IKKβ Kinase Promotes Stemness, Migration, and Invasion in KRAS-Driven Lung Adenocarcinoma Cells

 , ,
, ,  , and
, and
Abstract
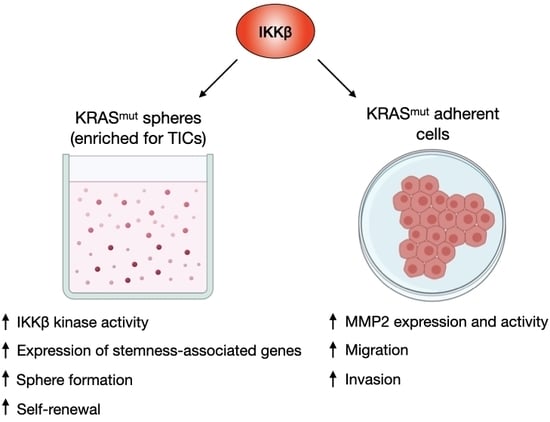
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
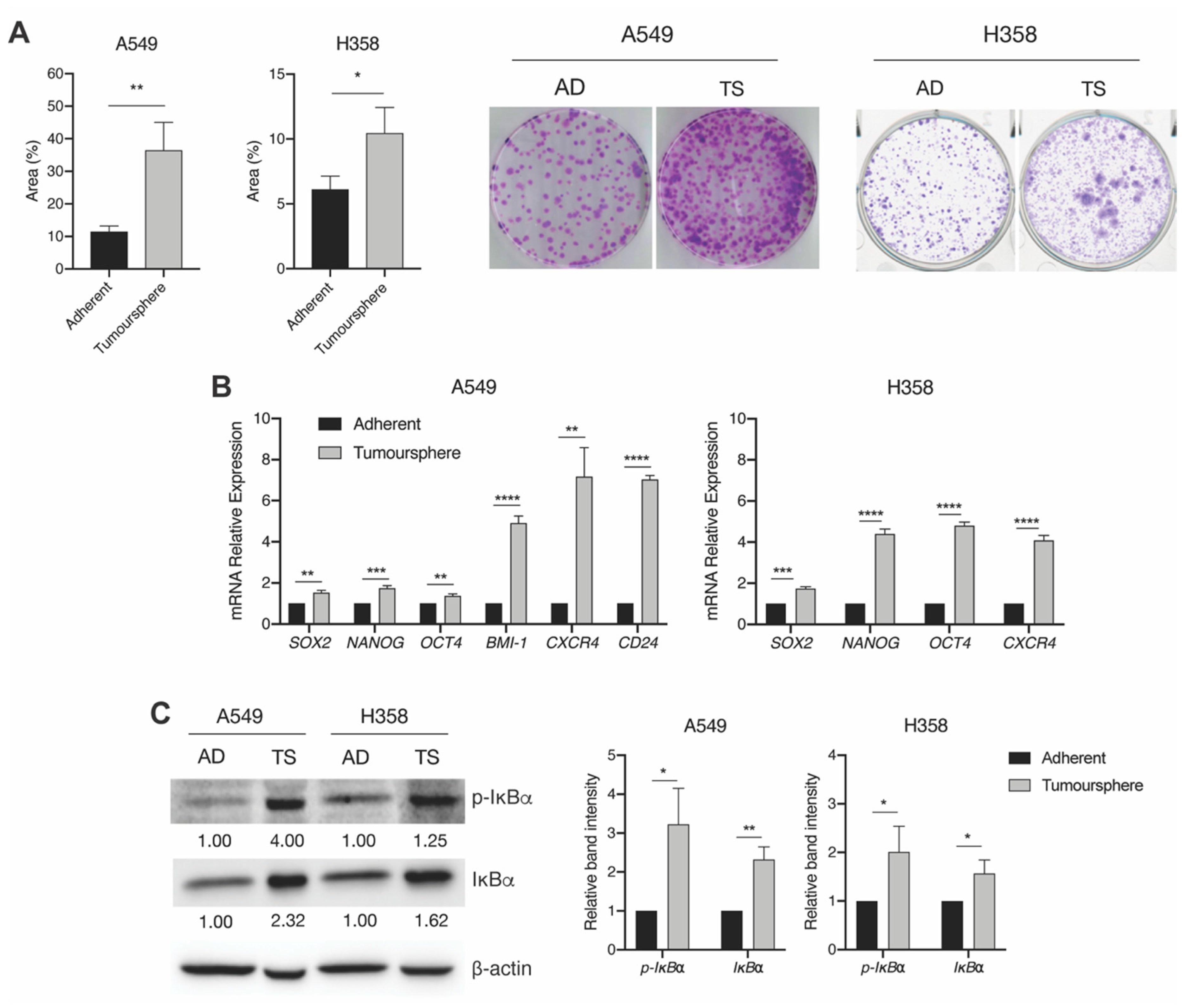
2.1. KRAS-Mutant Lung Tumoursphere-Derived Cells Exhibit Stemness Features and Increased IKKβ Kinase Activity
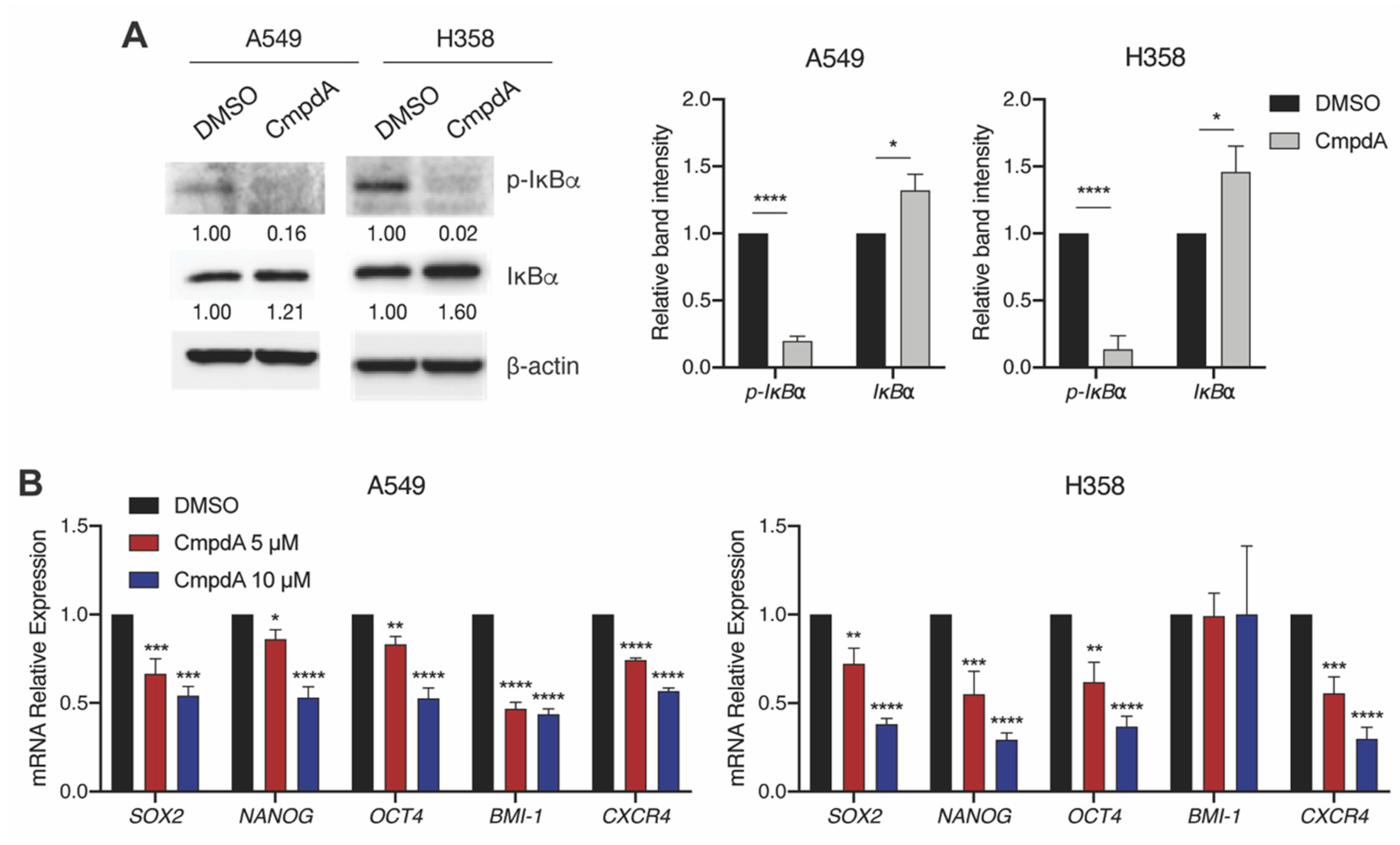
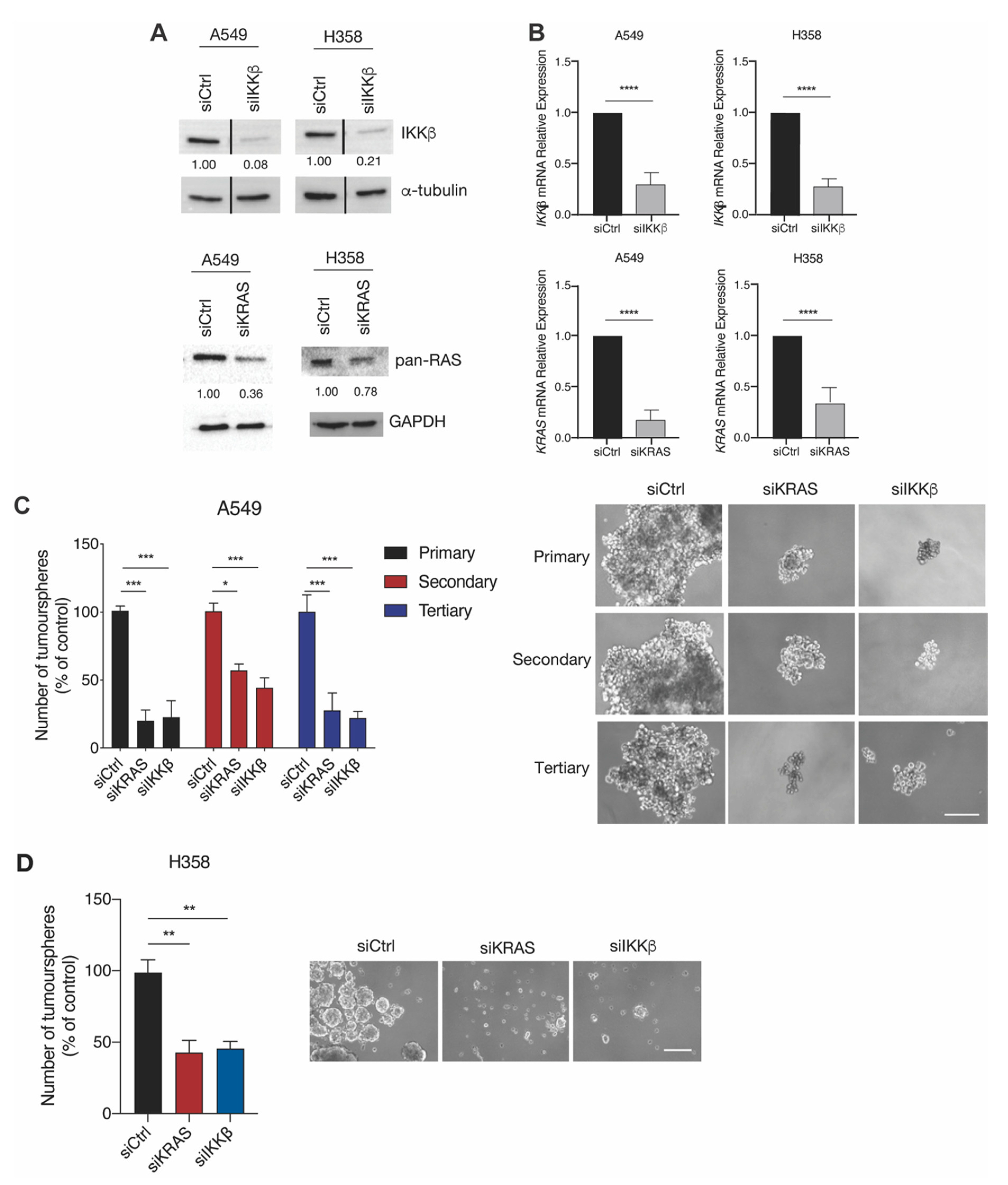
2.2. IKKβ Targeting in KRAS-Positive Lung Cancer Cells Reduces the Expression of Stemness-Associated Genes
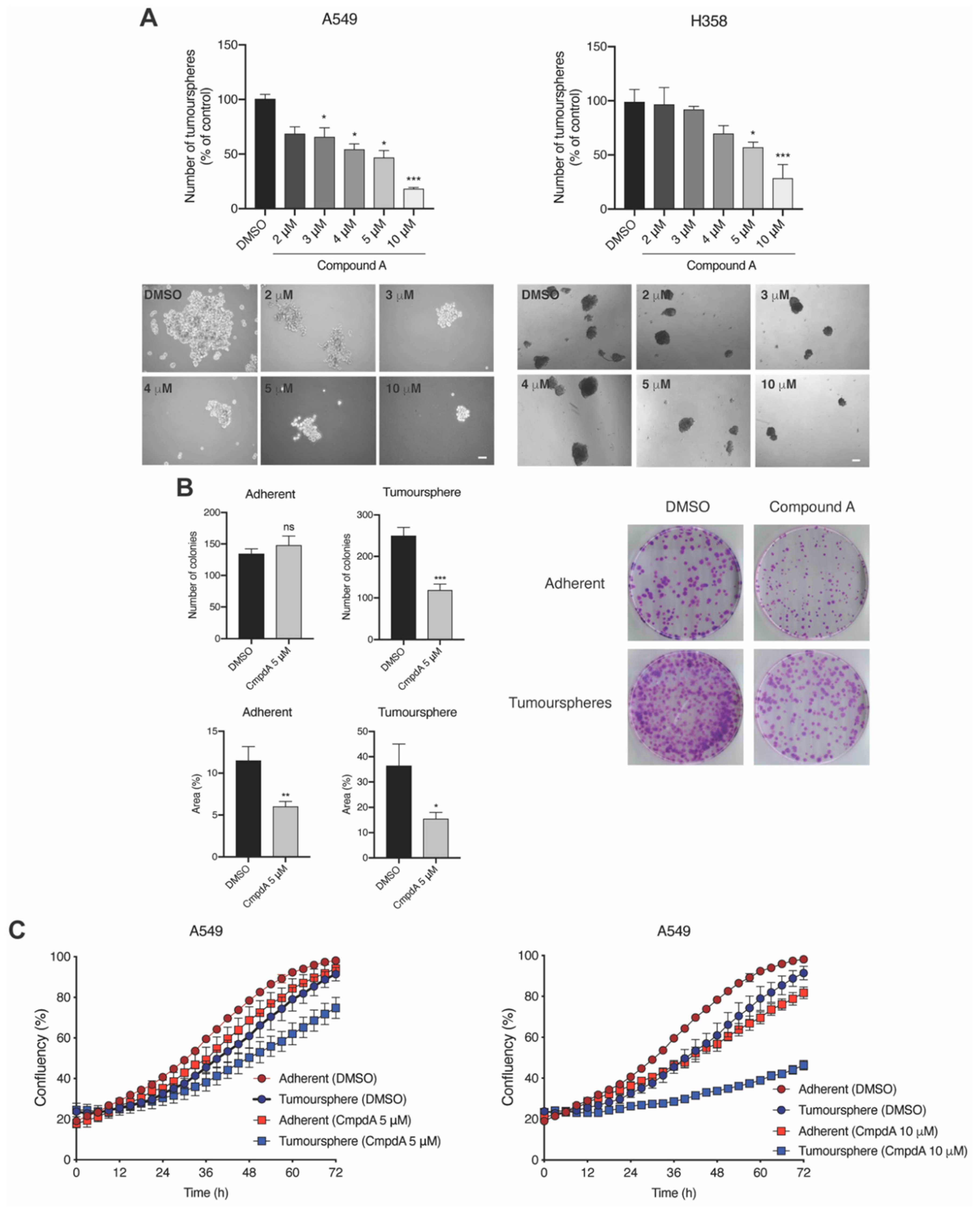
2.3. IKKβ Targeting Reduces Tumoursphere Formation, Self-Renewal, and Preferentially Impairs Proliferation of TIC-Enriched KRAS-Mutant Cells
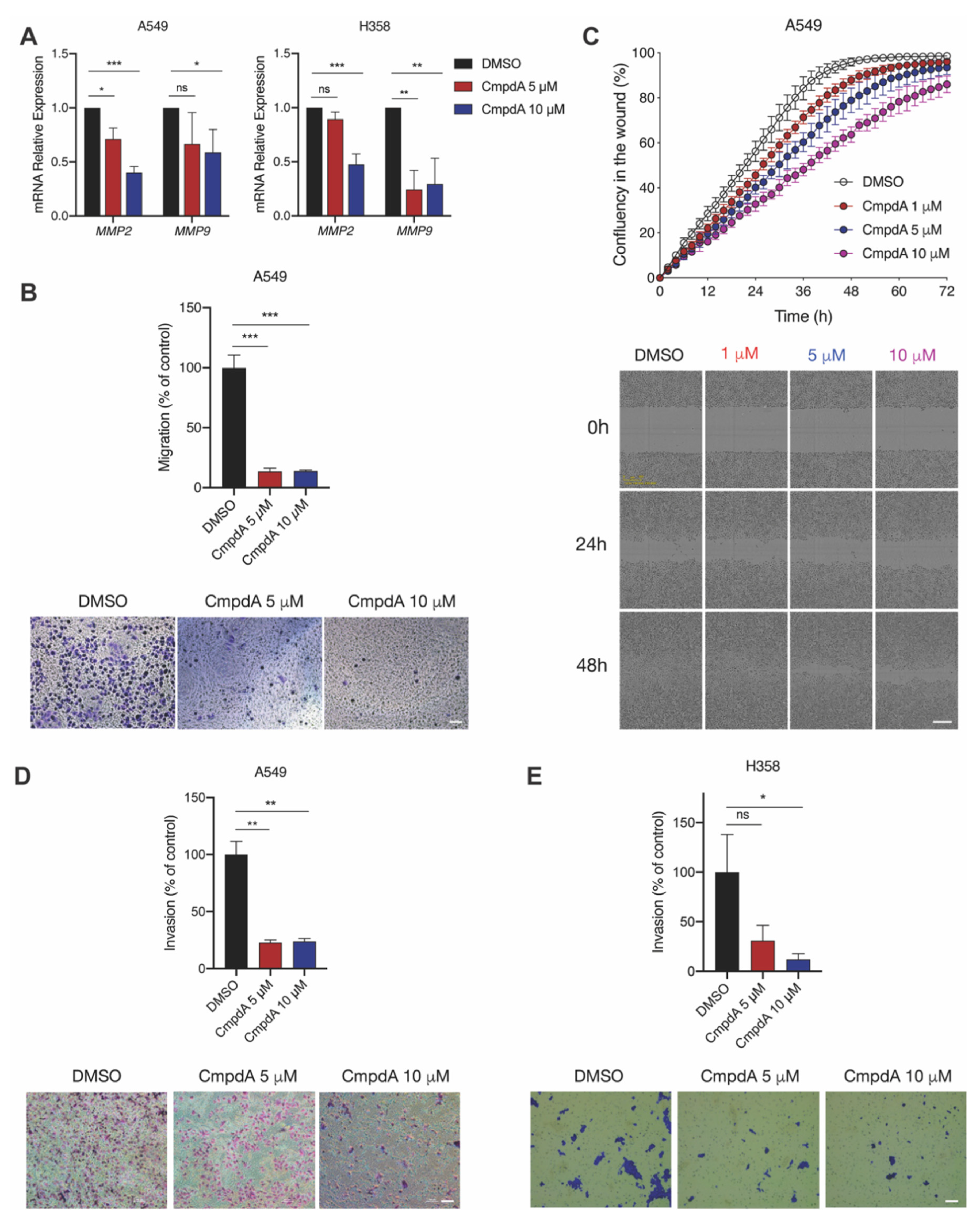
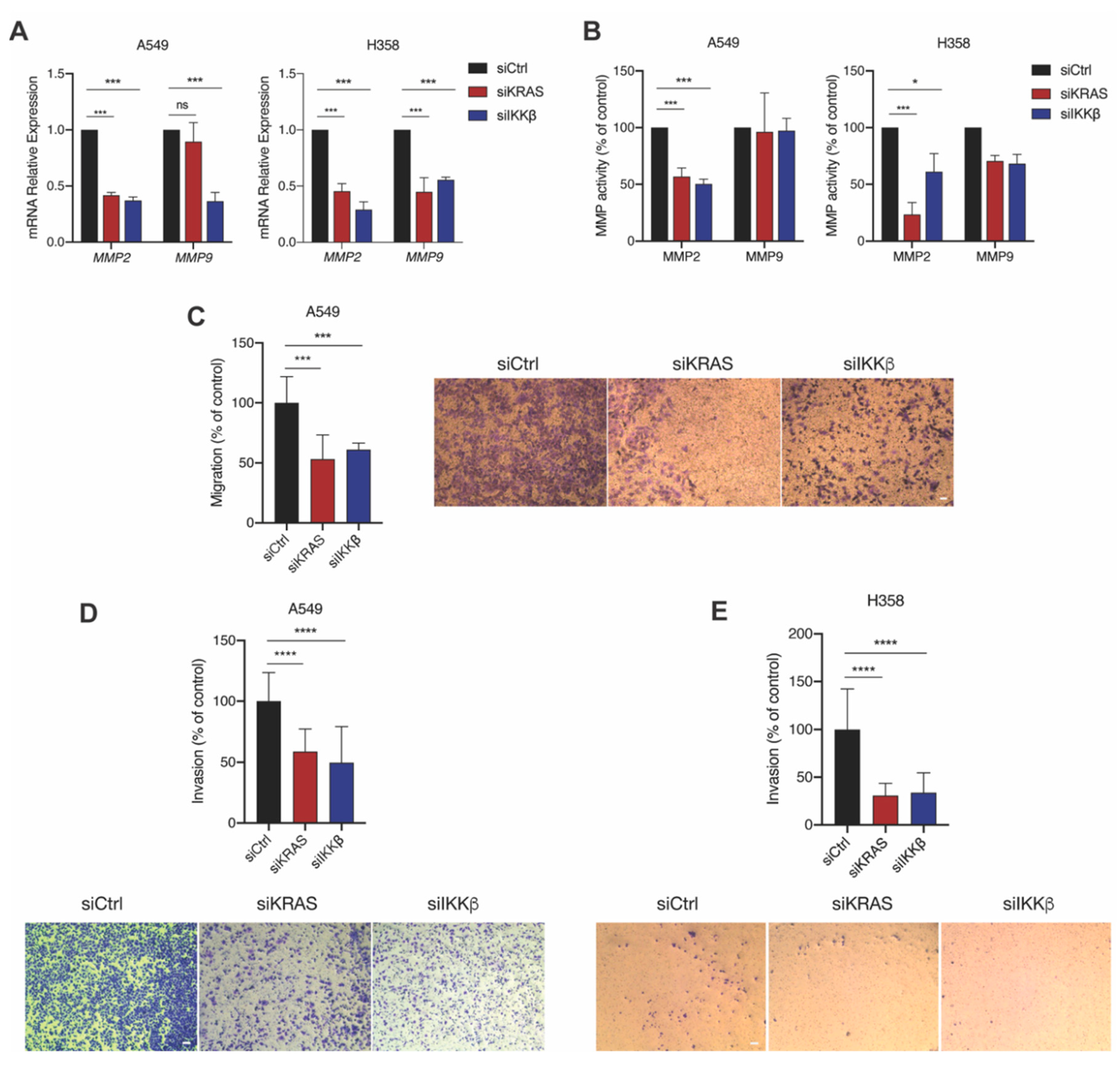
2.4. IKKβ Kinase Targeting Reduces KRAS-Mutant Lung Cell Migration and Invasion
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture Conditions
4.2. Tumoursphere Formation
4.3. siRNA Transfections
4.4. Clonogenic Assay
4.5. Quantitative Real-Time Polymerase Chain Reaction (qPCR)
4.6. Western Blotting
4.7. Cell Proliferation, Cell Death and Wound-Healing Assays
4.8. Transwell Migration Assays
4.9. Invasion Assay
4.10. Measurement of MMP2 and MMP9 Activity
4.11. Statistical Analysis
4.12. Data Availability
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BMI1 | B cell-specific Moloney murine leukaemia virus integration site 1 |
| CmpdA | Compound A |
| CD24 | Cluster of differentiation 24 |
| CXCR-4 | C-X-C chemokine receptor type 4 |
| DMSO | Dimethyl sulfoxide |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| IKKβ | Inhibitor of nuclear factor kappa-B kinase β |
| IκB | Inhibitor of nuclear factor kappa-B |
| MMP2 | Matrix metalloproteinase 2 |
| MMP9 | Matrix metalloproteinase 9 |
| NF-κB | Nuclear factor-kappa-light-chain-enhancer of activated B cells |
| OCT4 | Octamer-binding transcription factor 4 |
| SOX2 | SRY (sex determining region Y)-box 2 |
| siRNA | Small interfering RNA |
| TICs | Tumour-initiating cells |
References
