RB Regulates DNA Double Strand Break Repair Pathway Choice by Mediating CtIP Dependent End Resection




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
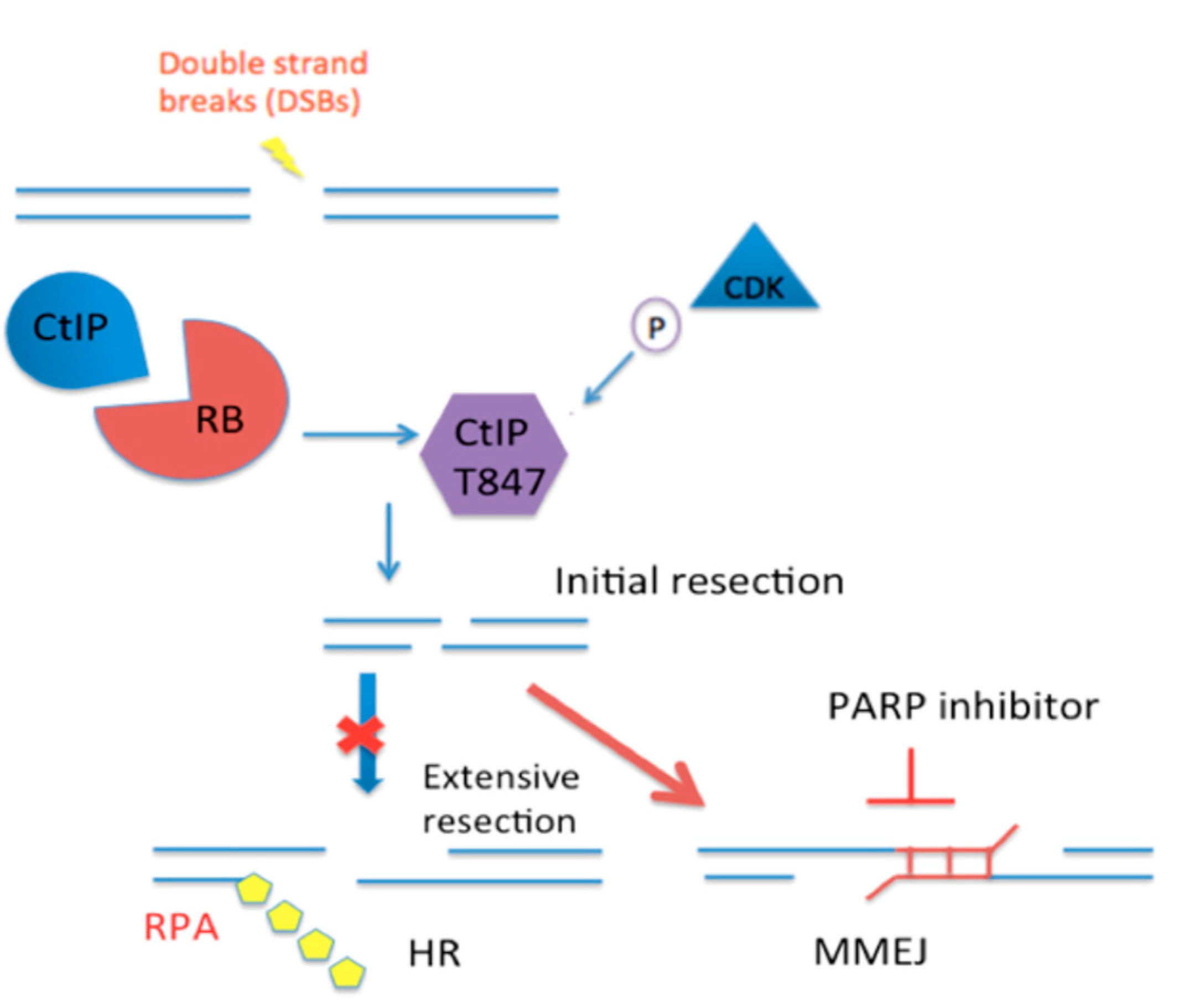
:1. Introduction
2. Results
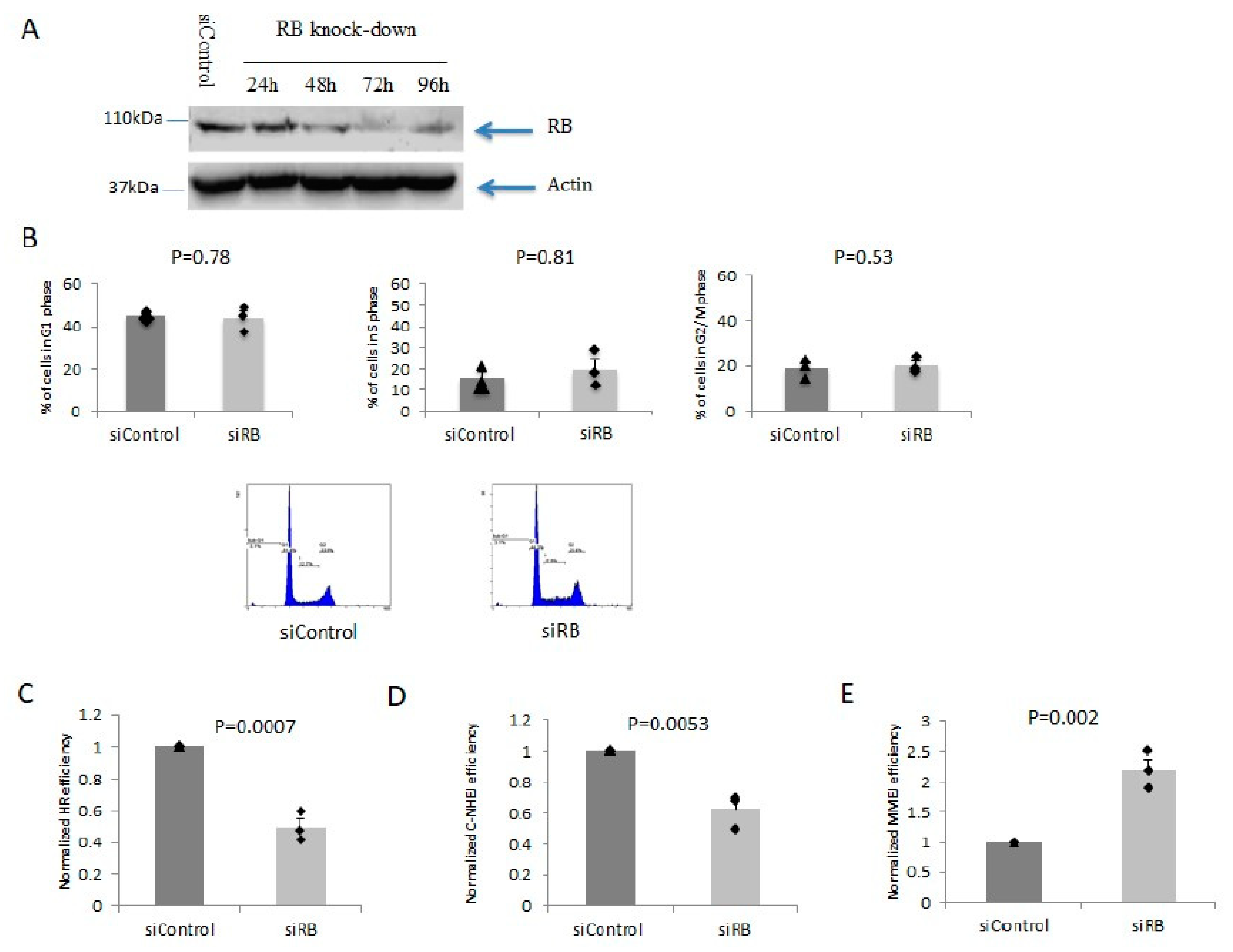
2.1. RB Depletion Caused Reduction in HR and C-NHEJ But Elevation in MMEJ Efficiencies
2.2. A Lower Degree of Resection Was Indirectly Detected in RB Knocked-Down Cells
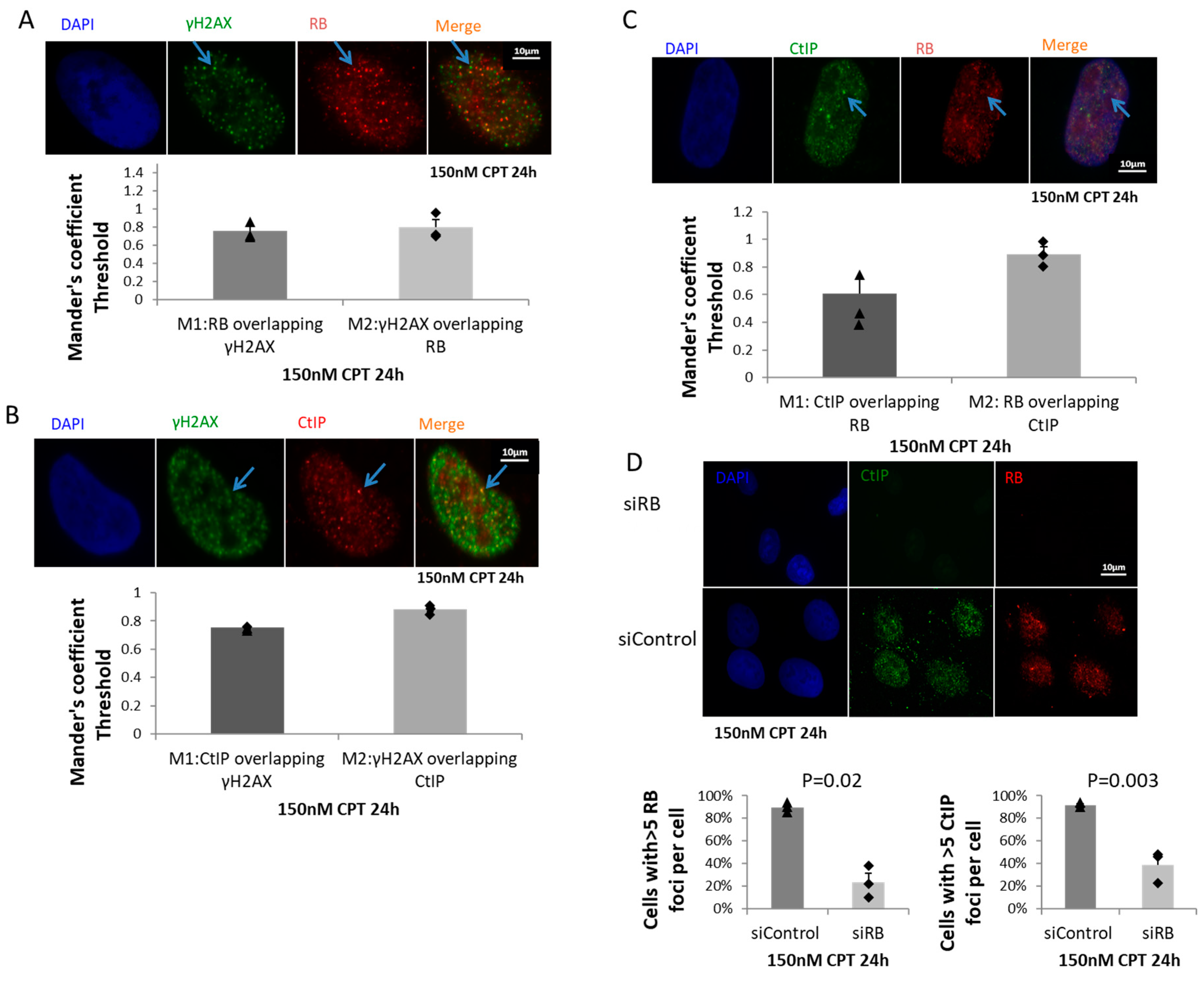
2.3. RB Regulated CtIP Foci Formation
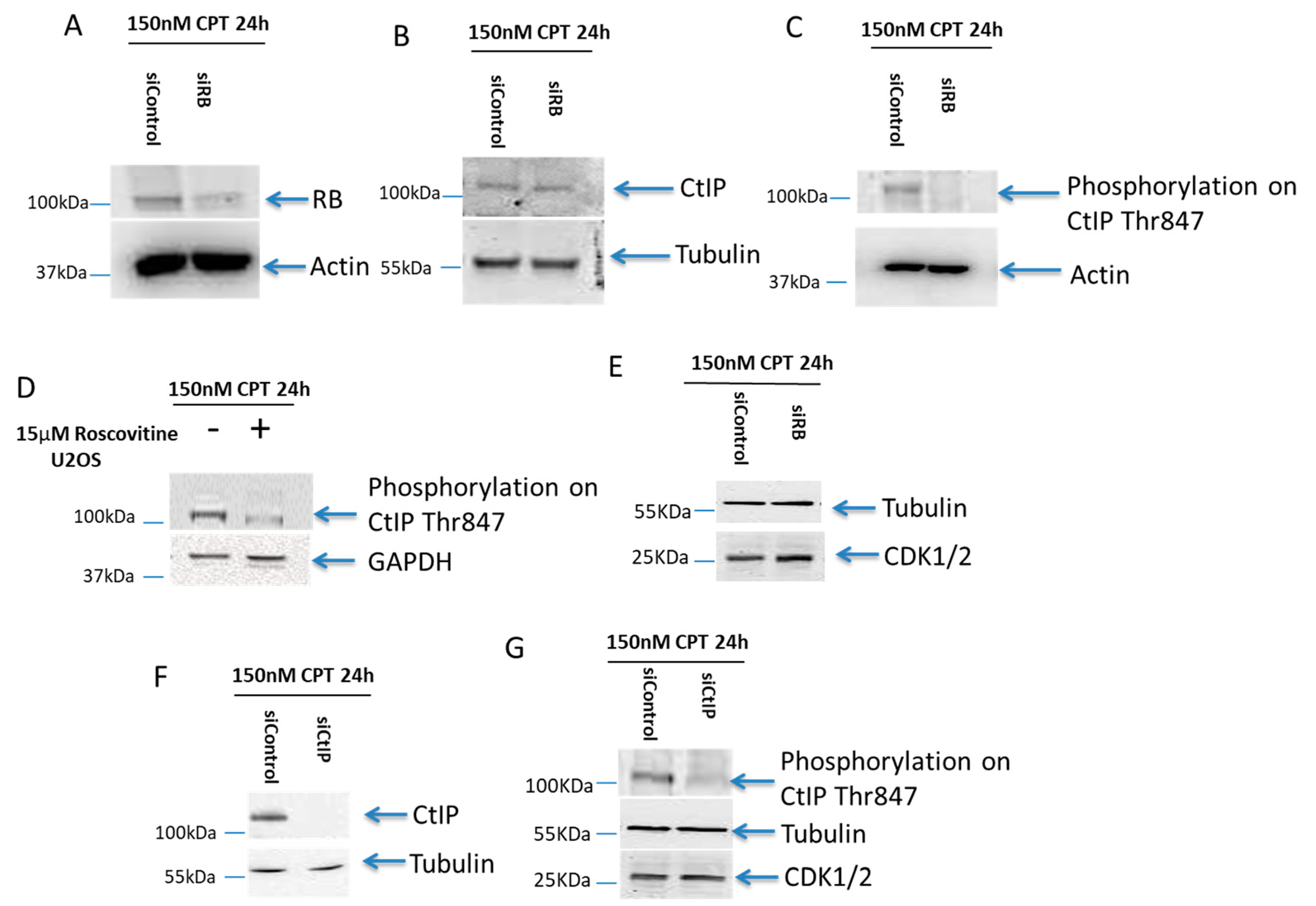
2.4. B Was Important for the T847 Phosphorylation on CtIP
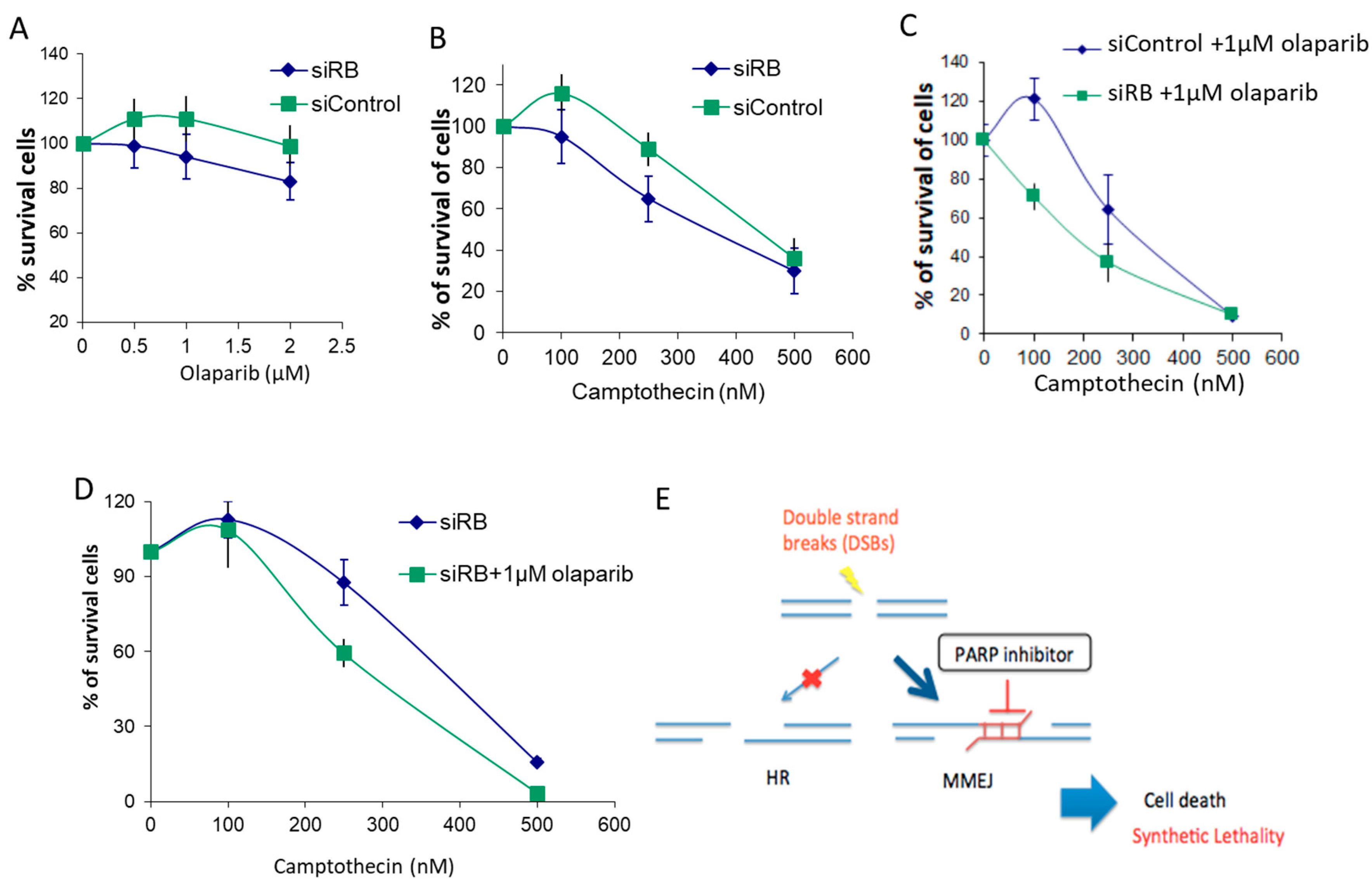
2.5. RB-Depleted Cells Were Hypersensitive to the Combined Treatment of PARP1 Inhibitor and Camptothecin
3. Discussion
4. Materials and Methods
4.1. Cell Culture and siRNA Transfection
4.2. HR, C-NHEJ and MMEJ Assays
4.3. Immunofluorescence
4.4. Cell Cycle Distribution Analysis
4.5. Western Blotting
4.6. MTT (Cell Viability Assay)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Harbour, J.W.; Dean, D.C. Rb function in cell-cycle regulation and apoptosis. Nat. Cell Biol. 2000, 2, E65–E67. [Google Scholar] [CrossRef]
- Stevaux, O.; Dyson, N.J. A revised picture of the E2F transcriptional network and RB function. Curr. Opin. Cell Biol. 2002, 14, 684–691. [Google Scholar] [CrossRef]
- Calo, E.; Quintero-Estades, J.A.; Danielian, P.S.; Nedelcu, S.; Berman, S.D.; Lees, J.A. Rb regulates fate choice and lineage commitment in vivo. Nat. Cell Biol. 2010, 466, 1110–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolay, B.N.; Danielian, P.S.; Kottakis, F.; Lapek, J.D., Jr.; Sanidas, I.; Miles, W.O.; Dehnad, M.; Tschöp, K.; Gierut, J.J.; Manning, A.L.; et al. Proteomic analysis