The Role of Herpes Simplex Virus Type 1 Infection in Demyelination of the Central Nervous System



1
Departamento de Biología Molecular, Universidad Autónoma de Madrid, Cantoblanco, 28049 Madrid, Spain
2
Centro de Biología Molecular Severo Ochoa, CSIC-UAM, Cantoblanco, 28049 Madrid, Spain
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(14), 5026; https://doi.org/10.3390/ijms21145026
Submission received: 30 June 2020
/
Revised: 14 July 2020
/
Accepted: 15 July 2020
/
Published: 16 July 2020
(This article belongs to the Special Issue Herpes Simplex Virus: From Reactivation to Assembly)
{kind=link}
{kind=link}
Abstract
:Herpes simplex type 1 (HSV-1) is a neurotropic virus that infects the peripheral and central nervous systems. After primary infection in epithelial cells, HSV-1 spreads retrogradely to the peripheral nervous system (PNS), where it establishes a latent infection in the trigeminal ganglia (TG). The virus can reactivate from the latent state, traveling anterogradely along the axon and replicating in the local surrounding tissue. Occasionally, HSV-1 may spread trans-synaptically from the TG to the brainstem, from where it may disseminate to higher areas of the central nervous system (CNS). It is not completely understood how HSV-1 reaches the CNS, although the most accepted idea is retrograde transport through the trigeminal or olfactory tracts. Once in the CNS, HSV-1 may induce demyelination, either as a direct trigger or as a risk factor, modulating processes such as remyelination, regulation of endogenous retroviruses, or molecular mimicry. In this review, we describe the current knowledge about the involvement of HSV-1 in demyelination, describing the pathways used by this herpesvirus to spread throughout the CNS and discussing the data that suggest its implication in demyelinating processes.
1. Introduction
Several neurotropic viruses may reach and infect the central nervous system (CNS) [1,2,3,4], including herpesviruses (herpes simplex virus type 1 (HSV-1), HSV-2, human cytomegalovirus (HCMV), and varicella zoster virus (VZV)), several arboviruses (West Nile, Japanese encephalitis, and chikungunya viruses), enteroviruses, henipaviruses, Ebola virus, and rabies virus [5]. These pathogens can cause a variety of nervous system diseases, such as encephalitis, flaccid paralysis, inflammatory immune disorders, or meningitis. Regarding the aetiology of demyelinating diseases (i.e., multiple sclerosis (MS)), several infectious agents, including viruses, bacteria, and protists, have been associated [6,7,8,9], in particular many viruses from the family Herpesviridae [10,11,12,13]. Epstein–Barr virus (EBV), human herpesvirus 6 (HH6), and HSV-1 have been linked to demyelinating diseases, although their role in these pathologies, and particularly in MS, is difficult to determine given their almost ubiquitous nature [11]. HSV-1 has also been involved in neurodegenerative disorders of the CNS [14,15,16,17].
It is not fully understood how HSV-1 reaches the CNS, although the most feasible explanation is retrograde transport through the olfactory or trigeminal tracts. It is also unknown whether herpes simplex encephalitis (HSE) is caused by the reactivation of the latent virus or primary infection, as both seem to be possible. Nevertheless, the poor correlation of HSE with primary infection suggests that HSE is more likely due to viral reactivation than to primo-infections [18]. However, latent HSV-1 has been demonstrated within several structures of the CNS, and the effects of infection with this virus in oligodendrocytes (OLs), the myelin-forming cells of the CNS, has also been reported.
In this review, we will describe the current knowledge about the involvement of HSV-1 in demyelination, discussing the pathways used by this herpesvirus to reach the CNS and the evidence implicating it in damage to OLs.
2. Herpes Simplex Type 1
HSV-1 is a double-stranded DNA herpesvirus belonging to the Alphaherpesvirinae subfamily [19]. It is an important neurotropic human pathogen that can infect also other species, especially non-human primates [20], as well as numerous cell types in vitro, although humans are the natural hosts [21]. HSV-1 is one of the most widely spread human viral pathogen, and around 67% of the global population have antibodies to this pathogen [22]. Primary infection takes place in epithelial cells and the virus is transmitted to new hosts via saliva. In this stage, HSV-1 typically causes labial and oral lesions, and although it may also cause genital herpes, the most common sexually transmitted type is herpes simplex virus type 2 (HSV-2) [23,24,25]. In addition, HSV-1 can cause severe pathologies such as encephalitis or keratoconjunctivitis [26]. HSE includes severe brain damage with hemorrhage, edema, and necrosis, and mostly affects the frontal and temporal lobes and the limbic system. It is generally considered that HSV-1 primary infections utilize oral routes of entry, given the common presentation of oral lesions. However, it has been argued that the acute oral lesions of human HSV-1 infections do not necessarily reflect oral host entry, and that the routes used for primary infection and reactivation are not necessarily the same [18].
After infection of epithelial cells, HSV-1 spreads to the peripheral nervous system (PNS), entering sensory neurons by fusion with the plasma membranes of their sensory terminals. Then, HSV-1 travels retrogradely to the cell body and establishes a latent infection in the trigeminal ganglia (TG) [27]. However, latent virus may also locate to CNS structures such as the olfactory bulb (OB), brainstem, or temporal cortex. During latency, the virus persists in the cell nucleus as an episome; the expression of lytic genes is repressed, and conversely the expression of latency-associated transcripts (LAT) begins. Periodically, HSV-1 may leave the latent state and reactivate, either spontaneously or in response to stimulation from immunosuppression, fever, ultraviolet light exposure, or injury to the tissues innervated by latently-infected neurons [19]. This process may lead to a recurrent lesion, but it may also proceed asymptomatically. Although reactivation is a complex process triggered by causes that are not fully understood, it has been demonstrated that the immune system plays a critical role, and in this regard host stress may lead to HSV-1 reactivation by increasing regulatory T cell (Treg) control of CD8+ T lymphocytes [28]. The roles of Tregs in the context of viral infections seem to be highly complex; Tregs may exert radically different roles depending on the infectious agent, the disease phase, or the genetic profile of the host, both suppressing antiviral immune responses and contributing to viral spread and establishment of latency, or conversely contributing to virus control [29]. Latency is also an epigenetically controlled process [19] in which changes induced by different stressors may trigger viral reactivation [30,31]. During reactivation, the virus travels anterogradely along the axon, replicating in the tissue of the dermatome innervated by the sensory neuron in which the virus established latency.
3. HSV-1 Infection of the CNS
HSV-1 may enter the CNS by two main routes: peripheral neurons and the bloodstream. Two cellular barriers (the blood–brain barrier and the blood–cerebrospinal fluid barrier) protect the CNS, separating it from the circulatory system [5,32]. However, other pathways to the CNS are available to pathogens, such as the olfactory system and the trigeminal nerve, which bypass the cellular barriers and provide a direct portal into the brain. Therefore, the trigeminal and olfactory nerves constitute direct routes to the brain that can evade the barriers imposed by the circulatory system [5].
The neurotropic character of HSV-1 has been known for almost a century, since experimental corneal infection produced encephalitis in rabbits, suggesting that the virus propagated through axons and synapses by invasive proliferation [33]. It is currently assumed that primary HSV-1 infection takes place in epithelial cells and subsequently reaches the PNS by direct cell-to-cell retrograde spread to the nerve endings of nearby sensory neurons (Figure 1A,1). During reactivation, HSV-1 virions travel by anterograde transport; that is, from the cell soma of the sensory neuron to the epithelial cells where the primary infection arose (Figure 1A,2). Another pathway for viral spread is the trans-synaptic route, e.g., from one neuron to an adjacent one across the synaptic cleft (Figure 1A,3). Unlike other neurotropic viruses that do not cross synapses, such as Moloney murine leukemia virus (MMLV), lentivirus, adeno-associated virus (AAV), or human adenovirus 5 (Ad5) [34], trans-synaptic spread is a major mode of HSV-1 propagation [35,36]. In fact, given their ability to spread bidirectionally along multi-synaptic pathways, herpesviruses, particularly HSV-1 and PRV, were the first to be widely used to trace neuronal circuits, providing information about connectivity between different areas of the brain [34,37,38,39]. Using HSV-1 as a trans-neuronal tracer, the virus has been observed to cross the synaptic space to label third- to fourth-order neurons [40].
After infection of sensory neurons, HSV-1 travels retrogradely to the cell bodies and establishes a latent infection in the TG (Figure 1B). All branches of the trigeminal nerve (ophthalmic, maxillary, and mandibular) may serve as portals for HSV-1 (Figure 1C), with entry from the oral and nasal epithelia or the cornea. Occasionally, the virus may infect the CNS if it spreads trans-synaptically from the TG to the trigeminal nucleus in the brainstem, from where it might disseminate to higher brain areas (Figure 1B). In fact, there are polysynaptic pathways from the brainstem to the thalamus and somatosensory cortex (Figure 1C) that might be hypothetically utilized by the virus. However, in human HSV-1 infections, viral antigens are mostly found in the olfactory pathways, the temporal cortex, and the limbic system, but not in higher brain areas related to the trigeminal projection pathways, such as the somatosensory cortex [41,42,43,44]. Therefore, those findings support the hypothesis of olfactory spread to the CNS in humans, although spread from the trigeminal nerve to the orbitofrontal and medial temporal lobes has been also proposed. In this regard, the meninges of the middle and anterior fossae are innervated by nerves derived from the TG, and the trigeminal nerve projects to the dura mater via the tentorial nerve, which arises from its ophthalmic division [45]. Spread of the virus to the anterior and middle fossae via tentorial nerves has been proposed to explain viral dissemination to frontal and temporal lobes [46]. However, if the trigeminal pathway was the main portal to the CNS, a higher rate of HSV-1 encephalitis affecting the brainstem would be expected [44]. Finally, the evidence is not sufficient to establish solid conclusions, and further studies are necessary to fully clarify this aspect regarding pathways of HSV-1 entry into the CNS.
