Impact of A134 and E218 Amino Acid Residues of Tropomyosin on Its Flexibility and Function

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
2.1. Stability of the A134L and E218L Tpm Molecules
2.2. Functional Studies
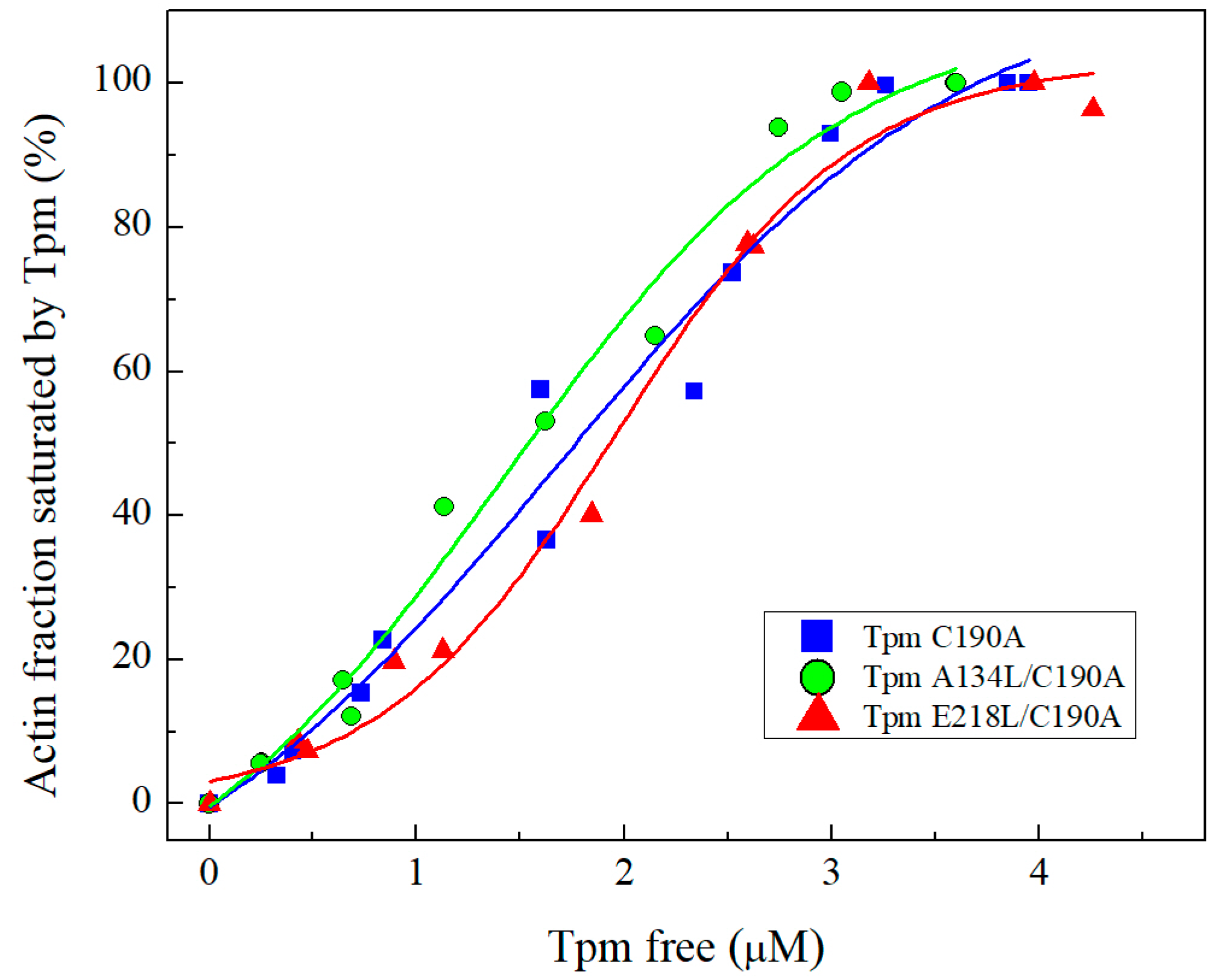
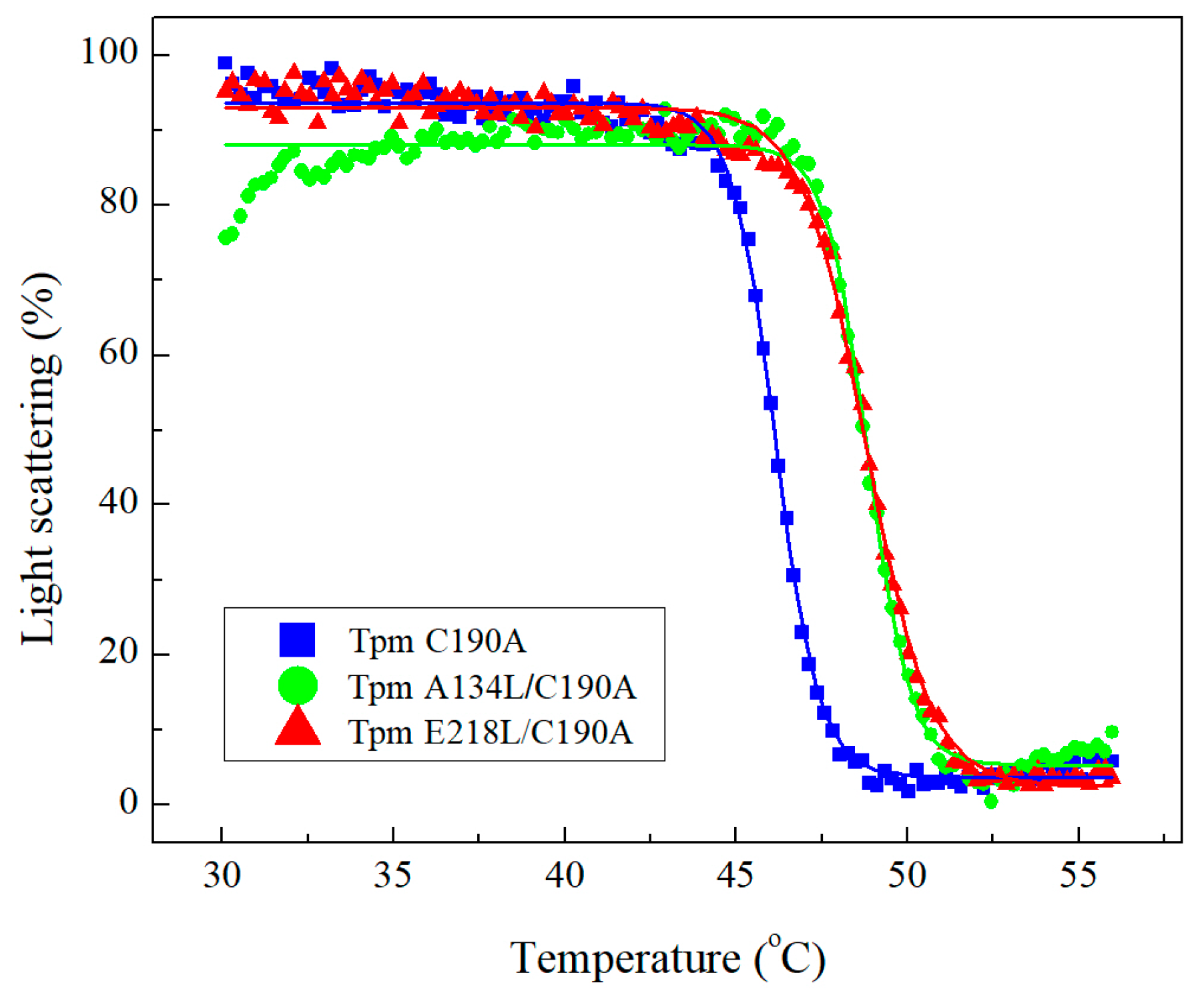
2.2.1. Co-Sedimentation Assay and Thermal Stability of Tpm-F-actin Complexes
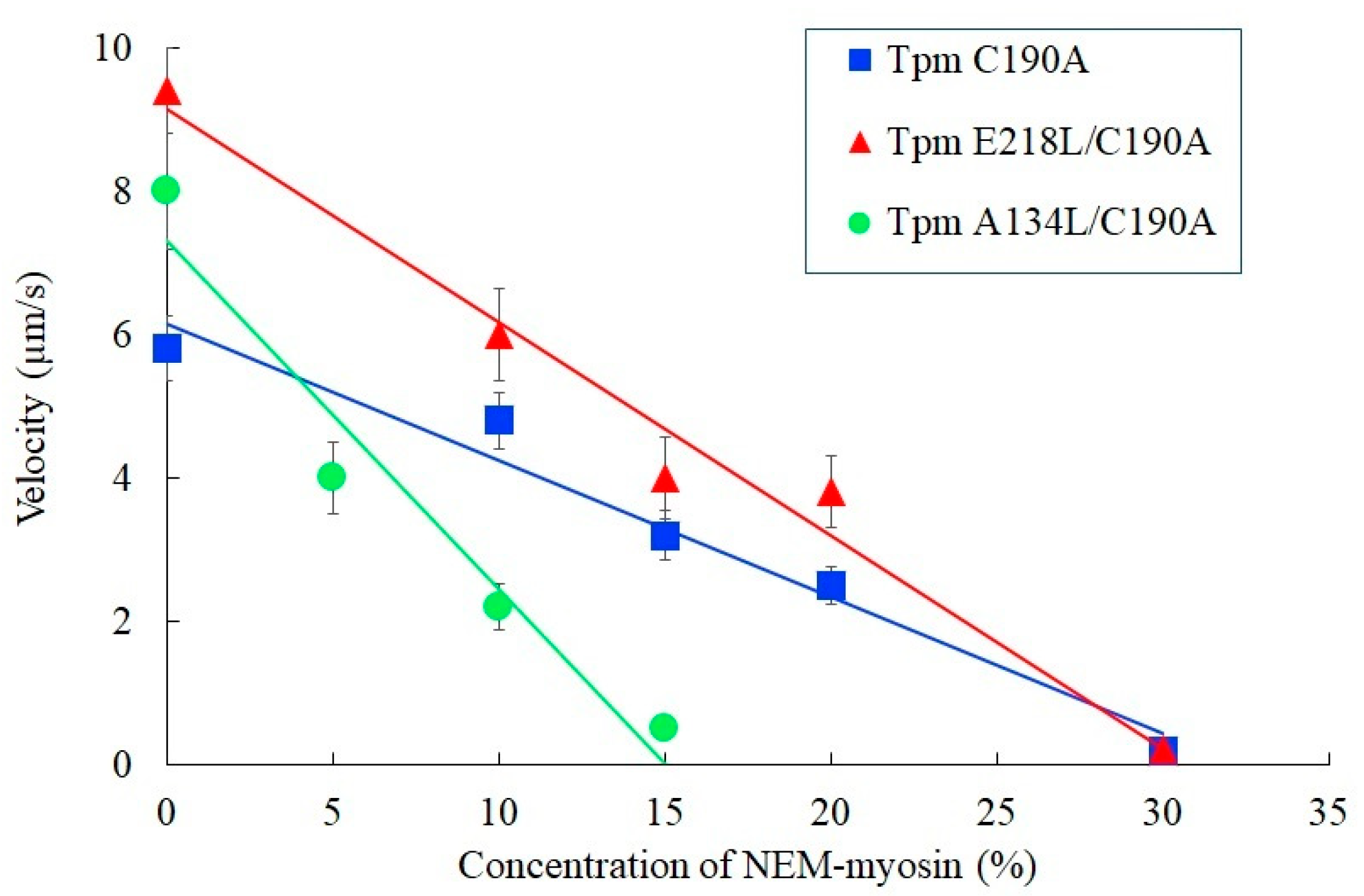
2.2.2. In Vitro Motility Assay
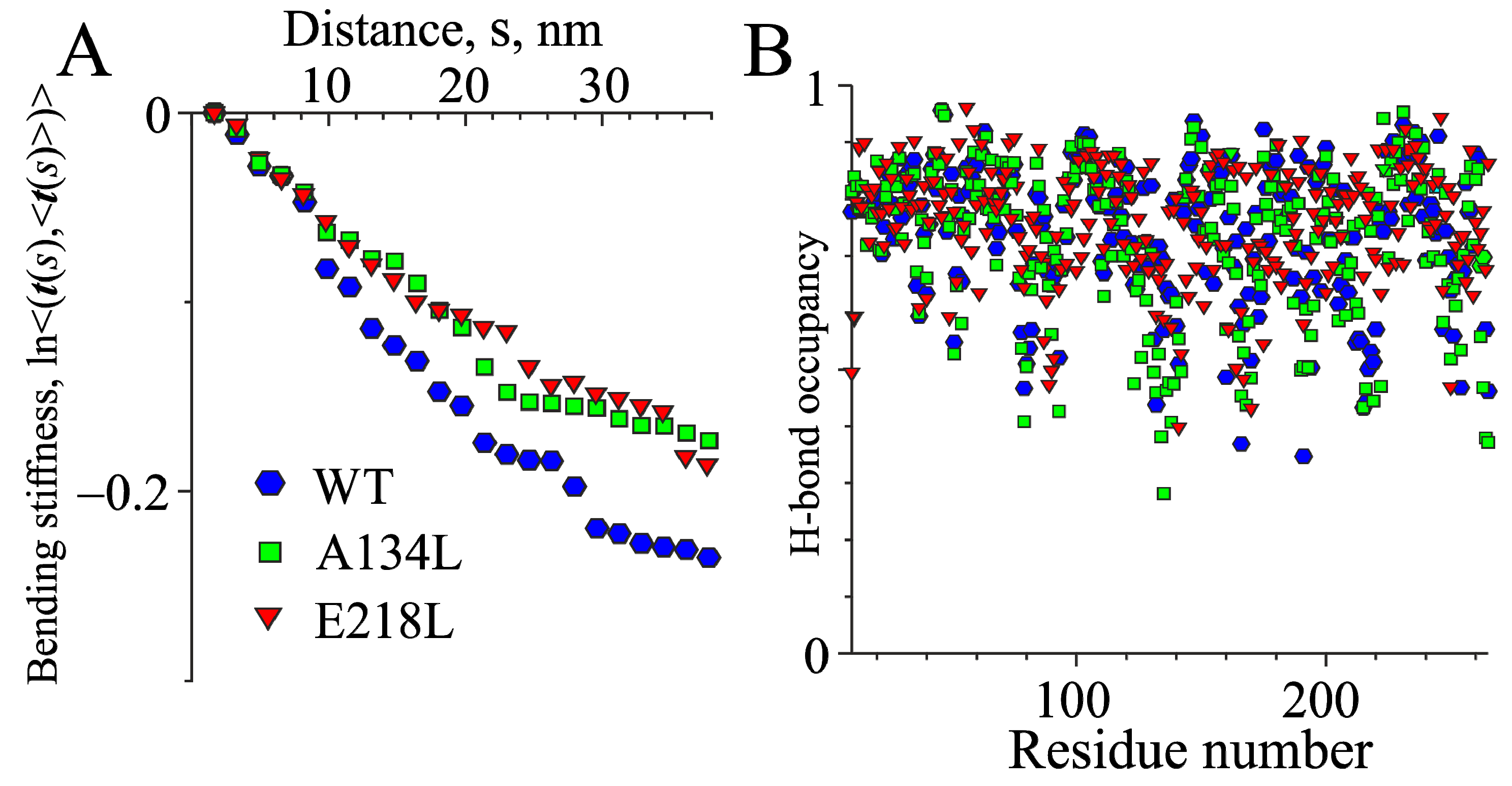
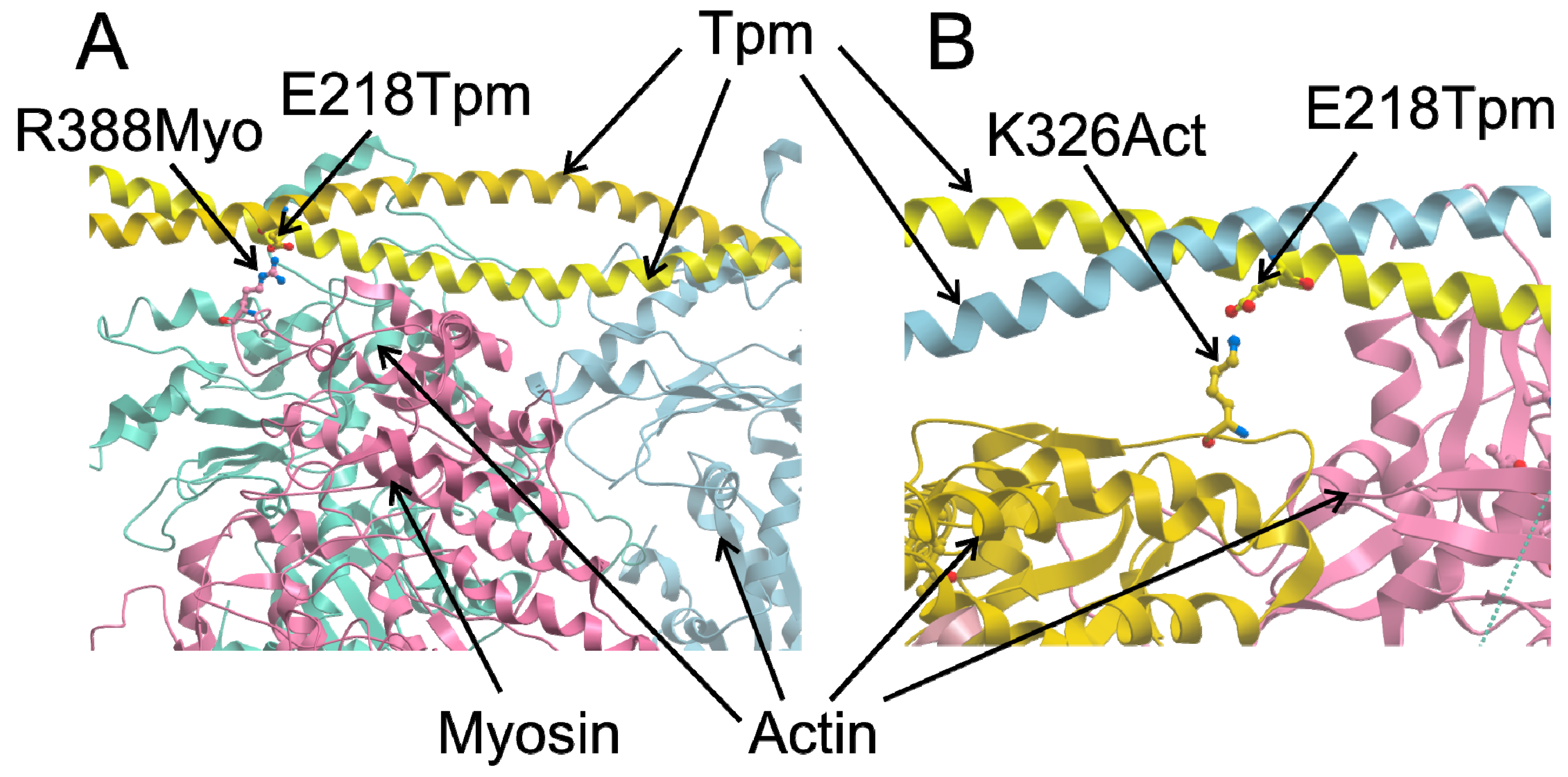
2.3. MD Simulations
3. Discussion
4. Materials and Methods
4.1. Protein Preparations
4.2. Trypsin Digestion
4.3. Differential Scanning Calorimetry (DSC)
4.4. Co-Sedimentation Assay
4.5. Temperature Dependences of Light Scattering
4.6. In Vitro Motility Assay
4.7. MD Simulation
4.8. Analysis of MD Trajectories
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Tpm | tropomyosin |
| F-actin | filamentous actin |
| DSC | differential scanning calorimetry |
| MD | molecular dynamics |
| h-bond | hydrogen bond |
References
- Gunning, P.; O’Neill, G.; Hardeman, E. Tropomyosin-based regulation of the actin cytoskeleton in time and space. Physiol. Rev. 2008, 88, 1–35. [Google Scholar] [CrossRef] [Green Version]
- Gunning, P.W.; Hardeman, E.C.; Lappalainen, P.; Mulvihill, D.P. Tropomyosin—Master regulator of actin filament function in the cytoskeleton. Cell Sci. 2015, 128, 2965–2974. [Google Scholar] [CrossRef] [Green Version]
- Dominguez, R. Tropomyosin: The gatekeeper’s view of the actin filament revealed. Biophys. J. 2011, 100, 797–808. [Google Scholar] [CrossRef] [Green Version]
- Khaitlina, S.Y. Tropomyosin as a Regulator of Actin Dynamics. Int. Rev. Cell Mol. Biol. 2015, 318, 255–291. [Google Scholar]
- Ostap, E.M. Tropomyosins as discriminators of myosin function. Adv. Exp. Med. Biol. 2008, 644, 273–282. [Google Scholar] [PubMed]