of pRb loss highlights a signature of decreased mitochondrial oxidative phosphorylation. Genes Dev. 2015, 29, 1875–1889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolay, B.N.; Gameiro, P.A.; Tschöp, K.; Korenjak, M.; Heilmann, A.M.; Asara, J.M.; Stephanopoulos, G.; Iliopoulos, O.; Dyson, N.J. Loss of RBF1 changes glutamine catabolism. Genes Dev. 2013, 27, 182–196. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, M.R.; Lane, A.N.; Robertson, B.; Kemp, S.R.M.; Liu, Y.; Hill, B.G.; Dean, D.C.; Clem, B.F. Control of glutamine metabolism by the tumor suppressor Rb. Oncogene 2014, 33, 556–566. [Google Scholar] [CrossRef] [Green Version]
- Dick, F.A.; Rubin, S.M. Molecular mechanisms underlying RB protein function. Nat. Rev. Mol. Cell Biol. 2013, 14, 297–306. [Google Scholar] [CrossRef] [Green Version]
- Ahrabi, S.; Sarkar, S.; Pfister, S.X.; Pirovano, G.; Higgins, G.S.; Porter, A.C.; Humphrey, T.C. A role for human homologous recombination factors in suppressing microhomology-mediated end joining. Nucleic Acids Res. 2016, 44, 5743–5757. [Google Scholar] [CrossRef] [Green Version]
- Cook, R.; Zoumpoulidou, G.; Luczynski, M.T.; Rieger, S.; Moquet, J.; Spanswick, V.J.; Hartley, J.A.; Rothkamm, K.; Huang, P.H.; Mittnacht, S. Direct Involvement of Retinoblastoma Family Proteins in DNA Repair by Non-homologous End-Joining. Cell Rep. 2015, 10, 2006–2018. [Google Scholar] [CrossRef] [Green Version]
- Lieber, M.R. The Mechanism of Double-Strand DNA Break Repair by the Nonhomologous DNA End-Joining Pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [Green Version]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle 2008, 7, 2902–2906. [Google Scholar] [CrossRef]
