Abnormal Lysosomal Positioning and Small Extracellular Vesicle Secretion in Arterial Stiffening and Calcification of Mice Lacking Mucolipin 1 Gene

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
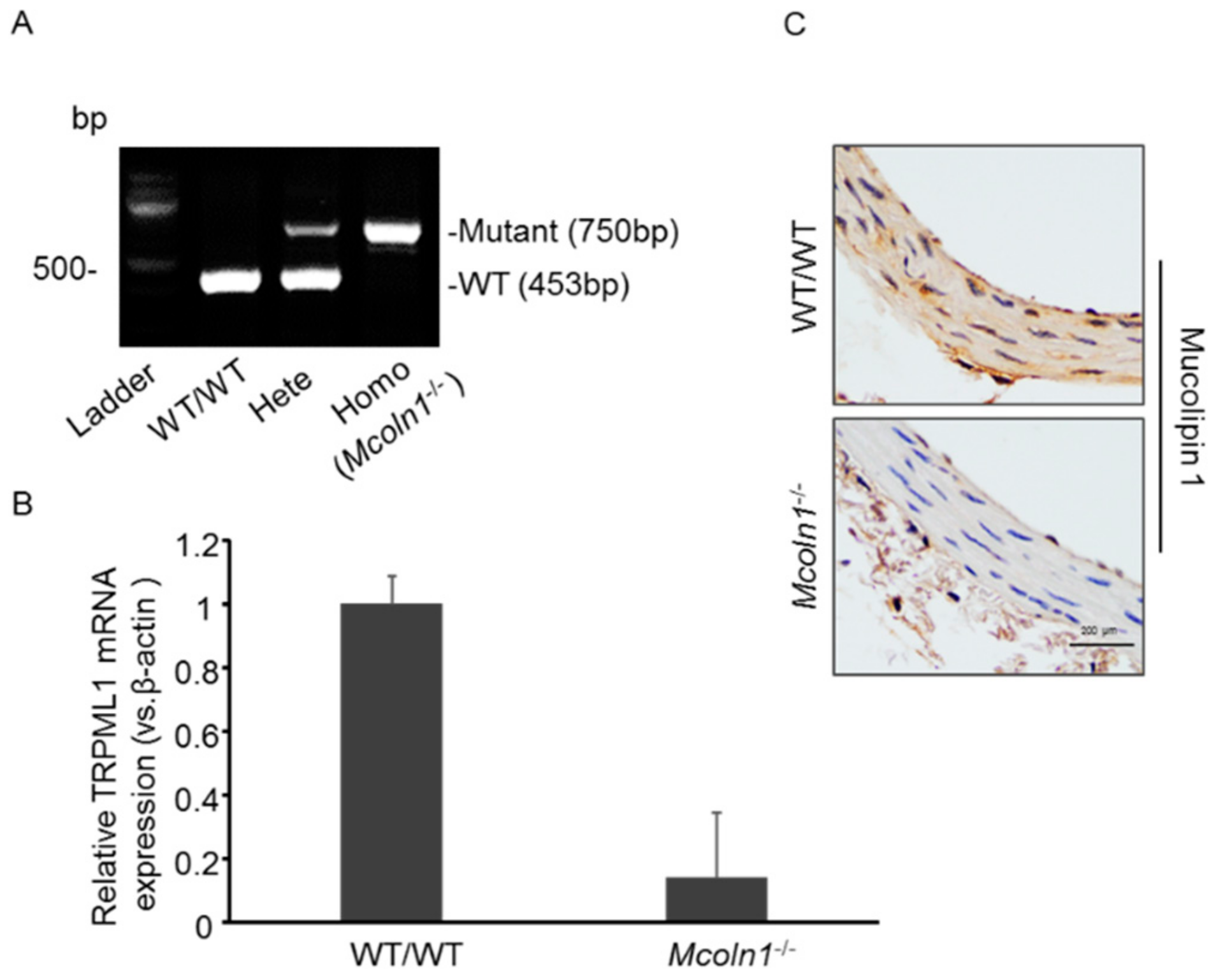
2.1. Characterization of Mucolipin 1 (Mcoln1) Gene KO Mice
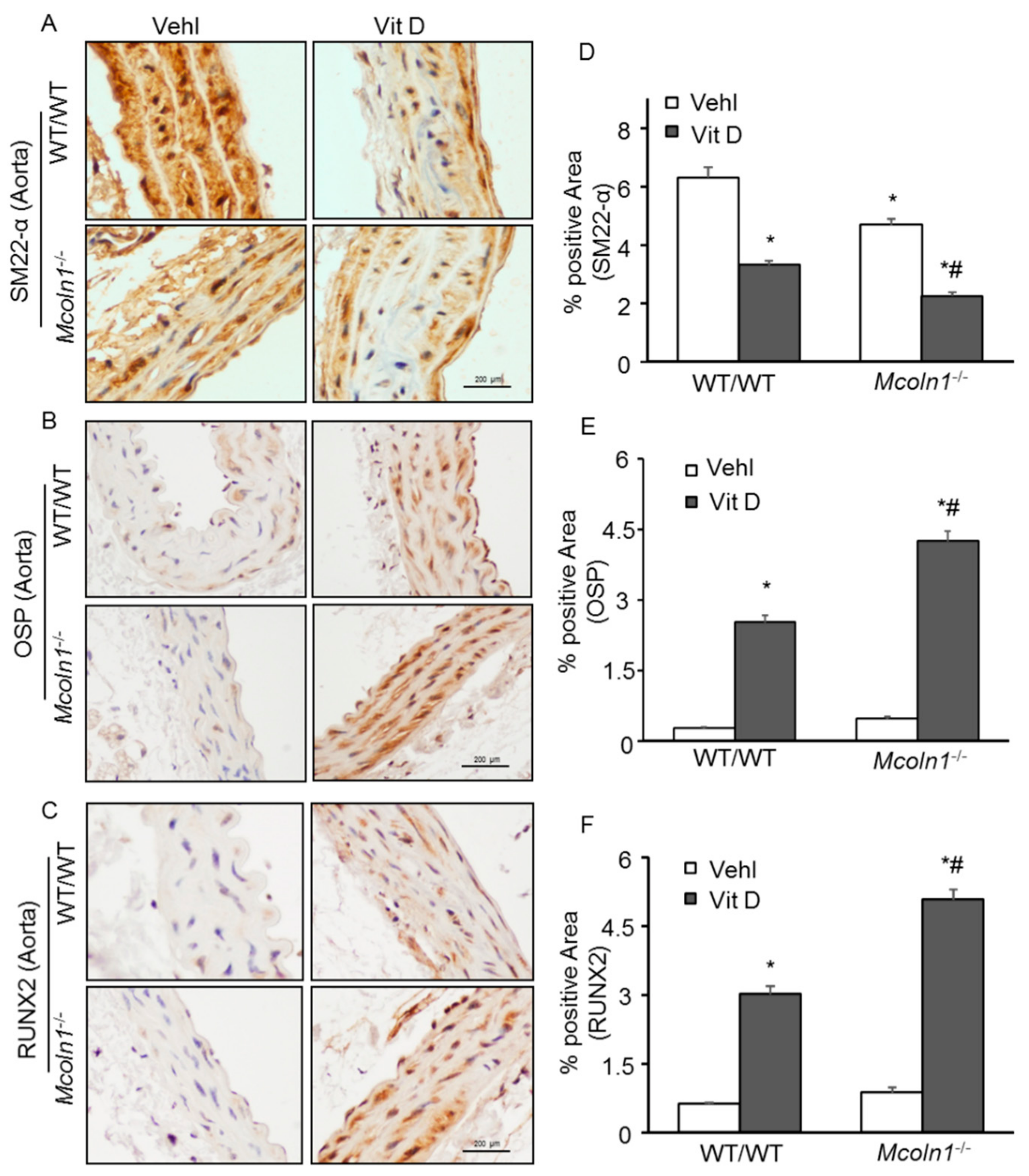
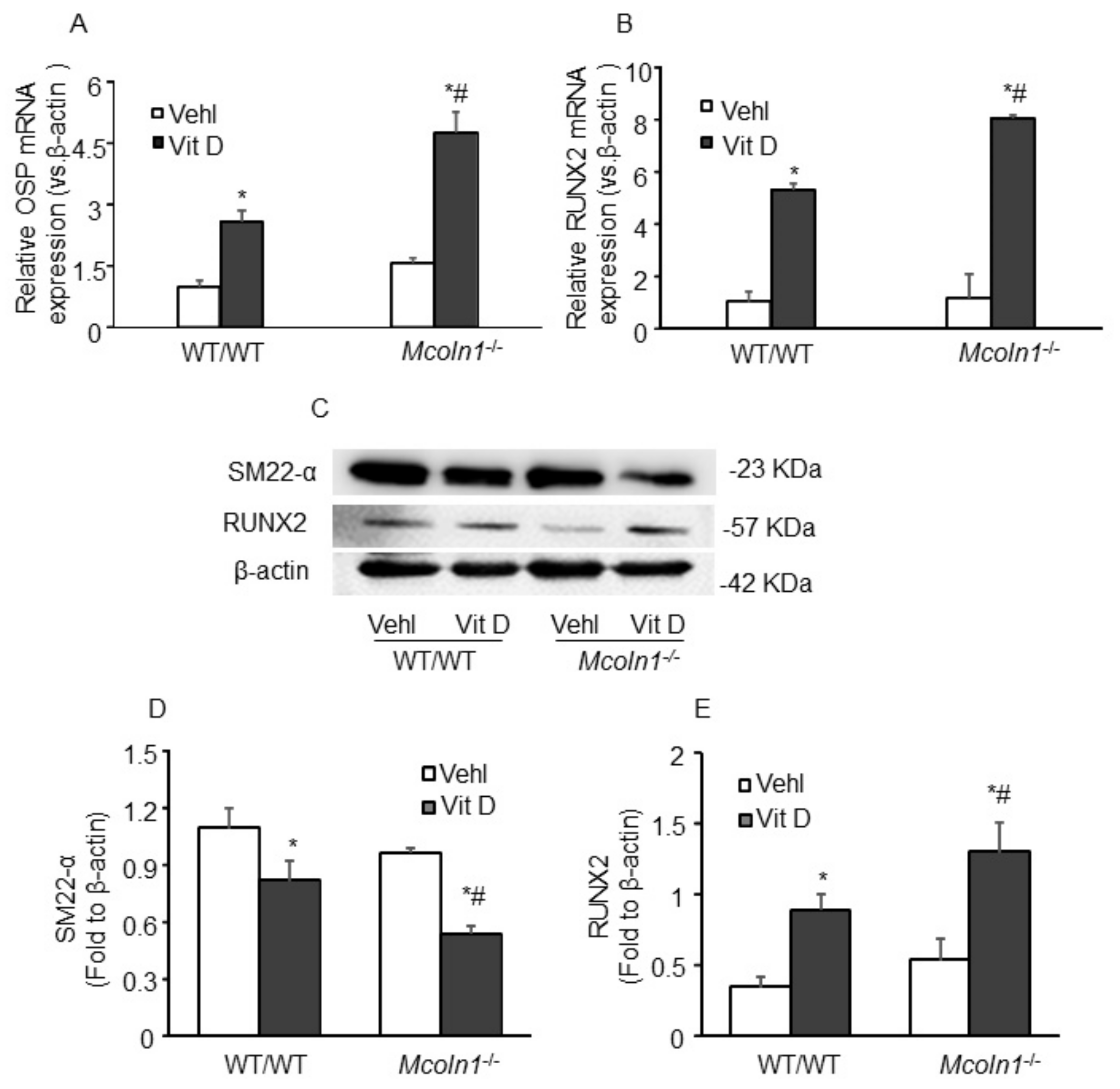
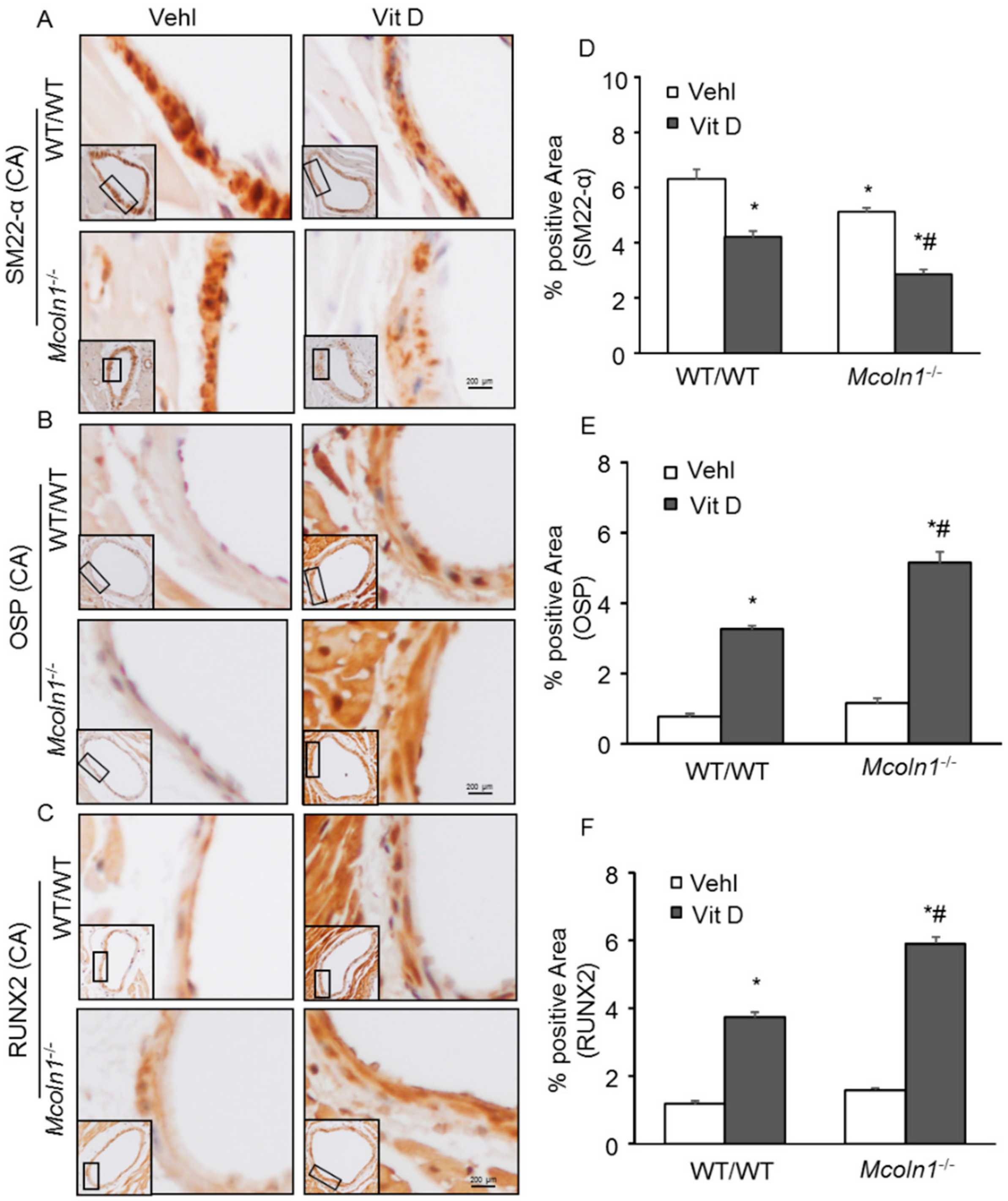
2.2. Mcoln1 Gene Deletion Enhanced Arterial Medial Calcification in Vit D-Treated Mice
2.3. Arterial Medial SMC Phenotypic Transition in Vit D-Treated Mcoln1−/− Mice
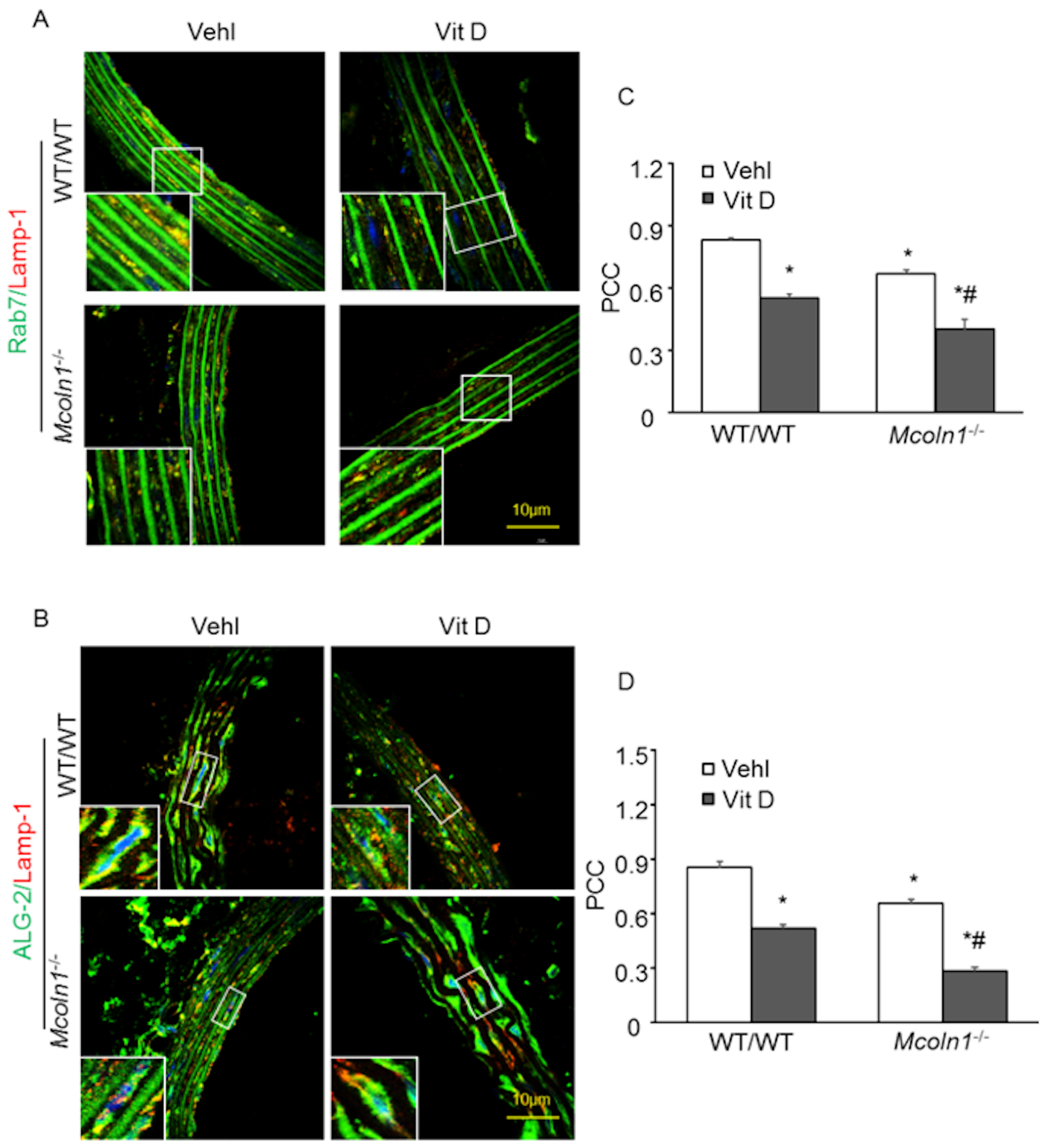
2.4. ALG-2 and Rab7 Interact with the Lysosome to Mediate Mcoln1-Dependent Lysosome Coupling in Aortic Medial SMCs in Vit D-Treated Mcoln1−/− Mice
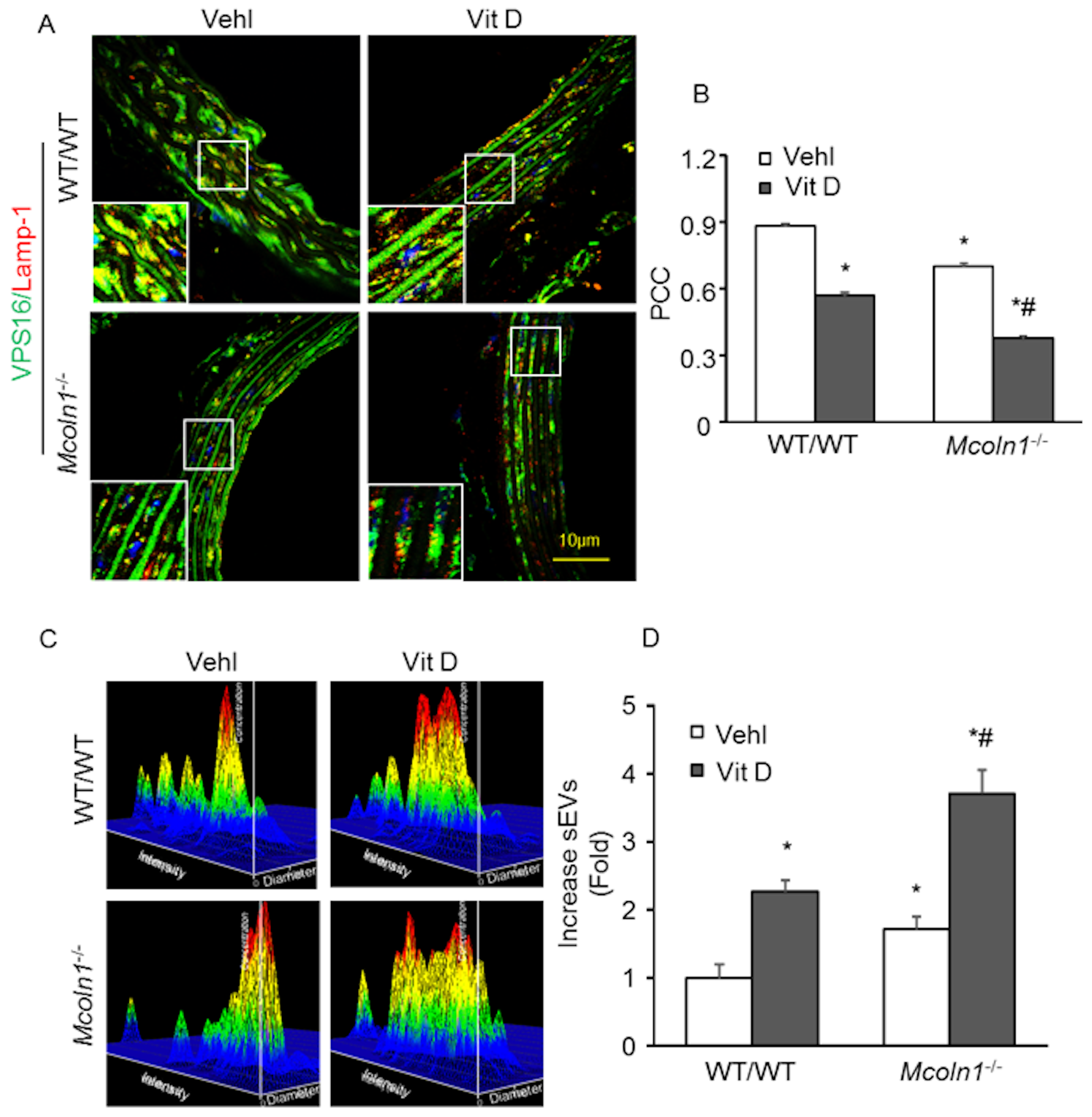
2.5. Mcoln1 Gene Deletion Elevated sEV Secretion by Inhibiting Lysosomes and MVB Interactions in the Arterial Medial Calcification
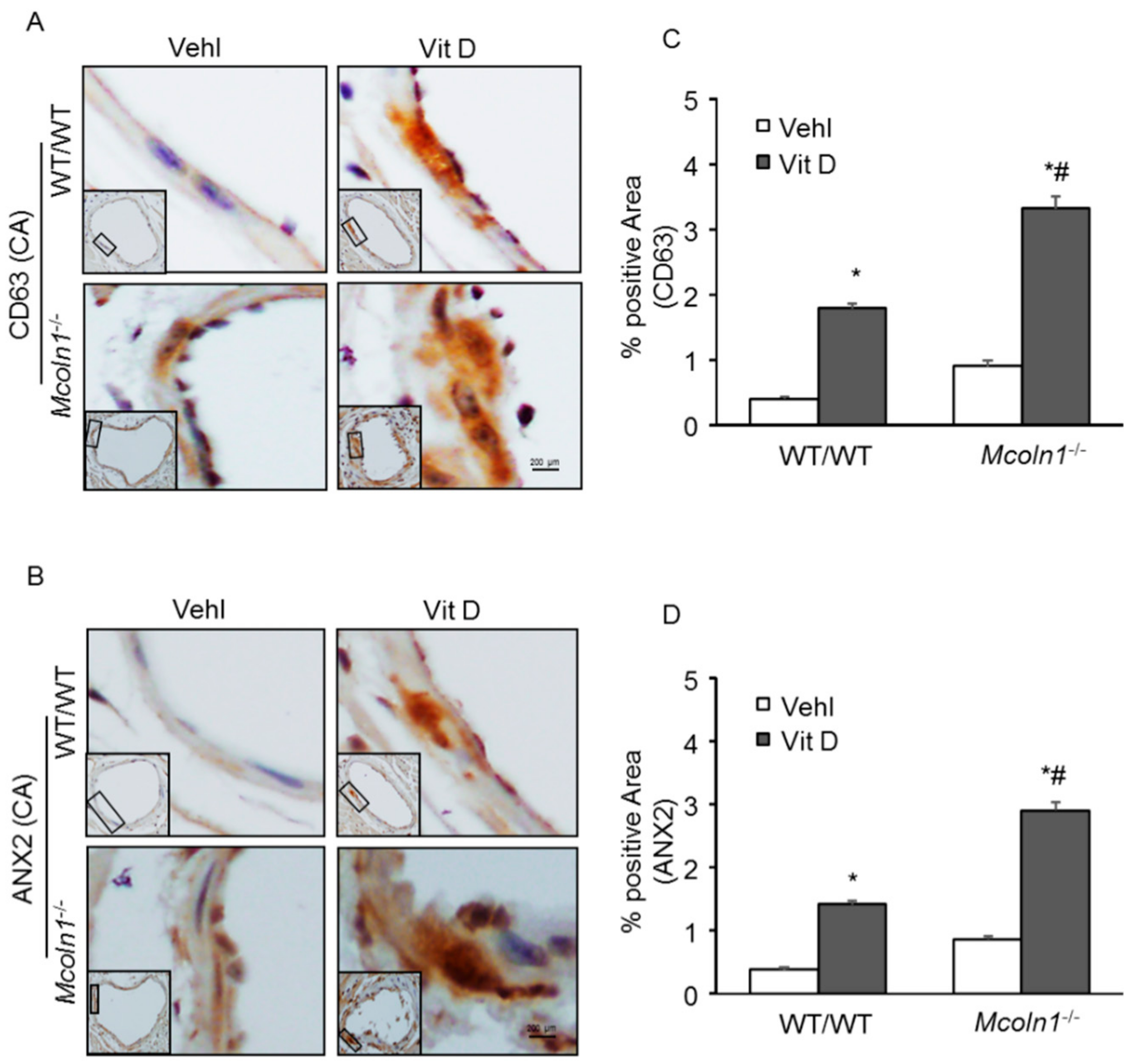
2.6. Role of Mcoln1 in sEV Release in the Arterial Medial Calcification
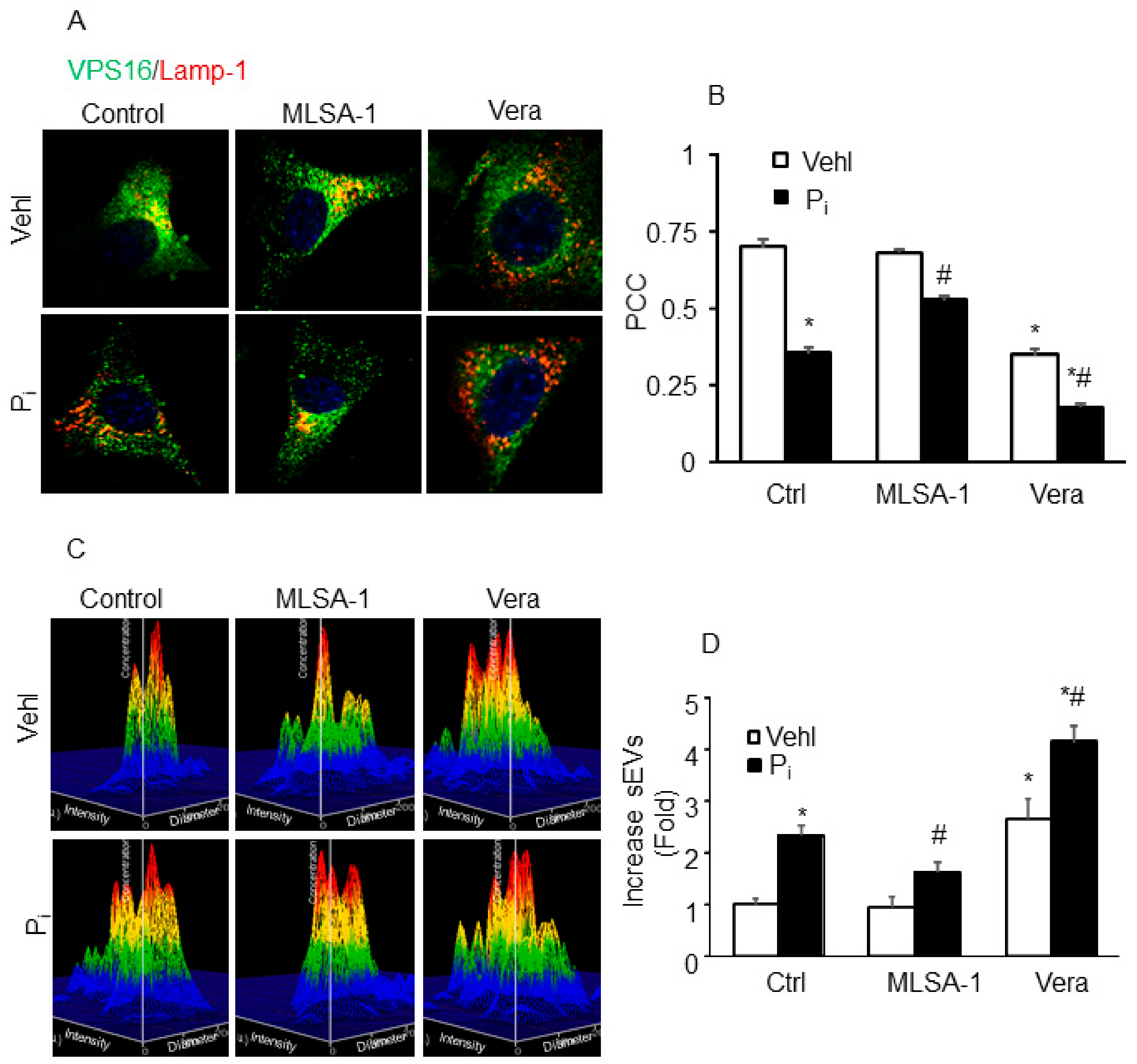
2.7. Lysosome–MVB Interactions and sEV Release Controlled by TRPML1 Signaling Pathway in Coronary Artery Smooth Muscle Cells (CASMCs)
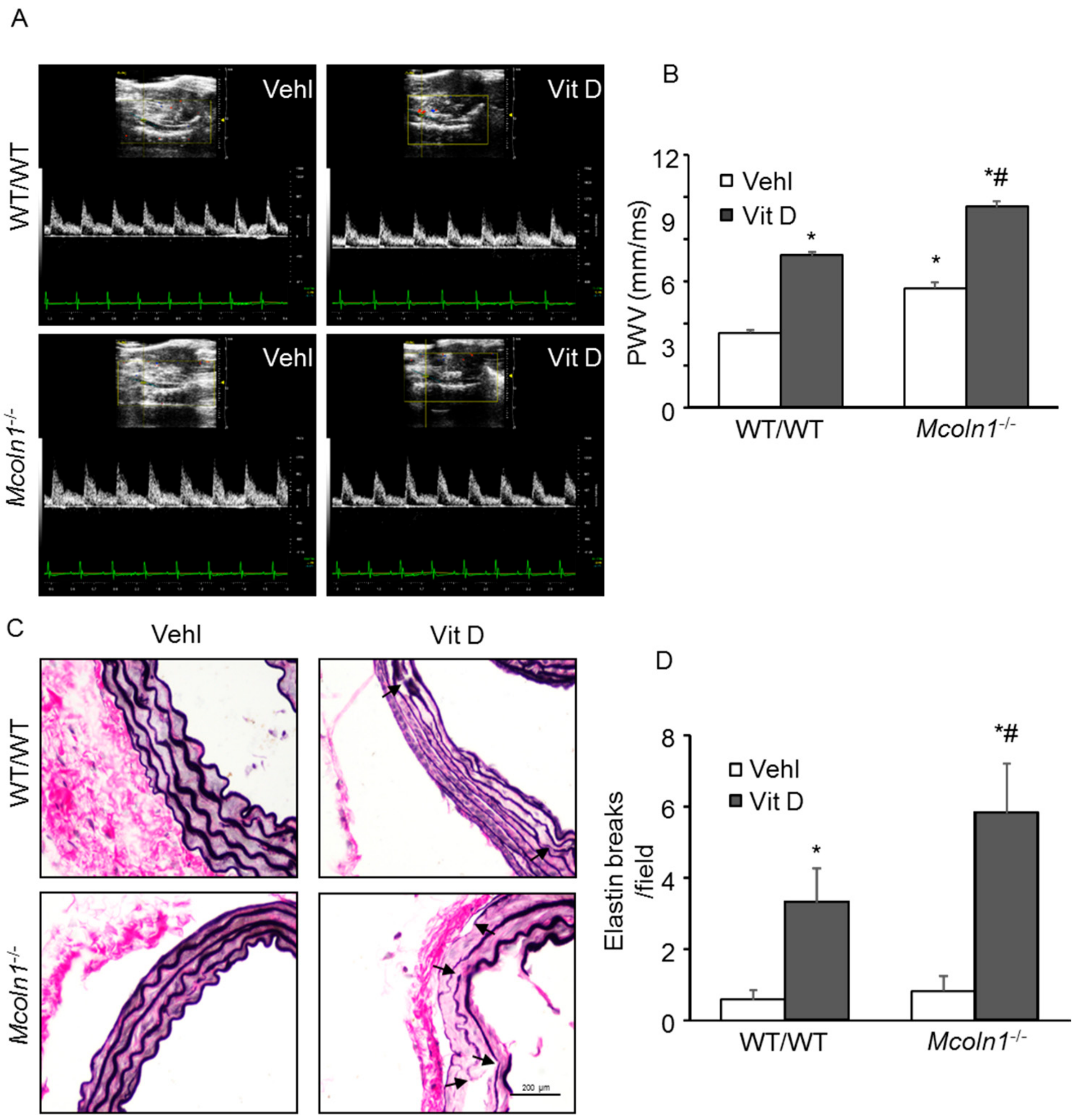
2.8. Mcoln1 Gene Deletion Accelerates Arterial Stiffness
3. Discussion
4. Material and Methods
4.1. Reagents and Antibodies
4.2. Primary Culture of Mouse CASMCs
4.3. Animal Model
4.4. Alizarin Red S Staining
4.5. Calcium Assay
4.6. Immunohistochemical Analyses
4.7. Immunofluorescent Staining
4.8. Nanoparticle Tracking Analysis (NTA)
4.9. Real Time-PCR Studies
4.10. Western Blot Analysis
4.11. Non-Invasive, in Vivo Measurement of Aortic Pulse Wave Velocity (PWV)
4.12. Elastin Staining
4.13. Statistical Analysis
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Speer, M.Y.; Giachelli, C.M. Regulation of cardiovascular calcification. Cardiovasc. Pathol. 2004, 13, 63–70. [Google Scholar] [CrossRef]
- Giachelli, C.M. Mechanisms of vascular calcification in uremia. Semin. Nephrol. 2004, 24, 401–402. [Google Scholar] [CrossRef] [PubMed]
- Burke, A.P.; Taylor, A.; Farb, A.; Malcom, G.T.; Virmani, R. Coronary calcification: insights from sudden coronary death victims. Z. Kardiol. 2000, 89, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Edmonds, M.E.; Morrison, N.; Laws, J.W.; Watkins, P.J. Medial arterial calcification and diabetic neuropathy. Br. Med. J. (Clin Res. Ed.). 1982, 284, 928–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demer, L.L.; Tintut, Y. Vascular calcification: Pathobiology of a multifaceted disease. Circulation 2008, 117, 2938–2948. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, J.L.; Joannides, A.J.; Skepper, J.N.; McNair, R.; Schurgers, L.J.; Proudfoot, D.; Jahnen Dechent, W.; Weissberg, P.L.; Shanahan, C.M. Human vascular smooth muscle cells undergo vesicle-mediated calcification in response to changes in extracellular calcium and phosphate concentrations: A potential mechanism for accelerated vascular calcification in ESRD. J. Am. Soc. Nephrol. 2004, 15, 2857–2867. [Google Scholar] [PubMed] [Green Version]
- Tkach, M.; Thery, C. Communication by Extracellular Vesicles: Where We Are and Where We Need to Go. Cell 2016, 164, 1226–1232. [Google Scholar] [CrossRef] [Green Version]
- Kapustin, A.N.; Chatrou, M.L.; Drozdov, I.; Zheng, Y.; Davidson, S.M.; Soong, D.; Furmanik, M.; Sanchis, P.; De Rosales, R.T.M.; Alvarez-Hernandez, D.; et al. Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ. Res. 2015, 116, 1312–1323. [Google Scholar] [CrossRef] [Green Version]
- Kapustin, A.N.; Schoppet, M.; Schurgers, L.J.; Reynolds, J.L.; McNair, R.; Heiss, A.; Jahnen-Dechent, W.; Hackeng, T.M.; Schlieper, G.; Harrison, P.; et al. Prothrombin Loading of Vascular Smooth Muscle Cell-Derived Exosomes Regulates Coagulation and Calcification. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e22–e32. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.X.; O’Neill, K.D.; Moe, S.M. Matrix vesicles induce calcification of recipient vascular smooth muscle cells through multiple signaling pathways. Kidney Int. 2018, 93, 343–354. [Google Scholar] [CrossRef] [Green Version]
- Kapustin, A.N.; Shanahan, C.M. Emerging roles for vascular smooth muscle cell exosomes in calcification and coagulation. J. Physiol. 2016, 594, 2905–2914. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, Y.H.; Jenkins, R.W.; Hannun, Y.A. Remodeling of cellular cytoskeleton by the acid sphingomyelinase/ceramide pathway. J. Cell Biol. 2008, 181, 335–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saftig, P.; Klumperman, J. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat. Rev. Mol. Cell Biol. 2009, 10, 623–635. [Google Scholar] [CrossRef]
- Luzio, J.P.; Pryor, P.R.; Bright, N.A. Lysosomes: fusion and function. Nat. Rev. Mol. Cell Biol. 2007, 8, 622–632. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Rydzewski, N.; Hider, A.; Zhang, X.; Yang, J.; Wang, W.; Gao, Q.; Cheng, X.; Xu, H. A molecular mechanism to regulate lysosome motility for lysosome positioning and tubulation. Nat. Cell Biol. 2016, 18, 404–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, X.; Shen, D.; Samie, M.; Xu, H. Mucolipins: Intracellular TRPML1-3 channels. FEBS Lett. 2010, 584, 2013–2021. [Google Scholar] [CrossRef] [Green Version]