- Ferrer, I.; Zugazagoitia, J.; Herbertz, S.; John, W.; Paz-Ares, L.; Schmid-Bindert, G. KRAS-Mutant non-small cell lung cancer: From biology to therapy. Lung Cancer 2018, 124, 53–64. [Google Scholar] [CrossRef] [Green Version]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef] [Green Version]
- Lito, P.; Solomon, M.; Li, L.S.; Hansen, R.; Rosen, N. Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science 2016, 351, 604–608. [Google Scholar] [CrossRef] [Green Version]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Papke, B.; Der, C.J. Drugging RAS: Know the enemy. Science 2017, 355, 1158–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visvader, J.E.; Lindeman, G.J. Cancer Stem cells: Current status and evolving complexities. Cell Stem Cell 2012, 10, 717–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eramo, A.; Lotti, F.; Sette, G.; Pilozzi, E.; Biffoni, M.; Di Virgilio, A.; Conticello, C.; Ruco, L.; Peschle, C.; de Maria, R.; et al. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008, 15, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Fessler, E.; Dijkgraaf, F.E.; De Sousa, E.; Melo, F.; Medema, J.P. Cancer stem cell dynamics in tumor progression and metastasis: Is the microenvironment to blame? Cancer Lett. 2013, 341, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.C.; Chen, H.C. p120RasGAP-mediated activation of c-Src is critical for oncogenic Ras to induce tumor invasion. Cancer Res. 2012, 72, 2405–2415. [Google Scholar] [CrossRef] [Green Version]
- Kim, R.K.; Suh, Y.; Yoo, K.C.; Cui, Y.H.; Kim, H.; Kim, M.J.; Gyu Kim, I.; Lee, S.J. Activation of KRAS promotes the mesenchymal features of basal-type breast cancer. Exp. Mol. Med. 2015, 47, e137. [Google Scholar] [CrossRef] [Green Version]
- Boutin, A.T.; Liao, W.T.; Wang, M.; Hwang, S.S.; Karpinets, T.V.; Cheung, H.; Chu, G.C.; Jiang, S.; Hu, J.; Chang, K.; et al. Oncogenic Kras drives invasion and maintains metastases in colorectal cancer. Genes Dev. 2017, 31, 370–382. [Google Scholar] [CrossRef] [Green Version]