- Hitchcock-DeGregori, S.E.; Barua, B. Tropomyosin Structure, Function, and Interactions: A Dynamic Regulator. Subcell. Biochem. 2017, 82, 253–284. [Google Scholar]
- Manstein, D.J.; Mulvihill, D.P. Tropomyosin-Mediated Regulation of Cytoplasmic Myosins. Traffic 2016, 17, 872–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nevzorov, I.A.; Levitsky, D.I. Tropomyosin: Double helix from the protein world. Biochemistry (Mosc.) 2011, 76, 1507–1527. [Google Scholar] [CrossRef] [PubMed]
- Lehman, W. Thin Filament Structure and the Steric Blocking Model. Compr. Physiol. 2016, 6, 1043–1069. [Google Scholar] [PubMed]
- Risi, C.; Eisner, J.; Belknap, B.; Heeley, D.H.; White, H.D.; Schroder, G.F.; Galkin, V.E. Ca(2+)-induced movement of tropomyosin on native cardiac thin filaments revealed by cryoelectron microscopy. Proc. Natl. Acad. Sci. USA 2017, 114, 6782–6787. [Google Scholar] [CrossRef] [Green Version]
- Wakabayashi, T. Mechanism of the calcium-regulation of muscle contraction—In pursuit of its structural basis. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2015, 91, 321–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruegg, J.C. Cardiac contractility: How calcium activates the myofilaments. Die Nat. 1998, 85, 575–582. [Google Scholar] [CrossRef] [PubMed]
- McKillop, D.F.; Geeves, M.A. Regulation of the interaction between actin and myosin subfragment 1: Evidence for three states of the thin filament. Biophys. J. 1993, 65, 693–701. [Google Scholar] [CrossRef] [Green Version]
- El-Mezgueldi, M. Tropomyosin dynamics. J. Muscle Res. Cell Motil. 2014, 35, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Perry, S.V. Vertebrate tropomyosin: Distribution, properties and function. J. Muscle Res. Cell Motil. 2001, 22, 5–49. [Google Scholar] [CrossRef]
- Barua, B. Periodicities designed in the tropomyosin sequence and structure define its functions. Bioarchitecture 2013, 3, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Mason, J.M.; Arndt, K.M. Coiled coil domains: Stability, specificity, and biological implications. Chembiochem. 2004, 5, 170–176. [Google Scholar] [CrossRef]