- Shen, D.; Wang, X.; Xu, H. Pairing phosphoinositides with calcium ions in endolysosomal dynamics: phosphoinositides control the direction and specificity of membrane trafficking by regulating the activity of calcium channels in the endolysosomes. Bioessays 2011, 33, 448–457. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Li, X.; Walsh, S.W.; Zhang, Y.; Abais, J.M.; Boini, K.M.; Li, P.L. Intracellular two-phase Ca2+ release and apoptosis controlled by TRP-ML1 channel activity in coronary arterial myocytes. Am. J. Physiol. Cell Physiol. 2013, 304, C458–C466. [Google Scholar] [CrossRef] [Green Version]
- Dayam, R.M.; Saric, A.; Shilliday, R.E.; Botelho, R.J. The Phosphoinositide-Gated Lysosomal Ca (2+) Channel, TRPML1, Is Required for Phagosome Maturation. Traffic 2015, 16, 1010–1026. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; McBrayer, M.K.; Wolfe, D.M.; Haslett, L.J.; Kumar, A.; Sato, Y.; Lie, P.P.Y.; Mohan, P.; Coffey, E.E.; Kompella, U.; et al. Presenilin 1 Maintains Lysosomal Ca(2+) Homeostasis via TRPML1 by Regulating vATPase-Mediated Lysosome Acidification. Cell Rep. 2015, 12, 1430–1444. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.S.; Bach, G.; Pagano, R.E. Abnormal transport along the lysosomal pathway in mucolipidosis, type IV disease. Proc. Natl. Acad. Sci. USA 1998, 95, 6373–6378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, A.; Schafer, J.; Upchurch, C.; Spooner, E.; Huynh, J.; Hernandez, S.; McLaughlin, B.; Oden, L.; Fares, H. Mucolipidosis type IV protein TRPML1-dependent lysosome formation. Traffic 2015, 16, 284–297. [Google Scholar] [CrossRef] [PubMed]
- Treusch, S.; Knuth, S.; Slaugenhaupt, S.A.; Goldin, E.; Grant, B.D.; Fares, H. Caenorhabditis elegans functional orthologue of human protein h-mucolipin-1 is required for lysosome biogenesis. Proc. Natl. Acad. Sci. USA 2004, 101, 4483–4488. [Google Scholar] [CrossRef] [Green Version]
- Bargal, R.; Bach, G. Mucolipidosis type IV: abnormal transport of lipids to lysosomes. J. Inherit. Metab. Dis. 1997, 20, 625–632. [Google Scholar] [CrossRef]
- Thompson, E.G.; Schaheen, L.; Dang, H.; Fares, H. Lysosomal trafficking functions of mucolipin-1 in murine macrophages. BMC Cell Biol. 2007, 8, 54. [Google Scholar] [CrossRef] [Green Version]
- Kiselyov, K.; Chen, J.; Rbaibi, Y.; Oberdick, D.; Tjon-Kon-Sang, S.; Shcheynikov, N.; Muallem, S.; Soyombo, A. TRP-ML1 is a lysosomal monovalent cation channel that undergoes proteolytic cleavage. J. Biol. Chem. 2005, 280, 43218–43223. [Google Scholar] [CrossRef] [Green Version]
