Heregulin Drives Endocrine Resistance by Altering IL-8 Expression in ER-Positive Breast Cancer

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
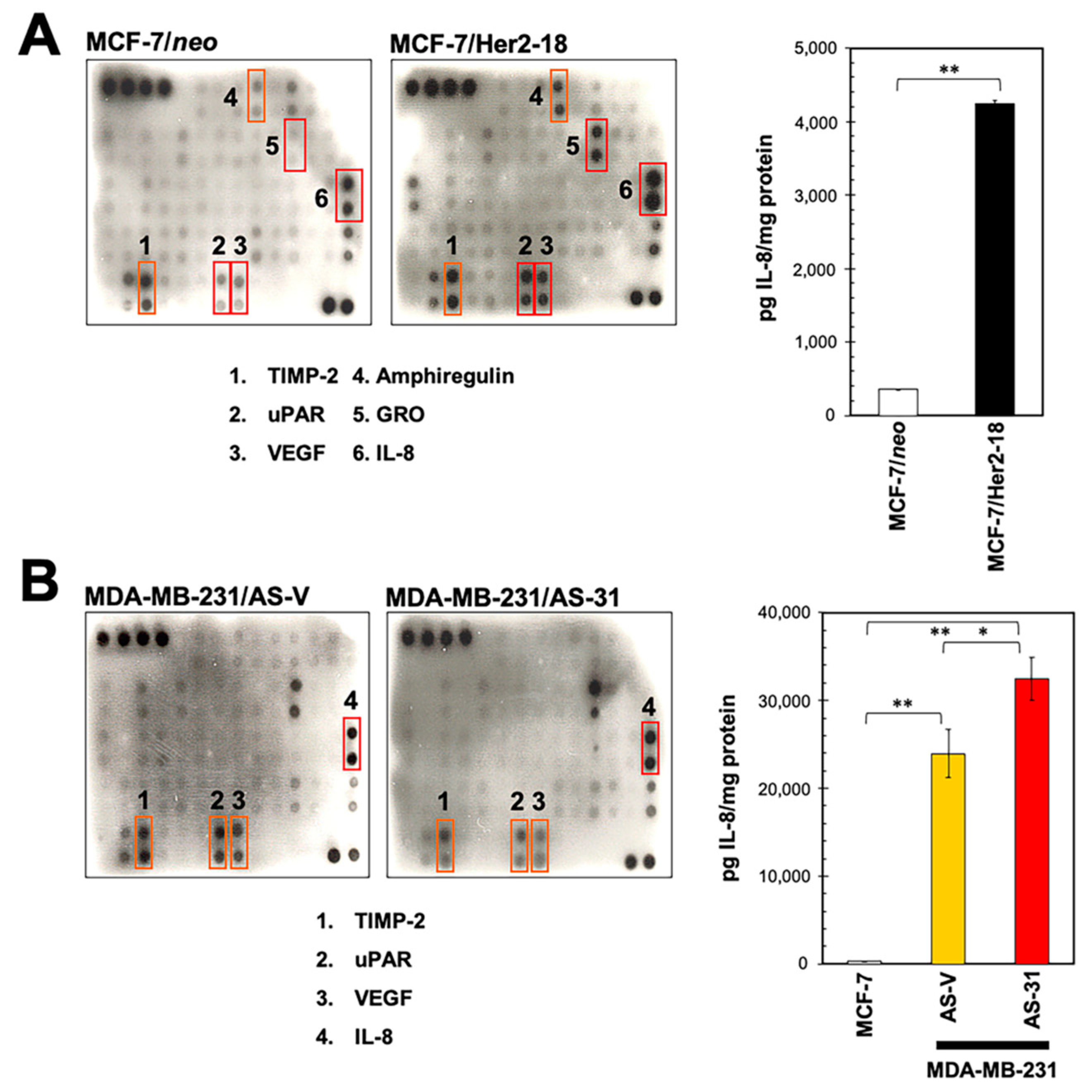
2.1. Identification of IL-8 as an HRG-Driven Cytokine Using Chemokine Antibody Array Technology
2.2. HRG Overexpression in HER2-Negative Breast Cancer Cells Qualitatively Phenocopies the IL-8 Cytokine Signature Driven by her2 Overexpression
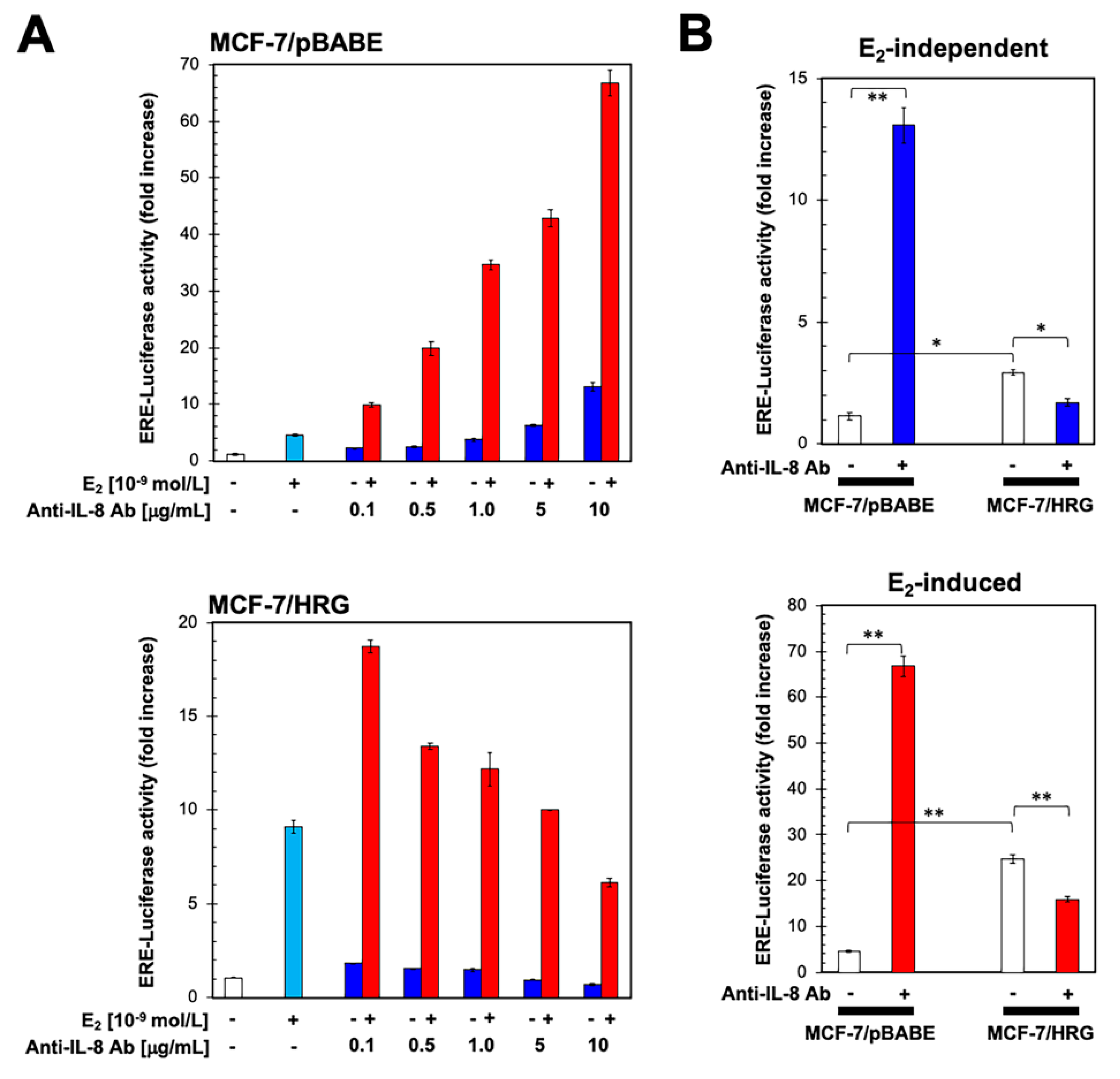
2.3. HRG-Driven Regulation of IL-8 Is ER-Dependent
2.4. Functional Blockade of IL-8 Regulates ER Transcriptional Activity in an HRG-Dependent Manner
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Lines and Culture Conditions
4.3. Conditioned Medium
4.4. Cytokine Antibody Arrays
4.5. IL-8 ELISA
4.6. ER Transcriptional Activity
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| HRG | Heregulin |
| ER | Estrogen Receptor |
| IL-8 | Interleukin-8 |
References
- McGuire, W.L. Endocrine therapy of breast cancer. Annu. Rev. Med. 1975, 26, 353–363. [Google Scholar] [CrossRef]
- Ring, A.; Dowsett, M. Mechanisms of tamoxifen resistance. Endocr. Relat. Cancer 2004, 11, 643–658. [Google Scholar] [CrossRef]
- Rondón-Lagos, M.; Villegas, V.E.; Rangel, N.; Sánchez, M.C.; Zaphiropoulos, P.G. Tamoxifen resistance: Emerging molecular targets. Int. J. Mol. Sci. 2016, 17, 1357. [Google Scholar]
- Creighton, C.J.; Massarweh, S.; Huang, S.; Tsimelzon, A.; Hilsenbeck, S.G.; Osborne, C.K.; Shou, J.; Malorni, L.; Schiff, R. Development of resistance to targeted therapies transforms the clinically associated molecular profile subtype of breast tumor xenografts. Cancer Res. 2008, 68, 7493–7501. [Google Scholar] [CrossRef] [Green Version]
- Arpino, G.; De Angelis, C.; Giuliano, M.; Giordano, A.; Falato, C.; De Laurentiis, M.; De Placido, S. Molecular mechanism and clinical implications of endocrine therapy resistance in breast cancer. Oncology 2009, 77, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Chang, J.; Fu, P. Endocrine therapy resistance in breast cancer: Current status, possible mechanisms and overcoming strategies. Future Med. Chem. 2015, 7, 1511–1519. [Google Scholar] [CrossRef] [Green Version]
- Haque, M.M.; Desai, K.V. Pathways to Endocrine Therapy Resistance in Breast Cancer. Front. Endocrinol. Lausanne 2019, 10, 573. [Google Scholar] [CrossRef] [PubMed]
