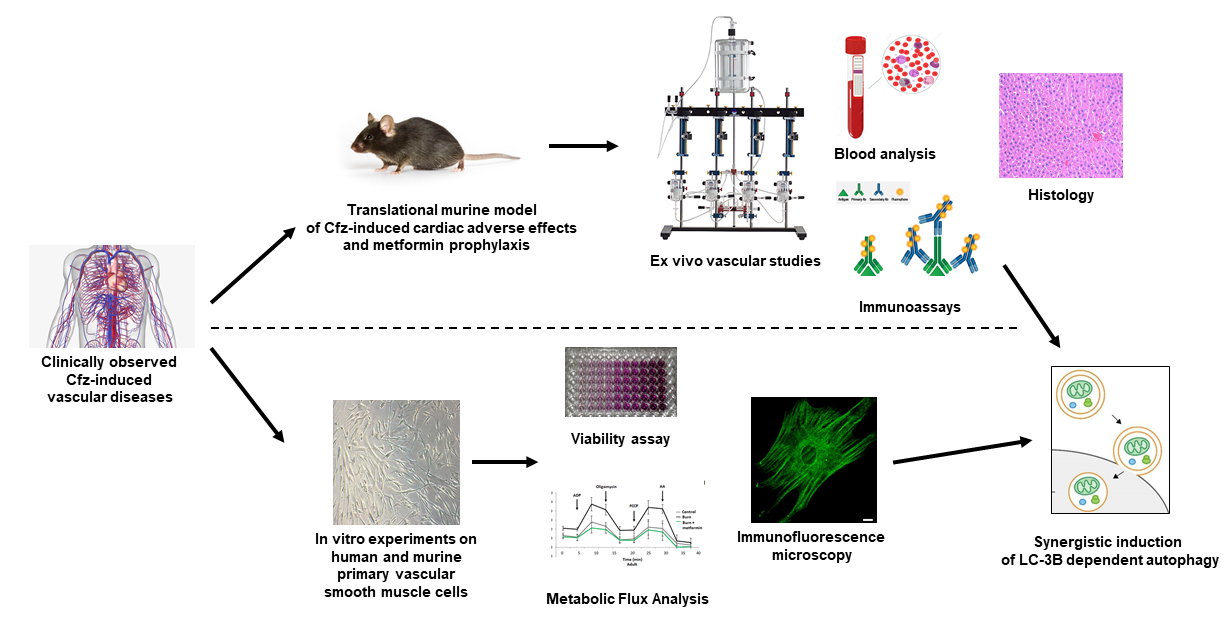
Investigating the Vascular Toxicity Outcomes of the Irreversible Proteasome Inhibitor Carfilzomib

 , , ,
, , , 
Abstract
:
1. Introduction
2. Results
2.1. Carfilzomib Led to an Acute Decrease in PGF2α-Induced Aortic Constriction and Leucocyte-Derived Oxidative Burst, Independently of Metformin Cotreatment
2.2. Carfilzomib Subacute Treatment Did not Induce Vascular Dysfunction, while Cotreatment With Metformin Led to an Increased Response to PGF2α
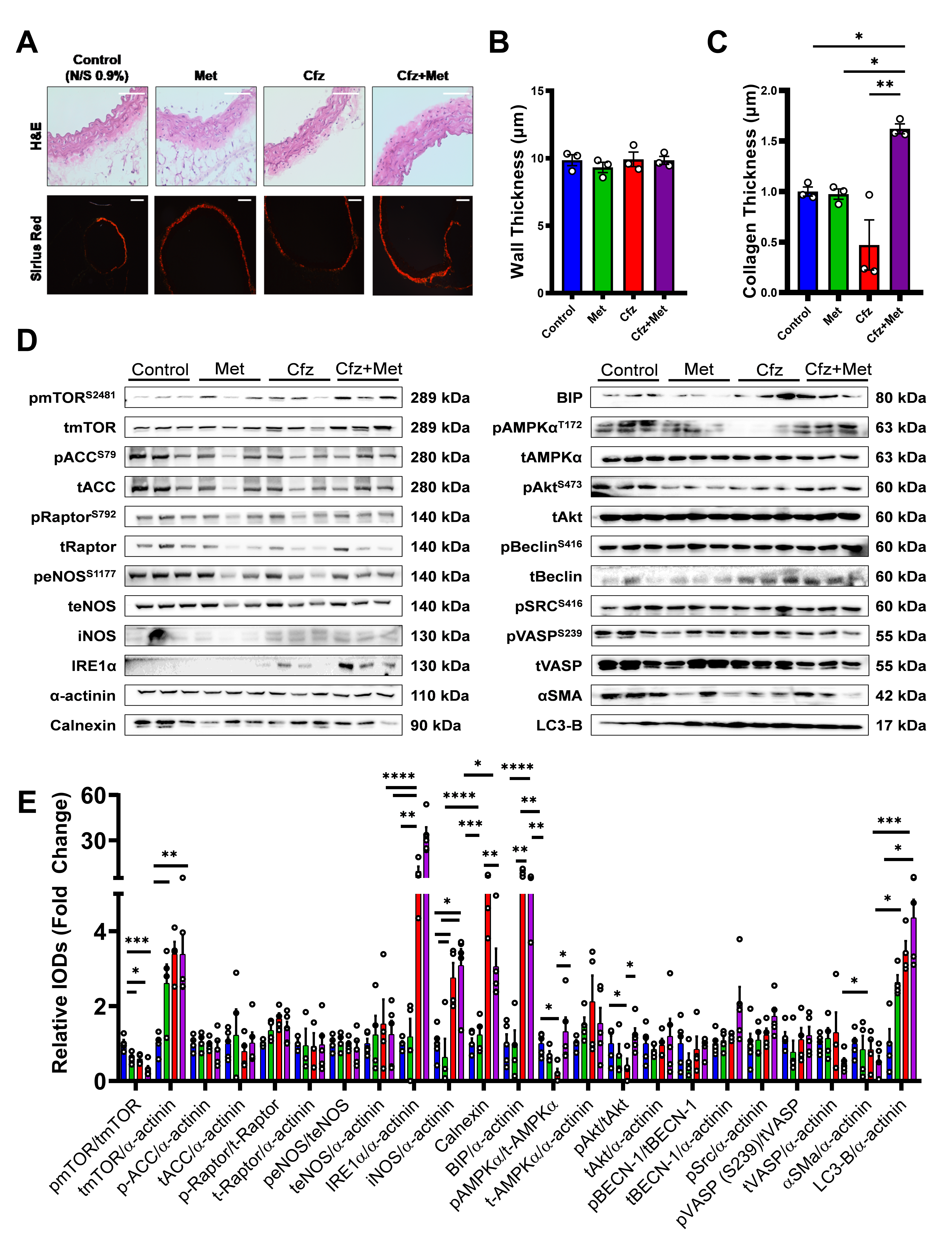
2.3. Coadministration of Carfilzomib and Metformin Led to an Enhanced Ampkα-Endoplasmic Reticulum Stress-Dependent Autophagy and a Decreased A-Smooth Muscle Cell Actin Expression in Vivo
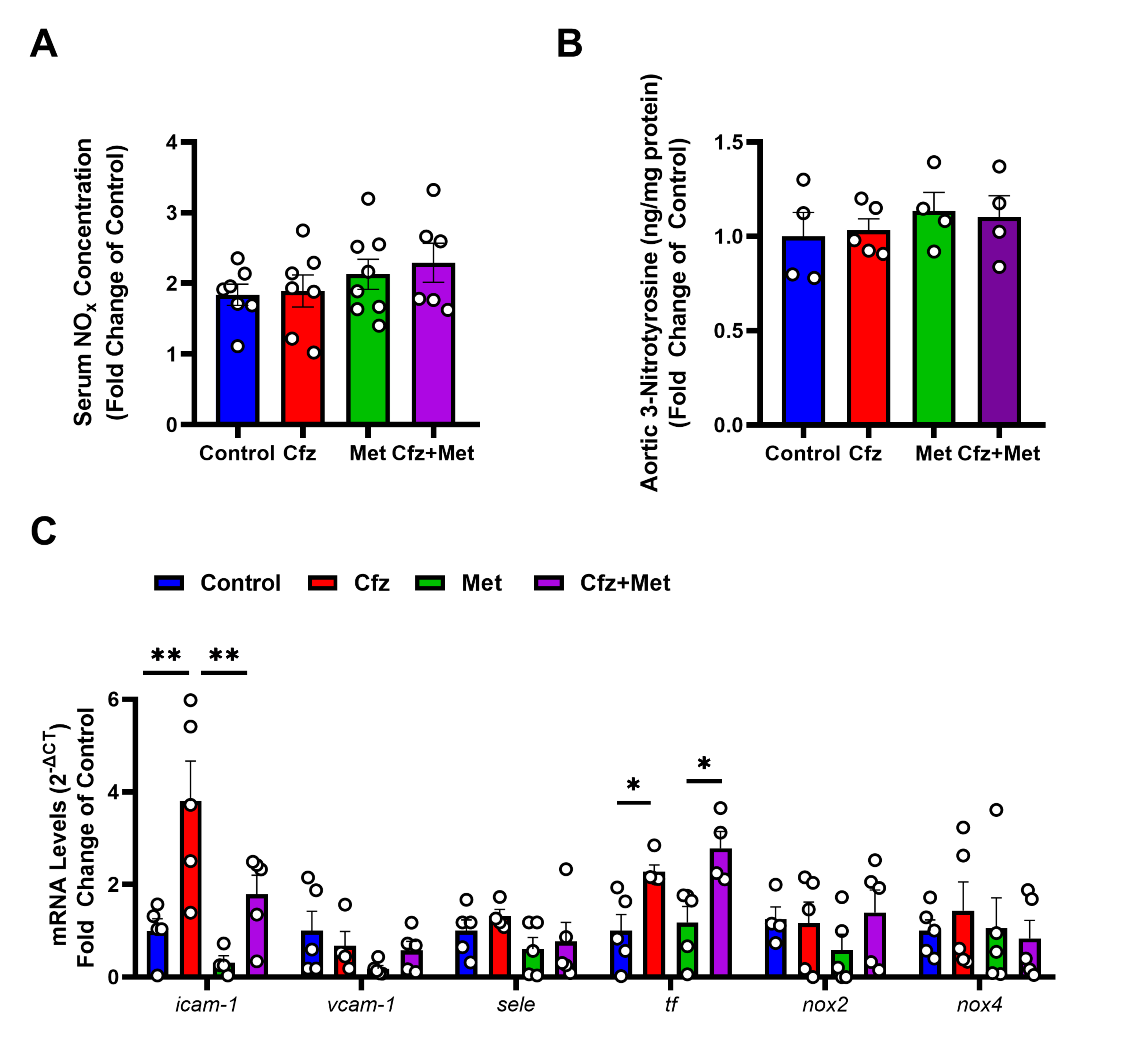
2.4. Carfilzomib Treatment Did Not Affect Endothelial Homeostasis and Did Not Induce Nitro-Oxidative Stress in the Murine Aortas
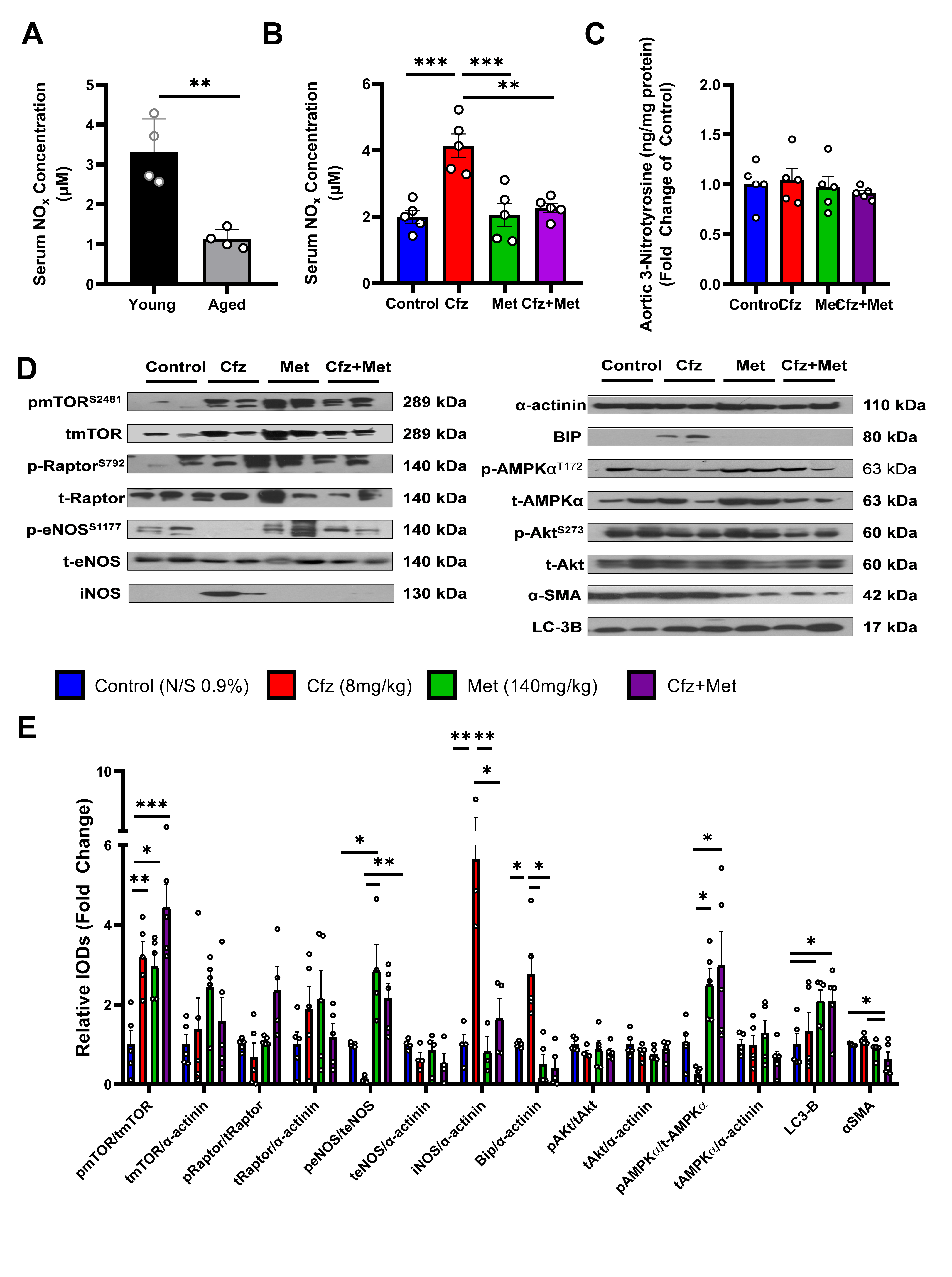
2.5. Carfilzomib Increases NOx Content in the Serum of the Aged Mice and Upregulates Inos and BIP in the Aortic Tissue. Coadministration of Carfilzomib and Metformin Leads to an Induction of LC3-B-Dependent Autophagy in the Aged Murine Aortas
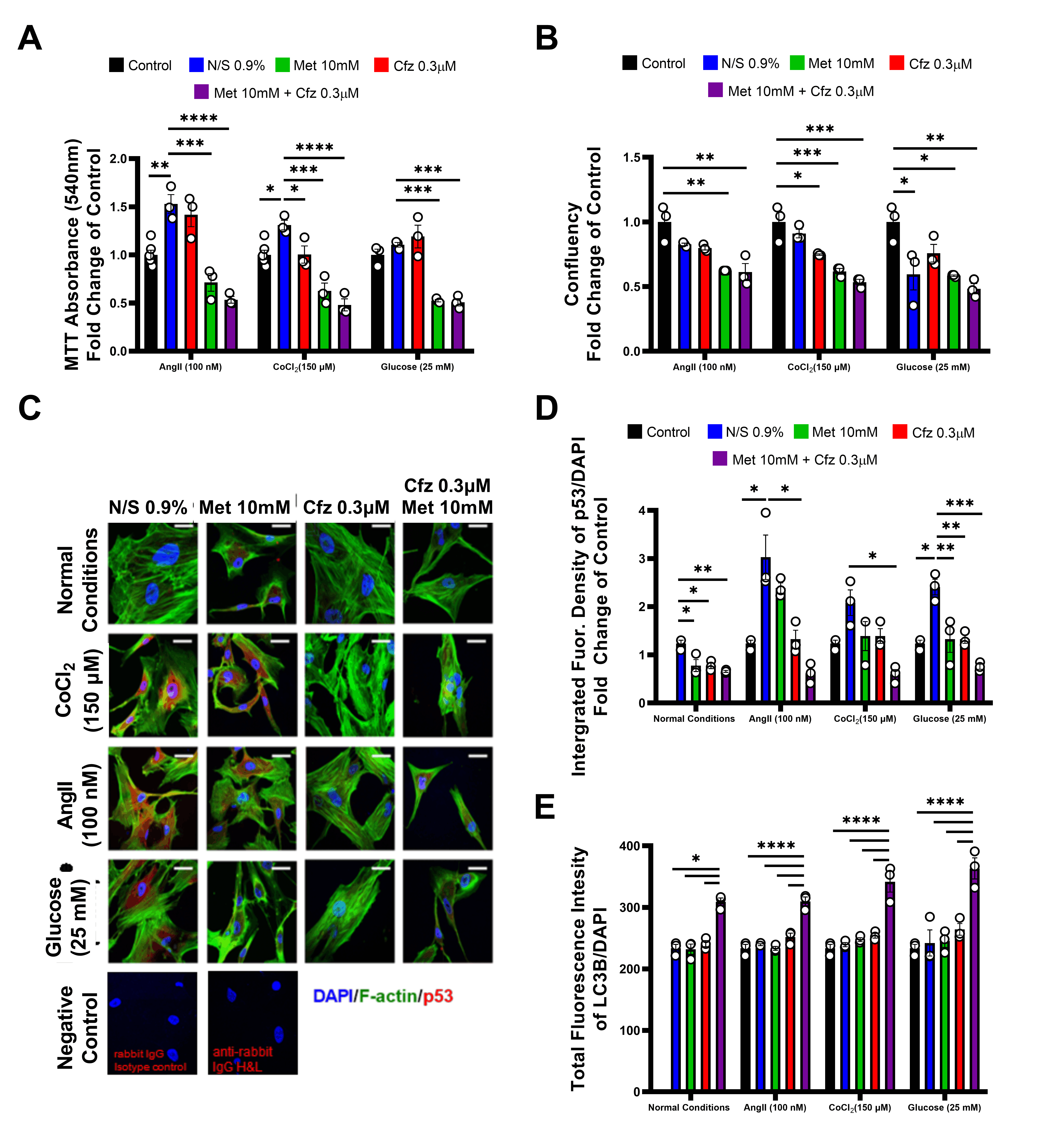
2.6. Carfilzomib and Metformin-Induced Cytotoxicity in Primary Murine Vascular Smooth Muscle Cells Mediated by a Cellular Metabolism Shift and LC3-B Dependent Autophagy
2.7. Carfilzomib and Metformin Led to Decreased Cellular Confluency in Senescent Human Aortic Smooth Muscle Cells, while Metformin Shifts Cellular Metabolism to Glycolysis
2.8. Carfilzomib and Metformin Induce Cytotoxicity on Senescent HAoSMCs in the Presence of Comorbidities in an LC-3B-Dependent Mechanism
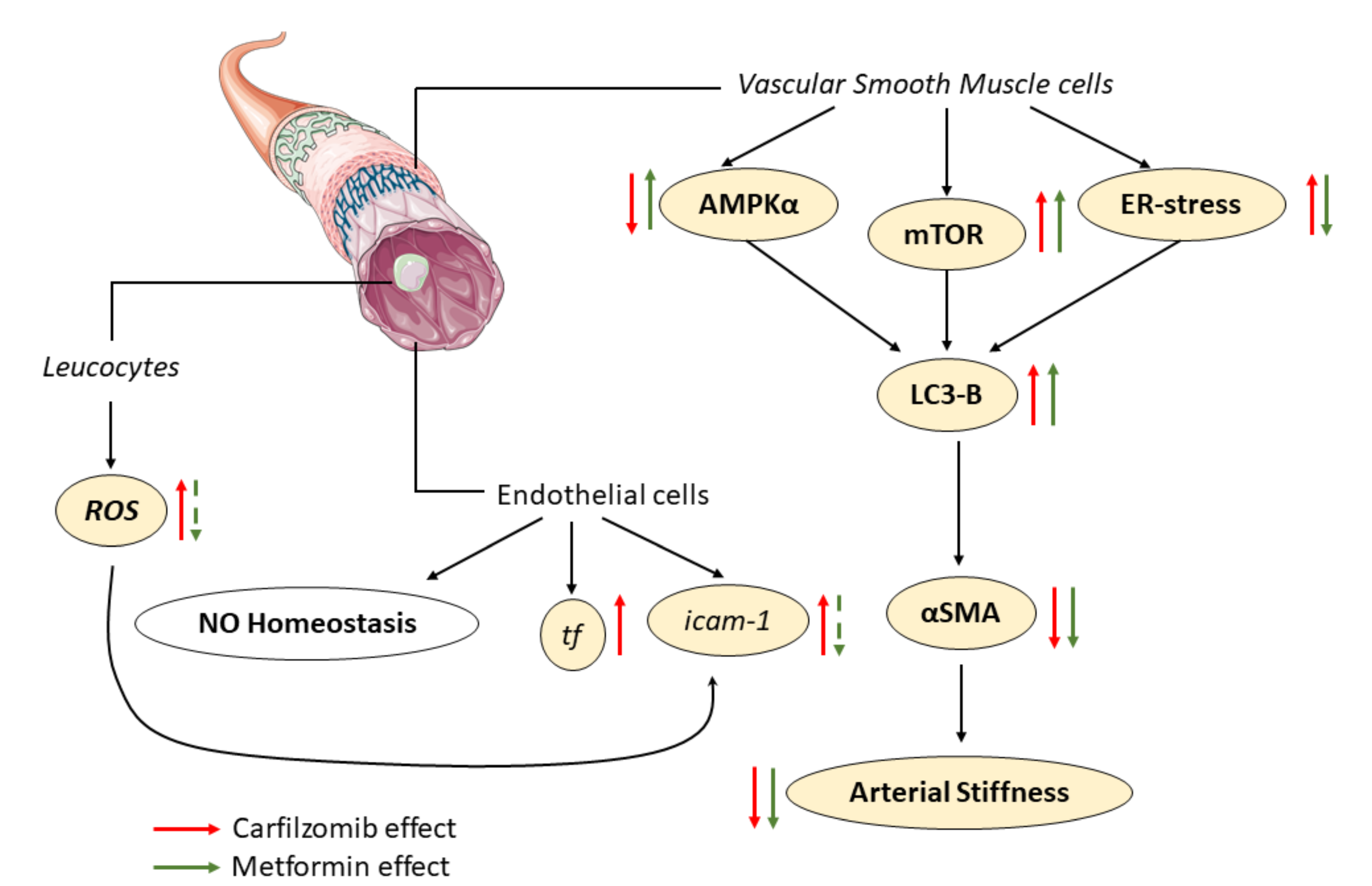
3. Discussion
4. Methods
4.1. Animals
4.2. Vascular Relaxation and Constriction Studies
4.3. Leucocyte Derived Oxidative Burst (ROS Release) Measurement
4.4. NO Metabolites (NOx) and 3-Nitrotyrosine Measurements
4.5. Histology
4.6. Western Blot Immunoblotting Analysis
4.7. Analysis of Endothelial Markers via Real-Time PCR
4.8. Primary Murine Vascular Smooth Muscle Cell (PrmVSMCs) Isolation and Human Aortic Smooth Muscle Cell Culture
4.9. In Vitro Study of the Cellular Metabolism—Oxygen Consumption Rate (OCR), Proton Efflux Rate (PER) and ATP Rate Measurements
4.10. Immunofluorescence Confocal Microscopy
4.11. Automated Immunofluorescence Microscopy
4.12. Statistics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dimopoulos, M.A.; Moreau, P.; Palumbo, A.; Joshua, D.; Pour, L.; Hajek, R.; Facon, T.; Ludwig, H.; Oriol, A.; Goldschmidt, H.; et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): A randomised, phase 3, open-label, multicentre study. Lancet Oncol. 2016, 17, 27–38. [Google Scholar] [CrossRef]
