Rhabdomyolysis-Induced AKI Was Ameliorated in NLRP3 KO Mice via Alleviation of Mitochondrial Lipid Peroxidation in Renal Tubular Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. NLRP3 KO Mice Were Protected from RIAKI
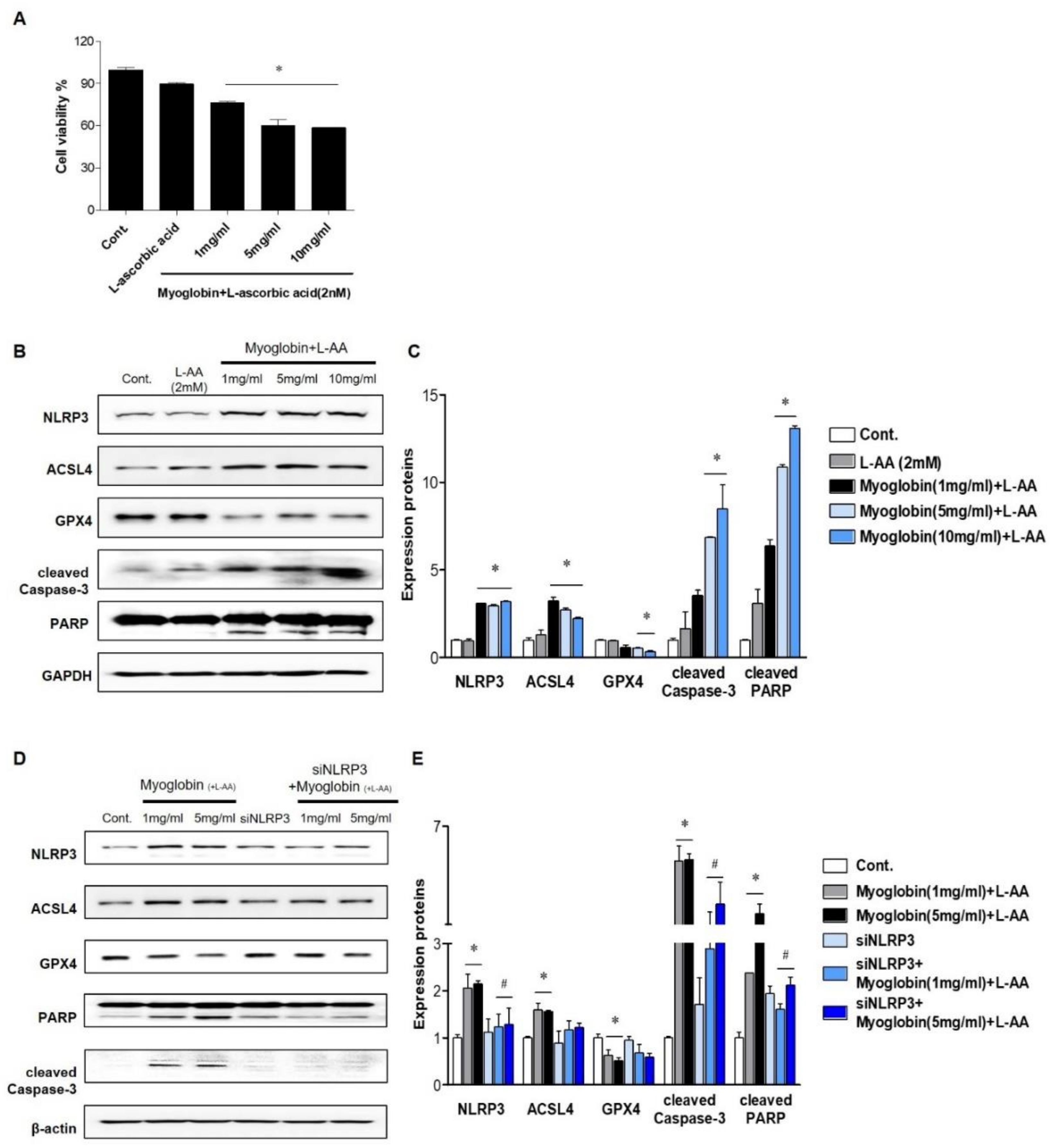
2.2. NLRP3 Depletion in Renal Tubular Cells Mitigated Myoglobin-Induced Ferroptosis and Apoptosis
2.3. NLRP3 Depletion in Renal Tubular Cells Attenuated Myoglobin-Induced Mitochondrial Injury
2.4. NLRP3 Depletion Contributed to Decreasing Myoglobin-Induced ROS Levels and Lipid Peroxidation
3. Discussion
4. Material and Methods
4.1. Animal Models
4.2. Blood Chemistry
4.3. Histopathology
4.4. TdT Mediated dUTP Nick End Labeling (TUNEL) Assay
4.5. Real-Time PCR
4.6. Western Blot
4.7. Cell Culture
4.8. Small Interfering RNA Knockdown Experiments
4.9. MTT Assay
4.10. Flow Cytometric Analysis
4.11. Immunocytochemistry
4.12. Lipid Peroxidation Product Assay
4.13. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Bosch, X.; Poch, E.; Grau, J.M. Rhabdomyolysis and acute kidney injury. N. Engl. J. Med. 2009, 361, 62–72. [Google Scholar] [CrossRef] [Green Version]
- Petejova, N.; Martinek, A. Acute kidney injury due to rhabdomyolysis and renal replacement therapy: A critical review. Crit. Care 2014, 18, 224. [Google Scholar] [CrossRef] [Green Version]
- Chatzizisis, Y.S.; Misirli, G.; Hatzitolios, A.I.; Giannoglou, G.D. The syndrome of rhabdomyolysis: Complications and treatment. Eur. J. Intern. Med. 2008, 19, 568–574. [Google Scholar] [CrossRef]
- Chander, V.; Chopra, K. Molsidomine, a nitric oxide donor and L-arginine protects against rhabdomyolysis-induced myoglobinuric acute renal failure. Biochim. Biophys. Acta 2005, 1723, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Zager, R.A. Studies of mechanisms and protective maneuvers in myoglobinuric acute renal injury. Lab. Investig. 1989, 60, 619–629. [Google Scholar] [PubMed]
- Zager, R.A.; Foerder, C.A. Effects of inorganic iron and myoglobin on in vitro proximal tubular lipid peroxidation and cytotoxicity. J. Clin. Investig. 1992, 89, 989–995. [Google Scholar] [CrossRef] [PubMed]
- Reeder, B.J.; Wilson, M.T. Hemoglobin and myoglobin associated oxidative stress: From molecular mechanisms to disease States. Curr. Med. Chem. 2005, 12, 2741–2751. [Google Scholar] [CrossRef]
- Guerrero-Hue, M.; Garcia-Caballero, C.; Palomino-Antolin, A.; Rubio-Navarro, A.; Vazquez-Carballo, C.; Herencia, C.; Martin-Sanchez, D.; Farre-Alins, V.; Egea, J.; Cannata, P.; et al. Curcumin reduces renal damage associated with rhabdomyolysis by decreasing ferroptosis-mediated cell death. FASEB J. 2019, 33, 8961–8975. [Google Scholar] [CrossRef]