- Aylon, Y.; Liefshitz, B.; Kupiec, M. The CDK regulates repair of double-strand breaks by homologous recombination during the cell cycle. EMBO J. 2004, 23, 4868–4875. [Google Scholar] [CrossRef] [Green Version]
- West, S.C. Molecular views of recombination proteins and their control. Nat. Rev. Mol. Cell Biol. 2003, 4, 435–445. [Google Scholar] [CrossRef]
- Symington, L.S. End Resection at Double-Strand Breaks: Mechanism and Regulation. Cold Spring Harb. Perspect. Biol. 2014, 6, a016436. [Google Scholar] [CrossRef] [Green Version]
- Huertas, P. DNA resection in eukaryotes: Deciding how to fix the break. Nat. Struct. Mol. Biol. 2010, 17, 11–16. [Google Scholar] [CrossRef] [Green Version]
- Gravel, S.; Chapman, J.R.; Magill, C.; Jackson, S.P. DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 2008, 22, 2767–2772. [Google Scholar] [CrossRef] [Green Version]
- Nimonkar, A.V.; Özsoy, A.Z.; Genschel, J.; Modrich, P.; Kowalczykowski, S.C. Human exonuclease 1 and BLM helicase interact to resect DNA and initiate DNA repair. Proc. Natl. Acad. Sci. USA 2008, 105, 16906–16911. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Nievera, C.J.; Lee, A.Y.-L.; Wu, X. Cell Cycle-dependent Complex Formation of BRCA1·CtIP·MRN Is Important for DNA Double-strand Break Repair. J. Biol. Chem. 2008, 283, 7713–7720. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Steinfeld, J.B.; Liang, F.; Chen, X.; Maranon, D.G.; Ma, C.J.; Kwon, Y.; Rao, T.; Wang, W.; Sheng, C.; et al. BRCA1–BARD1 promotes RAD51-mediated homologous DNA pairing. Nature 2017, 550, 360–365. [Google Scholar] [CrossRef]
- Davies, A.A.; Masson, J.-Y.; McIlwraith, M.J.; Stasiak, A.Z.; Stasiak, A.; Venkitaraman, A.R.; West, S.C. Role of BRCA2 in Control of the RAD51 Recombination and DNA Repair Protein. Mol. Cell 2001, 7, 273–282. [Google Scholar] [CrossRef]