- Loi, S.; Sotiriou, C.; Haibe-Kains, B.; Lallemand, F.; Conus, N.M.; Piccart, M.J.; Speed, T.P.; McArthur, G.A. Gene expression profiling identifies activated growth factor signaling in poor prognosis (Luminal-B) estrogen receptor positive breast cancer. BMC Med. Genomics 2009, 2, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tovey, S.; Dunne, B.; Witton, C.J.; Forsyth, A.; Cooke, T.G.; Bartlett, J.M. Can molecular markers predict when to implement treatment with aromatase inhibitors in invasive breast cancer? Clin. Cancer Res. 2005, 11, 4835–4842. [Google Scholar] [CrossRef] [Green Version]
- Tang, C.K.; Perez, C.; Grunt, T.; Waibel, C.; Cho, C.; Lupu, R. Involvement of heregulin-beta2 in the acquisition of the hormone-independent phenotype of breast cancer cells. Cancer Res. 1996, 56, 3350–3358. [Google Scholar]
- Saceda, M.; Grunt, T.W.; Colomer, R.; Lippman, M.E.; Lupu, R.; Martin, M.B. Regulation of estrogen receptor concentration and activity by an erbB/HER ligand in breast carcinoma cell lines. Endocrinology 1996, 137, 4322–4330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lupu, R.; Cardillo, M.; Cho, C.; Harris, L.; Hijazi, M.; Perez, C.; Rosenberg, K.; Yang, D.; Tang, C. The significance of heregulin in breast cancer tumor progression and drug resistance. Breast Cancer Res. Treat. 1996, 38, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Hutcheson, I.R.; Knowlden, J.M.; Hiscox, S.E.; Barrow, D.; Gee, J.M.; Robertson, J.F.; Ellis, I.O.; Nicholson, R.I. Heregulin beta1 drives gefitinib-resistant growth and invasion in tamoxifen-resistant MCF-7 breast cancer cells. Breast Cancer Res. 2007, 9, R50. [Google Scholar] [CrossRef] [Green Version]
- Hamburger, A.W. The role of ErbB3 and its binding partners in breast cancer progression and resistance to hormone and tyrosine kinase directed therapies. J. Mammary Gland Biol. Neoplasia 2008, 13, 225–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Lee, J.; Kim, C.; Choi, J.; Kim, A. Anti-cancer effect of metformin by suppressing signaling pathway of HER2 and HER3 in tamoxifen-resistant breast cancer cells. Tumour Biol. 2016, 37, 5811–5819. [Google Scholar] [CrossRef]
- Benz, C.C.; Scott, G.K.; Sarup, J.C.; Johnson, R.M.; Tripathy, D.; Coronado, E.; Shepard, H.M.; Osborne, C.K. Estrogen-dependent, tamoxifen-resistant tumorigenic growth of MCF-7 cells transfected with HER2/neu. Breast Cancer Res. Treat. 1992, 24, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Shou, J.; Massarweh, S.; Osborne, C.K.; Wakeling, A.E.; Ali, S.; Weiss, H.; Schiff, R. Mechanisms of tamoxifen resistance: Increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J. Natl. Cancer Inst. 2004, 96, 926–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massarweh, S.; Osborne, C.K.; Creighton, C.J.; Qin, L.; Tsimelzon, A.; Huang, S.; Weiss, H.; Rimawi, M.; Schiff, R. Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function. Cancer Res. 2008, 68, 826–833. [Google Scholar] [CrossRef] [Green Version]