- Palumbo, A.; Anderson, K. Multiple myeloma. N. Engl. J. Med. 2011, 364, 1046–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimopoulos, M.A.; Roussou, M.; Gavriatopoulou, M.; Psimenou, E.; Ziogas, D.; Eleutherakis-Papaiakovou, E.; Fotiou, D.; Migkou, M.; Kanellias, N.; Panagiotidis, I.; et al. Cardiac and renal complications of carfilzomib in patients with multiple myeloma. Blood Adv. 2017, 1, 449–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, C.; Bishnoi, R.; Jain, A.; Bejjanki, H.; Xiong, S.; Wang, Y.; Zou, F.; Moreb, J.S. Cardiotoxicity associated with carfilzomib: Systematic review and meta-analysis. Leuk. Lymphoma 2018, 59, 2557–2569. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Terpos, E.; Niesvizky, R.; Palumbo, A. Clinical characteristics of patients with relapsed multiple myeloma. Cancer Treat Rev. 2015, 41, 827–835. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Garcia, D.; Cornell, R.F.; Gailani, D.; Laubach, J.; Maglio, M.E.; Richardson, P.G.; Moslehi, J. Cardiovascular and Thrombotic Complications of Novel Multiple Myeloma Therapies: A Review. JAMA Oncol. 2017, 3, 980–988. [Google Scholar] [CrossRef]
- Jakubowiak, A.J.; DeCara, J.M.; Mezzi, K. Cardiovascular events during carfilzomib therapy for relapsed myeloma: Practical management aspects from two case studies. Hematology 2017, 22, 585–591. [Google Scholar] [CrossRef] [Green Version]
- Zamorano, J.L.; Lancellotti, P.; Rodriguez Munoz, D.; Aboyans, V.; Asteggiano, R.; Galderisi, M.; Habib, G.; Lenihan, D.J.; Lip, G.Y.H.; Lyon, A.R.; et al. 2016 ESC Position Paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC Committee for Practice Guidelines: The Task Force for cancer treatments and cardiovascular toxicity of the European Society of Cardiology (ESC). Eur. Heart J. 2016, 37, 2768–2801. [Google Scholar] [CrossRef]
- Chari, A.; Hajje, D. Case series discussion of cardiac and vascular events following carfilzomib treatment: Possible mechanism, screening, and monitoring. BMC Cancer 2014, 14, 915. [Google Scholar] [CrossRef] [Green Version]
- Imam, F.; Al-Harbi, N.O.; Al-Harbia, M.M.; Korashy, H.M.; Ansari, M.A.; Sayed-Ahmed, M.M.; Nagi, M.N.; Iqbal, M.; Khalid Anwer, M.; Kazmi, I.; et al. Rutin Attenuates Carfilzomib-Induced Cardiotoxicity Through Inhibition of NF-kappaB, Hypertrophic Gene Expression and Oxidative Stress. Cardiovasc. Toxicol. 2017, 17, 58–66. [Google Scholar] [CrossRef]