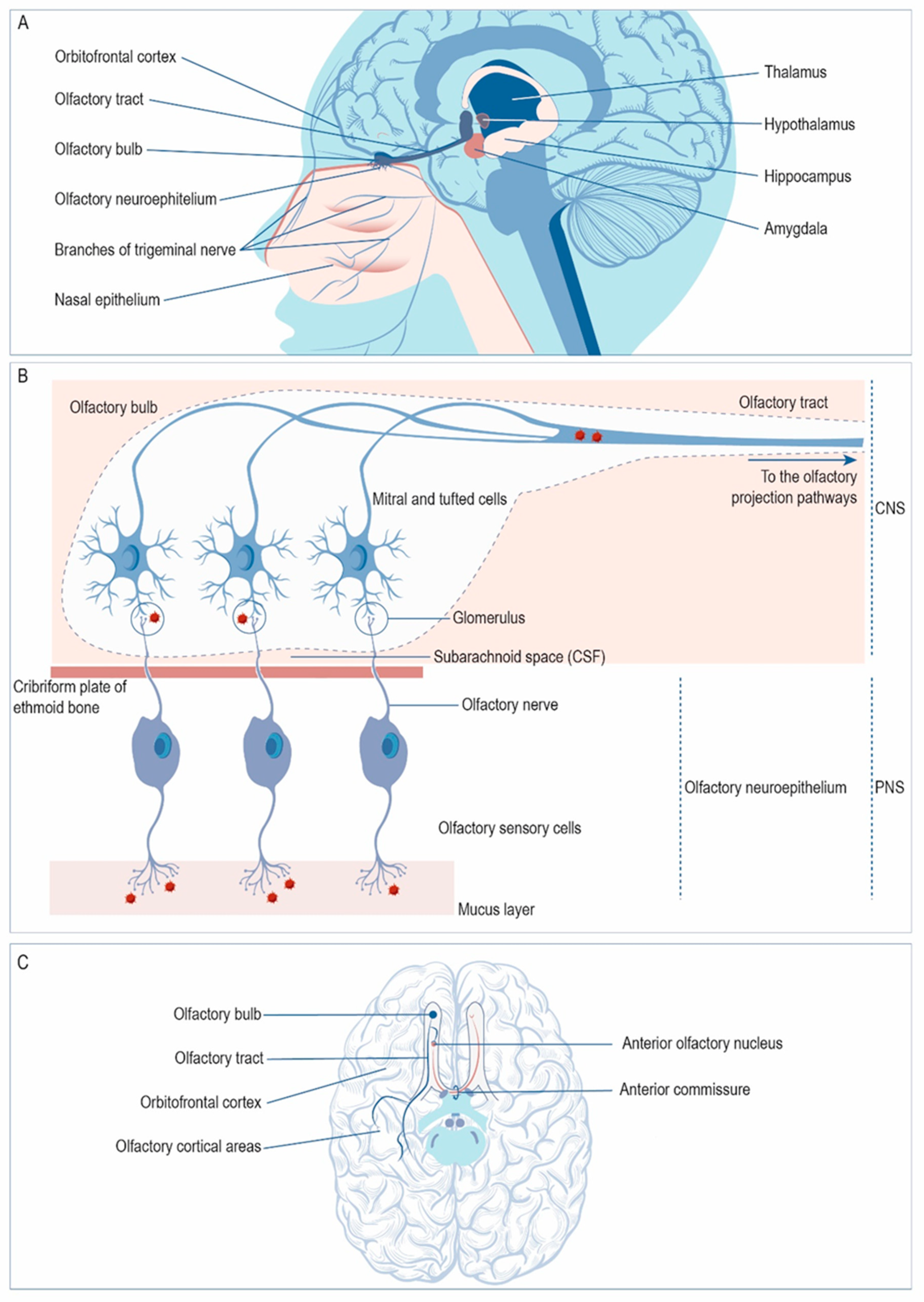
Besides the trigeminal nerve, HSV-1 may enter the CNS via the sensory neurons of the olfactory neuroepithelium (Figure 2A). From there, the virus may access the OB and then spread through the olfactory tract to reach limbic structures such as the hippocampus, amygdala, or orbitofrontal cortex (Figure 2A). In the olfactory neuroepithelium, the cilia of the olfactory sensory cells covering the upper nasal cavity provide a portal for viral entry (Figure 2B). The dendrites of these neurons are covered by a thin layer of mucus that protects them. The axons of these first-order olfactory sensory neurons gather in bundles that project through the cribriform plate of the ethmoid bone to reach the OB, forming the olfactory nerves. Once in the OB, the olfactory sensory neurons synapse with mitral and tufted cells in the glomeruli; the virus may infect these cells trans-synaptically at the glomeruli and spread along the olfactory tract towards the olfactory projection pathways. In the olfactory tract, HSV-1 may access the ipsilateral olfactory projection areas via the lateral olfactory stria. However, virus circulating along the medial olfactory stria may reach the contralateral olfactory structures via the anterior commissure (Figure 2C).
3.1. HSV-1 Receptors in the CNS
HSV-1 may enter cells by membrane fusion at the cell surface or by endocytosis [47,48,49]. Four glycoproteins (gB, gD, gH, and gL) are required for viral entry, and gC is involved in cell attachment, although it is not essential to enter cells [50,51]. Heparan sulphate proteoglycans (HSPGs) act as attachment receptors and gD interacts with nectin-1, herpes virus entry mediator (HVEM), and 3-O-sulphated heparan sulphate to enter cells. Cell receptors for gB are the paired immunoglobulin-like type 2 receptor alpha (PILRα), the myelin-associated glycoprotein (MAG), and the non-muscle myosin heavy chain IIA (NMHC-IIA) [47].
Heparan sulphate (HS) is a linear, negatively charged glycosaminoglycan (GAG) consisting of N-acetylglucosamine and glucuronic acid residues that may be O- or N-sulphated at different positions. This polysaccharide is highly expressed as HSPG on the surface and extracellular matrix of most animal cell types [52,53]. Since most human herpesviruses use them as cell attachment factors, the polarized distribution of HSPGs may modulate viral entry [54], although the distribution of entry receptors may also play a crucial role in influencing the polarity of infection. Nectin-1 has been observed abundantly beneath the HS-positive cilia of the sensory neurons in the olfactory neuroepithelium [18]. In most epithelia, HSPGs are mainly expressed in the basolateral domain [55]. However, in the olfactory epithelium, HSPGs and nectin-1 are also localized to the apical domain, thus facilitating viral binding and providing an important herpesvirus entry portal [56].
HSV-1 entry into neurons takes place by fusion between the viral envelope and the plasma membrane of the sensory neuron dendrites. Therefore, unlike other neurotropic viruses—such as rabies or polioviruses, which enter cells by endocytosis—HSV-1 and alphaherpesviruses in general deliver naked capsids into the host cytoplasm [57]. Then, nucleocapsids travel towards the microtubule organizing center (MTOC) by dynein-mediated retrograde transport and eventually reach the nucleus to implement viral replication. The tegument plays a crucial role in this process, since it participates in the recruitment of the dynein–dynactin complex [57]. To exit neurons and infect adjacent epithelial cells or higher-order neurons, HSV-1 travels along the axons by microtubule-directed anterograde transport. Two hypotheses, the so-called “separate” and “married” models, have been proposed for this transport. In the “separate” model, naked viral nucleocapsids lacking viral envelope and membrane glycoproteins are transported in axons, whereas in the “married” model, enveloped particles enclosed in vesicles are transported in the anterograde direction. It is not clear which process is used by HSV-1 to exit neurons, and it has been even proposed that both mechanisms may be used [58,59].
Nectin-1 expression has been reported in the limbic system, frontal association cortex, and OB in adult mice [60]. In fact, the distribution of this entry receptor in the brain undergoes spatiotemporal changes throughout development. Thus, in the newborn murine brain, expression of nectin-1 was mostly observed in the cerebral cortex, corpus callosum, and anterior commissure, whereas on the seventh postnatal day, this expression increased in limbic regions and decreased in association areas [60]. This developmental change in the distribution of viral receptors may explain the change in susceptibility to HSV-1 infection during brain development. In fact, HSV-1 encephalitis in adults typically affects the orbitofrontal and medial temporal lobes and the insula, whereas neonatal encephalitis shows a much more diffuse inflammation and necrosis [44,46]. Other studies in murine models have reported that nectin-1 mediates infection of dorsal root ganglia neurons [61] and the cornea [62].
In the human brain, nectin-1 has been has been detected in the cornea, neocortex, and hippocampus of formalin-fixed paraffin-embedded tissues from autopsies [63]. Expression of nectin-1 has been also shown to be necessary for CNS infection with HSV-2 and for triggering encephalitis [64]. Later reports confirmed that expression of both gB and gD receptors, specifically nectin-1, HVEM, NMHC-IIA, and possibly MAG, is higher in the hippocampus, suggesting that differential expression levels of these viral receptors in the adult hippocampus might be responsible for the susceptibility of this area to HSV-1 infection [65].
3.2. HSV-1 Entry Pathways
In vivo, herpesviruses have demonstrated tropism for the olfactory epithelium, but not the respiratory epithelium [5]. Thus, intranasal infection of mice with HSV-1 mostly targeted the olfactory neuroepithelium, although the respiratory epithelium was affected to some extent. From this site, the virus travelled to the TGs and then returned peripherally without causing significant neurological disease, nor spreading to the OB [18]. However, other studies in murine models have found HSV-1 in the OBs and higher brain regions after intranasal inoculation [40,66]. The brainstem is another main target of HSV-1 infection; after intranasal inoculation of mice with HSV-1, viral antigens were detected not only in neurons of the TG, but also in trigeminal nuclei and reticular formation, including the raphe nuclei and locus ceruleus [67]. In another study with intranasally infected mice, HSV-1 was detected in the trigeminal root entry zone in the brainstem and the OBs. Later, the virus spread to other brainstem nuclei, and in some mice to the thalamus and cerebellum. From the OBs, virus spread to olfactory projection areas such as the anterior olfactory nucleus, lateral olfactory tract, temporal lobe, or hippocampus [68]. On the other hand, following intracorneal inoculation, the virus was also detected in the OBs, possibly as a result of the passage of the virus to the olfactory neuroepithelium along the nasolacrimal duct [68]. In addition, direct inoculation of HSV-1 into the OB of rabbits led to an infection localized to the frontal and temporal cortices of the animals [69].
Another approach in animal models is the use of corneal inoculation to infect the ophthalmic branch of the trigeminal nerve. Using this route, the infection extends along the trigeminal pathways [70], olfactory system, and limbic structures, such as the entorhinal cortex [71]. Studies of HSV-1 spread using corneal infection of mice have also contributed significant information about viral dissemination. Early studies showed that corneal inoculation of HSV-1 triggers productive infection, not only in peripheral ganglia, but also in brain tissue, indicating viral spread from the PNS to the CNS [72]. More recent studies revealed that HSV-1 may spread to the OB as early as the trigeminal nerve following ocular infection, later inducing a chronic inflammatory response in the OB during latency [73]. Regarding mandibular division, inoculation of the virus into this branch of the trigeminal nerve produced encephalitis, affecting the temporal cortex and limbic system [74]. In addition to the olfactory neurons, in animals HSV-1 may also enter the CNS via the sensory neurons of the vomeronasal organ [75]. From here, the infection is transmitted to the accessory OB and then to the amygdala and hypothalamus [75].
It has also been demonstrated that the anterior commissure may serve as a pathway for contralateral spread of HSV-1. After nasal inoculation of rats, HSV-1 initially infected the right TG and the right OB, and then spread to the same structures on the contralateral side. That spread to the left hemisphere was shown to proceed via the anterior commissure [76]. In addition, that study suggested that HSV-1 used two pathways to spread in the brain. In the first one, the virus was internalized by neurons in the olfactory mucosa and transported to the mitral cells within the right OB. The virus then spread via the right olfactory tract and crossed the anterior commissure to finally reach the left olfactory tract, from where it travelled towards the left OB. The second route consisted of infection entering the right TG and infecting the medulla oblongata and the cerebellum. The hippocampus was infected either from the olfactory tract or the TG. The aquaporin 9 (AQP)-positive cells infected with HSV-1 in the anterior commissure were proposed as OLs, since staining of infected cells of the anterior commissure were negative for markers of astrocytes, microglia, or neurons, and the AQP9-positive cells morphologically resembled OLs. The virus might exploit these myelinating cells to spread rapidly, since a single OL may myelinate several axons [76].