- Lehman, W.; Rynkiewicz, M.J.; Moore, J.R. A new twist on tropomyosin binding to actin filaments: Perspectives on thin filament function, assembly and biomechanics. J. Muscle Res. Cell Motil. 2020, 41, 23–38. [Google Scholar] [CrossRef]
- Zheng, W.; Hitchcock-DeGregori, S.E.; Barua, B. Investigating the effects of tropomyosin mutations on its flexibility and interactions with filamentous actin using molecular dynamics simulation. J. Muscle Res. Cell Motil. 2016, 37, 131–147. [Google Scholar] [CrossRef]
- Hitchcock-DeGregori, S.E.; Singh, A. What makes tropomyosin an actin binding protein? A perspective. J. Struct. Biol. 2010, 170, 319–324. [Google Scholar] [CrossRef] [Green Version]
- Minakata, S.; Maeda, K.; Oda, N.; Wakabayashi, K.; Nitanai, Y.; Maeda, Y. Two-crystal structures of tropomyosin C-terminal fragment 176–273: Exposure of the hydrophobic core to the solvent destabilizes the tropomyosin molecule. Biophys. J. 2008, 95, 710–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ooi, T. Tryptic hydrolysis of tropomyosin. Biochemistry 1967, 6, 2433–2438. [Google Scholar] [CrossRef] [PubMed]
- Gorecka, A.; Drabikowski, W. Digestion of tropomyosin with trypsin. FEBS Lett. 1977, 75, 145–148. [Google Scholar] [CrossRef] [Green Version]
- Pato, M.D.; Mak, A.S.; Smillie, L.B. Fragments of rabbit striated muscle alpha-tropomyosin. I. Preparation and characterization. J. Biol. Chem. 1981, 256, 593–601. [Google Scholar] [PubMed]
- Brown, J.H.; Zhou, Z.; Reshetnikova, L.; Robinson, H.; Yammani, R.D.; Tobacman, L.S.; Cohen, C. Structure of the mid-region of tropomyosin: Bending and binding sites for actin. Proc. Natl. Acad. Sci. USA 2005, 102, 18878–18883. [Google Scholar] [CrossRef] [Green Version]