- Kim, R.J.; Kim, S.R.; Roh, K.J.; Park, S.B.; Park, J.R.; Kang, K.S.; Kong, G.; Tang, B.; Yang, Y.; Kohn, E.A.; et al. Ras activation contributes to the maintenance and expansion of Sca-1pos cells in a mouse model of breast cancer. Cancer Lett. 2010, 287, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Seguin, L.; Kato, S.; Franovic, A.; Camargo, M.F.; Lesperance, J.; Elliott, K.C.; Yebra, M.; Mielgo, A.; Lowy, A.M.; Husain, H.; et al. An integrin β3–KRAS–RalB complex drives tumour stemness and resistance to EGFR inhibition. Nat. Cell Biol. 2014, 16, 457–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.T.; Holderfield, M.; Galeas, J.; Delrosario, R.; To, M.D.; Balmain, A.; McCormick, F. K-Ras promotes tumorigenicity through suppression of Non-canonical Wnt signaling. Cell 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, S.A.; Justilien, V.; Jamieson, L.; Murray, N.R.; Fields, A.P. Protein kinase C Drives a NOTCH3-dependent stem-like phenotype in mutant KRAS lung adenocarcinoma. Cancer Cell 2016, 29, 367–378. [Google Scholar] [CrossRef] [Green Version]
- Weng, C.C.; Ding, P.Y.; Liu, Y.H.; Hawse, J.R.; Subramaniam, M.; Wu, C.C.; Lin, Y.C.; Chen, C.Y.; Hung, W.C.; Cheng, K.H.; et al. Mutant Kras-induced upregulation of CD24 enhances prostate cancer stemness and bone metastasis. Oncogene 2019, 38, 2005–2019. [Google Scholar] [CrossRef] [Green Version]
- Bassères, D.S.; Ebbs, A.; Levantini, E.; Baldwin, A.S. Requirement of the NF-κB subunit p65/RelA for K-Ras–induced lung tumorigenesis. Cancer Res. 2010, 70, 3537–3546. [Google Scholar] [CrossRef] [Green Version]
- Meylan, E.; Dooley, A.L.; Feldser, D.M.; Shen, L.; Turk, E.; Ouyang, C.; Jacks, T. Requirement for NF-κB signalling in a mouse model of lung adenocarcinoma. Nature 2009, 462, 104–107. [Google Scholar] [CrossRef]
- Bassères, D.S.; Baldwin, A.S. Nuclear factor-κB and inhibitor of κB kinase pathways in oncogenic initiation and progression. Oncogene 2006, 25, 6817–6830. [Google Scholar] [CrossRef] [Green Version]
- Rinkenbaugh, A.; Baldwin, A. The NF-κB pathway and cancer stem cells. Cells 2016, 5, 16. [Google Scholar] [CrossRef]