- Freund, A.; Chauveau, C.; Brouillet, J.P.; Lucas, A.; Lacroix, M.; Licznar, A.; Vignon, F.; Lazennec, G. IL-8 expression and its possible relationship with estrogen-receptor-negative status of breast cancer cells. Oncogene 2003, 22, 256–265. [Google Scholar] [CrossRef] [Green Version]
- Freund, A.; Jolivel, V.; Durand, S.; Kersual, N.; Chalbos, D.; Chavey, C.; Vignon, F.; Lazennec, G. Mechanisms underlying differential expression of interleukin-8 in breast cancer cells. Oncogene 2004, 23, 6105–6114. [Google Scholar] [CrossRef] [Green Version]
- Yao, C.; Lin, Y.; Chua, M.S.; Ye, C.S.; Bi, J.; Li, W.; Zhu, Y.F.; Wang, S.M. Interleukin-8 modulates growth and invasiveness of estrogen receptor-negative breast cancer cells. Int. J. Cancer 2007, 121, 1949–1957. [Google Scholar] [CrossRef]
- Chavey, C.; Mühlbauer, M.; Bossard, C.; Freund, A.; Durand, S.; Jorgensen, C.; Jobin, C.; Lazennec, G. Interleukin-8 expression is regulated by histone deacetylases through the nuclear factor-kappaB pathway in breast cancer. Mol. Pharmacol. 2008, 74, 1359–1366. [Google Scholar] [CrossRef] [PubMed]
- Todorović-Raković, N.; Milovanović, J. Interleukin-8 in breast cancer progression. J. Interferon Cytokine Res. 2013, 33, 563–570. [Google Scholar] [CrossRef]
- Milovanovic, J.; Todorovic-Rakovic, N.; Vujasinovic, T.; Abu Rabi, Z. Interleukin 8 in progression of hormone-dependent early breast cancer. J. Biosci. 2017, 42, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Milovanovic, J.; Todorovic-Rakovic, N.; Abu Rabi, Z. The role of interleukin 8 and matrix metalloproteinases 2 and 9 in breast cancer treated with tamoxifen. J. BUON 2017, 22, 628–637. [Google Scholar] [PubMed]
- Milovanović, J.; Todorović-Raković, N.; Radulovic, M. Interleukin-6 and interleukin-8 serum levels in prognosis of hormone-dependent breast cancer. Cytokine 2019, 118, 93–98. [Google Scholar] [CrossRef]
- Vazquez-Martin, A.; Colomer, R.; Menendez, J.A. Protein array technology to detect HER2 (erbB-2)-induced ’cytokine signature’ in breast cancer. Eur. J. Cancer 2007, 43, 1117–1124. [Google Scholar] [CrossRef] [PubMed]
- Atlas, E.; Cardillo, M.; Mehmi, I.; Zahedkargaran, H.; Tang, C.; Lupu, R. Heregulin is sufficient for the promotion of tumorigenicity and metastasis of breast cancer cells in vivo. Mol. Cancer Res. 2003, 1, 165–175. [Google Scholar] [PubMed]