- Nevzorov, I.A.; Nikolaeva, O.P.; Kainov, Y.A.; Redwood, C.S.; Levitsky, D.I. Conserved noncanonical residue Gly-126 confers instability to the middle part of the tropomyosin molecule. J. Biol. Chem. 2011, 286, 15766–15772. [Google Scholar] [CrossRef] [Green Version]
- Sumida, J.P.; Wu, E.; Lehrer, S.S. Conserved Asp-137 imparts flexibility to tropomyosin and affects function. J. Biol. Chem. 2008, 283, 6728–6734. [Google Scholar] [CrossRef] [Green Version]
- Matyushenko, A.M.; Artemova, N.V.; Shchepkin, D.V.; Kopylova, G.V.; Bershitsky, S.Y.; Tsaturyan, A.K.; Sluchanko, N.N.; Levitsky, D.I. Structural and functional effects of two stabilizing substitutions, D137L and G126R, in the middle part of alpha-tropomyosin molecule. FEBS J. 2014, 281, 2004–2016. [Google Scholar] [CrossRef] [Green Version]
- Shchepkin, D.V.; Matyushenko, A.M.; Kopylova, G.V.; Artemova, N.V.; Bershitsky, S.Y.; Tsaturyan, A.K.; Levitsky, D.I. Stabilization of the Central Part of Tropomyosin Molecule Alters the Ca2+-sensitivity of Actin-Myosin Interaction. Acta Nat. 2013, 5, 126–129. [Google Scholar] [CrossRef]
- Nitanai, Y.; Minakata, S.; Maeda, K.; Oda, N.; Maeda, Y. Crystal structures of tropomyosin: Flexible coiled-coil. Adv. Exp. Med. Biol. 2007, 592, 137–151. [Google Scholar]
- Donkervoort, S.; Papadaki, M.; de Winter, J.M.; Neu, M.B.; Kirschner JBolduc, V.; Yang, M.L.; Gibbons, M.A.; Hu, Y.; Dastgir, J.; Leach, M.E.; et al. TPM3 deletions cause a hypercontractile congenital muscle stiffness phenotype. Ann. Neurol. 2015, 78, 982–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehman, W.; Li, X.; Kiani, F.A.; Moore, J.R.; Campbell, S.G.; Fischer, S.; Rynkiewicz, M.J. Precise Binding of Tropomyosin on Actin Involves Sequence-Dependent Variance in Coiled-Coil Twisting. Biophys. J. 2018, 115, 1082–1092. [Google Scholar] [CrossRef] [Green Version]