- Ma, E.; Dupaigne, P.; Maloisel, L.; Guerois, R.; Le Cam, E.; Coïc, E. Rad52-Rad51 association is essential to protect Rad51 filaments against Srs2, but facultative for filament formation. eLife 2018, 7, e32744. [Google Scholar] [CrossRef]
- Szostak, J.W.; Orr-Weaver, T.L.; Rothstein, R.J.; Stahl, F.W. The double-strand-break repair model for recombination. Cell 1983, 33, 25–35. [Google Scholar] [CrossRef]
- Velez-Cruz, R.; Manickavinayaham, S.; Biswas, A.K.; Clary, R.W.; Premkumar, T.; Cole, F.; Johnson, D.G. RB localizes to DNA double-strand breaks and promotes DNA end resection and homologous recombination through the recruitment of BRG1. Genes Dev. 2016, 30, 2500–2512. [Google Scholar] [CrossRef] [Green Version]
- Marshall, A.E.; Roes, M.V.; Passos, D.T.; Deweerd, M.C.; Chaikovsky, A.C.; Sage, J.; Howlett, C.J.; Dick, F.A. RB1 Deletion in Retinoblastoma Protein Pathway-Disrupted Cells Results in DNA Damage and Cancer Progression. Mol. Cell. Biol. 2019, 39. [Google Scholar] [CrossRef] [Green Version]
- Manickavinayaham, S.; Vélez-Cruz, R.; Biswas, A.K.; Bedford, E.; Klein, B.J.; Kutateladze, T.G.; Liu, B.; Bedford, M.T.; Johnson, D.G. E2F1 acetylation directs p300/CBP-mediated histone acetylation at DNA double-strand breaks to facilitate repair. Nat. Commun. 2019, 10, 4951. [Google Scholar] [CrossRef]
- Knudsen, K.E.; Booth, D.; Naderi, S.; Sever-Chroneos, Z.; Fribourg, A.F.; Hunton, I.C.; Feramisco, J.R.; Wang, J.Y.J.; Knudsen, E.S. RB-Dependent S-Phase Response to DNA Damage. Mol. Cell. Biol. 2000, 20, 7751–7763. [Google Scholar] [CrossRef] [Green Version]
- Aparicio, T.; Baer, R.; Gautier, J. DNA double-strand break repair pathway choice and cancer. DNA Repair 2014, 19, 169–175. [Google Scholar] [CrossRef] [Green Version]