- Duran, A.; Linares, J.F.; Galvez, A.S.; Wikenheiser, K.; Flores, J.M.; Diaz-Meco, M.T.; Moscat, J. The signaling adaptor p62 is an important NF-κB mediator in tumorigenesis. Cancer Cell 2008, 13, 343–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassères, D.S.; Ebbs, A.; Cogswell, P.C.; Baldwin, A.S. IKK is a therapeutic target in KRAS-induced lung cancer with disrupted p53 activity. Genes Cancer 2014, 5, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, Y.; Yeddula, N.; Leblanc, M.; Ke, E.; Zhang, Y.; Oldfield, E.; Shaw, R.J.; Verma, I.M. Reduced cell proliferation by IKK2 depletion in a mouse lung-cancer model. Nat. Cell Biol. 2012, 14, 257–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carneiro-Lobo, T.C.; Scalabrini, L.C.; da Magalhães, L.S.; Cardeal, L.B.; Rodrigues, F.S.; dos Santos, E.O.; Baldwin, A.S.; Levantini, E.; Giordano, R.J.; Bassères, D.S.; et al. IKKβ targeting reduces KRAS-induced lung cancer angiogenesis in vitro and in vivo: A potential anti-angiogenic therapeutic target. Lung Cancer 2019, 130, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Bielenberg, D.R.; Zetter, B.R. The contribution of angiogenesis to the process of metastasis. Cancer J. 2015, 21, 267–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.H.; Yu, C.C.; Wang, B.Y.; Chang, W.W. Tumorsphere as an effective in vitro platform for screening anti-cancer stem cell drugs. Oncotarget 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziegelbauer, K.; Gantner, F.; Lukacs, N.W.; Berlin, A.; Fuchikami, K.; Niki, T.; Sakai, K.; Inbe, H.; Takeshita, K.; Ishimori, M.; et al. A selective novel low-molecular-weight inhibitor of I κ B kinase- β (IKK- β ) prevents pulmonary inflammation and shows broad anti-inflammatory activity. Br. J. Pharmacol. 2005, 145, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Brabletz, T.; Jung, A.; Spaderna, S.; Hlubek, F.; Kirchner, T. Migrating cancer stem cells—An integrated concept of malignant tumour progression. Nat. Rev. Cancer 2005, 5, 744–749. [Google Scholar] [CrossRef]
- Wu, Y.; Zhou, B.P. TNF-α/NF-κB/Snail pathway in cancer cell migration and invasion. Br. J. Cancer 2010, 102, 639–644. [Google Scholar] [CrossRef] [Green Version]
- Foda, H.D.; Zucker, S. Matrix metalloproteinases in cancer invasion, metastasis and angiogenesis. Drug Discov. Today 2001, 6, 478–482. [Google Scholar] [CrossRef]