- Efentakis, P.; Kremastiotis, G.; Varela, A.; Nikolaou, P.E.; Papanagnou, E.D.; Davos, C.H.; Tsoumani, M.; Agrogiannis, G.; Konstantinidou, A.; Kastritis, E.; et al. Molecular mechanisms of carfilzomib-induced cardiotoxicity in mice and the emerging cardioprotective role of metformin. Blood 2019, 133, 710–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatelopoulos, K.; Georgiopoulos, G.; Athanasouli, F.; Nikolaou, P.E.; Lykka, M.; Roussou, M.; Gavriatopoulou, M.; Laina, A.; Trakada, G.; Charakida, M.; et al. Reactive Vasodilation Predicts Mortality in Primary Systemic Light-Chain Amyloidosis. Circ. Res. 2019, 125, 744–758. [Google Scholar] [CrossRef] [PubMed]
- Nussenzweig, S.C.; Verma, S.; Finkel, T. The role of autophagy in vascular biology. Circ. Res. 2015, 116, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ibrahim, Y.F.; Das, D.; Zungu-Edmondson, M.; Shults, N.V.; Suzuki, Y.J. Carfilzomib reverses pulmonary arterial hypertension. Cardiovasc. Res. 2016, 110, 188–199. [Google Scholar] [CrossRef] [Green Version]
- Hao, B.; Xiao, Y.; Song, F.; Long, X.; Huang, J.; Tian, M.; Deng, S.; Wu, Q. Metformin-induced activation of AMPK inhibits the proliferation and migration of human aortic smooth muscle cells through upregulation of p53 and IFI16. Int. J. Mol. Med. 2018, 41, 1365–1376. [Google Scholar] [CrossRef]
- Mondaca-Ruff, D.; Riquelme, J.A.; Quiroga, C.; Norambuena-Soto, I.; Sanhueza-Olivares, F.; Villar-Fincheira, P.; Hernandez-Diaz, T.; Cancino-Arenas, N.; San Martin, A.; Garcia, L.; et al. Angiotensin II-Regulated Autophagy Is Required for Vascular Smooth Muscle Cell Hypertrophy. Front. Pharmacol. 2018, 9, 1553. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wang, H.M. Effects of cobalt chloride on phenotypes of normal human saphenous vein smooth muscle cells. Int. J. Clin. Exp. Med. 2014, 7, 4933–4941. [Google Scholar]
- Manea, A.; Manea, S.A.; Todirita, A.; Albulescu, I.C.; Raicu, M.; Sasson, S.; Simionescu, M. High-glucose-increased expression and activation of NADPH oxidase in human vascular smooth muscle cells is mediated by 4-hydroxynonenal-activated PPARalpha and PPARbeta/delta. Cell Tissue Res. 2015, 361, 593–604. [Google Scholar] [CrossRef]
- Duan, Q.; Song, P.; Ding, Y.; Zou, M.H. Activation of AMP-activated protein kinase by metformin ablates angiotensin II-induced endoplasmic reticulum stress and hypertension in mice in vivo. Br. J. Pharmacol. 2017, 174, 2140–2151. [Google Scholar] [CrossRef] [Green Version]
- Ma, W.Q.; Sun, X.J.; Wang, Y.; Zhu, Y.; Han, X.Q.; Liu, N.F. Restoring mitochondrial biogenesis with metformin attenuates beta-GP-induced phenotypic transformation of VSMCs into an osteogenic phenotype via inhibition of PDK4/oxidative stress-mediated apoptosis. Mol. Cell Endocrinol. 2019, 479, 39–53. [Google Scholar] [CrossRef]