In humans, the olfactory route seems to be highly relevant in symptomatic and asymptomatic HSV-1 infections; additionally, HSV-1 encephalitis in adults typically affects the limbic system. Immunostaining of HSV-1 in autopsied encephalitis cases revealed the presence of viral antigens in the temporal lobe, hippocampus, amygdala, olfactory cortex, insula, and cingulate gyrus. The virus was also found in glial cells of the olfactory tract [77]. Paradoxically, the entry of HSV-1 to the CNS via the trigeminal nerve is unclear, although the sensory neurons of the TG are the major site of latency [5].
3.3. HSV-1 Reactivation
Rabbits and mice have traditionally been the major animal models used to study HSV-1 infection, reactivation, and spread to the CNS [78]. To detect latent HSV-1 in the peripheral sensory TG or dorsal root ganglia, early approaches used co-cultivation of explants with permissive cells [19,79]. During latency, infectious virus cannot be detected after inoculating susceptible cells with ganglion homogenates, but it is possible after incubation of susceptible cells with intact ganglionic explants [19]. In contrast, infectious virus was rarely recovered from the CNS of latently infected animals after explant culture [80], although latent states can be detected in the sensory ganglia or CNS using PCR-based techniques [81].
In humans, several systemic factors such as emotional stress or UV light exposure have been implicated in HSV-1 reactivation. In experimental animal models, this process has been induced by different methods, such as axotomy, cold restrain stress, hyperthermia, hypothermia, corneal scarification, or immunosuppression [82,83]. It has also been demonstrated that epinephrine may induce HSV-1 reactivation in animal models [84,85,86], consistent with the role of stress in HSV-1 reactivation [87,88,89,90]. Iontophoresis is a process that uses an electrical current to transport ions or charged molecules into a tissue. Using this method, epinephrine can be directly conveyed into rabbit eyes to ensure its penetration into ocular tissues of the whole cornea without any trauma [91]. In an early study with the rabbit model, latently infected animals, after prior infection via the cornea, were submitted to reactivation using iontophoresis of epinephrine. Infectious HSV-1 was recovered from the superior cervical ganglion, the TG, and the ophthalmic branch of the trigeminal nerve, suggesting that in addition to TG, the superior cervical ganglion may also harbor HSV-1 during latency [92].
It was initially supposed that reactivation occurred in the sensory ganglia and that brain infection was originated by the spread of reactivated virus from those ganglia. However, later findings have cast doubt on that hypothesis. In a latent-infected mouse model, hyperthermic stress caused reactivation of HSV-1 in the brain earlier than in the TGs [93]. That study, which showed CNS reactivation in explants, suggested that recurrent brain infection might be prompted by the latent virus within the brain itself, not by the spread of the reactivated virus from the ganglia. Nevertheless, a more recent report had conflicting results—a study of reactivation in the TGs and the brainstem detected HSV-1 from both the TG and brainstem of latently infected mice following a reactivation stimulus, but a higher frequency of reactivation and increased infectious titer were recovered from the TGs [94]. Since both studies were developed with C57BL/6 mice, excluding the model as a source of discrepancy, the contradictory results are possibly due to differences in methodology. To directly compare the frequency of reactivation and the amount of infectious virus produced in the TG and brainstem after hyperthermia, Doll et al. used a quantitative assay in which recovery of infectious virus was related to tissue weight. In addition, they used a direct method to detect HSV reactivation in the brainstem, whereas Yao et al. used a two-step assay, which may imply a high contamination risk [94]. Further research will be necessary to fully clarify these conflicting results and to definitively determine the dynamics of HSV-1 reactivation in the nervous system.
4. HSV-1 and Demyelination
MS is an immune-mediated demyelinating and neurodegenerative disease of the CNS of unknown etiology, although several viruses are known to be involved in such demyelinating diseases [6,8,95,96]. The disease is multifactorial, influenced by genetic and environmental factors [97], and it is characterized by multifocal demyelinating lesions in both the white and gray matter [98] of the brain and spinal cord. These lesions can be associated with axon degeneration and synaptic loss. MS is typically multifocal and multiphasic (relapsing), and lesions are thought to be caused by infiltration of immune cells into the CNS [99].
One hallmark of MS is the presence of oligoclonal IgG bands (OCBs) in the cerebrospinal fluid (CSF) of patients. These OCBs, which indicate an anomalous intrathecal B-cell response, are found in the CSF of more than 95% of MS patients and cannot be detected in serum. OCBs are typically detected in inflammatory and infectious CNS disorders. In addition to MS, there are other pathologies with reported CSF OCBs, such as systemic lupus erythematosus, aseptic meningitis, HIV infection, and HSV-1 encephalitis [99]. It has been suggested that OCBs are directed against the infectious agent that causes the disease, and that MS might be triggered by an agent against which the antibody response in the brain and CSF was directed [100]. In fact, OCBs from patients with infectious CNS diseases have been proven to recognize the relevant infectious agent [101]. Regarding MS, OCBs directed against EBV and HHV-6 have been identified in patients [102]. OCBs directed against HSV-1 in the CSF of patients with MS has also been reported [103], although other studies did not find reactivity to HSV-1 antigens [104]. Regardless, the antibody activity of most OCBs remains unknown to date.
It is known that human oligodendrocytic cells are susceptible to HSV-1 in vitro [105]. In murine models, infected OLs were found in the mandibular division of the spinal trigeminal tract after infection of mice through cranial nerve XII [106]. Early studies with animal models showed latent TG infections and demyelinating lesions in mice intranasally infected with HSV-1 [107]. Later research also reported that mice infected with HSV-1 can develop lethal encephalitis or virus-induced CNS multifocal demyelinating lesions, with outcome affected by several factors, including the route of infection and mouse strain [108,109,110,111,112]. Demyelinating lesions have also been associated with HSV-1-induced facial nerve paralysis [113]. A recent study demonstrated a direct association between infection with HSV-1 and multifocal brain demyelination in a murine model [114]. Moreover, in that study, demyelination was followed by remyelination, although it was incomplete and the presence of scars was observed. As in studies with experimental animals, resistance to HSV-1 varies between primary cultures of human OLs and is donor-dependent [115]. Susceptibility to HSV-1 encephalitis may be caused at least partly by mutations in Toll-like receptors that decrease the intrinsic resistance of CNS cells (neurons and OLs in particular) to HSV-1 infection [116,117].
An association between HSV-1 infection and demyelination has been also suggested from studies with human patients. HSV was detected early in the CNS of an MS patient [118], and later HSV-1 was also isolated from the CSF of a patient during the first attack of MS [119]. Postmortem brain samples from 37 cases of MS were screened for HSV-1 and HSV-2, finding higher prevalence of HSV in MS patients compared to controls and in more active plaques than inactive plaques [120,121]. A case of acute MS preceded by varicella-zoster virus (VZV) infection was reported at the same time as intrathecal reactivation of HSV-1 and HHV-6 [122], and a coincident onset of HSV-1 encephalitis and MS has been also described [123]. In another study, patients of MS treated with valacyclovir showed a reduction in the number of new active demyelinating lesions and a decrease in the number of scans free of new active lesions [124]. HSV-1 may also play a role in triggering MS relapses during clinical acute attacks of MS, at least in the most frequent clinical presentation of the disease, the relapsing–remitting form [125]. HSV-1 DNA was detected by PCR in peripheral blood mononuclear cell (PBMC) samples from relapsing–remitting MS patients [126]. However, the involvement of HSV-1 in MS etiology is far from confirmed, and other investigators have proposed other herpesviruses as more plausible etiological agents [127,128,129], or even doubt the role of herpesviruses in the etiology of demyelinating diseases [130].
Viruses, specifically HSV-1, might not operate as unique causative agents, but rather as risk factors, and indeed genetic susceptibility and the immune system are also crucial in demyelinating pathologies. For instance, when a recombinant HSV-1 constitutively expressing interleukin-2 (IL-2) was inoculated into mice, it provoked CNS demyelination and optic neuropathy, whereas infection with recombinant viruses expressing IL-4, gamma interferon, IL-12p35, IL-12p40, or IL-12p70 did not induce this effect [131]. On the other hand, donor-dependent differences in resistance to infection with HSV-1 were established in primary cultures of human OLs [115].
4.1. HSV-1 and Endogenous Retroviruses
Endogenous retroviruses (ERVs) are vestiges of ancient retroviral infections that remain in the host genomes of all vertebrates. They make up around 8% of the human genome and their expression may be triggered by environmental factors, inducing pathogenesis under some circumstances. Several human ERV (HERV) transcripts and proteins have been identified in the CNS, often associated with neuroinflammation [132], and there is a solid epidemiological association between MS and the expression of ERVs, which are upregulated in the brains of MS patients compared to healthy controls [12,133,134,135,136]. The MS-associated retrovirus (MSRV), a member of the HERV-W family, has been frequently isolated from MS patients [137], and its presence in the CSF of these patients has been associated with a greater rate of disability and progression of the disease [138]. Herpesviruses have been implicated in regulation of the HERV-W family [139], and a role for HSV-1 in HERV-W [140] and HERV-K [141] expression has been reported. HERV-W–MSRV expression may be enhanced by HSV-1 in leptomeningeal cells [142,143]. HSV-1 can activate HERV-W in cells involved in MS pathogenesis, such as B cells, macrophages, microglia, and astrocytes [144], and may induce ERV proteins [145].
Syncytins are Env glycoproteins encoded by ERV genes that are involved in mammalian placental morphogenesis [146,147]. These proteins stimulate cell–to–cell fusion in a process analogous to viral entry, promoting the formation of syncytia [146], and can activate pro-inflammatory and autoimmune processes [148]. Syncytin-1, an Env glycoprotein encoded by the HERV-W env gene, plays a crucial role in placental trophoblastic formation and has an immunosuppressive role that impedes rejection of the fetus by the maternal immune system. However, this protein has also been associated with different pathogenic processes, triggering neuroimmune activation and OL damage [148]. In this regard, syncytin-1 may inhibit the differentiation of oligodendroglial precursors, thus hindering remyelination [149]. In astrocytes, overexpression of syncytin-1 (which is upregulated in glial cells in demyelinating lesions of MS patients) triggered the release of redox reactants, inducing neuroinflammation and death of OLs [150]. In this context, HSV-1 has been demonstrated to upregulate syncytin-1 [145], and therefore deepening the understanding of this role may greatly increase knowledge of the demyelinating processes.