- Atlas, E.; Bojanowski, K.; Mehmi, I.; Lupu, R. A deletion mutant of heregulin increases the sensitivity of breast cancer cells to chemotherapy without promoting tumorigenicity. Oncogene 2003, 22, 3441–3451. [Google Scholar] [CrossRef] [Green Version]
- Tsai, M.S.; Shamon-Taylor, L.A.; Mehmi, I.; Tang, C.K.; Lupu, R. Blockage of heregulin expression inhibits tumorigenicity and metastasis of breast cancer. Oncogene 2003, 22, 761–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vellon, L.; Menendez, J.A.; Lupu, R. AlphaVbeta3 integrin regulates heregulin (HRG)-induced cell proliferation and survival in breast cancer. Oncogene 2005, 24, 3759–3773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vellon, L.; Menendez, J.A.; Lupu, R. A bidirectional “alpha(v)beta(3) integrin-ERK1/ERK2 MAPK” connection regulates the proliferation of breast cancer cells. Mol. Carcinog. 2006, 45, 795–804. [Google Scholar] [CrossRef]
- Menendez, J.A.; Rubio, M.A.; Campisi, J.; Lupu, R. Heregulin, a new regulator of telomere length in human cells. Oncotarget 2015, 6, 39422–39436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eastman, B.M.; Jo, M.; Webb, D.L.; Takimoto, S.; Gonias, S.L. A transformation in the mechanism by which the urokinase receptor signals provides a selection advantage for estrogen receptor-expressing breast cancer cells in the absence of estrogen. Cell Signal. 2012, 24, 1847–1855. [Google Scholar] [CrossRef] [Green Version]
- Ciarloni, L.; Mallepell, S.; Brisken, C. Amphiregulin is an essential mediator of estrogen receptor alpha function in mammary gland development. Proc. Natl. Acad. Sci. USA 2007, 104, 5455–5460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, E.A.; Jenkins, E.C.; Lofgren, K.A.; Chandiramani, N.; Liu, H.; Aranda, E.; Barnett, M.; Kenny, P.A. Amphiregulin Is a Critical Downstream Effector of Estrogen Signaling in ERα-Positive Breast Cancer. Cancer Res. 2015, 75, 4830–4838. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Masri, S.; Phung, S.; Chen, S. The role of amphiregulin in exemestane-resistant breast cancer cells: Evidence of an autocrine loop. Cancer Res. 2008, 68, 2259–2265. [Google Scholar] [CrossRef] [Green Version]
- Dobrzycka, K.M.; Townson, S.M.; Jiang, S.; Oesterreich, S. Estrogen receptor corepressors—A role in human breast cancer? Endocr. Relat. Cancer 2003, 10, 517–536. [Google Scholar] [CrossRef] [Green Version]