- Omura, J.; Satoh, K.; Kikuchi, N.; Satoh, T.; Kurosawa, R.; Nogi, M.; Otsuki, T.; Kozu, K.; Numano, K.; Suzuki, K.; et al. Protective Roles of Endothelial AMP-Activated Protein Kinase Against Hypoxia-Induced Pulmonary Hypertension in Mice. Circ. Res. 2016, 119, 197–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, R.N.; Qu, W. Metformin inhibits LPS-induced inflammatory response in VSMCs by regulating TLR4 and PPAR-gamma. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 4988–4995. [Google Scholar] [PubMed]
- Song, Y.; Wu, Y.; Su, X.; Zhu, Y.; Liu, L.; Pan, Y.; Zhu, B.; Yang, L.; Gao, L.; Li, M. Activation of AMPK inhibits PDGF-induced pulmonary arterial smooth muscle cells proliferation and its potential mechanisms. Pharmacol. Res. 2016, 107, 117–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Guo, J.; Han, X.; Xue, M.; Wang, W.; Mi, L.; Sheng, Y.; Ma, C.; Wu, J.; Wu, X. Metformin represses the pathophysiology of AAA by suppressing the activation of PI3K/AKT/mTOR/autophagy pathway in ApoE(-/-) mice. Cell Biosci. 2019, 9, 68. [Google Scholar] [CrossRef]
- Koulaouzidis, G.; Lyon, A.R. Proteasome Inhibitors as a Potential Cause of Heart Failure. Heart Fail. Clin. 2017, 13, 289–295. [Google Scholar] [CrossRef]
- Sahni, A.; Thomasson, E.D.; Shah, R.; Sahni, S.K. Bortezomib Effects on Human Microvascular Endothelium in vitro. Pharmacology 2016, 98, 272–278. [Google Scholar] [CrossRef]
- Zhang, J.; Lu, W.; Chen, Y.; Jiang, Q.; Yang, K.; Li, M.; Wang, Z.; Duan, X.; Xu, L.; Tang, H.; et al. Bortezomib alleviates experimental pulmonary hypertension by regulating intracellular calcium homeostasis in PASMCs. Am. J. Physiol. Cell. Physiol. 2016, 311, C482–C497. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Lee, J.H.; Huh, J.W.; Kim, H.J.; Park, M.K.; Ro, J.Y.; Oh, Y.M.; Lee, S.D.; Lee, Y.S. Bortezomib alleviates experimental pulmonary arterial hypertension. Am. J. Respir. Cell Mol. Biol. 2012, 47, 698–708. [Google Scholar] [CrossRef]
- Ren, H.; Li, F.; Tian, C.; Nie, H.; Wang, L.; Li, H.H.; Zheng, Y. Inhibition of Proteasome Activity by Low-dose Bortezomib Attenuates Angiotensin II-induced Abdominal Aortic Aneurysm in Apo E(-/-) Mice. Sci. Rep. 2015, 5, 15730. [Google Scholar] [CrossRef]
- Chen-Scarabelli, C.; Corsetti, G.; Pasini, E.; Dioguardi, F.S.; Sahni, G.; Narula, J.; Gavazzoni, M.; Patel, H.; Saravolatz, L.; Knight, R.; et al. Spasmogenic Effects of the Proteasome Inhibitor Carfilzomib on Coronary Resistance, Vascular Tone and Reactivity. EBioMedicine 2017, 21, 206–212. [Google Scholar] [CrossRef] [Green Version]