- Zheng, W.; Barua, B.; Hitchcock-DeGregori, S.E. Probing the flexibility of tropomyosin and its binding to filamentous actin using molecular dynamics simulations. Biophys. J. 2013, 105, 1882–1892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doran, M.H.; Pavadai, E.; Rynkiewicz, M.J.; Walklate, J.; Bullitt, E.; Moore, J.R.; Regnier, M.; Geeves, M.A.; Lehman, W. Cryo-EM and Molecular Docking Shows Myosin Loop 4 Contacts Actin and Tropomyosin on Thin Filaments. Biophys. J. 2020, 119, 821–830. [Google Scholar] [CrossRef] [PubMed]
- Behrmann, E.; Muller, M.; Penczek, P.A.; Mannherz, H.G.; Manstein, D.J.; Raunser, S. Structure of the rigor actin-tropomyosin-myosin complex. Cell 2012, 150, 327–338. [Google Scholar] [CrossRef] [Green Version]
- Yamada, Y.; Namba, K.; Fujii, T. Cardiac muscle thin filament structures reveal calcium regulatory mechanism. Nat. Commun. 2020, 11, 153. [Google Scholar] [CrossRef]
- Whitby, F.G.; Phillips, G.N., Jr. Crystal structure of tropomyosin at 7 Angstroms resolution. Proteins 2000, 38, 49–59. [Google Scholar] [CrossRef]
- Kopylova, G.V.; Matyushenko, A.M.; Koubassova, N.A.; Shchepkin, D.V.; Bershitsky, S.Y.; Levitsky, D.I.; Tsaturyan, A.K. Functional outcomes of structural peculiarities of striated muscle tropomyosin. J. Muscle Res. Cell Motil. 2020, 41, 55–70. [Google Scholar] [CrossRef]
- Johnson, C.A.; Brooker, H.R.; Gyamfi, I.; O’Brien, J.; Ashley, B.; Brazier, J.E.; Dean, A.; Embling, J.; Grimsey, E.; Tomlinson, A.C.; et al. Temperature sensitive point mutations in fission yeast tropomyosin have long range effects on the stability and function of the actin-tropomyosin copolymer. Biochem. Biophys. Res. Commun. 2018, 506, 339–346. [Google Scholar] [CrossRef]
- Ly, S.; Lehrer, S.S. Long-range effects of familial hypertrophic cardiomyopathy mutations E180G and D175N on the properties of tropomyosin. Biochemistry 2012, 51, 6413–6420. [Google Scholar] [CrossRef] [Green Version]
- Monteiro, P.B.; Lataro, R.C.; Ferro, J.A.; Reinach, F.D.C. Functional alpha-tropomyosin produced in Escherichia coli. A dipeptide extension can substitute the amino-terminal acetyl group. J. Biol. Chem. 1994, 269, 10461–10466. [Google Scholar] [PubMed]
