Dual Role of Chondrocytes in Rheumatoid Arthritis: The Chicken and the Egg


 and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Chondrocytes in Normal Physiology
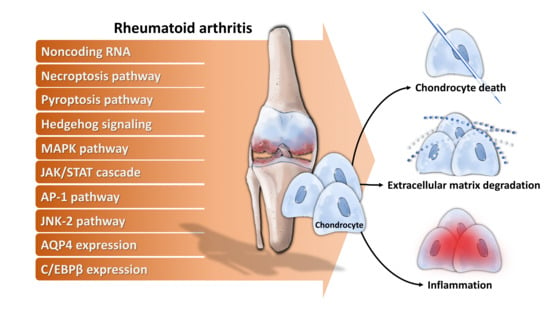
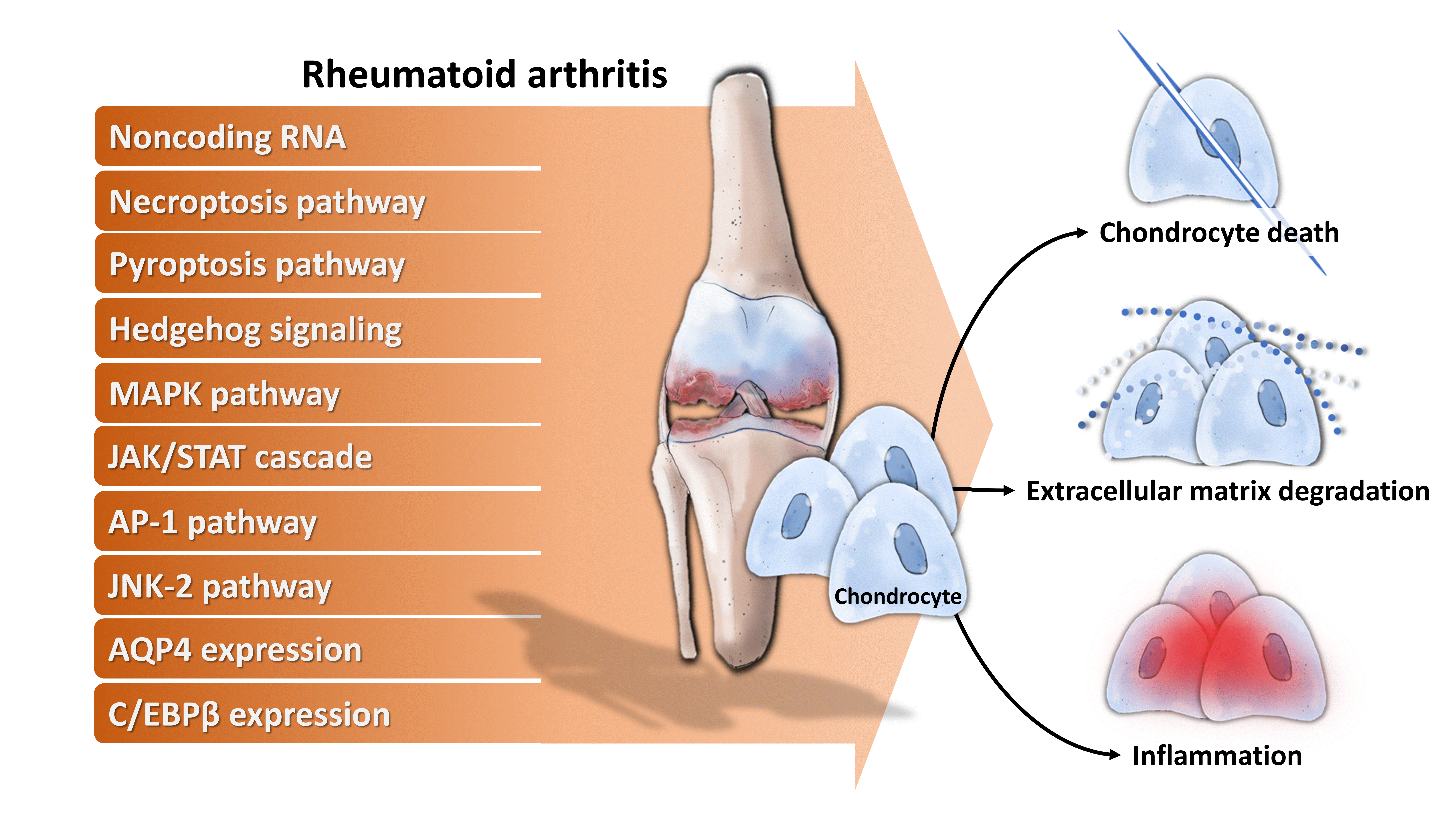
3. Chondrocytes in RA
3.1. Chondrocytes Acting as Target Cells in RA
3.2. Chondrocytes Acting as Effector Cells in RA
3.2.1. Chondrocytes Directly Involve in RA Through Releasing Multiple Enzymes of Extracellular Matrix Degradation, Facilitating Angiogenesis, Enhancing Inflammation and Immune Responses
3.2.2. Chondrocytes Indirectly Involve in RA Through Crosstalk with Related Cells
4. Molecular Mechanisms Underlying Chondrocytes Dysfunction in RA
- a)
- Noncoding RNA: For example, long noncoding RNA HOTAIR increases chondrocyte proliferation, decreases inflammatory cytokine from chondrocytes, and alleviates RA in the animal model [71], while micro RNA-23a (miR-23a) inhibits IL-17-mediated proinflammatory mediator expression via targeting IκB kinase α (IKKα) in articular chondrocytes [32]. Downregulated miR-26a is found in articular chondrocytes of RA rats, and upregulation of miR-26a reduces swelling and inflammation of joints, diminishes cartilage damage, apoptosis of chondrocytes, and inflammatory injury [72]. Moreover, miR-26a promotes proliferation and counterbalances apoptosis of inflammatory articular chondrocytes [72]. Expression level of miR-27b-3p is decreased in RA, and overexpression of miR-27b-3p significantly reduces the expression of pro-apoptotic protein caspase 3 and increases the expression of anti-apoptotic Bcl-2 in chondrocytes [73].
- b)
- Necroptosis pathway: Activation of necroptosis pathway molecules (receptor interacting protein (RIP) 1, RIP3 and mixed lineage kinase domain-like protein phosphorylation (p-MLKL)) are detected in adjuvant arthritis (AA) rat articular cartilage and RIP1 inhibitor necrostatin-1 (Nec-1) could reduce articular cartilage damage and necroinflammation in AA rats [74].