- Girault, I.; Bièche, I.; Lidereau, R. Role of estrogen receptor alpha transcriptional coregulators in tamoxifen resistance in breast cancer. Maturitas 2006, 54, 342–351. [Google Scholar] [CrossRef]
- van Agthoven, T.; Sieuwerts, A.M.; Veldscholte, J.; Meijer-van Gelder, M.E.; Smid, M.; Brinkman, A.; den Dekker, A.T.; Leroy, I.M.; van Ijcken, W.F.; Sleijfer, S.; et al. CITED2 and NCOR2 in anti-oestrogen resistance and progression of breast cancer. Br. J. Cancer 2009, 101, 1824–1832. [Google Scholar] [CrossRef] [Green Version]
- Légaré, S.; Basik, M. Minireview: The Link between ERα Corepressors and Histone Deacetylases in Tamoxifen Resistance in Breast Cancer. Mol. Endocrinol. 2016, 30, 965–976. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.J.; Joo, H.S.; Won, H.Y.; Min, K.W.; Kim, H.Y.; Son, T.; Oh, Y.H.; Lee, J.Y.; Kong, G. Role of RBP2-Induced ER and IGF1R-ErbB Signaling in Tamoxifen Resistance in Breast Cancer. J. Natl. Cancer Inst. 2018, 110. [Google Scholar] [CrossRef] [PubMed]
- Padró, M.; Louie, R.J.; Lananna, B.V.; Krieg, A.J.; Timmerman, L.A.; Chan, D.A. Genome-independent hypoxic repression of estrogen receptor alpha in breast cancer cells. BMC Cancer 2017, 17, 203. [Google Scholar] [CrossRef] [Green Version]
- Stoner, M.; Saville, B.; Wormke, M.; Dean, D.; Burghardt, R.; Safe, S. Hypoxia induces proteasome-dependent degradation of estrogen receptor alpha in ZR-75 breast cancer cells. Mol. Endocrinol. 2002, 16, 2231–2242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, J.M.; Kwon, H.Y.; Cho, J.Y.; Lee, Y.J. Estrogen and hypoxia regulate estrogen receptor alpha in a synergistic manner. Biochem. Biophys. Res. Commun. 2009, 378, 842–846. [Google Scholar] [CrossRef] [PubMed]
- Lonard, D.M.; Nawaz, Z.; Smith, C.L.; O′Malley, B.W. The 26S proteasome is required for estrogen receptor-alpha and coactivator turnover and for efficient estrogen receptor-alpha transactivation. Mol. Cell 2000, 5, 939–948. [Google Scholar] [CrossRef]
- Aceto, N.; Duss, S.; MacDonald, G.; Meyer, D.S.; Roloff, T.C.; Hynes, N.E.; Bentires-Alj, M. Co-expression of HER2 and HER3 receptor tyrosine kinases enhances invasion of breast cells via stimulation of interleukin-8 autocrine secretion. Breast Cancer Res. 2014, 14, R131. [Google Scholar] [CrossRef] [Green Version]
- Hinohara, K.; Kobayashi, S.; Kanauchi, H.; Shimizu, S.; Nishioka, K.; Tsuji, E.; Tada, K.; Umezawa, K.; Mori, M.; Ogawa, T.; et al. ErbB receptor tyrosine kinase/NF-κB signaling controls mammosphere formation in human breast cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 6584–6589. [Google Scholar] [CrossRef] [Green Version]