- Kocabaş, U.; Atalay, F.; Altay, H.; Altun, A.; Pehlivanoğlu, S. Acute Coronary Syndrome Associated with Carfilzomib Treatment. Indian J. Hematol. Blood Transfus. 2019, 36, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Pidkovka, N.A.; Cherepanova, O.A.; Yoshida, T.; Alexander, M.R.; Deaton, R.A.; Thomas, J.A.; Leitinger, N.; Owens, G.K. Oxidized phospholipids induce phenotypic switching of vascular smooth muscle cells in vivo and in vitro. Circ. Res. 2007, 101, 792–801. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.A.; Merenick, B.L.; Ding, M.; Fetalvero, K.M.; Rzucidlo, E.M.; Kozul, C.D.; Brown, D.J.; Chiu, H.Y.; Shyu, M.; Drapeau, B.L.; et al. Rapamycin promotes vascular smooth muscle cell differentiation through insulin receptor substrate-1/phosphatidylinositol 3-kinase/Akt2 feedback signaling. J. Biol. Chem. 2007, 282, 36112–36120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tai, S.; Hu, X.Q.; Peng, D.Q.; Zhou, S.H.; Zheng, X.L. The roles of autophagy in vascular smooth muscle cells. Int. J. Cardiol. 2016, 211, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.R.; Bae, Y.H.; Bae, S.K.; Choi, K.S.; Yoon, K.H.; Koo, T.H.; Jang, H.O.; Yun, I.; Kim, K.W.; Kwon, Y.G.; et al. Visfatin enhances ICAM-1 and VCAM-1 expression through ROS-dependent NF-kappaB activation in endothelial cells. Biochim. Biophys. Acta. 2008, 1783, 886–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirchhofer, D.; Tschopp, T.B.; Hadvary, P.; Baumgartner, H.R. Endothelial-Cells Stimulated with Tumor-Necrosis-Factor-Alpha Express Varying Amounts of Tissue Factor Resulting in Inhomogenous Fibrin Deposition in a Native Blood-Flow System - Effects of Thrombin Inhibitors. J. Clin. Invest. 1994, 93, 2073–2083. [Google Scholar] [CrossRef] [Green Version]
- Qaqish, I.; Schlam, I.M.; Chakkera, H.A.; Fonseca, R.; Adamski, J. Carfilzomib: A cause of drug associated thrombotic microangiopathy. Transfus. Apher. Sci. 2016, 54, 401–404. [Google Scholar] [CrossRef]
- Sullivan, M.R.; Danilov, A.V.; Lansigan, F.; Dunbar, N.M. Carfilzomib associated thrombotic microangiopathy initially treated with therapeutic plasma exchange. J. Clin. Apher. 2015, 30, 308–310. [Google Scholar] [CrossRef]
- Luo, F.; Das, A.; Chen, J.; Wu, P.; Li, X.; Fang, Z. Metformin in patients with and without diabetes: A paradigm shift in cardiovascular disease management. Cardiovasc. Diabetol. 2019, 18, 54. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Xu, Y.; Zhu, J.; Li, H.; Zhang, J.; Yang, G.; Sun, Z. Metformin Prevents Progression of Experimental Pulmonary Hypertension via Inhibition of Autophagy and Activation of Adenosine Monophosphate-Activated Protein Kinase. J. Vasc. Res. 2019, 56, 117–128. [Google Scholar] [CrossRef]