- Truong, L.N.; Li, Y.; Shi, L.Z.; Hwang, P.Y.-H.; He, J.; Razavian, N.; Berns, M.W.; Wu, X. Microhomology-mediated End Joining and Homologous Recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc. Natl. Acad. Sci. USA 2013, 110, 7720–7725. [Google Scholar] [CrossRef] [Green Version]
- Mateos-Gomez, P.A.; Gong, F.; Nair, N.; Miller, K.M.; Lazzerini-Denchi, E.; Sfeir, A. Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination. Nature 2015, 518, 254–257. [Google Scholar] [CrossRef] [Green Version]
- Audebert, M.; Salles, B.; Calsou, P. Involvement of Poly(ADP-ribose) Polymerase-1 and XRCC1/DNA Ligase III in an Alternative Route for DNA Double-strand Breaks Rejoining. J. Biol. Chem. 2004, 279, 55117–55126. [Google Scholar] [CrossRef] [Green Version]
- Sfeir, A.; Symington, L.S. Microhomology-Mediated End Joining: A Back-up Survival Mechanism or Dedicated Pathway? Trends Biochem. Sci. 2015, 40, 701–714. [Google Scholar] [CrossRef] [Green Version]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef] [Green Version]
- Bennardo, N.; Cheng, A.; Huang, N.; Stark, J.M. Alternative-NHEJ Is a Mechanistically Distinct Pathway of Mammalian Chromosome Break Repair. PLoS Genet. 2008, 4, e1000110. [Google Scholar] [CrossRef] [Green Version]
- Stiff, T.; O’Driscoll, M.; Rief, N.; Iwabuchi, K.; Löbrich, M.; Jeggo, P.A. ATM and DNA-PK Function Redundantly to Phosphorylate H2AX after Exposure to Ionizing Radiation. Cancer Res. 2004, 64, 2390–2396. [Google Scholar] [CrossRef] [Green Version]
- Anand, R.; Ranjha, L.; Cannavo, E.; Cejka, P. Phosphorylated CtIP Functions as a Co-factor of the MRE11-RAD50-NBS1 Endonuclease in DNA End Resection. Mol. Cell 2016, 64, 940–950. [Google Scholar] [CrossRef] [Green Version]