4.2. HSV-1 and Molecular Mimicry
Another mechanism that has been associated with HSV-1-related demyelination is molecular mimicry. In this process, the peptides of a pathogen are similar to those of the host organism, triggering activation of autoreactive immune cells in susceptible individuals. Viruses may induce autoimmunity by molecular mimicry [151,152], and several viral peptides have been shown to activate autoreactive T cells [153]. The triggering of autoimmunity by HSV-1 infection was demonstrated when an epitope expressed by the capsid protein was recognized by autoreactive T cells targeting corneal antigens in a murine model of autoimmune herpes stromal keratitis [154]. Mimicry between an epitope shared by the HSV-1 glycoprotein gB and a brain-specific factor has also been reported, supporting the hypothesis that viral infections may prompt the production of self-reactive CSF antibodies [155].
After a HSE episode, a complex immune cellular and humoral response starts. Cytotoxic T lymphocytes lyse cells infected by HSV-1 and the production of inflammatory cytokines (predominantly by Toll-like receptor [TLR] 2) commences [156]. The presence of cytokines such as IL-6, interferon gamma, or TNF alpha, which are detected in the serum and CSF of HSE patients, indicates a strong immune response and suggests that inflammation may contribute to the pathological impact of the viral infection [156]. However, autoimmunity may be another effect triggered by infection. In fact, HSV-1 infection may induce antibodies against neurotransmitter receptors. After an episode of encephalitis caused by HSV-1, around 10–20% of patients may experience a relapse syndrome known as post–herpes simplex encephalitis (PHSE) [157]. This syndrome is immune-mediated, and many patients acquire antibodies against the ionotropic glutamate NMDA receptor (NMDAR). In addition to NMDAR, PHSE may trigger the release of antibodies against GABA A and AMPA receptors, or other still unidentified antigens [157,158,159]. However, although a link between HSV-1 and anti-NMDAR encephalitis has been found, it is not completely clear whether or not molecular mimicry is the responsible mechanism [160]. Regarding ocular HSV-1 infection, it has been proposed that virus infection might result in unmasking of corneal autoantigens, leading to chronic autoreactive T cell stimulation. Alternatively, autoimmunity might be explained by a process of molecular mimicry triggered by a viral peptide sharing reactivity to the unmasked corneal autoantigen. Thus, the initial antiviral response against the virus might subsequently become sustained by autoreactive Taggressor cells [157].
4.3. HSV-1: A Role in Remyelination Impairment?
Primary demyelination in the CNS is a process by which a direct injury to OLs harms the myelin sheath. On the contrary, in secondary demyelination (Wallerian degeneration), the myelin sheath degenerates as a result of primary axonal loss [161]. After demyelinating processes, a mechanism of remyelination starts, which aims to repair the damaged myelin. This process is critically regulated by numerous intracellular signaling pathways [162,163] and depends on generation of new mature OLs derived from a population of adult CNS precursor cells (adult OL precursor or progenitor cells (OPCs)). Remyelinating OLs can derive from the adult subventricular zone (SVZ), a region which may be a source of remyelinating OLs during MS, although the contribution of cells derived from that region might be small compared to the local sources [161].
It has been suggested that infection of OPCs with HHV-6 might impair remyelination [164]. In response to demyelinating damage, OPCs proliferate and migrate to the lesion site, where they differentiate into myelinating OLs and wrap damaged axons with new myelin sheaths. A hypothetical viral infection of OPCs leading to impairment of differentiation or migration might affect remyelination in patients with demyelinating diseases [164]. In this context, HSV-1 might exert a similar role, as it has been proven to infect OPCs in vitro, although the infection increases along with differentiation [165]. Therefore, infection of a population of OPCs by HSV-1 during remyelinating processes might affect this process, resulting in impairment of remyelination.
5. Conclusions
Several viruses are involved in demyelinating diseases such as MS, an immune-mediated demyelinating disease of the CNS of unknown etiology. This disorder is influenced by genetic and environmental factors. Among them, a viral trigger or risk factor has been considered possible. OLs, the myelin-forming cells of the CNS, are highly susceptible to HSV-1 infection in vitro. In animal models, this virus may induce CNS multifocal demyelinating lesions, while studies with human patients have suggested an association between HSV-1 infection and demyelination. However, HSV-1 may act as a risk factor for MS progression rather than as a causative agent. Processes such as molecular mimicry, regulation of ERVs, or remyelination might be mediated by this neurotropic pathogen.
Author Contributions
Conceptualization, R.B.-M.; writing—original draft preparation, R.B.-M.; writing—review and editing, R.B.-M. and J.A.L.-G.; infographics design, R.B.-M. and S.A.; project administration, J.A.L.-G.; funding acquisition, J.A.L.-G. All authors have read and agreed to the published version of the manuscript.
Funding
Financial support for the study was provided by Fundación Severo Ochoa-Aeromédica Canaria. The funders had no role in the decision to publish or preparation of the manuscript.
Acknowledgments
We thank Mateo Casas for infographics. The professional editing service NB Revisions was used for technical preparation of the text prior to submission.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Koyuncu, O.O.; Hogue, I.B.; Enquist, L.W. Virus infections in the nervous system. Cell Host Microbe 2013, 13, 379–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGAVERN, D.B.; Kang, S.S. Illuminating viral infections in the nervous system. Nat. Rev. Immunol. 2011, 11, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Bharucha, T.; Houlihan, C.F.; Bharucha, T. Herpesvirus Infections of the Central Nervous System. Semin. Neurol. 2019, 39, 369–382. [Google Scholar] [CrossRef]
- Kakooza-Mwesige, A.; Tshala-Katumbay, D.; Juliano, S. Viral infections of the central nervous system in Africa. Brain Res. Bull. 2019, 145, 2–17. [Google Scholar] [CrossRef]
- Dando, S.J.; Mackay-Sim, A.; Norton, R.E.; Currie, B.J.; John, J.S.; Ekberg, J.A.; Batzloff, M.; Ulett, G.C.; Beacham, I.R. Pathogens Penetrating the Central Nervous System: Infection Pathways and the Cellular and Molecular Mechanisms of Invasion. Clin. Microbiol. Rev. 2014, 27, 691–726. [Google Scholar] [CrossRef] [Green Version]
- Virtanen, J.O.; Jacobson, S. Viruses and multiple sclerosis. CNS Neurol. Disord. Drug Targets 2012, 11, 528–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libbey, J.E.; Cusick, M.F.; Fujinami, R.S. Role of pathogens in multiple sclerosis. Int. Rev. Immunol. 2014, 33, 266–283. [Google Scholar] [CrossRef]
- Johnson, R.T.; Major, E.O. Infectious Demyelinating Diseases. In Myelin Biology and Disorders; Elsevier: Amsterdam, The Netherlands, 2004; Volume 2, pp. 953–983. [Google Scholar]
- Kakalacheva, K.; Münz, C.; Lünemann, J.D. Viral triggers of multiple sclerosis. Biochim. Biophys. Acta 2011, 1812, 132–140. [Google Scholar] [CrossRef]
- Venkatesan, A.; Johnson, R.T. I nfections and multiple sclerosis. Handb. Clin. Neurol. 2014, 122, 151–171. [Google Scholar] [CrossRef]
- Simmons, A. Herpesvirus and multiple sclerosis. Herpes J. IHMF 2001, 8, 60–63. [Google Scholar]
- Alvarez-Lafuente, R.; García-Montojo, M.; Heras, V.D.L.; Dominguez-Mozo, M.I.; Bartolome, M.; Martín, M.S.B.; Arroyo, R. Herpesviruses and human endogenous retroviral sequences in the cerebrospinal fluid of multiple sclerosis patients. Mult. Scler. 2008, 14, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Giraudon, P.; Bernard, A. Chronic viral infections of the central nervous system: Aspects specific to multiple sclerosis. Rev. Neurol. 2009, 165, 789–795. [Google Scholar] [CrossRef] [PubMed]
- Duarte, L.F.; Farías, M.A.; Álvarez, D.M.; Bueno, S.M.; Riedel, C.A.; Gonzalez, P.A. Herpes Simplex Virus Type 1 Infection of the Central Nervous System: Insights Into Proposed Interrelationships With Neurodegenerative Disorders. Front. Cell. Neurosci. 2019, 13, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itzhaki, R.F. Corroboration of a Major Role for Herpes Simplex Virus Type 1 in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 324. [Google Scholar] [CrossRef] [Green Version]
- Kristen, H.; Sastre, I.; Muñoz-Galdeano, T.; Recuero, M.; Aldudo, J.; Bullido, M.J. The lysosome system is severely impaired in a cellular model of neurodegeneration induced by HSV-1 and oxidative stress. Neurobiol. Aging 2018, 68, 5–17. [Google Scholar] [CrossRef]
- Mangold, C.; Szpara, M.L. Persistent Infection with Herpes Simplex Virus 1 and Alzheimer’s Disease—A Call to Study How Variability in Both Virus and Host may Impact Disease. Viruses 2019, 11, 966. [Google Scholar] [CrossRef] [Green Version]
- Shivkumar, M.; Milho, R.; May, J.S.; Nicoll, M.P.; Efstathiou, S.; Stevenson, P.G. Herpes Simplex Virus 1 Targets the Murine Olfactory Neuroepithelium for Host Entry. J. Virol. 2013, 87, 10477–10488. [Google Scholar] [CrossRef]
- Roizman, B.; Knipe, D.M.; Whitley, R. Herpes simplex viruses. In Fields Virology; Howley, D.M.K.A.P.M., Ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; pp. 2501–2601. [Google Scholar]