- Quinlan, M.P.; Quatela, S.E.; Philips, M.R.; Settleman, J. Activated kras, but not Hras or Nras, may initiate tumors of endodermal origin via stem cell expansion. Mol. Cell. Biol. 2008, 28, 2659–2674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Casimiro, M.C.; Wang, C.; Shirley, L.A.; Jiao, X.; Katiyar, S.; Ju, X.; Li, Z.; Yu, Z.; Zhou, J.; et al. p21 CIP1 attenuates Ras- and c-Myc-dependent breast tumor epithelial mesenchymal transition and cancer stem cell-like gene expression in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 19035–19039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, R.; Li, Y.; Zhang, A.; Wang, B.; Xu, Y.; Xu, W.; Zhao, Y.; Luo, F.; Liu, Q. The acquisition of cancer stem cell-like properties and neoplastic transformation of human keratinocytes induced by arsenite involves epigenetic silencing of let-7c via Ras/NF-κB. Toxicol. Lett. 2014, 227, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Shibuya, K.; Sato, A.; Seino, S.; Suzuki, S.; Seino, M.; Kitanaka, C. Targeting the K-Ras-JNK axis eliminates cancer stem-like cells and prevents pancreatic tumor formation. Oncotarget 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Polyak, K.; Weinberg, R.A. Transitions between epithelial and mesenchymal states: Acquisition of malignant and stem cell traits. Nat. Rev. Cancer 2009, 9, 265–273. [Google Scholar] [CrossRef]
- Lock, R.; Kenific, C.M.; Leidal, A.M.; Salas, E.; Debnath, J. Autophagy-dependent production of secreted factors facilitates oncogenic RAS-driven invasion. Cancer Discov. 2014, 4, 466–479. [Google Scholar] [CrossRef] [Green Version]
- Win, H.Y.; Acevedo-Duncan, M. Atypical protein kinase C phosphorylates IKKαβ in transformed non-malignant and malignant prostate cell survival. Cancer Lett. 2008, 270, 302–311. [Google Scholar] [CrossRef]
- Villalonga, P.; Lopez-Alcala, C.; Bosch, M.; Chiloeches, A.; Rocamora, N.; Gil, J.; Marais, R.; Marshall, C.J.; Bachs, O.; Agell, N.; et al. Calmodulin binds to K-Ras, but not to H- or N-Ras, and modulates its downstream aignaling. Mol. Cell. Biol. 2001, 21, 7345–7354. [Google Scholar] [CrossRef] [Green Version]