- Wang, G.Y.; Bi, Y.G.; Liu, X.D.; Zhao, Y.; Han, J.F.; Wei, M.; Zhang, Q.Y. Autophagy was involved in the protective effect of metformin on hyperglycemia-induced cardiomyocyte apoptosis and Connexin43 downregulation in H9c2 cells. Int. J. Med. Sci. 2017, 14, 698–704. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Xu, W.; Yan, Z.; Zhao, W.; Mi, J.; Li, J.; Yan, H. Metformin induces autophagy and G0/G1 phase cell cycle arrest in myeloma by targeting the AMPK/mTORC1 and mTORC2 pathways. J. Exp. Clin. Cancer Res. 2018, 37, 63. [Google Scholar] [CrossRef]
- Xie, S.A.; Zhang, T.; Wang, J.; Zhao, F.; Zhang, Y.P.; Yao, W.J.; Hur, S.S.; Yeh, Y.T.; Pang, W.; Zheng, L.S.; et al. Matrix stiffness determines the phenotype of vascular smooth muscle cell in vitro and in vivo: Role of DNA methyltransferase 1. Biomaterials 2018, 155, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Karsdal, M.A.; Nielsen, S.H.; Leeming, D.J.; Langholm, L.L.; Nielsen, M.J.; Manon-Jensen, T.; Siebuhr, A.; Gudmann, N.S.; Ronnow, S.; Sand, J.M.; et al. The good and the bad collagens of fibrosis - Their role in signaling and organ function. Adv. Drug Deliv. Rev. 2017, 121, 43–56. [Google Scholar] [CrossRef]
- Ungvari, Z.; Tarantini, S.; Donato, A.J.; Galvan, V.; Csiszar, A. Mechanisms of Vascular Aging. Circ. Res. 2018, 123, 849–867. [Google Scholar] [CrossRef]
- Abdellatif, M.; Sedej, S.; Carmona-Gutierrez, D.; Madeo, F.; Kroemer, G. Autophagy in Cardiovascular Aging. Circ. Res. 2018, 123, 803–824. [Google Scholar] [CrossRef]
- De Meyer, G.R.; Grootaert, M.O.; Michiels, C.F.; Kurdi, A.; Schrijvers, D.M.; Martinet, W. Autophagy in vascular disease. Circ. Res. 2015, 116, 468–479. [Google Scholar] [CrossRef]
- Brandt, M.; Giokoglu, E.; Garlapati, V.; Bochenek, M.L.; Molitor, M.; Hobohm, L.; Schonfelder, T.; Munzel, T.; Kossmann, S.; Karbach, S.H.; et al. Pulmonary Arterial Hypertension and Endothelial Dysfunction Is Linked to NADPH Oxidase-Derived Superoxide Formation in Venous Thrombosis and Pulmonary Embolism in Mice. Oxid. Med. Cell. Longev. 2018, 2018, 1860513. [Google Scholar] [CrossRef]
- Thompson, R.M.; Dytfeld, D.; Reyes, L.; Robinson, R.M.; Smith, B.; Manevich, Y.; Jakubowiak, A.; Komarnicki, M.; Przybylowicz-Chalecka, A.; Szczepaniak, T.; et al. Glutaminase inhibitor CB-839 synergizes with carfilzomib in resistant multiple myeloma cells. Oncotarget 2017, 8, 35863–35876. [Google Scholar] [CrossRef] [Green Version]
- Rosano, G.M.; Vitale, C. Metabolic Modulation of Cardiac Metabolism in Heart Failure. Card Fail Rev. 2018, 4, 99–103. [Google Scholar] [CrossRef]