- Shen, H.; Kreisel, D.; Goldstein, D.R. Processes of sterile inflammation. J. Immunol. 2013, 191, 2857–2863. [Google Scholar] [CrossRef]
- Chen, G.Y.; Nunez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef] [Green Version]
- Homsi, E.; Janino, P.; de Faria, J.B. Role of caspases on cell death, inflammation, and cell cycle in glycerol-induced acute renal failure. Kidney Int. 2006, 69, 1385–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homsi, E.; Ribeiro-Alves, M.A.; Lopes de Faria, J.B.; Dias, E.P. Interleukin-6 stimulates tubular regeneration in rats with glycerol-induced acute renal failure. Nephron 2002, 92, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Komada, T.; Usui, F.; Kawashima, A.; Kimura, H.; Karasawa, T.; Inoue, Y.; Kobayashi, M.; Mizushina, Y.; Kasahara, T.; Taniguchi, S.; et al. Role of NLRP3 Inflammasomes for Rhabdomyolysis-induced Acute Kidney Injury. Sci. Rep. 2015, 5, 10901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.M.; Kim, Y.G.; Kim, D.J.; Park, S.H.; Jeong, K.H.; Lee, Y.H.; Lim, S.J.; Lee, S.H.; Moon, J.Y. Inflammasome-Independent Role of NLRP3 Mediates Mitochondrial Regulation in Renal Injury. Front. Immunol. 2018, 9, 2563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, H.; Vilaysane, A.; Lau, A.; Stahl, M.; Morampudi, V.; Bondzi-Simpson, A.; Platnich, J.M.; Bracey, N.A.; French, M.C.; Beck, P.L.; et al. NLRP3 regulates a non-canonical platform for caspase-8 activation during epithelial cell apoptosis. Cell Death Differ. 2016, 23, 1331–1346. [Google Scholar] [CrossRef] [PubMed]
- Zager, R.A.; Burkhart, K. Myoglobin toxicity in proximal human kidney cells: Roles of Fe, Ca2+, H2O2, and terminal mitochondrial electron transport. Kidney Int. 1997, 51, 728–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shigeoka, A.A.; Mueller, J.L.; Kambo, A.; Mathison, J.C.; King, A.J.; Hall, W.F.; Correia Jda, S.; Ulevitch, R.J.; Hoffman, H.M.; McKay, D.B. An inflammasome-independent role for epithelial-expressed Nlrp3 in renal ischemia-reperfusion injury. J. Immunol. 2010, 185, 6277–6285. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Wang, L.; Jiang, N.; Mou, S.; Zhang, M.; Gu, L.; Shao, X.; Wang, Q.; Qi, C.; Li, S.; et al. NLRP3 inflammasome mediates contrast media-induced acute kidney injury by regulating cell apoptosis. Sci. Rep. 2016, 6, 34682. [Google Scholar] [CrossRef]
- Yamagishi, H.; Yokoo, T.; Imasawa, T.; Mitarai, T.; Kawamura, T.; Utsunomiya, Y. Genetically modified bone marrow-derived vehicle cells site specifically deliver an anti-inflammatory cytokine to inflamed interstitium of obstructive nephropathy. J. Immunol. 2001, 166, 609–616. [Google Scholar] [CrossRef]