- Dagia, N.M.; Agarwal, G.; Kamath, D.V.; Chetrapal-Kunwar, A.; Gupte, R.D.; Jadhav, M.G.; Dadarkar, S.S.; Trivedi, J.; Kulkarni-Almeida, A.A.; Kharas, F.; et al. A preferential p110α/γ PI3K inhibitor attenuates experimental inflammation by suppressing the production of proinflammatory mediators in a NF-κB-dependent manner. Am. J. Physiol. Physiol. 2010, 298, C929–C941. [Google Scholar] [CrossRef]
- Kumar, M.; Allison, D.F.; Baranova, N.N.; Wamsley, J.J.; Katz, A.J.; Bekiranov, S.; Jones, D.R.; Mayo, M.W. NF-κB regulates mesenchymal transition for the induction of non-small cell lung cancer initiating cells. PLoS ONE 2013, 8, e68597. [Google Scholar] [CrossRef] [Green Version]
- Kendellen, M.F.; Bradford, J.W.; Lawrence, C.L.; Clark, K.S.; Baldwin, A.S. Canonical and non-canonical NF-κB signaling promotes breast cancer tumor-initiating cells. Oncogene 2014, 33, 1297–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Cao, F.; Bai, L.; Liu, Y.; Xie, J.; Wang, W.; Si, Q.; Yang, J.; Chang, A.; Liu, D.; et al. IKK enforces a LIN28B/TCF7L2 positive feedback loop that promotes cancer cell stemness and metastasis. Cancer Res. 2015, 75, 1725–1735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battula, V.L.; Nguyen, K.; Sun, J.; Pitner, M.K.; Yuan, B.; Bartholomeusz, C.; Hail, N.; Andreeff, M. IKK inhibition by BMS-345541 suppresses breast tumorigenesis and metastases by targeting GD2+ cancer stem cells. Oncotarget 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.S.; Kim, D.A.; Chung, H.; Park, I.H.; Kim, B.H.; Oh, E.S.; Kang, D.H. Screening of breast cancer stem cell inhibitors using a protein kinase inhibitor library. Cancer Cell Int. 2017, 17, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Lapidus, R.G.; Liu, P.; Choi, E.Y.; Adediran, S.; Hussain, A.; Wang, X.; Liu, X.; Dan, H.C. Targeting IκB kinase β/NF-κB signaling in human prostate cancer by a novel IκB kinase β inhibitor CmpdA. Mol. Cancer Ther. 2016, 15, 1504–1514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinkenbaugh, A.L.; Cogswell, P.C.; Calamini, B.; Dunn, D.E.; Persson, A.I.; Weiss, W.A.; Lo, D.C.; Baldwin, A.S. IKK/NF signaling contributes to glioblastoma stem cell