [Green Version]
- Tan, W.; Zhu, S.; Cao, J.; Zhang, L.; Li, W.; Liu, K.; Zhong, J.; Shang, C.; Chen, Y. Inhibition of MMP-2 Expression Enhances the Antitumor Effect of Sorafenib in Hepatocellular Carcinoma by Suppressing the PI3K/AKT/mTOR Pathway. Oncol. Res. 2017, 25, 1543–1553. [Google Scholar] [CrossRef]
- Koch, M.; Krieger, M.L.; Stölting, D.; Brenner, N.; Beier, M.; Jaehde, U.; Wiese, M.; Royer, H.-D.; Bendas, G. Overcoming chemotherapy resistance of ovarian cancer cells by liposomal cisplatin: Molecular mechanisms unveiled by gene expression profiling. Biochem. Pharmacol. 2013, 85, 1077–1090. [Google Scholar] [CrossRef]
- Piva, M.B.R.; Jakubzig, B.; Bendas, G. Integrin Activation Contributes to Lower Cisplatin Sensitivity in MV3 Melanoma Cells by Inducing the Wnt Signalling Pathway. Cancers 2017, 9, 125. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wantoch von Rekowski, K.; König, P.; Henze, S.; Schlesinger, M.; Zawierucha, P.; Januchowski, R.; Bendas, G. Insight into Cisplatin-Resistance Signaling of W1 Ovarian Cancer Cells Emerges mTOR and HSP27 as Targets for Sensitization Strategies. Int. J. Mol. Sci. 2020, 21, 9240. https://doi.org/10.3390/ijms21239240
Wantoch von Rekowski K, König P, Henze S, Schlesinger M, Zawierucha P, Januchowski R, Bendas G. Insight into Cisplatin-Resistance Signaling of W1 Ovarian Cancer Cells Emerges mTOR and HSP27 as Targets for Sensitization Strategies. International Journal of Molecular Sciences. 2020; 21(23):9240. https://doi.org/10.3390/ijms21239240
Chicago/Turabian StyleWantoch von Rekowski, Kathleen, Philipp König, Svenja Henze, Martin Schlesinger, Piotr Zawierucha, Radosław Januchowski, and Gerd Bendas. 2020. "Insight into Cisplatin-Resistance Signaling of W1 Ovarian Cancer Cells Emerges mTOR and HSP27 as Targets for Sensitization Strategies" International Journal of Molecular Sciences 21, no. 23: 9240. https://doi.org/10.3390/ijms21239240