- c)
- Pyroptosis pathway: Extracellular acidosis, which accompanies joint inflammation of RA, significantly increases the expression of acid-sensing ion channel 1a (ASIC1a), IL-1β, IL-18, apoptosis-associated speck-like protein (ASC), neuronal apoptosis inhibitor protein, class 2 transcription activator, of the major histocomplex, heterokaryon incompatibility and telomerase-associated protein 1 (NACHT), leucine-rich repeat (LRR) and PYRIN domain (PYD) domains-containing protein 3 (NLRP3) and caspase-1 and mediates chondrocyte pyroptosis [75,76].
- d)
- Hedgehog signaling: Expression of hedgehog signal pathway (Shh, Ptch1, Smo, Gli1) in articular cartilage is associated with the severity of cartilage damage in rats with adjuvant-induced arthritis, and hedgehog signal inhibition promotes ECM production [77].
- e)
- MAPK pathway: TNF-α activates mitogen-activated kinase (MEK)/ extracellular regulated kinase (ERK) pathway and subsequent early growth response 1 (Egr1) DNA binding activity, which are required for TNF-α regulated catabolic and anabolic gene expression of chondrocytes [78]. Furthermore, acidosis also acts via ASIC1a, leading to intracellular Ca2+ elevation, ERK phosphorylation, culminating in articular chondrocyte apoptosis [79]. MAPK pathway also contributes to IL-1β-stimulated MMP-13 production in RA chondrocytes [80].
- f)
- JAK/STAT cascade: IL-6 could enhance acid-induced articular chondrocyte apoptosis, which might partially be involved in regulating the activation of ASIC1a-dependent JAK/STAT pathway [29].
- g)
- AP-1 pathway: Stromal cell-derived factor (SDF)-1, significantly higher in RA, acts through CXCR4 to activate ERK and the downstream transcription factors (c-Fos and c-Jun), resulting in the activation of AP-1 on the MMP promoter and contributing to MMP secretion of chondrocytes [81].
- h)
- JNK-2 pathway: IL-1 signals via TRAF-6/TAK-1/MKK-4/JNK-2 axis to cause JNK-2-dependent shedding of LRP-1 and subsequent ADAMTS-5-mediated aggrecanolysis [82].Membrane protein: Overexpression of membrane protein aquaporin 4 (AQP4) in articular chondrocytes exacerbates chondrocyte dysfunction of adjuvant-induced arthritis in rats [83].
- i)
- Intracellular protein: C/EBPβ mediates expression of MMP-13 in human articular chondrocytes in inflammatory arthritis [84].
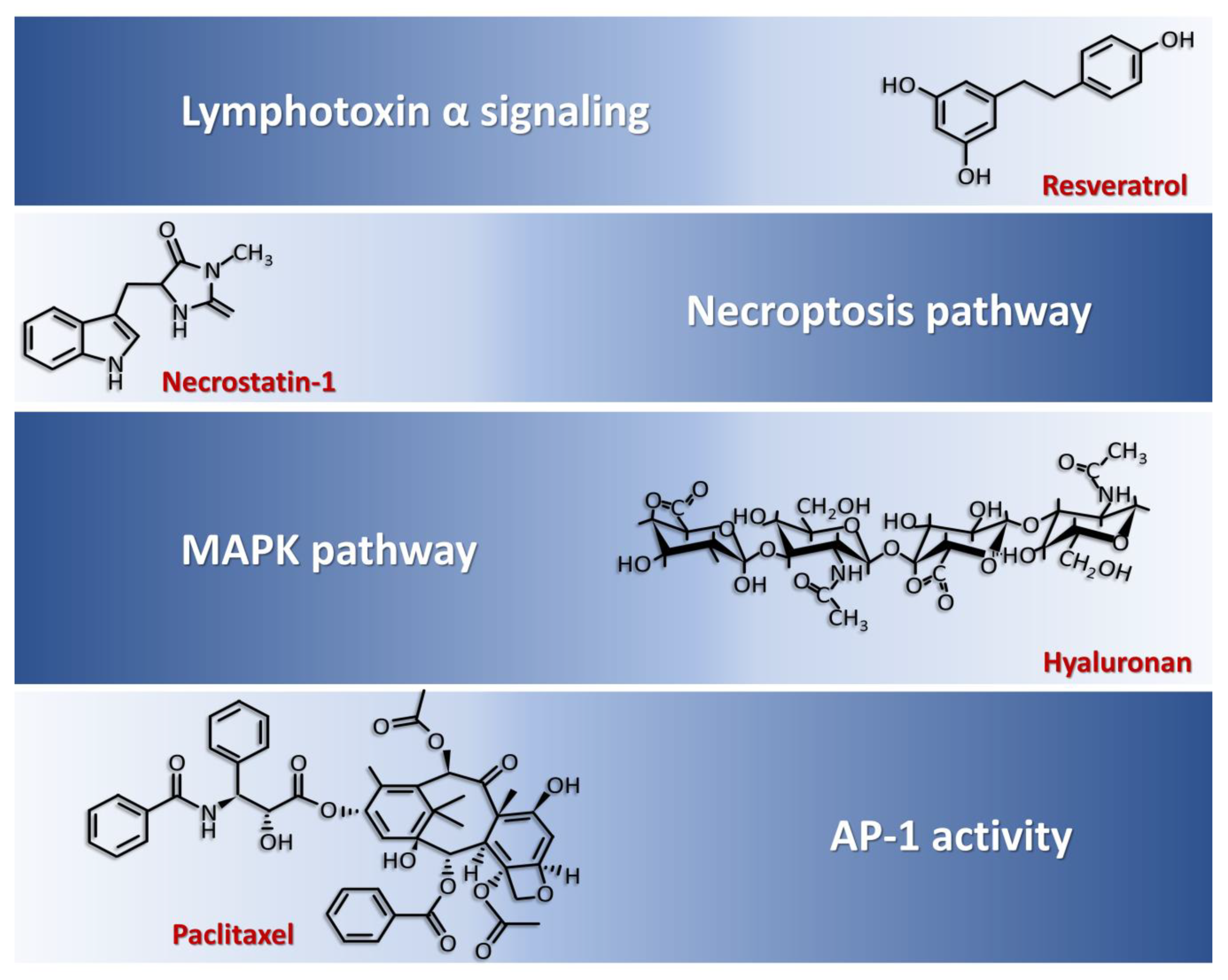
5. Inhibitors of Chondrocyte Dysfunction in RA
6. Relationship Between Current Treatment of RA and Chondrocytes
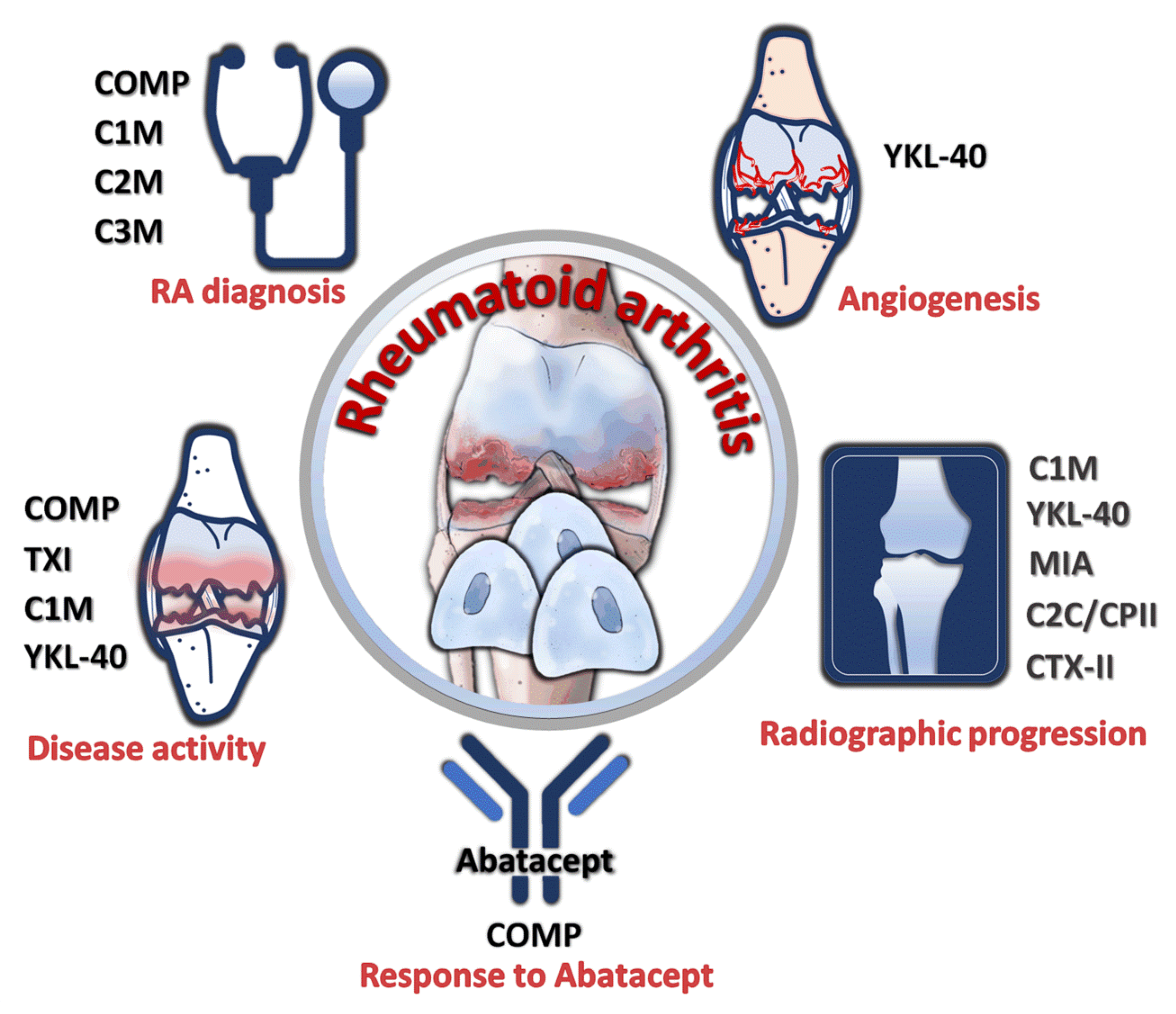
7. Utility of Chondrocyte Products as Diagnostic and Prognostic Markers of RA
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Karouzakis, E.; Neidhart, M.; Gay, R.E.; Gay, S. Molecular and cellular basis of rheumatoid joint destruction. Immunol. Lett. 2006, 106, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Tang, Y.; Song, B.; Yu, M.; Li, Q.; Zhang, C.; Hou, J.; Yang, R. Nomenclature clarification: Synovial fibroblasts and synovial mesenchymal stem cells. Stem Cell Res. Ther. 2019, 10, 260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Liu, Y.; Wang, C.; Xia, W.R.; Zheng, J.Y.; Yang, J.; Liu, B.; Liu, J.Q.; Liu, L.F. Succinate induces synovial angiogenesis in rheumatoid arthritis through metabolic remodeling and HIF-1α/VEGF axis. Free Radic. Biol. Med. 2018, 126, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S. RANKL is a therapeutic target of bone destruction in rheumatoid arthritis. F1000Research 2019, 8, 533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, X.; Zhang, L.; Wei, W. Regulatory B cells in inflammatory diseases and tumor. Int. Immunopharmacol. 2019, 67, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Di Benedetto, P.; Ruscitti, P.; Vadasz, Z.; Toubi, E.; Giacomelli, R. Macrophages with regulatory functions, a possible new therapeutic perspective in autoimmune diseases. Autoimmun. Rev. 2019, 18, 102369. [Google Scholar] [CrossRef]
- Hu, X.X.; Wu, Y.J.; Zhang, J.; Wei, W. T-cells interact with B cells, dendritic cells, and fibroblast-like synoviocytes as hub-like key cells in rheumatoid arthritis. Int. Immunopharmacol. 2019, 70, 428–434. [Google Scholar] [CrossRef]
- Yasuda, T. Cartilage destruction by matrix degradation products. Mod. Rheumatol. 2006, 16, 197–205. [Google Scholar] [CrossRef]
- Adán, N.; Guzmán-Morales, J.; Ledesma-Colunga, M.G.; Perales-Canales, S.I.; Quintanar-Stéphano, A.; López-Barrera, F.; Méndez, I.; Moreno-Carranza, B.; Triebel, J.; Binart, N.; et al. Prolactin promotes cartilage survival and attenuates inflammation in inflammatory arthritis. J. Clin. Invest. 2013, 123, 3902–3913. [Google Scholar] [CrossRef]