- Pu, J.; Guardia, C.M.; Keren-Kaplan, T.; Bonifacino, J.S. Mechanisms and functions of lysosome positioning. J. Cell Sci. 2016, 129, 4329–4339. [Google Scholar] [CrossRef] [Green Version]
- Futter, C.E.; Pearse, A.; Hewlett, L.J.; Hopkins, C.R. Multivesicular endosomes containing internalized EGF-EGF receptor complexes mature and then fuse directly with lysosomes. J. Cell Biol. 1996, 132, 1011–1023. [Google Scholar] [CrossRef]
- Hessvik, N.P.; Llorente, A. Current knowledge on exosome biogenesis and release. Cell Mol. Life Sci. 2018, 75, 193–208. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.S.; Muallem, S.; Kim, S.H.; Kwon, K.B.; Kim, M.S. Exosomal release through TRPML1-mediated lysosomal exocytosis is required for adipogenesis. Biochem. Biophys Res. Commun. 2019, 510, 409–415. [Google Scholar] [CrossRef]
- Li, G.; Huang, D.; Hong, J.; Bhat, O.M.; Yuan, X.; Li, P.L. Control of lysosomal TRPML1 channel activity and exosome release by acid ceramidase in mouse podocytes. Am. J. Physiol. Cell Physiol. 2019, 317, C481–C491. [Google Scholar] [CrossRef] [PubMed]
- Hutcheson, J.D.; Goettsch, C.; Bertazzo, S.; Maldonado, N.; Ruiz, J.L.; Goh, W.; Yabusaki, K.; Faits, T.; Bouten, C.; Franck, G.; et al. Genesis and growth of extracellular-vesicle-derived microcalcification in atherosclerotic plaques. Nat. Mater. 2016, 15, 335–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, Z.; Xie, J.; Yu, A.S.; Stock, J.; Du, J.; Yue, L. Role of TRP channels in the cardiovascular system. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H157–H182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanahan, C.M.; Crouthamel, M.H.; Kapustin, A.; Giachelli, C.M. Arterial calcification in chronic kidney disease: key roles for calcium and phosphate. Circ. Res. 2011, 109, 697–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akers, J.C.; Gonda, D.; Kim, R.; Carter, B.S.; Chen, C.C. Biogenesis of extracellular vesicles (EV): exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. J. Neurooncol. 2013, 113, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, H.A.; Smith, J.C.; Davies, J.S. Noninvasive assessment of preclinical atherosclerosis. Vasc. Health Risk Manag. 2006, 2, 19–30. [Google Scholar] [CrossRef] [Green Version]
- Ju, X.; Ijaz, T.; Sun, H.; Lejeune, W.; Vargas, G.; Shilagard, T.; RecinosIII, A.; Milewicz, D.M.; Brasier, A.R.; Tilton, R.G. IL-6 regulates extracellular matrix remodeling associated with aortic dilation in a fibrillin-1 hypomorphic mgR/mgR mouse model of severe Marfan syndrome. J. Am. Heart Assoc. 2014, 3, e000476. [Google Scholar] [CrossRef] [Green Version]