- Sartori, A.A.; Lukas, C.; Coates, J.; Mistrik, M.; Fu, S.; Bartek, J.; Baer, R.; Lukas, J.; Jackson, S.P. Human CtIP promotes DNA end resection. Nature 2007, 450, 2954. [Google Scholar] [CrossRef] [Green Version]
- Caron, M.-C.; Sharma, A.K.; O’Sullivan, J.; Myler, L.R.; Ferreira, M.T.; Rodrigue, A.; Coulombe, Y.; Ethier, C.; Gagné, J.-P.; Langelier, M.-F.; et al. Poly(ADP-ribose) polymerase-1 antagonizes DNA resection at double-strand breaks. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Fusco, C.; Reymond, A.; Zervos, A.S. Molecular Cloning and Characterization of a Novel Retinoblastoma-Binding Protein. Genomics 1998, 51, 351–358. [Google Scholar] [CrossRef]
- Wang, J.-L.; Wang, X.; Wang, H.; Iliakis, G.; Wang, Y. CHK1-Regulated S-phase Checkpoint Response Reduces Camptothecin Cytotoxicity. Cell Cycle 2002, 1, 273–278. [Google Scholar] [CrossRef] [Green Version]
- Huertas, P.; Jackson, S.P. Human CtIP Mediates Cell Cycle Control of DNA End Resection and Double Strand Break Repair. J. Biol. Chem. 2009, 284, 9558–9565. [Google Scholar] [CrossRef] [Green Version]
- Dick, F.A.; Goodrich, D.W.; Sage, J.; Dyson, N.J. Non-canonical functions of the RB protein in cancer. Nat. Rev. Cancer 2018, 18, 442–451. [Google Scholar] [CrossRef]
- Liu, F.; Lee, W.-H. CtIP Activates Its Own and Cyclin D1 Promoters via the E2F/RB Pathway during G1/S Progression. Mol. Cell. Biol. 2006, 26, 3124–3134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barton, O.; Naumann, S.C.; Diemer-Biehs, R.; Künzel, J.; Steinlage, M.; Conrad, S.; Makharashvili, N.; Wang, J.; Feng, L.; Lopez, B.S.; et al. Polo-like kinase 3 regulates CtIP during DNA double-strand break repair in G1. J. Cell Biol. 2014, 206, 877–894. [Google Scholar] [CrossRef]