- Lehrer, S.S.; Ly, S.; Fuchs, F. Tropomyosin is in a reduced state in rat cardiac muscle. J. Muscle Res. Cell Motil. 2011, 32, 63–64. [Google Scholar] [CrossRef] [PubMed]
- Lehrer, S.S.; Ly, S.; Fuchs, F. Tropomyosin is in a reduced state in rabbit psoas muscle. J. Muscle Res. Cell Motil. 2011, 32, 19–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matyushenko, A.M.; Artemova, N.V.; Sluchanko, N.N.; Levitsky, D.I. Effects of two stabilizing substitutions, D137L and G126R, in the middle part of alpha-tropomyosin on the domain structure of its molecule. Biophys. Chem. 2015, 196, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Spudich, J.A.; Watt, S. The regulation of rabbit skeletal muscle contraction. I. Biochemical studies of the interaction of the tropomyosin-troponin complex with actin and the proteolytic fragments of myosin. J. Biol. Chem. 1971, 246, 4866–4871. [Google Scholar] [PubMed]
- Margossian, S.S.; Lowey, S. Preparation of myosin and its subfragments from rabbit skeletal muscle. Methods Enzymol. 1982, 85 Pt B, 55–71. [Google Scholar]
- Potter, J.D. Preparation of troponin and its subunits. Methods Enzymol. 1982, 85, 241–263. [Google Scholar]
- Nefedova, V.V.; Marchenko, M.A.; Kleymenov, S.Y.; Datskevich, P.N.; Levitsky, D.I.; Matyushenko, A.M. Thermal unfolding of various human non-muscle isoforms of tropomyosin. Biochem. Biophys. Res. Commun. 2019, 514, 613–617. [Google Scholar] [CrossRef]
- Matyushenko, A.M.; Koubassova, N.A.; Shchepkin, D.V.; Kopylova, G.V.; Nabiev, S.R.; Nikitina, L.V.; Bershitsky, S.Y.; Levitsky, D.I.; Tsaturyan, A.K. The effects of cardiomyopathy-associated mutations in the head-to-tail overlap junction of alpha-tropomyosin on its properties and interaction with actin. Int. J. Biol. Macromol. 2019, 125, 1266–1274. [Google Scholar] [CrossRef]