- Katoh, Y.; Periasamy, M. Growth and differentiation of smooth muscle cells during vascular development. Trends Cardiovasc. Med. 1996, 6, 100–106. [Google Scholar] [CrossRef]
- Giachelli, C.M.; Liaw, L.; Murry, C.E.; Schwartz, S.M.; Almeida, M. Osteopontin expression in cardiovascular diseases. Ann. N. Y. Acad Sci. 1995, 760, 109–126. [Google Scholar] [CrossRef]
- Steitz, S.A.; Speer, M.Y.; Curinga, G.; Yang, H.Y.; Haynes, P.; Aebersold, R.; Schinke, T.; Karsenty, G.; Giachelli, C.M. Smooth muscle cell phenotypic transition associated with calcification: upregulation of Cbfa1 and downregulation of smooth muscle lineage markers. Circ. Res. 2001, 89, 1147–1154. [Google Scholar] [CrossRef]
- Shroff, R.C.; McNair, R.; Figg, N.; Skepper, J.N.; Schurgers, L.; Gupta, A.; Hiorns, M.; Donald, A.E.; Deanfield, J.; Rees, J.; et al. Dialysis accelerates medial vascular calcification in part by triggering smooth muscle cell apoptosis. Circulation. 2008, 118, 1748–1757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, X.P.; Cheng, X.; Mills, E.; Delling, M.; Wang, F.; Kurz, T.; Xu, H. The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel. Nature 2008, 455, 992–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Ren, D. Lysosomal physiology. Annu. Rev. Physiol. 2015, 77, 57–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietrich, A.; Chubanov, V.; Kalwa, H.; Rost, B.R.; Gudermann, T. Cation channels of the transient receptor potential superfamily: their role in physiological and pathophysiological processes of smooth muscle cells. Pharmacol. Ther. 2006, 112, 744–760. [Google Scholar] [CrossRef]
- Sullivan, M.N.; Earley, S. TRP channel Ca(2+) sparklets: fundamental signals underlying endothelium-dependent hyperpolarization. Am. J. Physiol. Cell Physiol. 2013, 305, C999–C1008. [Google Scholar] [CrossRef]
- Dietrich, A.; Mederos, Y.S.M.; Gollasch, M.; Gross, V.; Storch, U.; Dubrovska, G.; Obst, M.; Yildirim, E.; Salanova, B.; Kalwa, H.; et al. Increased vascular smooth muscle contractility in TRPC6-/- mice. Mol. Cell Biol. 2005, 25, 6980–6989. [Google Scholar] [CrossRef] [Green Version]
- Bergdahl, A.; Gomez, M.F.; Wihlborg, A.K.; Erlinge, D.; Eyjolfson, A.; Xu, S.Z.; Beech, D.J.; Dreja, K.; Hellstrand, P. Plasticity of TRPC expression in arterial smooth muscle: Correlation with store-operated Ca2+ entry. Am. J. Physiol. Cell Physiol. 2005, 288, C872–C880. [Google Scholar] [CrossRef] [Green Version]