- Wang, H.; Qiu, Z.; Liu, B.; Wu, Y.; Ren, J.; Liu, Y.; Zhao, Y.; Wang, Y.; Hao, S.; Li, Z.; et al. PLK1 targets CtIP to promote microhomology-mediated end joining. Nucleic Acids Res. 2018, 46, 10724–10739. [Google Scholar] [CrossRef]
- Deng, S.K.; Gibb, B.; De Almeida, M.J.; Greene, E.C.; Symington, L.S. RPA antagonizes microhomology-mediated repair of DNA double-strand breaks. Nat. Struct. Mol. Biol. 2014, 21, 405–412. [Google Scholar] [CrossRef] [Green Version]
- McVey, M. RPA puts the brakes on MMEJ. Nat. Struct. Mol. Biol. 2014, 21, 348–349. [Google Scholar] [CrossRef]
- Nakaya, R.; Takaya, J.; Onuki, T.; Moritani, M.; Nozaki, N.; Ishimi, Y. Identification of proteins that may directly interact with human RPA. J. Biochem. 2010, 148, 539–547. [Google Scholar] [CrossRef]
- Dimaras, H.; Corson, T.W.; Cobrinik, D.; White, A.; Zhao, J.; Munier, F.L.; Abramson, D.H.; Shields, C.L.; Chantada, G.L.; Njuguna, F.; et al. Retinoblastoma. Nat. Rev. Dis. Prim. 2015, 1, 15021. [Google Scholar] [CrossRef] [Green Version]
- Woods, D.; Turchi, J.J. Chemotherapy induced DNA damage response: Convergence of drugs and pathways. Cancer Biol. Ther. 2013, 14, 379–389. [Google Scholar] [CrossRef]
- Vega-Stromberg, T. Chemotherapy-induced Secondary Malignancies. J. Infus. Nurs. 2003, 26, 353–361. [Google Scholar] [CrossRef]
- Kleinerman, R.A.; Yu, C.-L.; Little, M.P.; Li, Y.; Abramson, D.; Seddon, J.; Tucker, M.A. Variation of Second Cancer Risk by Family History of Retinoblastoma Among Long-Term Survivors. J. Clin. Oncol. 2012, 30, 950–957. [Google Scholar] [CrossRef] [Green Version]
- Vesela, E.; Chroma, K.; Turi, Z.; Mistrik, M. Common Chemical Inductors of Replication Stress: Focus on Cell-Based Studies. Biomolecules 2017, 7, 19. [Google Scholar] [CrossRef] [Green Version]