- Azab, W.; Dayaram, A.; Greenwood, A.D.; Osterrieder, N. How Host Specific Are Herpesviruses? Lessons from Herpesviruses Infecting Wild and Endangered Mammals. Annu. Rev. Virol. 2018, 5, 53–68. [Google Scholar] [CrossRef]
- A Karasneh, G.; Shukla, D. Herpes simplex virus infects most cell types in vitro: Clues to its success. Virol. J. 2011, 8, 481. [Google Scholar] [CrossRef] [Green Version]
- Looker, K.J.; Magaret, A.S.; May, M.T.; Turner, K.M.E.; Vickerman, P.; Gottlieb, S.L.; Newman, L.M. Global and Regional Estimates of Prevalent and Incident Herpes Simplex Virus Type 1 Infections in 2012. PLoS ONE 2015, 10, e0140765. [Google Scholar] [CrossRef] [Green Version]
- Horowitz, R.; Aierstuck, S.; Williams, E.A.; Melby, B. Herpes Simplex Virus Infection in a University Health Population: Clinical Manifestations, Epidemiology, and Implications. J. Am. Coll. Health 2010, 59, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, D.I.; Bellamy, A.R.; Hook, E.W.; Levin, M.J.; Wald, A.; Ewell, M.G.; Wolff, P.A.; Deal, C.D.; Heineman, T.C.; Dubin, G.; et al. Epidemiology, Clinical Presentation, and Antibody Response to Primary Infection With Herpes Simplex Virus Type 1 and Type 2 in Young Women. Clin. Infect. Dis. 2013, 56, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Wald, A.; Corey, L. Persistence in the population: Epidemiology, transmission. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Whitley, R.J. Herpes simplex encephalitis: Adolescents and adults. Antivir. Res. 2006, 71, 141–148. [Google Scholar] [CrossRef]
- Roizman, B.; Zhou, G.; Du, T. Checkpoints in productive and latent infections with herpes simplex virus 1: Conceptualization of the issues. J. Neurovirol. 2011, 17, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Geng, S.; Suo, Y.; Wei, X.; Cai, Q.; Wu, B.; Zhou, X.; Shi, Y.; Wang, B. Critical Role of Regulatory T Cells in the Latency and Stress-Induced Reactivation of HSV-1. Cell Rep. 2018, 25, 2379–2389. [Google Scholar] [CrossRef] [PubMed]
- Ciurkiewicz, M.; Herder, V.; Beineke, A. Beneficial and Detrimental Effects of Regulatory T Cells in Neurotropic Virus Infections. Int. J. Mol. Sci. 2020, 21, 1705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bloom, D.C.; Giordani, N.V.; Kwiatkowski, D.L. Epigenetic regulation of latent HSV-1 gene expression. Biochim. Biophys. Acta 2010, 1799, 246–256. [Google Scholar] [CrossRef]
- Cliffe, A.R.; Arbuckle, J.H.; Vogel, J.L.; Geden, M.J.; Rothbart, S.B.; Cusack, C.L.; Strahl, B.D.; Kristie, T.M.; Deshmukh, M. Neuronal Stress Pathway Mediating a Histone Methyl/Phospho Switch Is Required for Herpes Simplex Virus Reactivation. Cell Host Microbe 2015, 18, 649–658. [Google Scholar] [CrossRef] [Green Version]
- Swanson, P.; McGAVERN, D.B. Portals of Viral Entry into the Central Nervous System. In The Blood Brain Barrier in Health and Disease Volume 2: Pathophysiology and Pathology; Dorovini-Zis, K., Ed.; CRC Press: Boca Raton, FL, USA, 2015; Volume 2. [Google Scholar]
- Goodpasture, E.W.; Teague, O. Transmission of the Virus of Herpes Febrilis along Nerves in experimentally infected Rabbits. J. Med. Res. 1923, 44, 139. [Google Scholar]
- Nassi, J.J.; Cepko, C.L.; Born, R.; Beier, K.T. Neuroanatomy goes viral! Front. Neuroanat. 2015, 9, 80. [Google Scholar] [CrossRef] [Green Version]
- Enquist, L. Five Questions about Viral Trafficking in Neurons. PLoS Pathog. 2012, 8, e1002472. [Google Scholar] [CrossRef] [Green Version]
- Miller, K.D.; Schnell, M.J.; Rall, G.F. Keeping it in check: Chronic viral infection and antiviral immunity in the brain. Nat. Rev. Neurosci. 2016, 17, 766–776. [Google Scholar] [CrossRef] [PubMed]
- Card, J.; Enquist, L.W. Transneuronal Circuit Analysis with Pseudorabies Viruses. Curr. Protoc. Neurosci. 2014, 68, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callaway, E.M. Transneuronal circuit tracing with neurotropic viruses. Curr. Opin. Neurobiol. 2008, 18, 617–623. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Liu, T.; Dong, Y.; Kondoh, K.; Lu, Z. Trans-synaptic Neural Circuit-Tracing with Neurotropic Viruses. Neurosci. Bull. 2019, 35, 909–920. [Google Scholar] [CrossRef]
- McLean, J.H.; Shipley, M.T.; Bernstein, D.I. Golgi-like, transneuronal retrograde labelling with CNS injections of herpes simplex virus type. Brain Res. Bull. 1989, 22, 867–881. [Google Scholar] [CrossRef]
- Damasio, A.R.; Van Hoesen, G.W. The limbic system and the localisation of herpes simplex encephalitis. J. Neurol. Neurosurg. Psychiatry 1985, 48, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Sokolov, A.A.; Reincke, M. Herpes Simplex Encephalitis Affecting the Entire Limbic System. Mayo Clin. Proc. 2012, 87, e69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyler, K.L. Update on herpes simplex encephalitis. Rev. Neurol. Dis. 2004, 1, 169–178. [Google Scholar]
- Bradshaw, M.J.; Venkatesan, A. Herpes Simplex Virus-1 Encephalitis in Adults: Pathophysiology, Diagnosis, and Management. Neurotherapeutics 2016, 13, 493–508. [Google Scholar] [CrossRef]
- Lee, S.H.; Shin, K.J.; Koh, K.S.; Song, W.C. Visualization of the tentorial innervation of human dura mater. J. Anat. 2017, 231, 683–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, L.E.; Johnson, R.T. An explanation for the localization of herpes simplex encephalitis? Ann. Neurol. 1979, 5, 2–5. [Google Scholar] [CrossRef] [PubMed]
- Agelidis, A.M.; Shukla, D. Cell entry mechanisms of HSV: What we have learned in recent years. Future Virol. 2015, 10, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Heldwein, E.E.; Krummenacher, C. Entry of herpesviruses into mammalian cells. Cell. Mol. Life Sci. 2008, 65, 1653–1668. [Google Scholar] [CrossRef] [PubMed]
- Nicola, A. Herpesvirus Entry into Host Cells Mediated by Endosomal Low pH. Traffic 2016, 17, 965–975. [Google Scholar] [CrossRef] [Green Version]
- Reske, A.; Pollara, G.; Krummenacher, C.; Chain, B.; Katz, D.R. Understanding HSV-1 entry glycoproteins. Rev. Med. Virol. 2007, 17, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Cairns, T.M.; Huang, Z.-Y.; Whitbeck, J.C.; De Leon, M.P.; Lou, H.; Wald, A.; Krummenacher, C.; Eisenberg, R.J.; Cohen, G.H. Dissection of the Antibody Response against Herpes Simplex Virus Glycoproteins in Naturally Infected Humans. J. Virol. 2014, 88, 12612–12622. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, C.D.; Shukla, D. The importance of heparan sulfate in herpesvirus infection. Virol. Sin. 2008, 23, 383–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarrazin, S.; Lamanna, W.C.; Esko, J.D. Heparan Sulfate Proteoglycans. Cold Spring Harb. Perspect. Boil. 2011, 3, a004952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, D.; Spear, P.G. Herpesviruses and heparan sulfate: An intimate relationship in aid of viral entry. J. Clin. Investig. 2001, 108, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Caplan, M.J.; Stow, J.L.; Newman, A.P.; Madri, J.; Anderson, H.C.; Farquhar, M.G.; Palade, G.E.; Jamieson, J.D. Dependence on pH of polarized sorting of secreted proteins. Nature 1987, 329, 632–635. [Google Scholar] [CrossRef]
- Milho, R.; Frederico, B.; Efstathiou, S.; Stevenson, P.G. A Heparan-Dependent Herpesvirus Targets the Olfactory Neuroepithelium for Host Entry. PLoS Pathog. 2012, 8, e1002986. [Google Scholar] [CrossRef] [PubMed]
- Diwaker, D.; Wilson, D.W. Microtubule-Dependent Trafficking of Alphaherpesviruses in the Nervous System: The Ins and Outs. Viruses 2019, 11, 1165. [Google Scholar] [CrossRef] [Green Version]
- Negatsch, A.; Granzow, H.; Maresch, C.; Klupp, B.G.; Fuchs, W.; Teifke, J.P.; Mettenleiter, T.C. Ultrastructural Analysis of Virion Formation and Intraaxonal Transport of Herpes Simplex Virus Type 1 in Primary Rat Neurons. J. Virol. 2010, 84, 13031–13035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wisner, T.W.; Sugimoto, K.; Howard, P.W.; Kawaguchi, Y.; Johnson, D.C. Anterograde Transport of Herpes Simplex Virus Capsids in Neurons by both Separate and Married Mechanisms. J. Virol. 2011, 85, 5919–5928. [Google Scholar] [CrossRef] [PubMed]
- Horváth, S.; Prandovszky, E.; Kis, Z.; Krummenacher, C.; Eisenberg, R.J.; Cohen, G.H.; Janka, Z.; Toldi, J. Spatiotemporal changes of the herpes simplex virus entry receptor nectin-1 in murine brain during postnatal development. J. Neurovirol. 2006, 12, 161–170. [Google Scholar] [CrossRef]
- Richart, S.M.; Simpson, S.A.; Krummenacher, C.; Whitbeck, J.C.; Pizer, L.I.; Cohen, G.H.; Eisenberg, R.J.; Wilcox, C.L. Entry of Herpes Simplex Virus Type 1 into Primary Sensory Neurons In Vitro Is Mediated by Nectin-1/HveC. J. Virol. 2003, 77, 3307–3311. [Google Scholar] [CrossRef]
- Karaba, A.; Kopp, S.J.; Longnecker, R. Herpesvirus Entry Mediator and Nectin-1 Mediate Herpes Simplex Virus 1 Infection of the Murine Cornea. J. Virol. 2011, 85, 10041–10047. [Google Scholar] [CrossRef] [Green Version]
- Prandovszky, E.; Horváth, S.; Gellért, L.; Kovács, S.K.; Janka, Z.; Toldi, J.; Shukla, D.; Vályi–Nagy, T. Nectin-1 (HveC) is expressed at high levels in neural subtypes that regulate radial migration of cortical and cerebellar neurons of the developing human and murine brain. J. Neurovirol. 2008, 14, 164–172. [Google Scholar] [CrossRef]
- Kopp, S.J.; Banisadr, G.; Glajch, K.; Maurer, U.E.; Grünewald, K.; Miller, R.J.; Osten, P.; Spear, P.G. Infection of neurons and encephalitis after intracranial inoculation of herpes simplex virus requires the entry receptor nectin-1. Proc. Natl. Acad. Sci. USA 2009, 106, 17916–17920. [Google Scholar] [CrossRef]