- Kumar, B.; Dreja, K.; Shah, S.S.; Cheong, A.; Xu, S.Z.; Sukumar, P.; Naylor, J.; Forte, A.; Cipollaro, M.; McHugh, D.; et al. Upregulated TRPC1 channel in vascular injury in vivo and its role in human neointimal hyperplasia. Circ. Res. 2006, 98, 557–563. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, Y.; Watanabe, H.; Murakami, M.; Ohba, T.; Radovanovic, M.; Ono, K.; Iijima, T.; Ito, H. Involvement of transient receptor potential canonical 1 (TRPC1) in angiotensin II-induced vascular smooth muscle cell hypertrophy. Atherosclerosis 2007, 195, 287–296. [Google Scholar] [CrossRef]
- Jongsma, M.L.; Berlin, I.; Wijdeven, R.H.; Janssen, L.; Janssen, G.M.; Garstka, M.A.; Janssen, H.; Mensink, M.; van Veelen, P.A.; Spaapen, R.M.; et al. An ER-Associated Pathway Defines Endosomal Architecture for Controlled Cargo Transport. Cell. 2016, 166, 152–166. [Google Scholar] [CrossRef] [Green Version]
- Cabukusta, B.; Neefjes, J. Mechanisms of lysosomal positioning and movement. Traffic. 2018, 19, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Jordens, I.; Fernandez-Borja, M.; Marsman, M.; Dusseljee, S.; Janssen, L.; Calafat, J.; Janssen, H.; Wubbolts, R.; Neefjes, J. The Rab7 effector protein RILP controls lysosomal transport by inducing the recruitment of dynein-dynactin motors. Curr. Biol. 2001, 11, 1680–1685. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Zhang, X.; Gao, Q.; Xu, H. TRPML1: an ion channel in the lysosome. Handb. Exp. Pharmacol. 2014, 222, 631–645. [Google Scholar] [PubMed]
- Kowal, J.; Tkach, M.; Thery, C. Biogenesis and secretion of exosomes. Curr. Opin. Cell Biol. 2014, 29, 116–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, O.M.; Li, G.; Yuan, X.; Huang, D.; Gulbins, E.; Kukreja, R.C.; Li, P.L. Arterial Medial Calcification through Enhanced small Extracellular Vesicle Release in Smooth Muscle-Specific Asah1 Gene Knockout Mice. Sci. Rep. 2020, 10, 1645. [Google Scholar] [CrossRef] [PubMed]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomal accumulation of anticancer drugs triggers lysosomal exocytosis. Oncotarget 2017, 8, 45117–45132. [Google Scholar] [CrossRef] [Green Version]
- Miao, Y.; Li, G.; Zhang, X.; Xu, H.; Abraham, S.N. A TRP Channel Senses Lysosome Neutralization by Pathogens to Trigger Their Expulsion. Cell 2015, 161, 1306–1319. [Google Scholar] [CrossRef] [Green Version]
- Samie, M.A.; Xu, H. Lysosomal exocytosis and lipid storage disorders. J. Lipid Res. 2014, 55, 995–1009. [Google Scholar] [CrossRef] [Green Version]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef] [Green Version]