- Hirose, T.; Fukuma, Y.; Takeshita, A.; Nishida, K. The role of lymphotoxin-α in rheumatoid arthritis. Inflamm. Res. 2018, 67, 495–501. [Google Scholar] [CrossRef]
- Guo, Q.; Wang, Y.; Xu, D.; Nossent, J.; Pavlos, N.J.; Xu, J. Rheumatoid arthritis: Pathological mechanisms and modern pharmacologic therapies. Bone Res. 2018, 6, 15. [Google Scholar] [CrossRef] [PubMed]
- Rhee, D.K.; Marcelino, J.; Baker, M.; Gong, Y.; Smits, P.; Lefebvre, V.; Jay, G.D.; Stewart, M.; Wang, H.; Warman, M.L.; et al. The secreted glycoprotein lubricin protects cartilage surfaces and inhibits synovial cell overgrowth. J. Clin. Invest. 2005, 115, 622–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gleghorn, J.P.; Jones, A.R.; Flannery, C.R.; Bonassar, L.J. Boundary mode lubrication of articular cartilage by recombinant human lubricin. J. Orthop. Res. 2009, 27, 771–777. [Google Scholar] [CrossRef]
- Al-Sharif, A.; Jamal, M.; Zhang, L.X.; Larson, K.; Schmidt, T.A.; Jay, G.D.; Elsaid, K.A. Lubricin/proteoglycan 4 binding to CD44 receptor: A mechanism of the suppression of proinflammatory cytokine-induced synoviocyte proliferation by lubricin. Arthritis Rheumatol. 2015, 67, 1503–1513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateen, S.; Moin, S.; Shahzad, S.; Khan, A.Q. Level of inflammatory cytokines in rheumatoid arthritis patients: Correlation with 25-hydroxy vitamin D and reactive oxygen species. PLoS ONE 2017, 12, e0178879. [Google Scholar] [CrossRef]
- Bunte, K.; Beikler, T. Th17 Cells and the IL-23/IL-17 Axis in the pathogenesis of periodontitis and immune-mediated inflammatory diseases. Int. J. Mol. Sci. 2019, 20, 3394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polasik, K.; Piotrowska, E.; Lipińska, B.; Witkowski, J.M.; Bryl, E.; Tukaj, S. Vitamin D status in patients with rheumatoid arthritis: A correlation analysis with disease activity and progression, as well as serum IL-6 levels. Acta Biochim. Pol. 2017, 64, 667–670. [Google Scholar] [CrossRef]
- Noack, M.; Miossec, P. Selected cytokine pathways in rheumatoid arthritis. Semin. Immunopathol. 2017, 39, 365–383. [Google Scholar] [CrossRef]
- Schuerwegh, A.J.; Dombrecht, E.J.; Stevens, W.J.; Van Offel, J.F.; Bridts, C.H.; De Clerck, L.S. Influence of pro-inflammatory (IL-1α, IL-6, TNF-α, IFN-γ) and anti-inflammatory (IL-4) cytokines on chondrocyte function. Osteoarthritis Cartilage 2003, 11, 681–687. [Google Scholar] [CrossRef] [Green Version]
- Polzer, K.; Schett, G.; Zwerina, J. The lonely death: Chondrocyte apoptosis in TNF-induced arthritis. Autoimmunity 2007, 40, 333–336. [Google Scholar] [CrossRef]
- Yatsugi, N.; Tsukazaki, T.; Osaki, M.; Koji, T.; Yamashita, S.; Shindo, H. Apoptosis of articular chondrocytes in rheumatoid arthritis and osteoarthritis: Correlation of apoptosis with degree of cartilage destruction and expression of apoptosis-related proteins of p53 and c-myc. J. Orthop. Sci. 2000, 5, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Okuma-Yoshioka, C.; Seto, H.; Kadono, Y.; Hikita, A.; Oshima, Y.; Kurosawa, H.; Nakamura, K.; Tanaka, S. Tumor necrosis factor-α inhibits chondrogenic differentiation of synovial fibroblasts through p38 mitogen activating protein kinase pathways. Mod. Rheumatol. 2008, 18, 366–378. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, H.; Kawaguchi, Y.; Harigai, M.; Hara, M.; Saito, S.; Yamaguchi, T.; Shimada, K.; Kawamoto, M.; Tomatsu, T.; Kamatani, N. Increased CD40 expression on articular chondrocytes from patients with rheumatoid arthritis: Contribution to production of cytokines and matrix metalloproteinases. J. Rheumatol. 2004, 31, 1506–1512. [Google Scholar] [PubMed]
- Andreas, K.; Lübke, C.; Häupl, T.; Dehne, T.; Morawietz, L.; Ringe, J.; Kaps, C.; Sittinger, M. Key regulatory molecules of cartilage destruction in rheumatoid arthritis: An in vitro study. Arthritis Res. Ther. 2008, 10, R9. [Google Scholar] [CrossRef] [Green Version]
- Peck, Y.; Leom, L.T.; Low, P.F.P.; Wang, D.A. Establishment of an in vitro three-dimensional model for cartilage damage in rheumatoid arthritis. J. Tissue Eng. Regen. Med. 2018, 12, e237–e249. [Google Scholar] [CrossRef]
- Steinhagen, J.; Bruns, J.; Niggemeyer, O.; Fuerst, M.; Rüther, W.; Schünke, M.; Kurz, B. Perfusion culture system: Synovial fibroblasts modulate articular chondrocyte matrix synthesis in vitro. Tissue Cell 2010, 42, 151–157. [Google Scholar] [CrossRef]
- Barksby, H.E.; Hui, W.; Wappler, I.; Peters, H.H.; Milner, J.M.; Richards, C.D.; Cawston, T.E.; Rowan, A.D. Interleukin-1 in combination with oncostatin M up-regulates multiple genes in chondrocytes: Implications for cartilage destruction and repair. Arthritis Rheum. 2006, 54, 540–550. [Google Scholar] [CrossRef]