- Selitrennik, M.; Lev, S. PYK2 integrates growth factor and cytokine receptors signaling and potentiates breast cancer invasion via a positive feedback loop. Oncotarget 2015, 6, 22214–22226. [Google Scholar] [CrossRef] [Green Version]
- Galien, R.; Garcia, T. Estrogen receptor impairs interleukin-6 expression by preventing protein binding on the NF-kappaB site. Nucleic Acids Res. 1997, 25, 2424–2429. [Google Scholar] [CrossRef] [Green Version]
- Speirs, V.; Kerin, M.J.; Walton, D.S.; Newton, C.J.; Desai, S.B.; Atkin, S.L. Direct activation of oestrogen receptor-alpha by interleukin-6 in primary cultures of breast cancer epithelial cells. Br. J. Cancer 2000, 82, 1312–1316. [Google Scholar] [CrossRef]
- Sasser, A.K.; Sullivan, N.J.; Studebaker, A.W.; Hendey, L.F.; Axel, A.E.; Hall, B.M. Interleukin-6 is a potent growth factor for ER-alpha-positive human breast cancer. FASEB J. 2007, 21, 3763–3770. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, N.J.; Sasser, A.K.; Axel, A.E.; Vesuna, F.; Raman, V.; Ramirez, N.; Oberyszyn, T.M.; Hall, B.M. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene 2009, 28, 2940–2947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casneuf, T.; Axel, A.E.; King, P.; Alvarez, J.D.; Werbeck, J.L.; Verhulst, T.; Verstraeten, K.; Hall, B.M.; Sasser, A.K. Interleukin-6 is a potential therapeutic target in interleukin-6 dependent, estrogen receptor-α-positive breast cancer. Breast Cancer 2016, 8, 13–27. [Google Scholar]
- Yamaguchi, N.; Nakayama, Y.; Yamaguchi, N. Down-regulation of Forkhead box protein A1 (FOXA1) leads to cancer stem cell-like properties in tamoxifen-resistant breast cancer cells through induction of interleukin-6. J. Biol. Chem. 2017, 292, 8136–8148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, J.; Li, J.; Fu, L.; Gai, J.; Guan, J.; Li, Q. SIRT4 enhances the sensitivity of ER-positive breast cancer to tamoxifen by inhibiting the IL-6/STAT3 signal pathway. Cancer Med. 2019, 8, 7086–7097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, X.; Jeselsohn, R.; Pereira, R.; Hollingsworth, E.F.; Creighton, C.J.; Li, F.; Shea, M.; Nardone, A.; De Angelis, C.; Heiser, L.M.; et al. FOXA1 overexpression mediates endocrine resistance by altering the ER transcriptome and IL-8 expression in ER-positive breast cancer. Proc. Natl. Acad. Sci. USA 2016, 113, E6600–E6609. [Google Scholar] [CrossRef] [Green Version]