- Gong, W.; Mao, S.; Yu, J.; Song, J.; Jia, Z.; Huang, S.; Zhang, A. NLRP3 deletion protects against renal fibrosis and attenuates mitochondrial abnormality in mouse with 5/6 nephrectomy. Am. J. Physiol. Renal Physiol. 2016, 310, F1081–F1088. [Google Scholar] [CrossRef] [Green Version]
- Ludwig-Portugall, I.; Bartok, E.; Dhana, E.; Evers, B.D.; Primiano, M.J.; Hall, J.P.; Franklin, B.S.; Knolle, P.A.; Hornung, V.; Hartmann, G.; et al. An NLRP3-specific inflammasome inhibitor attenuates crystal-induced kidney fibrosis in mice. Kidney Int. 2016, 90, 525–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahzad, K.; Bock, F.; Dong, W.; Wang, H.; Kopf, S.; Kohli, S.; Al-Dabet, M.M.; Ranjan, S.; Wolter, J.; Wacker, C.; et al. Nlrp3-inflammasome activation in non-myeloid-derived cells aggravates diabetic nephropathy. Kidney Int. 2015, 87, 74–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, R.S.; Zhou, J.J.; Feng, Y.Y.; Shi, M.; Guo, F.; Gou, S.J.; Salerno, S.; Ma, L.; Fu, P. Pharmacological Inhibition of Macrophage Toll-like Receptor 4/Nuclear Factor-kappa B Alleviates Rhabdomyolysis-induced Acute Kidney Injury. Chin. Med. J. 2017, 130, 2163–2169. [Google Scholar] [CrossRef] [PubMed]
- Kundert, F.; Platen, L.; Iwakura, T.; Zhao, Z.; Marschner, J.A.; Anders, H.J. Immune mechanisms in the different phases of acute tubular necrosis. Kidney Res. Clin. Pract. 2018, 37, 185–196. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Wang, X.; Chun, J.; Vilaysane, A.; Clark, S.; French, G.; Bracey, N.A.; Trpkov, K.; Bonni, S.; Duff, H.J.; et al. Inflammasome-independent NLRP3 augments TGF-beta signaling in kidney epithelium. J. Immunol. 2013, 190, 1239–1249. [Google Scholar] [CrossRef] [Green Version]
- Bracey, N.A.; Gershkovich, B.; Chun, J.; Vilaysane, A.; Meijndert, H.C.; Wright, J.R., Jr.; Fedak, P.W.; Beck, P.L.; Muruve, D.A.; Duff, H.J. Mitochondrial NLRP3 protein induces reactive oxygen species to promote Smad protein signaling and fibrosis independent from the inflammasome. J. Biol. Chem. 2014, 289, 19571–19584. [Google Scholar] [CrossRef] [Green Version]
- Anders, H.J.; Suarez-Alvarez, B.; Grigorescu, M.; Foresto-Neto, O.; Steiger, S.; Desai, J.; Marschner, J.A.; Honarpisheh, M.; Shi, C.; Jordan, J.; et al. The macrophage phenotype and inflammasome component NLRP3 contributes to nephrocalcinosis-related chronic kidney disease independent from IL-1-mediated tissue injury. Kidney Int. 2018, 93, 656–669. [Google Scholar] [CrossRef] [Green Version]