- Meszaros, E.C.; Dahoud, W.; Mesiano, S.; Malemud, C.J. Blockade of recombinant human IL-6 by tocilizumab suppresses matrix metalloproteinase-9 production in the C28/I2 immortalized human chondrocyte cell line. Integr. Mol. Med. 2015, 2, 304–310. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Wu, X.; Wang, Z.; Ge, J.; Chen, F. Interleukin-6 enhances acid-induced apoptosis via upregulating acid-sensing ion channel 1a expression and function in rat articular chondrocytes. Int. Immunopharmacol. 2015, 29, 748–760. [Google Scholar] [CrossRef] [Green Version]
- Tortorella, M.D.; Malfait, A.M.; Deccico, C.; Arner, E. The role of ADAM-TS4 (aggrecanase-1) and ADAM-TS5 (aggrecanase-2) in a model of cartilage degradation. Osteoarthr. Cartil. 2001, 9, 539–552. [Google Scholar] [CrossRef] [Green Version]
- Koshy, P.J.; Lundy, C.J.; Rowan, A.D.; Porter, S.; Edwards, D.R.; Hogan, A.; Clark, I.M.; Cawston, T.E. The modulation of matrix metalloproteinase and ADAM gene expression in human chondrocytes by interleukin-1 and oncostatin M: A time-course study using real-time quantitative reverse transcription-polymerase chain reaction. Arthritis Rheum. 2002, 46, 961–967. [Google Scholar] [CrossRef]
- Hu, J.; Zhai, C.; Hu, J.; Li, Z.; Fei, H.; Wang, Z.; Fan, W. MiR-23a inhibited IL-17-mediated proinflammatory mediators expression via targeting IKKα in articular chondrocytes. Int. Immunopharmacol. 2017, 43, 1–6. [Google Scholar] [CrossRef]
- Dreier, R.; Wallace, S.; Fuchs, S.; Bruckner, P.; Grässel, S. Paracrine interactions of chondrocytes and macrophages in cartilage degradation: Articular chondrocytes provide factors that activate macrophage-derived pro-gelatinase B (pro-MMP-9). J. Cell Sci. 2001, 114, 3813–3822. [Google Scholar]
- Otero, M.; Goldring, M.B. Cells of the synovium in rheumatoid arthritis. Chondrocytes. Arthritis Res. Ther. 2007, 9, 220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulai, J.I.; Chen, H.; Im, H.J.; Kumar, S.; Hanning, C.; Hegde, P.S.; Loeser, R.F. NF-κB mediates the stimulation of cytokine and chemokine expression by human articular chondrocytes in response to fibronectin fragments. J. Immunol. 2005, 174, 5781–5788. [Google Scholar] [CrossRef] [Green Version]
- Yasuda, T.; Kakinuma, T.; Julovi, S.M.; Yoshida, M.; Hiramitsu, T.; Akiyoshi, M.; Nakamura, T. COOH-terminal heparin-binding fibronectin fragment induces nitric oxide production in rheumatoid cartilage through CD44. Rheumatology 2004, 43, 1116–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malemud, C.J. Chondrocyte apoptosis in rheumatoid arthritis: Is preventive therapy possible? Immunotherapy 2015, 1, 102. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.Y.; Kim, W.U.; Cho, M.L.; Lee, S.K.; Youn, J.; Kim, S.I.; Yoo, W.H.; Park, J.H.; Min, J.K.; Lee, S.H.; et al. Enhanced T cell proliferative response to type II collagen and synthetic peptide CII (255-274) in patients with rheumatoid arthritis. Arthritis Rheum. 1999, 42, 2085–2093. [Google Scholar] [CrossRef] [Green Version]
- Ohnishi, Y.; Tsutsumi, A.; Sakamaki, T.; Sumida, T. T cell epitopes of type II collagen in HLA-DRB1*0101 or DRB1*0405-positive Japanese patients with rheumatoid arthritis. Int. J. Mol. Med. 2003, 11, 331–335. [Google Scholar] [CrossRef]
- Steenvoorden, M.M.; Bank, R.A.; Ronday, H.K.; Toes, R.E.; Huizinga, T.W.; DeGroot, J. Fibroblast-like synoviocyte-chondrocyte interaction in cartilage degradation. Clin. Exp. Rheumatol. 2007, 25, 239–245. [Google Scholar]
- Korb-Pap, A.; Stratis, A.; Mühlenberg, K.; Niederreiter, B.; Hayer, S.; Echtermeyer, F.; Stange, R.; Zwerina, J.; Pap, T.; Pavenstädt, H.; et al. Early structural changes in cartilage and bone are required for the attachment and invasion of inflamed synovial tissue during destructive inflammatory arthritis. Ann. Rheum. Dis. 2012, 71, 1004–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Calatrava, M.J.; Prieto-Potín, I.; Roman-Blas, J.A.; Tardio, L.; Largo, R.; Herrero-Beaumont, G. RANKL synthesized by articular chondrocytes contributes to juxta-articular bone loss in chronic arthritis. Arthritis Res. Ther. 2012, 14, R149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, A.Z.; Wang, J.C.; Fisher, G.W.; Diamond, H.S. Interleukin-1β-stimulated invasion of articular cartilage by rheumatoid synovial fibroblasts is inhibited by antibodies to specific integrin receptors and by collagenase inhibitors. Arthritis Rheum. 1997, 40, 1298–1307. [Google Scholar] [CrossRef] [PubMed]
- Barlič, A.; Žigon, S.; Blejec, A.; Kregar Velikonja, N. Gene expression of cultured human chondrocytes as a model for assessing neutralization efficacy of soluble TNFα by TNFα antagonists. Biologicals 2015, 43, 171–180. [Google Scholar] [CrossRef]