- Magalhaes, S.; Goodfellow, B.J.; Nunes, A. Aging and Proteins: What Does Proteostasis Have to Do with Age? Curr. Mol. Med. 2018, 18, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Saha, T.; Kar, R.K.; Sa, G. Structural and sequential context of p53: A review of experimental and theoretical evidence. Prog. Biophys. Mol. Biol. 2015, 117, 250–263. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, I.; Minamino, T. Physiological and pathological cardiac hypertrophy. J. Mol. Cell Cardiol. 2016, 97, 245–262. [Google Scholar] [CrossRef] [PubMed]
- Kilkenny, C.; Browne, W.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Animal research: Reporting in vivo experiments: The ARRIVE guidelines. Brit. J. Pharmacol. 2010, 160, 1577–1579. [Google Scholar] [CrossRef]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. Osteoarthr. Cartil. 2012, 20, 256–260. [Google Scholar] [CrossRef] [Green Version]
- Kossmann, S.; Lagrange, J.; Jackel, S.; Jurk, K.; Ehlken, M.; Schonfelder, T.; Weihert, Y.; Knorr, M.; Brandt, M.; Xia, N.; et al. Platelet-localized FXI promotes a vascular coagulation-inflammatory circuit in arterial hypertension. Sci. Transl. Med. 2017, 9, 375. [Google Scholar] [CrossRef]
- Schuler, R.; Efentakis, P.; Wild, J.; Lagrange, J.; Garlapati, V.; Molitor, M.; Kossmann, S.; Oelze, M.; Stamm, P.; Li, H.; et al. T Cell-Derived IL-17A Induces Vascular Dysfunction via Perivascular Fibrosis Formation and Dysregulation of (.)NO/cGMP Signaling. Oxid. Med. Cell Longev. 2019, 2019, 6721531. [Google Scholar] [CrossRef] [Green Version]
- Nikolaou, P.E.; Efentakis, P.; Qourah, F.A.; Femmino, S.; Makridakis, M.; Kanaki, Z.; Varela, A.; Tsoumani, M.; Davos, C.H.; Dimitriou, C.A.; et al. Chronic Empaglifozin treatment reduces myocardial infarct size in non-diabetic mice through STAT-3 mediated protection on microvascular endothelial cells and reduction of oxidative stress. Antioxid. Redox Signal. 2020. (Online ahead of print). [Google Scholar] [CrossRef]
- Adhikari, N.; Shekar, K.C.; Staggs, R.; Win, Z.; Steucke, K.; Lin, Y.W.; Wei, L.N.; Alford, P.; Hall, J.L. International Society of Cardiovascular Translational, R. Guidelines for the isolation and characterization of murine vascular smooth muscle cells. A report from the International Society of Cardiovascular Translational Research. J. Cardiovasc. Transl. Res. 2015, 8, 158–163. [Google Scholar] [CrossRef] [Green Version]
- Efentakis, P.; Varela, A.; Chavdoula, E.; Sigala, F.; Sanoudou, D.; Tenta, R.; Gioti, K.; Kostomitsopoulos, N.; Papapetropoulos, A.; Tasouli, A.; et al. Levosimendan prevents doxorubicin-induced cardiotoxicity in time- and dose dependent manner: Implications for inotropy. Cardiovasc. Res. 2019, 116, 576–591. [Google Scholar] [CrossRef]