- Lei, P.; Bai, T.; Sun, Y. Mechanisms of Ferroptosis and Relations with Regulated Cell Death: A Review. Front. Physiol. 2019, 10, 139. [Google Scholar] [CrossRef] [Green Version]
- Reeder, B.J.; Sharpe, M.A.; Kay, A.D.; Kerr, M.; Moore, K.; Wilson, M.T. Toxicity of myoglobin and haemoglobin: Oxidative stress in patients with rhabdomyolysis and subarachnoid haemorrhage. Biochem. Soc. Trans. 2002, 30, 745–748. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, S.S.; Jung, M.H.; Yeo, H.D.; Kim, H.J.; Yang, J.I.; Roh, G.S.; Chang, S.H.; Park, D.J. N-acetylcysteine attenuates glycerol-induced acute kidney injury by regulating MAPKs and Bcl-2 family proteins. Nephrol. Dial. Transplant. 2010, 25, 1435–1443. [Google Scholar] [CrossRef] [Green Version]
- Nishida, K.; Watanabe, H.; Ogaki, S.; Kodama, A.; Tanaka, R.; Imafuku, T.; Ishima, Y.; Chuang, V.T.; Toyoda, M.; Kondoh, M.; et al. Renoprotective effect of long acting thioredoxin by modulating oxidative stress and macrophage migration inhibitory factor against rhabdomyolysis-associated acute kidney injury. Sci. Rep. 2015, 5, 14471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, M.; Zhong, H.; Xia, L.; Tao, Y.; Yin, H. Pathophysiology of mitochondrial lipid oxidation: Role of 4-hydroxynonenal (4-HNE) and other bioactive lipids in mitochondria. Free Radic. Biol. Med. 2017, 111, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Missiroli, S.; Patergnani, S.; Caroccia, N.; Pedriali, G.; Perrone, M.; Previati, M.; Wieckowski, M.R.; Giorgi, C. Mitochondria-associated membranes (MAMs) and inflammation. Cell Death Dis. 2018, 9, 329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, S.J.; Kim, S.-m.; Lee, S.-h.; Moon, J.-Y.; Hwang, H.S.; Kim, J.S.; Park, S.-H.; Jeong, K.H.; Kim, Y.G. Rhabdomyolysis-Induced AKI Was Ameliorated in NLRP3 KO Mice via Alleviation of Mitochondrial Lipid Peroxidation in Renal Tubular Cells. Int. J. Mol. Sci. 2020, 21, 8564. https://doi.org/10.3390/ijms21228564
Song SJ, Kim S-m, Lee S-h, Moon J-Y, Hwang HS, Kim JS, Park S-H, Jeong KH, Kim YG. Rhabdomyolysis-Induced AKI Was Ameliorated in NLRP3 KO Mice via Alleviation of Mitochondrial Lipid Peroxidation in Renal Tubular Cells. International Journal of Molecular Sciences. 2020; 21(22):8564. https://doi.org/10.3390/ijms21228564
Chicago/Turabian StyleSong, Seok Jong, Su-mi Kim, Sang-ho Lee, Ju-Young Moon, Hyeon Seok Hwang, Jin Sug Kim, Seon-Hwa Park, Kyung Hwan Jeong, and Yang Gyun Kim. 2020. "Rhabdomyolysis-Induced AKI Was Ameliorated in NLRP3 KO Mice via Alleviation of Mitochondrial Lipid Peroxidation in Renal Tubular Cells" International Journal of Molecular Sciences 21, no. 22: 8564. https://doi.org/10.3390/ijms21228564