- Feng, X.; Shi, Y.; Xu, L.; Peng, Q.; Wang, F.; Wang, X.; Sun, W.; Lu, Y.; Tsao, B.P.; Zhang, M.; et al. Modulation of IL-6 induced RANKL expression in arthritic synovium by a transcription factor SOX5. Sci. Rep. 2016, 6, 32001. [Google Scholar] [CrossRef] [Green Version]
- Mihara, M.; Moriya, Y.; Kishimoto, T.; Ohsugi, Y. Interleukin-6 (IL-6) induces the proliferation of synovial fibroblastic cells in the presence of soluble IL-6 receptor. Br. J. Rheumatol. 1995, 34, 321–325. [Google Scholar] [CrossRef]
- Koch, A.E.; Volin, M.V.; Woods, J.M.; Kunkel, S.L.; Connors, M.A.; Harlow, L.A.; Woodruff, D.C.; Burdick, M.D.; Strieter, R.M. Regulation of angiogenesis by the C-X-C chemokines interleukin-8 and epithelial neutrophil activating peptide 78 in the rheumatoid joint. Arthritis Rheum. 2001, 44, 31–40. [Google Scholar] [CrossRef]
- Long, D.; Blake, S.; Song, X.Y.; Lark, M.; Loeser, R.F. Human articular chondrocytes produce IL-7 and respond to IL-7 with increased production of matrix metalloproteinase-13. Arthritis Res. Ther. 2008, 10, R23. [Google Scholar] [CrossRef] [Green Version]
- Cai, L.; Xu, H.; Zhang, H.; Zhang, L.; Wang, G.; Nie, H. Blockade of IL-7Rα alleviates collagen-induced arthritis via inhibiting Th1 cell differentiation and CD4+ T cell migration. Mol. Immunol. 2016, 79, 83–91. [Google Scholar] [CrossRef]
- Churchman, S.M.; El-Jawhari, J.J.; Burska, A.N.; Parmar, R.; Goëb, V.; Conaghan, P.G.; Emery, P.; Ponchel, F. Modulation of peripheral T-cell function by interleukin-7 in rheumatoid arthritis. Arthritis Res. Ther. 2014, 16, 511. [Google Scholar] [CrossRef] [Green Version]
- Pickens, S.R.; Chamberlain, N.D.; Volin, M.V.; Pope, R.M.; Talarico, N.E.; Mandelin, A.M.; Shahrara, S. Characterization of interleukin-7 and interleukin-7 receptor in the pathogenesis of rheumatoid arthritis. Arthritis Rheum. 2011, 63, 2884–2893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartgring, S.A.; Bijlsma, J.W.; Lafeber, F.P.; van Roon, J.A. Interleukin-7 induced immunopathology in arthritis. Ann. Rheum. Dis. 2006, 65, iii69–iii74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buhrmann, C.; Shayan, P.; Aggarwal, B.B.; Shakibaei, M. Evidence that TNF-β (lymphotoxin α) can activate the inflammatory environment in human chondrocytes. Arthritis Res. Ther. 2013, 15, R202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calmon-Hamaty, F.; Combe, B.; Hahne, M.; Morel, J. Lymphotoxin α stimulates proliferation and pro-inflammatory cytokine secretion of rheumatoid arthritis synovial fibroblasts. Cytokine 2011, 53, 207–214. [Google Scholar] [CrossRef]
- Iwamoto, T.; Okamoto, H.; Iikuni, N.; Takeuchi, M.; Toyama, Y.; Tomatsu, T.; Kamatani, N.; Momohara, S. Monocyte chemoattractant protein-4 (MCP-4)/CCL13 is highly expressed in cartilage from patients with rheumatoid arthritis. Rheumatology 2006, 45, 421–424. [Google Scholar] [CrossRef] [Green Version]
- Iwamoto, T.; Okamoto, H.; Kobayashi, S.; Ikari, K.; Toyama, Y.; Tomatsu, T.; Kamatani, N.; Momohara, S. A role of monocyte chemoattractant protein-4 (MCP-4)/CCL13 from chondrocytes in rheumatoid arthritis. FEBS J. 2007, 274, 4904–4912. [Google Scholar] [CrossRef]
- Campbell, I.K.; Piccoli, D.S.; Roberts, M.J.; Muirden, K.D.; Hamilton, J.A. Effects of tumor necrosis factor α and β on resorption of human articular cartilage and production of plasminogen activator by human articular chondrocytes. Arthritis Rheum. 1990, 33, 542–552. [Google Scholar] [CrossRef]
- Serratì, S.; Margheri, F.; Chillà, A.; Neumann, E.; Müller-Ladner, U.; Benucci, M.; Fibbi, G.; Del Rosso, M. Reduction of in vitro invasion and in vivo cartilage degradation in a SCID mouse model by loss of function of the fibrinolytic system of rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2011, 63, 2584–2594. [Google Scholar] [CrossRef] [Green Version]
- Lotz, M.; Moats, T.; Villiger, P.M. Leukemia inhibitory factor is expressed in cartilage and synovium and can contribute to the pathogenesis of arthritis. J. Clin. Invest. 1992, 90, 888–896. [Google Scholar] [CrossRef]
- Nguyen, H.N.; Noss, E.H.; Mizoguchi, F.; Huppertz, C.; Wei, K.S.; Watts, G.F.M.; Brenner, M.B. Autocrine loop involving IL-6 family member LIF, LIF receptor, and STAT4 drives sustained fibroblast production of inflammatory mediators. Immunity 2017, 46, 220–232. [Google Scholar] [CrossRef] [Green Version]
- Momohara, S.; Okamoto, H.; Yamanaka, H. Chondrocyte of rheumatoid arthritis serve as a source of intra-articular acute-phase serum amyloid A protein. Clin. Chim. Acta 2008, 398, 155–156. [Google Scholar] [CrossRef]