- Matyushenko, A.M.; Shchepkin, D.V.; Susorov, D.S.; Nefedova, V.V.; Kopylova, G.V.; Berg, V.Y.; Kleymenov, S.Y.; Levitsky, D.I. Structural and functional properties of alphabeta-heterodimers of tropomyosin with myopathic mutations Q147P and K49del in the beta-chain. Biochem. Biophys. Res. Commun. 2019, 508, 934–939. [Google Scholar] [CrossRef]
- Matyushenko, A.M.; Shchepkin, D.V.; Kopylova, G.V.; Bershitsky, S.Y.; Koubassova, N.A.; Tsaturyan, A.K.; Levitsky, D.I. Functional role of the core gap in the middle part of tropomyosin. FEBS J. 2018, 285, 871–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kremneva, E.; Boussouf, S.; Nikolaeva, O.; Maytum, R.; Geeves, M.A.; Levitsky, D.I. Effects of two familial hypertrophic cardiomyopathy mutations in alpha-tropomyosin, Asp175Asn and Glu180Gly, on the thermal unfolding of actin-bound tropomyosin. Biophys. J. 2004, 87, 3922–3933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mashanov, G.I.; Molloy, J.E. Automatic detection of single fluorophores in live cells. Biophys. J. 2007, 92, 2199–2211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haeberle, J.R. Calponin decreases the rate of cross-bridge cycling and increases maximum force production by smooth muscle myosin in an in vitro motility assay. J. Biol. Chem. 1994, 269, 12424–12431. [Google Scholar]
- Shchepkin, D.V.; Nabiev, S.R.; Kopylova, G.V.; Matyushenko, A.M.; Levitsky, D.I.; Bershitsky, S.Y.; Tsaturyan, A.K. Cooperativity of myosin interaction with thin filaments is enhanced by stabilizing substitutions in tropomyosin. J. Muscle Res. Cell Motil. 2017, 38, 183–191. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Mackerell, A.D., Jr.; Feig, M.; Brooks, C.L., 3rd. Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004, 25, 1400–1415. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [Green Version]
- Li, X.E.; Lehman, W.; Fischer, S. The relationship between curvature, flexibility and persistence length in the tropomyosin coiled-coil. J. Struct. Biol. 2010, 170, 313–318. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tpm | Tm a (°C) | ΔHcal b (Kj*mol−1) | Total ΔHcal b (Kj*mol−1) | ΔHcal (% of total) | Tdiss c (°C) |