- Bhat, O.M.; Yuan, X.; Cain, C.; Salloum, F.N.; Li, P.L. Medial calcification in the arterial wall of smooth muscle cell-specific Smpd1 transgenic mice: A ceramide-mediated vasculopathy. J. Cell Mol. Med. 2020, 24, 539–553. [Google Scholar] [CrossRef] [Green Version]
- Agouni, A.; Lagrue-Lak-Hal, A.H.; Ducluzeau, P.H.; Mostefai, H.A.; Draunet-Busson, C.; Leftheriotis, G.; Heymes, C.; Martinez, M.C.; Andriantsitohaina, R. Endothelial dysfunction caused by circulating microparticles from patients with metabolic syndrome. Am. J. Pathol. 2008, 173, 1210–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.M.; Su, C.; Wang, Y.; Huang, Y.J.; Yang, Z.; Chen, L.; Wu, F.; Xu, S.Y.; Tao, J. Elevated circulating endothelial microparticles and brachial-ankle pulse wave velocity in well-controlled hypertensive patients. J. Hum. Hypertens. 2009, 23, 307–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nozaki, T.; Sugiyama, S.; Koga, H.; Sugamura, K.; Ohba, K.; Matsuzawa, Y.; Sumida, H.; Matsui, K.; Jinnouchi, H.; Ogawa, H. Significance of a multiple biomarkers strategy including endothelial dysfunction to improve risk stratification for cardiovascular events in patients at high risk for coronary heart disease. J. Am. Coll. Cardiol. 2009, 54, 601–608. [Google Scholar] [CrossRef] [Green Version]
- Koga, H.; Sugiyama, S.; Kugiyama, K.; Watanabe, K.; Fukushima, H.; Tanaka, T.; Sakamoto, T.; Yoshimura, W.; Jinnouchi, H.; Ogawa, H. Elevated levels of VE-cadherin-positive endothelial microparticles in patients with type 2 diabetes mellitus and coronary artery disease. J. Am. Coll. Cardiol. 2005, 45, 1622–1630. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Feng, B.; Li, X.; Ni, Y.; Luo, Y. Plasma endothelial microparticles and their correlation with the presence of hypertension and arterial stiffness in patients with type 2 diabetes. J. Clin. Hypertens (Greenwich). 2012, 14, 455–460. [Google Scholar] [CrossRef]
- Shroff, R.C.; Shanahan, C.M. The vascular biology of calcification. Semin. Dial. 2007, 20, 103–109. [Google Scholar] [CrossRef]
- Mackey, R.H.; Venkitachalam, L.; Sutton-Tyrrell, K. Calcifications, arterial stiffness and atherosclerosis. Adv. Cardiol. 2007, 44, 234–244. [Google Scholar]
- Earley, S.; Brayden, J.E. Transient receptor potential channels in the vasculature. Physiol. Rev. 2015, 95, 645–690. [Google Scholar] [CrossRef] [Green Version]
- Tu, H.; Zhang, D.; Li, Y.L. Cellular and Molecular Mechanisms Underlying Arterial Baroreceptor Remodeling in Cardiovascular Diseases and Diabetes. Neurosci. Bull. 2019, 35, 98–112. [Google Scholar] [CrossRef]