- Connolly, M.; Mullan, R.H.; McCormick, J.; Matthews, C.; Sullivan, O.; Kennedy, A.; FitzGerald, O.; Poole, A.R.; Bresnihan, B.; Veale, D.J.; et al. Acute-phase serum amyloid A regulates tumor necrosis factor α and matrix turnover and predicts disease progression in patients with inflammatory arthritis before and after biologic therapy. Arthritis Rheum. 2012, 64, 1035–1045. [Google Scholar] [CrossRef]
- Mullan, R.H.; Bresnihan, B.; Golden-Mason, L.; Markham, T.; O’Hara, R.; FitzGerald, O.; Veale, D.J.; Fearon, U. Acute-phase serum amyloid A stimulation of angiogenesis, leukocyte recruitment, and matrix degradation in rheumatoid arthritis through an NF-κB-dependent signal transduction pathway. Arthritis Rheum. 2006, 54, 105–114. [Google Scholar] [CrossRef]
- Connolly, M.; Marrelli, A.; Blades, M.; McCormick, J.; Maderna, P.; Godson, C.; Mullan, R.; FitzGerald, O.; Bresnihan, B.; Pitzalis, C.; et al. Acute serum amyloid A induces migration, angiogenesis, and inflammation in synovial cells in vitro and in a human rheumatoid arthritis/SCID mouse chimera model. J. Immunol. 2010, 184, 6427–6437. [Google Scholar] [CrossRef] [Green Version]
- Neidhart, M.; Zaucke, F.; von Knoch, R.; Jüngel, A.; Michel, B.A.; Gay, R.E.; Gay, S. Galectin-3 is induced in rheumatoid arthritis synovial fibroblasts after adhesion to cartilage oligomeric matrix protein. Ann. Rheum. Dis. 2005, 64, 419–424. [Google Scholar] [CrossRef]
- Arad, U.; Madar-Balakirski, N.; Angel-Korman, A.; Amir, S.; Tzadok, S.; Segal, O.; Menachem, A.; Gold, A.; Elkayam, O.; Caspi, D. Galectin-3 is a sensor-regulator of toll-like receptor pathways in synovial fibroblasts. Cytokine 2015, 73, 30–35. [Google Scholar] [CrossRef]
- Ryu, J.H.; Chae, C.S.; Kwak, J.S.; Oh, H.; Shin, Y.; Huh, Y.H.; Lee, C.G.; Park, Y.W.; Chun, C.H.; Kim, Y.M.; et al. Hypoxia-inducible factor-2α is an essential catabolic regulator of inflammatory rheumatoid arthritis. PLoS Biol. 2014, 12, e1001881. [Google Scholar]
- Huh, Y.H.; Lee, G.; Song, W.H.; Koh, J.T.; Ryu, J.H. Crosstalk between FLS and chondrocytes is regulated by HIF-2α-mediated cytokines in arthritis. Exp. Mol. Med. 2015, 47, e197. [Google Scholar] [CrossRef] [Green Version]
- Huh, Y.H.; Lee, G.; Lee, K.B.; Koh, J.T.; Chun, J.S.; Ryu, J.H. HIF-2α-induced chemokines stimulate motility of fibroblast-like synoviocytes and chondrocytes into the cartilage-pannus interface in experimental rheumatoid arthritis mouse models. Arthritis Res. Ther. 2015, 17, 302. [Google Scholar] [CrossRef] [Green Version]
- Ostensen, M.; Veiby, O.P.; Raiss, R.; Hagen, A.; Pahle, J. Responses of normal and rheumatic human articular chondrocytes cultured under various experimental conditions in agarose. Scand. J. Rheumatol. 1991, 20, 172–182. [Google Scholar] [CrossRef]
- Zhang, H.J.; Wei, Q.F.; Wang, S.J.; Zhang, H.J.; Zhang, X.Y.; Geng, Q.; Cui, Y.H.; Wang, X.H. LncRNA HOTAIR alleviates rheumatoid arthritis by targeting miR-138 and inactivating NF-κB pathway. Int. Immunopharmacol. 2017, 50, 283–290. [Google Scholar] [CrossRef]
- Jiang, L.; Cao, S. Role of microRNA-26a in cartilage injury and chondrocyte proliferation and apoptosis in rheumatoid arthritis rats by regulating expression of CTGF. J. Cell Physiol. 2019. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, S.; Chen, P.; Yang, B.; Yang, J.; Liu, R.; Li, J.; Xia, D. MicroRNA-27b-3p inhibits apoptosis of chondrocyte in rheumatoid arthritis by targeting HIPK2. Artif. Cells Nanomed. Biotechnol. 2019, 47, 1766–1771. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zhu, C.J.; Zhu, F.; Dai, B.B.; Song, S.J.; Wang, Z.Q.; Feng, Y.-B.; Ge, J.-F.; Zhou, R.-P.; Chen, F.-H. Necrostatin-1 ameliorates adjuvant arthritis rat articular chondrocyte injury via inhibiting ASIC1a-mediated necroptosis. Biochem. Biophys. Res. Commun. 2018, 504, 843–850. [Google Scholar] [CrossRef]
- Bobkov, V.A.; Brylenkova, T.N.; Kopilov, E.I.; Mitskaia, S.G.; Kazakova, N.I. Changes in the acid-base status of the synovial fluid in rheumatoid arthritis patients. Terapevticheskii Arkhiv 1999, 71, 20–22. [Google Scholar]
- Wu, X.; Ren, G.; Zhou, R.; Ge, J.; Chen, F.H. The role of Ca2+ in acid-sensing ion channel 1a-mediated chondrocyte pyroptosis in rat adjuvant arthritis. Lab. Invest. 2019, 99, 499–513. [Google Scholar] [CrossRef]
- Li, R.; Cai, L.; Hu, C.M.; Wu, T.N.; Li, J. Expression of hedgehog signal pathway in articular cartilage is associated with the severity of cartilage damage in rats with adjuvant-induced arthritis. J. Inflamm. 2015, 12, 24. [Google Scholar] [CrossRef] [Green Version]