maintenance. Oncotarget 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- An, J.; Wu, M.; Xin, X.; Lin, Z.; Li, X.; Zheng, Q.; Gui, X.; Li, T.; Pu, H.; Li, H.; et al. Inflammatory related gene IKKα, IKKβ, IKKγ cooperates to determine liver cancer stem cells progression by altering telomere via heterochromatin protein 1-HOTAIR axis. Oncotarget 2016, 7, 50131. [Google Scholar] [CrossRef] [Green Version]
- Zakaria, N.; Yusoff, N.M.; Zakaria, Z.; Widera, D.; Yahaya, B.H. Inhibition of NF-κB signaling reduces the stemness characteristics of lung cancer stem cells. Front. Oncol. 2018, 7, 166. [Google Scholar] [CrossRef] [Green Version]
- Page, A.; Navarro, M.; Suárez-Cabrera, C.; Bravo, A.; Ramirez, A. Context-dependent role of IKKβ in cancer. Genes 2017, 8, 376. [Google Scholar] [CrossRef] [Green Version]
- Fusella, F.; Seclì, L.; Busso, E.; Krepelova, A.; Moiso, E.; Rocca, S.; Conti, L.; Annaratone, L.; Rubinetto, C.; Mello-Grand, M.; et al. The IKK/NF-κB signaling pathway requires Morgana to drive breast cancer metastasis. Nat. Commun. 2017, 8, 1636. [Google Scholar] [CrossRef]
- Huber, M.A.; Maier, H.J.; Alacakaptan, M.; Wiedemann, E.; Braunger, J.; Boehmelt, G.; Madwed, J.B.; Young, E.R.R.; Marshall, D.R.; Pehamberger, H.; et al. BI 5700, a selective chemical inhibitor of I B Kinase 2, specifically suppresses epithelial-mesenchymal transition and metastasis in mouse models of tumor progression. Genes Cancer 2010, 1, 101–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, C.; Zhang, N.; Feng, Y.; Cao, J.; Chen, X.; Liu, B. Aspirin inhibits IKK-β-mediated prostate cancer cell invasion by targeting matrix metalloproteinase-9 and urokinase-type plasminogen activator. Cell. Physiol. Biochem. 2017, 41, 1313–1324. [Google Scholar] [CrossRef] [PubMed]
- Waugh, D.J.J.; Wilson, C. The interleukin-8 pathway in cancer. Clin. Cancer Res. 2008, 14, 6735–6741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singha, B.; Gatla, H.R.; Phyo, S.; Patel, A.; Chen, Z.S.; Vancurova, I. IKK inhibition increases bortezomib effectiveness in ovarian cancer. Oncotarget 2015, 6. [Google Scholar] [CrossRef]