- Bosco, E.E.; Mayhew, C.N.; Hennigan, R.F.; Sage, J.; Jacks, T.; Knudsen, E.S. RB signaling prevents replication-dependent DNA double-strand breaks following genotoxic insult. Nucleic Acids Res. 2004, 32, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Chaudhuri, A.R.; Hashimoto, Y.; Herrador, R.; Neelsen, K.J.; Fachinetti, D.; Bermejo, R.; Cocito, A.; Costanzo, V.; Lopes, M. Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat. Struct. Mol. Biol. 2012, 19, 417–423. [Google Scholar] [CrossRef]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef]
- Colicchia, V.; Petroni, M.; Guarguaglini, G.; Sardina, F.; Sahún-Roncero, M.; Carbonari, M.; Ricci, B.; Heil, C.; Capalbo, C.; Belardinilli, F.; et al. PARP inhibitors enhance replication stress and cause mitotic catastrophe in MYCN-dependent neuroblastoma. Oncogene 2017, 36, 4682–4691. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, Y.; Yam, J.C.; Tham, C.C.; Pang, C.P.; Chu, W.K. RB Regulates DNA Double Strand Break Repair Pathway Choice by Mediating CtIP Dependent End Resection. Int. J. Mol. Sci. 2020, 21, 9176. https://doi.org/10.3390/ijms21239176
Jiang Y, Yam JC, Tham CC, Pang CP, Chu WK. RB Regulates DNA Double Strand Break Repair Pathway Choice by Mediating CtIP Dependent End Resection. International Journal of Molecular Sciences. 2020; 21(23):9176. https://doi.org/10.3390/ijms21239176
Chicago/Turabian StyleJiang, Yuning, Jason C. Yam, Clement C. Tham, Chi Pui Pang, and Wai Kit Chu. 2020. "RB Regulates DNA Double Strand Break Repair Pathway Choice by Mediating CtIP Dependent End Resection" International Journal of Molecular Sciences 21, no. 23: 9176. https://doi.org/10.3390/ijms21239176