- Rockel, J.S.; Bernier, S.M.; Leask, A. Egr-1 inhibits the expression of extracellular matrix genes in chondrocytes by TNFα-induced MEK/ERK signalling. Arthritis Res. Ther. 2009, 11, R8. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.P.; Ni, W.L.; Dai, B.B.; Wu, X.S.; Wang, Z.S.; Xie, Y.Y.; Wang, Z.Q.; Yang, W.J.; Ge, J.F.; Hu, W.; et al. ASIC2a overexpression enhances the protective effect of PcTx1 and APETx2 against acidosis-induced articular chondrocyte apoptosis and cytotoxicity. Gene 2018, 642, 230–240. [Google Scholar] [CrossRef]
- Julovi, S.M.; Ito, H.; Nishitani, K.; Jackson, C.J.; Nakamura, T. Hyaluronan inhibits matrix metalloproteinase-13 in human arthritic chondrocytes via CD44 and P38. J. Orthop. Res. 2011, 29, 258–264. [Google Scholar] [CrossRef]
- Chiu, Y.C.; Yang, R.S.; Hsieh, K.H.; Fong, Y.C.; Way, T.D.; Lee, T.S.; Wu, H.C.; Fu, W.M.; Tang, C.H. Stromal cell-derived factor-1 induces matrix metalloprotease-13 expression in human chondrocytes. Mol. Pharmacol. 2007, 72, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Ismail, H.M.; Yamamoto, K.; Vincent, T.L.; Nagase, H.; Troeberg, L.; Saklatvala, J. Interleukin-1 acts via the JNK-2 signaling pathway to induce aggrecan degradation by human chondrocytes. Arthritis Rheumatol. 2015, 67, 1826–1836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, L.; Lei, C.; Li, R.; Chen, W.N.; Hu, C.M.; Chen, X.Y.; Li, C.M. Overexpression of aquaporin 4 in articular chondrocytes exacerbates the severity of adjuvant-induced arthritis in rats: An in vivo and in vitro study. J. Inflamm. 2017, 14, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashida, M.; Okazaki, K.; Fukushi, J.; Sakamoto, A.; Iwamoto, Y. CCAAT/enhancer binding protein β mediates expression of matrix metalloproteinase 13 in human articular chondrocytes in inflammatory arthritis. Arthritis Rheum. 2009, 60, 708–716. [Google Scholar] [CrossRef] [PubMed]
- Buhrmann, C.; Popper, B.; Aggarwal, B.B.; Shakibaei, M. Resveratrol downregulates inflammatory pathway activated by lymphotoxin α (TNF-β) in articular chondrocytes: Comparison with TNF-α. PLoS ONE 2017, 12, e0186993. [Google Scholar] [CrossRef]
- Corrêa, M.G.; Pires, P.R.; Ribeiro, F.V.; Pimentel, S.P.; Cirano, F.R.; Napimoga, M.H.; Casati, M.Z.; Casarin, R.C.V. Systemic treatment with resveratrol reduces the progression of experimental periodontitis and arthritis in rats. PLoS ONE 2018, 13, e0204414. [Google Scholar] [CrossRef] [Green Version]
- Khojah, H.M.; Ahmed, S.; Abdel-Rahman, M.S.; Elhakeim, E.H. Resveratrol as an effective adjuvant therapy in the management of rheumatoid arthritis: A clinical study. Clin. Rheumatol. 2018, 37, 2035–2042. [Google Scholar] [CrossRef]
- Yasuda, T. Comparison of hyaluronan effects among normal, osteoarthritis, and rheumatoid arthritis cartilages stimulated with fibronectin fragment. Biomed. Res. 2010, 31, 63–69. [Google Scholar] [CrossRef] [Green Version]
- Yasuda, T. Hyaluronan inhibits p38 mitogen-activated protein kinase via the receptors in rheumatoid arthritis chondrocytes stimulated with fibronectin fragment. Clin. Rheumatol. 2010, 29, 1259–1267. [Google Scholar] [CrossRef]
- Hui, A.; Min, W.X.; Tang, J.; Cruz, T.F. Inhibition of activator protein 1 activity by paclitaxel suppresses interleukin-1-induced collagenase and stromelysin expression by bovine chondrocytes. Arthritis Rheum. 1998, 41, 869–876. [Google Scholar] [CrossRef]
- Saito, S.; Kotake, S. Is there evidence in support of the use of intra-articular hyaluronate in treating rheumatoid arthritis of the knee? A meta-analysis of the published literature. Mod. Rheumatol. 2009, 19, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.C.; Lee, S.H.; Lin, H.Y.; Liu, F.W.; Chiou, H.J.; Chan, R.C.; Chou, C.L. Short-term effect of ultrasound-guided low-molecular-weight hyaluronic acid injection on clinical outcomes and imaging changes in patients with rheumatoid arthritis of the ankle and foot joints. A randomized controlled pilot trial. Mod. Rheumatol. 2017, 27, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.L.; Ang, D.C.; Almeida-Porada, G. Targeting mesenchymal stromal cells/pericytes (MSCs) with pulsed electromagnetic field (PEMF) has the potential to treat rheumatoid arthritis. Front. Immunol. 2019, 10, 266. [Google Scholar] [CrossRef] [PubMed]
- Kastrinaki, M.C.; Papadaki, H.A. Mesenchymal stromal cells in rheumatoid arthritis: Biological properties and clinical applications. Curr. Stem Cell Res. Ther. 2009, 4, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.S.; Jung, Y.H.; Cho, M.Y.; Yeo, J.E.; Choi, Y.J.; Kim, Y.I.; Koh, Y.G. Co-culture with human synovium-derived mesenchymal stem cells inhibits inflammatory activity and increases cell proliferation of sodium nitroprusside-stimulated chondrocytes. Biochem. Biophys. Res. Commun. 2014, 447, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Costello, R.; David, T.; Jani, M. Impact of adverse events associated with medications in the treatment and prevention of rheumatoid arthritis. Clin. Ther. 2019, 41, 1376–1396. [Google Scholar] [CrossRef] [PubMed]