- Lathe, R.; Haas, J.G. Distribution of cellular HSV-1 receptor expression in human brain. J. Neurovirol. 2017, 23, 376–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boggian, I.; Buzzacaro, E.; Calistri, A.; Calvi, P.; Cavaggioni, A.; Mucignat-Caretta, C.; Palu, G. Asymptomatic herpes simplex type 1 virus infection of the mouse brain. J. Neurovirol. 2000, 6, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Kristensson, K.; Nennesmo, I.; Persson, L.; Lycke, E. Neuron to neuron transmission of herpes simplex virus. J. Neurol. Sci. 1982, 54, 149–156. [Google Scholar] [CrossRef]
- Tomlinson, A.; Esiri, M. Herpes simplex encephalitis. J. Neurol. Sci. 1983, 60, 473–484. [Google Scholar] [CrossRef]
- Schlitt, M.; Lakeman, A.D.; Wilson, E.R.; To, A.; Acoff, R.W.; Harsh, G.R.; Whitley, R.J. A Rabbit Model of Focal Herpes Simplex Encephalitis. J. Infect. Dis. 1986, 153, 732–735. [Google Scholar] [CrossRef]
- Knotts, F.B.; Cook, M.L.; Stevens, J.G. Pathogenesis of Herpetic Encephalitis in Mice after Ophthalmic Inoculation. J. Infect. Dis. 1974, 130, 16–27. [Google Scholar] [CrossRef]
- Stroop, W.G.; Rock, D.L.; Fraser, N.W. Localization of herpes simplex virus in the trigeminal and olfactory systems of the mouse central nervous system during acute and latent infections by in situ hybridization. Lab. Investig. 1984, 51, 27–38. [Google Scholar]
- Cabrera, C.V.; Wohlenberg, C.; Openshaw, H.; Méndez, M.R.; Puga, A.; Notkins, A.L. Herpes simplex virus DNA sequences in the CNS of latently infected mice. Nature 1980, 288, 288–290. [Google Scholar] [CrossRef]
- Menendez, C.; Carr, D.J.J. Herpes simplex virus-1 infects the olfactory bulb shortly following ocular infection and exhibits a long-term inflammatory profile in the form of effector and HSV-1-specific T cells. J. Neuroinflammation 2017, 14, 124. [Google Scholar] [CrossRef]
- Barnett, E.M.; Jacobsen, G.; Evans, G.; Cassell, M.; Perlman, S. Herpes Simplex Encephalitis in the Temporal Cortex and Limbic System after Trigeminal Nerve Inoculation. J. Infect. Dis. 1994, 169, 782–786. [Google Scholar] [CrossRef]
- Mori, I.; Goshima, F.; Ito, H.; Koide, N.; Yoshida, T.; Yokochi, T.; Kimura, Y.; Nishiyama, Y. The vomeronasal chemosensory system as a route of neuroinvasion by herpes simplex virus. Virolpgy 2005, 334, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Jennische, E.; E Eriksson, C.; Lange, S.; Trybala, E.; Bergström, T. The anterior commissure is a pathway for contralateral spread of herpes simplex virus type 1 after olfactory tract infection. J. Neurovirol. 2015, 21, 129–147. [Google Scholar] [CrossRef] [PubMed]
- Esiri, M.M. Herpes simplex encephalitis. An immunohistological study of the distribution of viral antigen within the brain. J. Neurol. Sci. 1982, 54, 209–226. [Google Scholar] [CrossRef]
- Kollias, C.M.; Huneke, R.B.; Wigdahl, B.; Jennings, S.R. Animal models of herpes simplex virus immunity and pathogenesis. J. Neurovirol. 2014, 21, 8–23. [Google Scholar] [CrossRef]
- Stevens, J.G.; Cook, M.L. Latent Herpes Simplex Virus in Spinal Ganglia of Mice. Science 1971, 173, 843–845. [Google Scholar] [CrossRef]
- Rock, D.L.; Fraser, N.W. Detection of HSV-1 genome in central nervous system of latently infected mice. Nature 1983, 302, 523–525. [Google Scholar] [CrossRef]
- Mehta, A.; Maggioncalda, J.; Bagasra, O.; Thikkavarapu, S.; Saikumari, P.; Valyi-Nagy, T.; Fraser, N.W.; Block, T.M. In situ DNA PCR and RNA hybridization detection of herpes simplex virus sequences in trigeminal gangliaof latently infected mice. Virology 1995, 206, 633–640. [Google Scholar] [CrossRef]
- Suzich, J.B.; Cliffe, A.R. Strength in diversity: Understanding the pathways to herpes simplex virus reactivation. Virology 2018, 522, 81–91. [Google Scholar] [CrossRef]
- Huang, W.; Xie, P.; Xu, M.; Li, P.; Zao, G. The Influence of Stress Factors on the Reactivation of Latent Herpes Simplex Virus Type 1 in Infected Mice. Cell Biochem. Biophys. 2011, 61, 115–122. [Google Scholar] [CrossRef]
- Rootman, D.S.; Haruta, Y.; Hill, J.M. Reactivation of HSV-1 in primates by transcorneal iontophoresis of adrenergic agents. Investig. Ophthalmol. Vis. Sci. 1990, 31, 597–600. [Google Scholar]
- Willey, D.E.; Trousdale, M.D.; Nesburn, A.B. Reactivation of murine latent HSV infection by epinephrine iontophoresis. Investig. Ophthalmol. Vis. Sci. 1984, 25, 945–950. [Google Scholar]
- Webre, J.M.; Hill, J.M.; Nolan, N.M.; Clement, C.; McFerrin, H.E.; Bhattacharjee, P.S.; Hsia, S.V.; Neumann, N.M.; Foster, T.P.; Lukiw, W.J.; et al. Rabbit and Mouse Models of HSV-1 Latency, Reactivation, and Recurrent Eye Diseases. J. Biomed. Biotechnol. 2012, 2012, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Luo, Z.; Li, W.; Li, X.; Dallmann, R.; Kurihara, H.; Li, Y.-F.; He, R.-R. Disturbed Yin–Yang balance: Stress increases the susceptibility to primary and recurrent infections of herpes simplex virus type 1. Acta Pharm. Sin. B 2020, 10, 383–398. [Google Scholar] [CrossRef] [PubMed]
- Blondeau, J.M.; Aoki, F.Y.; Glavin, G.B. Stress-induced reactivation of latent herpes simplex virus infection in rat lumbar dorsal root ganglia. J. Psychosom. Res. 1993, 37, 843–849. [Google Scholar] [CrossRef]
- Ashcraft, K.A.; Bonneau, R.H. Psychological stress exacerbates primary vaginal herpes simplex virus type 1 (HSV-1) infection by impairing both innate and adaptive immune responses. Brain Behav. Immun. 2008, 22, 1231–1240. [Google Scholar] [CrossRef] [PubMed]
- Chida, Y.; Mao, X. Does psychosocial stress predict symptomatic herpes simplex virus recurrence? A meta-analytic investigation on prospective studies. Brain Behav. Immun. 2009, 23, 917–925. [Google Scholar] [CrossRef] [PubMed]
- Kwon, B.S.; Gangarosa, L.P.; Burch, K.D.; Deback, J.; Hill, J.M. Induction of ocular herpes simplex virus shedding by iontophoresis of epinephrine into rabbit cornea. Investig. Ophthalmol. Vis. Sci. 1981, 21, 442–449. [Google Scholar]
- Shimomura, Y.; Dudley, J.B.; Gangarosa, L.P.; Hill, J.M. HSV-1 quantitation from rabbit neural tissues after epinephrine-induced reactivation. Investig. Ophthalmol. Vis. Sci. 1985, 26, 121–125. [Google Scholar]
- Yao, H.-W.; Ling, P.; Tung, Y.-Y.; Hsu, S.-M.; Chen, S.-H. In Vivo Reactivation of Latent Herpes Simplex Virus 1 in Mice Can Occur in the Brain before Occurring in the Trigeminal Ganglion. J. Virol. 2014, 88, 11264–11270. [Google Scholar] [CrossRef]
- Doll, J.R.; Thompson, R.L.; Sawtell, N.M. Infectious Herpes Simplex Virus in the Brain Stem Is Correlated with Reactivation in the Trigeminal Ganglia. J. Virol. 2019. [Google Scholar] [CrossRef]
- Donati, D. Viral infections and multiple sclerosis. Drug Discov. Today Dis. Model. 2020. [Google Scholar] [CrossRef] [PubMed]
- Tarlinton, R.E.; Martynova, E.; Rizvanov, A.; Khaiboullina, S.; Verma, S. Role of Viruses in the Pathogenesis of Multiple Sclerosis. Viruses 2020, 12, 643. [Google Scholar] [CrossRef] [PubMed]
- O’Gorman, C.; Lucas, R.M.; Taylor, B. Environmental Risk Factors for Multiple Sclerosis: A Review with a Focus on Molecular Mechanisms. Int. J. Mol. Sci. 2012, 13, 11718–11752. [Google Scholar] [CrossRef] [Green Version]
- Calabrese, M.; Magliozzi, R.; Ciccarelli, O.; Geurts, J.J.G.; Reynolds, R.; Martin, R. Exploring the origins of grey matter damage in multiple sclerosis. Nat. Rev. Neurosci. 2015, 16, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Deisenhammer, F.; Zetterberg, H.; Fitzner, B.; Zettl, U.K. The Cerebrospinal Fluid in Multiple Sclerosis. Front. Immunol. 2019, 10, 726. [Google Scholar] [CrossRef]
- Owens, G.P.; Gilden, D.; Burgoon, M.P.; Yu, X.; Bennett, J.L. Viruses and multiple sclerosis. Neuroscientist 2011, 17, 659–676. [Google Scholar] [CrossRef]
- Martin, R.; Martens, U.; Sticht-Groh, V.; Dörries, R.; Krüger, H. Persistent intrathecal secretion of oligoclonal, Borrelia burgdorferi-specific IgG in chronic meningoradiculomyelitis. J. Neurol. 1988, 235, 229–233. [Google Scholar] [CrossRef]
- Virtanen, J.O.; Wohler, J.; Fenton, K.; Reich, D.S.; Jacobson, S. Oligoclonal bands in multiple sclerosis reactive against two herpesviruses and association with magnetic resonance imaging findings. Mult. Scler. J. 2014, 20, 27–34. [Google Scholar] [CrossRef]
- Rostrom, B.; Link, H.; Laurenzi, M.A.; Kam-Hansen, S.; Norrby, E.; Wahren, B. Viral antibody activity of oligoclonal and polyclonal immunoglobulins synthesized within the central nervous system in multiple sclerosis. Ann. Neurol. 1981, 9, 569–574. [Google Scholar] [CrossRef]
- Virtanen, J.O.; Pietiläinen-Nicklén, J.; Uotila, L.; Färkkilä, M.; Vaheri, A.; Koskiniemi, M. Intrathecal human herpesvirus 6 antibodies in multiple sclerosis and other demyelinating diseases presenting as oligoclonal bands in cerebrospinal fluid. J. Neuroimmunol. 2011, 237, 93–97. [Google Scholar] [CrossRef]
- Bello-Morales, R.; Fedetz, M.; Alcina, A.; Tabarés, E.; Bosch, A.L. High susceptibility of a human oligodendroglial cell line to herpes simplex type 1 infection. J. Neurovirol. 2005, 11, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Ugolini, G.; Kuypers, H.; Simmons, A. Retrograde transneuronal transfer of Herpes simplex virus type 1 (HSV 1) from motoneurones. Brain Res. 1987, 422, 242–256. [Google Scholar] [CrossRef]
- Kristensson, K.; Svennerholm, B.; Persson, L.; Vahlne, A.; Lycke, E. Latent herpes simplex virus trigeminal ganglionic infection in mice and demyelination in the central nervous system. J. Neurol. Sci. 1979, 43, 253–263. [Google Scholar] [CrossRef]
- Kastrukoff, L.F.; Lau, A.S.; Kim, S.U. Multifocal CNS demyelination following peripheral inoculation with herpes simplex virus type 1. Ann. Neurol. 1987, 22, 52–59. [Google Scholar] [CrossRef]
- Kastrukoff, L.F.; Lau, A.S.; Thomas, E.E. The effect of mouse strain on herpes simplex virus type 1 (HSV-1) infection of the central nervous system (CNS). Herpesviridae 2012, 3, 4. [Google Scholar] [CrossRef] [Green Version]