|---|---|---|---|---|---|
| C190A | 1155 | 46.14 ± 0.02 | |||
| domain 1 | 33.7 | 100 | 8.7 | ||
| domain 2 | 41.5 | 640 | 55.4 | ||
| domain 3 | 49.7 | 415 | 35.9 | ||
| A134L/C190A | 1235 | 48.84 ± 0.04 | |||
| domain 1 | 35.3 | 275 | 22.3 | ||
| domain 2 | 42.7 | 600 | 48.6 | ||
| domain 3 | 51.0 | 360 | 29.1 | ||
| E218L/C190A | 1141 | 48.80 ± 0.03 | |||
| domain 1 | 41.6 | 210 | 18.4 | ||
| domain 2 | 48.6 | 661 | 57.9 | ||
| domain 3 | 54.9 | 270 | 23.7 |
| Tpm | Vmax, µm/s ± SD | pCa50 ± SD | n ± SD |
|---|---|---|---|
| C190A | 5.8 ± 0.2 | 6.13 ± 0.01 | 3.1 ± 0.2 |
| A134L/C190A | 8.0 ± 0.8 | 5.89 ± 0.07 | 1.3 ± 0.2 |
| E218L/C190A | 9.4 ± 0.1 | 6.30 + 0.01 | 3.5 + 0.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marchenko, M.A.; Nefedova, V.V.; Yampolskaya, D.S.; Kopylova, G.V.; Shchepkin, D.V.; Bershitsky, S.Y.; Koubassova, N.A.; Tsaturyan, A.K.; Levitsky, D.I.; Matyushenko, A.M. Impact of A134 and E218 Amino Acid Residues of Tropomyosin on Its Flexibility and Function. Int. J. Mol. Sci. 2020, 21, 8720. https://doi.org/10.3390/ijms21228720
Marchenko MA, Nefedova VV, Yampolskaya DS, Kopylova GV, Shchepkin DV, Bershitsky SY, Koubassova NA, Tsaturyan AK, Levitsky DI, Matyushenko AM. Impact of A134 and E218 Amino Acid Residues of Tropomyosin on Its Flexibility and Function. International Journal of Molecular Sciences. 2020; 21(22):8720. https://doi.org/10.3390/ijms21228720
Chicago/Turabian StyleMarchenko, Marina A., Victoria V. Nefedova, Daria S. Yampolskaya, Galina V. Kopylova, Daniil V. Shchepkin, Sergey Y. Bershitsky, Natalia A. Koubassova, Andrey K. Tsaturyan, Dmitrii I. Levitsky, and Alexander M. Matyushenko. 2020. "Impact of A134 and E218 Amino Acid Residues of Tropomyosin on Its Flexibility and Function" International Journal of Molecular Sciences 21, no. 22: 8720. https://doi.org/10.3390/ijms21228720