- Singh, J.K.; Simões, B.M.; Howell, S.J.; Farnie, G.; Clarke, R.B. Recent advances reveal IL-8 signaling as a potential key to targeting breast cancer stem cells. Breast Cancer Res. 2013, 15, 210. [Google Scholar] [CrossRef] [Green Version]
- Singh, J.K.; Farnie, G.; Bundred, N.J.; Simões, B.M.; Shergill, A.; Landberg, G.; Howell, S.J.; Clarke, R.B. Targeting CXCR1/2 significantly reduces breast cancer stem cell activity and increases the efficacy of inhibiting HER2 via HER2-dependent and -independent mechanisms. Clin. Cancer Res. 2013, 19, 643–656. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, R.; Narasanna, A.; Wang, S.E.; Liu, S.; Chakrabarty, A.; Balko, J.M.; González-Angulo, A.M.; Mills, G.B.; Penuel, E.; Winslow, J.; et al. Trastuzumab has preferential activity against breast cancers driven by HER2 homodimers. Cancer Res. 2011, 71, 1871–1882. [Google Scholar] [CrossRef] [Green Version]
- Charafe-Jauffret, E.; Ginestier, C.; Iovino, F.; Wicinski, J.; Cervera, N.; Finetti, P.; Hur, M.H.; Diebel, M.E.; Monville, F.; Dutcher, J.; et al. Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Res. 2009, 69, 1302–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginestier, C.; Liu, S.; Diebel, M.E.; Korkaya, H.; Luo, M.; Brown, M.; Wicinski, J.; Cabaud, O.; Charafe-Jauffret, E.; Birnbaum, D.; et al. CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J. Clin. Investig. 2010, 120, 485–497. [Google Scholar] [CrossRef]
- Korkaya, H.; Liu, S.; Wicha, M.S. Regulation of cancer stem cells by cytokine networks: Attacking cancer’s inflammatory roots. Clin. Cancer Res. 2011, 17, 6125–6129. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papadimitropoulou, A.; Vellon, L.; Atlas, E.; Steen, T.V.; Cuyàs, E.; Verdura, S.; Espinoza, I.; Menendez, J.A.; Lupu, R. Heregulin Drives Endocrine Resistance by Altering IL-8 Expression in ER-Positive Breast Cancer. Int. J. Mol. Sci. 2020, 21, 7737. https://doi.org/10.3390/ijms21207737
Papadimitropoulou A, Vellon L, Atlas E, Steen TV, Cuyàs E, Verdura S, Espinoza I, Menendez JA, Lupu R. Heregulin Drives Endocrine Resistance by Altering IL-8 Expression in ER-Positive Breast Cancer. International Journal of Molecular Sciences. 2020; 21(20):7737. https://doi.org/10.3390/ijms21207737
Chicago/Turabian StylePapadimitropoulou, Adriana, Luciano Vellon, Ella Atlas, Travis Vander Steen, Elisabet Cuyàs, Sara Verdura, Ingrid Espinoza, Javier A. Menendez, and Ruth Lupu. 2020. "Heregulin Drives Endocrine Resistance by Altering IL-8 Expression in ER-Positive Breast Cancer" International Journal of Molecular Sciences 21, no. 20: 7737. https://doi.org/10.3390/ijms21207737