- Kastrukoff, L.F.; Lau, A.S.; Leung, G.Y.; Walker, D.; Thomas, E.E. Herpes Simplex Virus Type I (HSV l)-lnduced Multifocal Central Nervous System (CNS) Demyelination in Mice. J. Neuropathol. Exp. Neurol. 1992, 51, 432–439. [Google Scholar] [CrossRef]
- Kristensson, K.; Svennerholm, B.; Vahlne, A.; Nilheden, E.; Persson, L.; Lycke, E. Virus-induced demyelination in herpes simplex virus-infected mice. J. Neurol. Sci. 1982, 53, 205–216. [Google Scholar] [CrossRef]
- Lee, D.H.; Zandian, M.; Kuo, J.; Mott, K.R.; Chen, S.; Arditi, M.; Ghiasi, H. Suppression of IL-12p70 formation by IL-2 or following macrophage depletion causes T-cell autoreactivity leading to CNS demyelination in HSV-1-infected mice. PLoS Pathog. 2017, 13, e1006401. [Google Scholar] [CrossRef] [Green Version]
- Wakisaka, H.; Hato, N.; Honda, N.; Takahashi, H.; Kisaki, H.; Murakami, S.; Gyo, K.; Mominoki, K.; Kobayashi, N.; Matsuda, S. Demyelination Associated with HSV-1-Induced Facial Paralysis. Exp. Neurol. 2002, 178, 68–79. [Google Scholar] [CrossRef]
- Boukhvalova, M.; Mortensen, E.; Mbaye, A.; Lopez, D.; Kastrukoff, L.; Blanco, J.C.G. Herpes Simplex Virus 1 Induces Brain Inflammation and Multifocal Demyelination in the Cotton Rat Sigmodon hispidus. J. Virol. 2019. [Google Scholar] [CrossRef]
- Kastrukoff, L.F.; Kim, S.U. Oligodendrocytes from human donors differ in resistance to herpes simplex virus 1 (HSV-1). Glia 2002, 38, 87–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leib, D.A. Herpes Simplex Virus Encephalitis: Toll-Free Access to the Brain. Cell Host Microbe 2012, 12, 731–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lafaille, F.G.; Pessach, I.M.; Zhang, S.-Y.; Ciancanelli, M.J.; Herman, M.; Abhyankar, A.; Ying, S.-W.; Keros, S.; Goldstein, P.A.; Mostoslavsky, G.; et al. Impaired intrinsic immunity to HSV-1 in human iPSC-derived TLR3-deficient CNS cells. Nature 2012, 491, 769–773. [Google Scholar] [CrossRef] [PubMed]
- Gudnadóttir, M.; Helgadóttir, H.; Bjarnason, O.; Jonsdottir, K. Virus isolated from the brain of a patient with multiple sclerosis. Exp. Neurol. 1964, 9, 85–95. [Google Scholar] [CrossRef]
- Bergström, T.; Andersen, O.; Vahlne, A. Isolation of herpes simplex virus type 1 during first attack of multiple sclerosis. Ann. Neurol. 1989, 26, 283–285. [Google Scholar] [CrossRef]
- Sanders, V.J.; Felisan, S.; Waddell, A.; Tourtellotte, W.W. Detection of Herpesviridae in postmortem multiple sclerosis brain tissue and controls by polymerase chain reaction. J. Neurovirol. 1996, 2, 249–258. [Google Scholar] [CrossRef]
- Sanders, V.J.; Waddell, A.E.; Felisan, S.L.; Li, X.; Conrad, A.J.; Tourtellotte, W.W. Herpes Simplex Virus in Postmortem Multiple Sclerosis Brain Tissue. Arch. Neurol. 1996, 53, 125–133. [Google Scholar] [CrossRef]
- Ferrò, M.T.; Franciotta, D.; Prelle, A.; Bestetti, A.; Cinque, P. Active intrathecal herpes simplex virus type 1 (HSV-1) and human herpesvirus-6 (HHV-6) infection at onset of multiple sclerosis. J. Neurovirol. 2012, 18, 437–440. [Google Scholar] [CrossRef]
- Buscarinu, M.C.; Fornasiero, A.; Romano, S.; Ferraldeschi, M.; Renié, R.; Trasimeni, G.; Salvetti, M.; Ristori, G. Coincident onset of multiple sclerosis and Herpes simplex virus 1 encephalitis: A case report. Mult. Scler. Demyelinating Disord. 2017, 2, 6. [Google Scholar] [CrossRef]
- Bech, E.; Lycke, J.; Gadeberg, P.; Hansen, H.; Malmeström, C.; Andersen, O.; Christensen, T.; Ekholm, S.; Haahr, S.; Hollsberg, P.; et al. A randomized, double-blind, placebo-controlled MRI study of anti-herpes virus therapy in MS. Neurology 2002, 58, 31–36. [Google Scholar] [CrossRef]
- Ferrante, P.; Mancuso, R.; Pagani, E.; Guerini, F.R.; Calvo, M.G.; Saresella, M.; Speciale, L.; Caputo, D. Molecular evidences for a role of HSV-1 in multiple sclerosis clinical acute attack. J. Neurovirol. 2000, 6, S109. [Google Scholar] [PubMed]
- Pietropaolo, V.; Fioriti, D.; Mischitelli, M.; Anzivino, E.; Santini, M.; Millefiorini, E.; Di Rezze, S.; Degener, A.M. Detection of human herpesviruses and polyomaviruses DNA in a group of patients with relapsing-remitting multiple sclerosis. New Microbiol. 2005, 28, 199–203. [Google Scholar] [PubMed]
- Ascherio, A.; Munger, K. Environmental risk factors for multiple sclerosis. Part I: The role of infection. Ann. Neurol. 2007, 61, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Correale, J.; Gaitan, M.I. Multiple sclerosis and environmental factors: The role of vitamin D, parasites, and Epstein-Barr virus infection. Acta Neurol. Scand. 2015, 132, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Leibovitch, E.C.; Jacobson, S. Viruses in chronic progressive neurologic disease. Mult. Scler. 2018, 24, 48–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, L.; McQuaid, S.; Cosby, S. Detection of herpes simplex virus (types 1 and 2) and human herpesvirus 6 DNA in human brain tissue by polymerase chain reaction. Clin. Diagn. Virol. 1996, 6, 33–40. [Google Scholar] [CrossRef]
- Mott, K.R.; Zandian, M.; Allen, S.J.; Ghiasi, H. Role of Interleukin-2 and Herpes Simplex Virus 1 in Central Nervous System Demyelination in Mice. J. Virol. 2013, 87, 12102–12109. [Google Scholar] [CrossRef] [Green Version]
- Antony, J.M.; DesLauriers, A.M.; Bhat, R.K.; Ellestad, K.K.; Power, C. Human endogenous retroviruses and multiple sclerosis: Innocent bystanders or disease determinants? Biochim. Biophys. Acta 2011, 1812, 162–176. [Google Scholar] [CrossRef]
- Morandi, E.; Tanasescu, R.; Tarlinton, R.E.; Constantinescu, C.S.; Zhang, W.; Tench, C.; Gran, B. The association between human endogenous retroviruses and multiple sclerosis: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0172415. [Google Scholar] [CrossRef]
- Christensen, T. Association of human endogenous retroviruses with multiple sclerosis and possible interactions with herpes viruses. Rev. Med. Virol. 2005, 15, 179–211. [Google Scholar] [CrossRef]
- Christensen, T. Human endogenous retroviruses in the aetiology of MS. Acta Neurol. Scand. 2017, 136, 18–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kriesel, J.D.; Bhetariya, P.J.; Chan, B.K.; Wilson, T.; Fischer, K.F. Enrichment of Retroviral Sequences in Brain Tissue from Patients with Severe Demyelinating Diseases. J. Emerg. Dis. Virol. 2017. [Google Scholar] [CrossRef]
- Perron, H.; Garson, J.A.; Bedin, F.; Besème, F.; Paranhos-Baccalà, G.; Komurian-Pradel, F.; Mallet, F.; Tuke, P.W.; Voisset, C.; Blond, J.L.; et al. Molecular identification of a novel retrovirus repeatedly isolated from patients with multiple sclerosis. The Collaborative Research Group on Multiple Sclerosis. Proc. Natl. Acad. Sci. USA 1997, 94, 7583–7588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sotgiu, S.; Mameli, G.; Serra, C.; Zarbo, I.; Arru, G.; Dolei, A. Multiple sclerosis-associated retrovirus and progressive disability of multiple sclerosis. Mult. Scler. 2010, 16, 1248–1251. [Google Scholar] [CrossRef]
- Küry, P.; Nath, A.; Creange, A.; Dolei, A.; Marche, P.N.; Gold, J.; Giovannoni, G.; Hartung, H.-P.; Perron, H. Human Endogenous Retroviruses in Neurological Diseases. Trends Mol. Med. 2018, 24, 379–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.J.; Kwun, H.J.; Kim, H.S.; Jang, K.L. Activation of the human endogenous retrovirus W long terminal repeat by herpes simplex virus type 1 immediate early protein 1. Mol. Cells 2003, 15, 75–80. [Google Scholar]
- Kwun, H.J.; Han, H.J.; Lee, W.J.; Kim, H.S.; Jang, K.L. Transactivation of the human endogenous retrovirus K long terminal repeat by herpes simplex virus type 1 immediate early protein 0. Virus Res. 2002, 86, 93–100. [Google Scholar] [CrossRef]
- Perron, H.; Geny, C.; Laurent, A.; Mouriquand, C.; Pellat, J.; Perret, J.; Seigneurin, J. Leptomeningeal cell line from multiple sclerosis with reverse transcriptase activity and viral particles. Res. Virol. 1989, 140, 551–561. [Google Scholar] [CrossRef]
- Perron, H.; Suh, M.; Lalande, B.; Gratacap, B.; Laurent, A.; Stoebner, P.; Seigneurin, J.M. Herpes simplex virus ICP0 and ICP4 immediate early proteins strongly enhance expression of a retrovirus harboured by a leptomeningeal cell line from a patient with multiple sclerosis. J. Gen. Virol. 1993, 74, 65–72. [Google Scholar] [CrossRef]
- Marrodan, M.; Alessandro, L.; Farez, M.F.; Correale, J. The role of infections in multiple sclerosis. Mult. Scler. 2019, 25, 891–901. [Google Scholar] [CrossRef]
- Ruprecht, K.; Obojes, K.; Wengel, V.; Gronen, F.; Kim, K.S.; Perron, H.; Schneider-Schaulies, J.; Rieckmann, P. Regulation of human endogenous retrovirus W protein expression by herpes simplex virus type 1: Implications for multiple sclerosis. J. Neurovirol. 2006, 12, 65–71. [Google Scholar] [CrossRef]
- Johnson, W.E. Origins and evolutionary consequences of ancient endogenous retroviruses. Nat. Rev. Microbiol. 2019, 17, 355–370. [Google Scholar] [CrossRef] [PubMed]
- Mi, S.; Lee, X.; Li, X.-P.; Veldman, G.M.; Finnerty, H.; Racie, L.; LaVallie, E.; Tang, X.-Y.; Edouard, P.; Howes, S.; et al. Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nature 2000, 403, 785–789. [Google Scholar] [CrossRef]
- Wang, X.; Huang, J.; Zhu, F. Human Endogenous Retroviral Envelope Protein Syncytin-1 and Inflammatory Abnormalities in Neuropsychological Diseases. Front. Psychol. 2018, 9, 442. [Google Scholar] [CrossRef] [PubMed]
- Kremer, D.; Schichel, T.; Förster, M.; Tzekova, N.; Bernard, C.; Van Der Valk, P.; Van Horssen, J.; Hartung, H.-P.; Perron, H.; Küry, P. Human endogenous retrovirus type W envelope protein inhibits oligodendroglial precursor cell differentiation. Ann. Neurol. 2013, 74, 721–732. [Google Scholar] [CrossRef]