- Price, P.A.; Buckley, J.R.; Williamson, M.K. The amino bisphosphonate ibandronate prevents vitamin D toxicity and inhibits vitamin D-induced calcification of arteries, cartilage, lungs and kidneys in rats. J. Nutr. 2001, 131, 2910–2915. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Bhat, O.M.; Meng, N.; Lohner, H.; Li, P.L. Protective Role of Autophagy in Nlrp3 Inflammasome Activation and Medial Thickening of Mouse Coronary Arteries. Am. J. Pathol. 2018, 188, 2948–2959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Rosa, L.R.; Perrone, L.; Nielsen, M.S.; Calissano, P.; Andersen, O.M.; Matrone, C. Y682 G Mutation of Amyloid Precursor Protein Promotes Endo-Lysosomal Dysfunction by Disrupting APP-SorLA Interaction. Front. Cell Neurosci. 2015, 9, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, X.; Bhat, O.M.; Lohner, H.; Zhang, Y.; Li, P.L. Endothelial acid ceramidase in exosome-mediated release of NLRP3 inflammasome products during hyperglycemia: Evidence from endothelium-specific deletion of Asah1 gene. Biochim. Biophys Acta Mol. Cell Biol. Lipids. 2019, 1864, 158532. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Bhat, O.M.; Lohner, H.; Li, N.; Zhang, Y.; Li, P.L. Inhibitory effects of growth differentiation factor 11 on autophagy deficiency-induced dedifferentiation of arterial smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H345–H356. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Khandelwal, A.R.; Wu, X.; Xu, Z.; Yu, W.; Chen, C.; Zhao, W.; Yang, J.; Qin, Z.; Weisbrod, R.M.; et al. Pro-atherogenic role of smooth muscle Nox4-based NADPH oxidase. J. Mol. Cell Cardiol. 2016, 92, 30–40. [Google Scholar] [CrossRef] [Green Version]










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhat, O.M.; Yuan, X.; Camus, S.; Salloum, F.N.; Li, P.-L. Abnormal Lysosomal Positioning and Small Extracellular Vesicle Secretion in Arterial Stiffening and Calcification of Mice Lacking Mucolipin 1 Gene. Int. J. Mol. Sci. 2020, 21, 1713. https://doi.org/10.3390/ijms21051713
Bhat OM, Yuan X, Camus S, Salloum FN, Li P-L. Abnormal Lysosomal Positioning and Small Extracellular Vesicle Secretion in Arterial Stiffening and Calcification of Mice Lacking Mucolipin 1 Gene. International Journal of Molecular Sciences. 2020; 21(5):1713. https://doi.org/10.3390/ijms21051713
Chicago/Turabian StyleBhat, Owais M., Xinxu Yuan, Sarah Camus, Fadi N. Salloum, and Pin-Lan Li. 2020. "Abnormal Lysosomal Positioning and Small Extracellular Vesicle Secretion in Arterial Stiffening and Calcification of Mice Lacking Mucolipin 1 Gene" International Journal of Molecular Sciences 21, no. 5: 1713. https://doi.org/10.3390/ijms21051713