- Lu, Y.; Li, S.; Wu, H.; Bian, Z.; Xu, J.; Gu, C.; Chen, X.; Yang, D. Beneficial effects of astragaloside IV against angiotensin II-induced mitochondrial dysfunction in rat vascular smooth muscle cells. Int. J. Mol. Med. 2015, 36, 1223–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westmeier, D.; Solouk-Saran, D.; Vallet, C.; Siemer, S.; Docter, D.; Gotz, H.; Mann, L.; Hasenberg, A.; Hahlbrock, A.; Erler, K.; et al. Nanoparticle decoration impacts airborne fungal pathobiology. Proc. Natl. Acad. Sci. USA 2018, 115, 7087–7092. [Google Scholar] [CrossRef] [Green Version]
- Brandt, M.; Garlapati, V.; Oelze, M.; Sotiriou, E.; Knorr, M.; Kroller-Schon, S.; Kossmann, S.; Schonfelder, T.; Morawietz, H.; Schulz, E.; et al. NOX2 amplifies acetaldehyde-mediated cardiomyocyte mitochondrial dysfunction in alcoholic cardiomyopathy. Sci. Rep. 2016, 6, 32554. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Forward | Reverse | Product Length | Tm |
|---|---|---|---|---|
| Mus musculus intercellular adhesion molecule 1 (icam1) | AGTCCGCTGTGCTTTGAGAA | CTCTCCGGAAACGAATACACG | 80 | 59.8/58.8 |
| Mus musculus vascular cell adhesion molecule 1 (vcam1) | TCTTTATGTCAACGTTGCCCCC | ACTTGAGCAGGTCAGGTTCAC | 97 | 61.1/60.2 |
| Mus musculus E-selectin | TCTGCAGTTCTGACGTGTGG | AGTGCAACTACCAAGGGACG | 98 | 60.2/60.0 |
| Mus musculus coagulation factor III (F3) | CGTGAAGGATGTGACCTGGG | TCACAAGTTGGTCTCCGTCC | 89 | 60.4/59.6 |
| Mus musculus glyceraldehyde 3-phosphate dehydrogenase (gadph) | CCCAGCTTAGGTTCATCAGGT | GCCAAATCCGTTCACACCG | 87 | 59.4/59.7 |
| Mus musculus nadph oxidase 2 (NOx2) | AAGTTCGCTGGAAACCCTCC | GCCAAAACCGAACCAACCTC | 88 | 60.3/60.0 |
| Mus musculus nadph oxidase 2 (NOx4) | CACCAAATGTTGGGCGATTGT | CAGGACTGTCCGGCACATAG | 60.0/60.2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Efentakis, P.; Doerschmann, H.; Witzler, C.; Siemer, S.; Nikolaou, P.-E.; Kastritis, E.; Stauber, R.; Dimopoulos, M.A.; Wenzel, P.; Andreadou, I.; et al. Investigating the Vascular Toxicity Outcomes of the Irreversible Proteasome Inhibitor Carfilzomib. Int. J. Mol. Sci. 2020, 21, 5185. https://doi.org/10.3390/ijms21155185
Efentakis P, Doerschmann H, Witzler C, Siemer S, Nikolaou P-E, Kastritis E, Stauber R, Dimopoulos MA, Wenzel P, Andreadou I, et al. Investigating the Vascular Toxicity Outcomes of the Irreversible Proteasome Inhibitor Carfilzomib. International Journal of Molecular Sciences. 2020; 21(15):5185. https://doi.org/10.3390/ijms21155185
Chicago/Turabian StyleEfentakis, Panagiotis, Hendrik Doerschmann, Claudius Witzler, Svenja Siemer, Panagiota-Efstathia Nikolaou, Efstathios Kastritis, Roland Stauber, Meletios Athanasios Dimopoulos, Philip Wenzel, Ioanna Andreadou, and et al. 2020. "Investigating the Vascular Toxicity Outcomes of the Irreversible Proteasome Inhibitor Carfilzomib" International Journal of Molecular Sciences 21, no. 15: 5185. https://doi.org/10.3390/ijms21155185