- Antony, J.; Van Marle, G.; Opii, W.; Butterfield, D.A.; Mallet, F.; Yong, V.W.; Wallace, J.L.; Deacon, R.M.; Warren, K.; Power, C. Human endogenous retrovirus glycoprotein-mediated induction of redox reactants causes oligodendrocyte death and demyelination. Nat. Neurosci. 2004, 7, 1088–1095. [Google Scholar] [CrossRef]
- Fujinami, R.; Oldstone, M. Amino acid homology between the encephalitogenic site of myelin basic protein and virus: Mechanism for autoimmunity. Science 1985, 230, 1043–1045. [Google Scholar] [CrossRef]
- Geginat, J.; Paroni, M.; Pagani, M.; Galimberti, D.; De Francesco, R.; Scarpini, E.; Abrignani, S. The Enigmatic Role of Viruses in Multiple Sclerosis: Molecular Mimicry or Disturbed Immune Surveillance? Trends Immunol. 2017, 38, 498–512. [Google Scholar] [CrossRef] [PubMed]
- Wucherpfennig, K.W.; Strominger, J.L. Molecular mimicry in T cell-mediated autoimmunity: Viral peptides activate human T cell clones specific for myelin basic protein. Cell 1995, 80, 695–705. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.S.; Granucci, F.; Yeh, L.; Schaffer, P.A.; Cantor, H. Molecular Mimicry by Herpes Simplex Virus-Type 1: Autoimmune Disease After Viral Infection. Science 1998, 279, 1344–1347. [Google Scholar] [CrossRef]
- Cortese, I.; Capone, S.; Luchetti, S.; Cortese, R.; Nicosia, A. Cross-reactive phage-displayed mimotopes lead to the discovery of mimicry between HSV-1 and a brain-specific protein. J. Neuroimmunol. 2001, 113, 119–128. [Google Scholar] [CrossRef]
- Gnann, J.W., Jr.; Whitley, R.J. Herpes Simplex Encephalitis: An Update. Curr. Infect. Dis. Rep. 2017, 19, 13. [Google Scholar] [CrossRef] [PubMed]
- Armangue, T.; Spatola, M.; Vlagea, A.; Mattozzi, S.; Cárceles-Cordon, M.; Martinez-Heras, E.; Llufriu, S.; Muchart, J.; Erro, M.E.; Abraira, L.; et al. Frequency, symptoms, risk factors, and outcomes of autoimmune encephalitis after herpes simplex encephalitis: A prospective observational study and retrospective analysis. Lancet Neurol. 2018, 17, 760–772. [Google Scholar] [CrossRef]
- Armangue, T.; Moris, G.; Cantarín-Extremera, V.; Conde, C.E.; Rostasy, K.; Erro, M.E.; Portilla-Cuenca, J.C.; Turón-Viñas, E.; Málaga, I.; Muñoz-Cabello, B.; et al. Autoimmune post-herpes simplex encephalitis of adults and teenagers. Neurol. 2015, 85, 1736–1743. [Google Scholar] [CrossRef] [Green Version]
- Nóbrega, P.R.; Pitombeira, M.S.; Mendes, L.S.; Krueger, M.B.; Santos, C.F.; Morais, N.M.D.M.; Simabukuro, M.M.; Maia, F.M.; Braga-Neto, P. Clinical Features and Inflammatory Markers in Autoimmune Encephalitis Associated With Antibodies Against Neuronal Surface in Brazilian Patients. Front. Neurol. 2019, 10, 472. [Google Scholar] [CrossRef] [PubMed]
- Sinmaz, N.; Nguyen, T.; Tea, F.; Dale, R.C.; Brilot, F. Mapping autoantigen epitopes: Molecular insights into autoantibody-associated disorders of the nervous system. J. Neuroinflammation 2016, 13, 219. [Google Scholar] [CrossRef] [Green Version]
- Franklin, R.J.; Ffrench-Constant, C. Remyelination in the CNS: From biology to therapy. Nature reviews. Neuroscience 2008, 9, 839–855. [Google Scholar] [CrossRef]
- Sabo, J.K.; Cate, H.T. Signalling Pathways that Inhibit the Capacity of Precursor Cells for Myelin Repair. Int. J. Mol. Sci. 2013, 14, 1031–1049. [Google Scholar] [CrossRef]
- Gaesser, J.M.; Fyffe-Maricich, S.L. Intracellular signaling pathway regulation of myelination and remyelination in the CNS. Exp. Neurol. 2016, 283, 501–511. [Google Scholar] [CrossRef] [Green Version]
- Mayer-Pröschel, M.; Hogestyn, J.M.; Mock, D.J. Contributions of neurotropic human herpesviruses herpes simplex virus 1 and human herpesvirus 6 to neurodegenerative disease pathology. Neural Regen. Res. 2018, 13, 211–221. [Google Scholar] [CrossRef]
- Bello-Morales, R.; Crespillo, A.J.; García, B.; Dorado, L.; Ángel Martin, B.; Tabarés, E.; Krummenacher, C.; De Castro, F.; Bosch, A.L. The Effect of Cellular Differentiation on HSV-1 Infection of Oligodendrocytic Cells. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Spread of HSV-1 to the CNS via the trigeminal nerve. (A) HSV-1 may pass from the epithelia to the peripheral nervous system (PNS) by cell-to-cell spread between epithelial cells and nerve endings of sensory neurons that innervate them (1). The virus travels along the axon by retrograde transport to the cell soma of the sensory neuron, located in the trigeminal ganglion (TG). Conversely, HSV-1 can travel back by anterograde transport to the epithelial cells where the primary infection took place (2). The virus can also spread trans-synaptically, crossing the synaptic cleft (3). (B) After infection of epithelial cells, HSV-1 spreads to the PNS, entering sensory neurons by fusion with the plasma membrane of its nerve terminals. Then, HSV-1 travels retrogradely to the cell body and establishes a latent infection in the TG. Afterwards, the virus may enter the central nervous system (CNS) if it spreads trans-synaptically to the brainstem, from where it might spread to higher brain areas. (C) Spread to the CNS may take place through the three branches of the trigeminal nerve: ophthalmic, maxillary and mandibular. From here, the virus can access the trigeminal nucleus and other brain structures. (Structures are schematically represented and they are not drawn to scale).
Figure 1.
Spread of HSV-1 to the CNS via the trigeminal nerve. (A) HSV-1 may pass from the epithelia to the peripheral nervous system (PNS) by cell-to-cell spread between epithelial cells and nerve endings of sensory neurons that innervate them (1). The virus travels along the axon by retrograde transport to the cell soma of the sensory neuron, located in the trigeminal ganglion (TG). Conversely, HSV-1 can travel back by anterograde transport to the epithelial cells where the primary infection took place (2). The virus can also spread trans-synaptically, crossing the synaptic cleft (3). (B) After infection of epithelial cells, HSV-1 spreads to the PNS, entering sensory neurons by fusion with the plasma membrane of its nerve terminals. Then, HSV-1 travels retrogradely to the cell body and establishes a latent infection in the TG. Afterwards, the virus may enter the central nervous system (CNS) if it spreads trans-synaptically to the brainstem, from where it might spread to higher brain areas. (C) Spread to the CNS may take place through the three branches of the trigeminal nerve: ophthalmic, maxillary and mandibular. From here, the virus can access the trigeminal nucleus and other brain structures. (Structures are schematically represented and they are not drawn to scale).

Figure 2.
Spread of herpes simplex virus type 1 (HSV-1) to the central nervous system (CNS) via the olfactory nerve. (A) HSV-1 may enter the CNS via the olfactory neuroepithelium. From there, the virus may reach the olfactory bulb and then spread through the olfactory tract to reach limbic structures, such as the hippocampus, amygdala, or orbitofrontal cortex. (B) HSV-1 may reach the olfactory bulb infecting olfactory sensory cells, whose axons cross the ethmoid bone through the cribriform plate. These neurons form synapses with mitral and tufted cells in the glomeruli. The virus may infect these cells trans-synaptically at the glomeruli and spread towards the olfactory projection pathways. (C) Once in the olfactory tract, the virus may access the ipsilateral projection areas, such as the orbitofrontal cortex, via the lateral olfactory stria (in blue), or they may reach the contralateral olfactory structures through the anterior commissure via the medial olfactory stria (in red). (Structures are schematically represented and are not drawn to scale).
Figure 2.
Spread of herpes simplex virus type 1 (HSV-1) to the central nervous system (CNS) via the olfactory nerve. (A) HSV-1 may enter the CNS via the olfactory neuroepithelium. From there, the virus may reach the olfactory bulb and then spread through the olfactory tract to reach limbic structures, such as the hippocampus, amygdala, or orbitofrontal cortex. (B) HSV-1 may reach the olfactory bulb infecting olfactory sensory cells, whose axons cross the ethmoid bone through the cribriform plate. These neurons form synapses with mitral and tufted cells in the glomeruli. The virus may infect these cells trans-synaptically at the glomeruli and spread towards the olfactory projection pathways. (C) Once in the olfactory tract, the virus may access the ipsilateral projection areas, such as the orbitofrontal cortex, via the lateral olfactory stria (in blue), or they may reach the contralateral olfactory structures through the anterior commissure via the medial olfactory stria (in red). (Structures are schematically represented and are not drawn to scale).

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bello-Morales, R.; Andreu, S.; López-Guerrero, J.A. The Role of Herpes Simplex Virus Type 1 Infection in Demyelination of the Central Nervous System. Int. J. Mol. Sci. 2020, 21, 5026. https://doi.org/10.3390/ijms21145026
AMA Style
Bello-Morales R, Andreu S, López-Guerrero JA. The Role of Herpes Simplex Virus Type 1 Infection in Demyelination of the Central Nervous System. International Journal of Molecular Sciences. 2020; 21(14):5026. https://doi.org/10.3390/ijms21145026
Chicago/Turabian StyleBello-Morales, Raquel, Sabina Andreu, and José Antonio López-Guerrero. 2020. "The Role of Herpes Simplex Virus Type 1 Infection in Demyelination of the Central Nervous System" International Journal of Molecular Sciences 21, no. 14: 5026. https://doi.org/10.3390/ijms21145026
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.