- Deryugina, E.I.; Quigley, J.P. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 2006, 25, 9–34. [Google Scholar] [CrossRef]
- Hobbs, G.A.; Der, C.J.; Rossman, K.L. RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 2016, 129, 1287–1292. [Google Scholar] [CrossRef] [Green Version]
- Liao, J.; Wolfman, J.C.; Wolfman, A. K-Ras regulates the steady-state expression of matrix metalloproteinase 2 in fibroblasts. J. Biol. Chem. 2003, 278, 31871–31878. [Google Scholar] [CrossRef] [Green Version]
- Vreka, M.; Lilis, I.; Papageorgopoulou, M.; Giotopoulou, G.A.; Lianou, M.; Giopanou, I.; Kanellakis, N.I.; Spella, M.; Agalioti, T.; Armenis, V.; et al. IκB kinase α is required for development and progression of KRAS -mutant lung adenocarcinoma. Cancer Res. 2018, 78, 2939–2951. [Google Scholar] [CrossRef] [Green Version]
- Song, N.Y.; Zhu, F.; Wang, Z.; Willette-Brown, J.; Xi, S.; Sun, Z.; Su, L.; Wu, X.; Ma, B.; Nussinov, R.; et al. IKKα inactivation promotes Kras-initiated lung adenocarcinoma development through disrupting major redox regulatory pathways. Proc. Natl. Acad. Sci. USA 2018, 115, E812–E821. [Google Scholar] [CrossRef] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodrigues, F.S.; Miranda, V.S.; Carneiro-Lobo, T.C.; Scalabrini, L.C.; Kruspig, B.; Levantini, E.; Murphy, D.J.; Bassères, D.S. IKKβ Kinase Promotes Stemness, Migration, and Invasion in KRAS-Driven Lung Adenocarcinoma Cells. Int. J. Mol. Sci. 2020, 21, 5806. https://doi.org/10.3390/ijms21165806
Rodrigues FS, Miranda VS, Carneiro-Lobo TC, Scalabrini LC, Kruspig B, Levantini E, Murphy DJ, Bassères DS. IKKβ Kinase Promotes Stemness, Migration, and Invasion in KRAS-Driven Lung Adenocarcinoma Cells. International Journal of Molecular Sciences. 2020; 21(16):5806. https://doi.org/10.3390/ijms21165806
Chicago/Turabian StyleRodrigues, Felipe Silva, Vanessa Silva Miranda, Tatiana Correa Carneiro-Lobo, Luiza Coimbra Scalabrini, Björn Kruspig, Elena Levantini, Daniel J. Murphy, and Daniela Sanchez Bassères. 2020. "IKKβ Kinase Promotes Stemness, Migration, and Invasion in KRAS-Driven Lung Adenocarcinoma Cells" International Journal of Molecular Sciences 21, no. 16: 5806. https://doi.org/10.3390/ijms21165806