- Andreas, K.; Häupl, T.; Lübke, C.; Ringe, J.; Morawietz, L.; Wachtel, A.; Sittinger, M.; Kaps, C. Antirheumatic drug response signatures in human chondrocytes: Potential molecular targets to stimulate cartilage regeneration. Arthritis Res. Ther. 2009, 11, R15. [Google Scholar] [CrossRef] [Green Version]
- Vuolteenaho, K.; Kujala, P.; Moilanen, T.; Moilanen, E. Aurothiomalate and hydroxychloroquine inhibit nitric oxide production in chondrocytes and in human osteoarthritic cartilage. Scand. J. Rheumatol. 2005, 34, 475–479. [Google Scholar] [CrossRef]
- Žigon-Branc, S.; Jeras, M.; Blejec, A.; Barlič, A. Applicability of human osteoarthritic chondrocytes for in vitro efficacy testing of anti-TNFα drugs. Biologicals 2017, 45, 96–101. [Google Scholar] [CrossRef]
- Endo, W.; Arito, M.; Sato, T.; Kurokawa, M.S.; Omoteyama, K.; Iizuka, N.; Okamoto, K.; Suematsu, N.; Nakamura, H.; Beppu, M.; et al. Effects of sulfasalazine and tofacitinib on the protein profile of articular chondrocytes. Mod. Rheumatol. 2014, 24, 844–850. [Google Scholar] [CrossRef] [PubMed]
- Palmer, G.; Burger, D.; Mezin, F.; Magne, D.; Gabay, C.; Dayer, J.M.; Guerne, P.A. The active metabolite of leflunomide, A77 1726, increases the production of IL-1 receptor antagonist in human synovial fibroblasts and articular chondrocytes. Arthritis Res. Ther. 2004, 6, R181–R189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abramson, S.B.; Amin, A. Blocking the effects of IL-1 in rheumatoid arthritis protects bone and cartilage. Rheumatology 2002, 41, 972–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, M.V.C.; Adrait, A.; Baillet, A.; Trocme, C.; Gottenberg, J.E.; Gaudin, P. Identification of cartilage oligomeric matrix protein as biomarker predicting abatacept response in rheumatoid arthritis patients with insufficient response to a first anti-TNFα treatment. Joint Bone Spine 2019, 86, 401–403. [Google Scholar] [CrossRef] [PubMed]
- Sakthiswary, R.; Rajalingam, S.; Hussein, H.; Sridharan, R.; Asrul, A.W. Cartilage oligomeric matrix protein (COMP) in rheumatoid arthritis and its correlation with sonographic knee cartilage thickness and disease activity. Clin. Rheumatol. 2017, 36, 2683–2688. [Google Scholar] [CrossRef]
- Liu, F.; Wang, X.; Zhang, X.; Ren, C.; Xin, J. Role of Serum cartilage oligomeric matrix protein (COMP) in the diagnosis of rheumatoid arthritis (RA): A case-control study. J. Int. Med. Res. 2016, 44, 940–949. [Google Scholar] [CrossRef]
- Ben Achour, W.; Bouaziz, M.; Mechri, M.; Zouari, B.; Bahlous, A.; Abdelmoula, L.; Laadhar, L.; Sellami, M.; Sahli, H.; Cheour, E. A cross sectional study of bone and cartilage biomarkers: Correlation with structural damage in rheumatoid arthritis. Libyan J. Med. 2018, 13, 1512330. [Google Scholar] [CrossRef] [Green Version]
- Maijer, K.I.; Gudmann, N.S.; Karsdal, M.A.; Gerlag, D.M.; Tak, P.P.; Bay-Jensen, A.C. Neo-epitopes—Fragments of cartilage and connective tissue degradation in early rheumatoid arthritis and unclassified arthritis. PLoS ONE 2016, 11, e0149329. [Google Scholar] [CrossRef] [Green Version]
- Siebuhr, A.S.; Bay-Jensen, A.C.; Leeming, D.J.; Plat, A.; Byrjalsen, I.; Christiansen, C.; van de Heijde, D.; Karsdal, M.A. Serological identification of fast progressors of structural damage with rheumatoid arthritis. Arthritis Res. Ther. 2013, 15, R86. [Google Scholar] [CrossRef] [Green Version]
- Väänänen, T.; Vuolteenaho, K.; Kautiainen, H.; Nieminen, R.; Möttönen, T.; Hannonen, P.; Korpela, M.; Kauppi, M.J.; Laiho, K.; Kaipiainen-Seppänen, O.; et al. Glycoprotein YKL-40: A potential biomarker of disease activity in rheumatoid arthritis during intensive treatment with csDMARDs and infliximab. Evidence from the randomised controlled NEO-RACo trial. PLoS ONE 2017, 12, e0183294. [Google Scholar] [CrossRef] [Green Version]
- Jafari-Nakhjavani, M.R.; Ghorbanihaghjo, A.; Bagherzadeh-Nobari, B.; Malek-Mahdavi, A.; Rashtchizadeh, N. Serum YKL-40 levels and disease characteristics in patients with rheumatoid arthritis. Caspian J. Intern. Med. 2019, 10, 92–97. [Google Scholar]
- Johansen, J.S.; Kirwan, J.R.; Price, P.A.; Sharif, M. Serum YKL-40 concentrations in patients with early rheumatoid arthritis: Relation to joint destruction. Scand. J. Rheumatol. 2001, 30, 297–304. [Google Scholar] [PubMed]
- Li, T.M.; Liu, S.C.; Huang, Y.H.; Huang, C.C.; Hsu, C.J.; Tsai, C.H.; Wang, S.W.; Tang, C.H. YKL-40-induced inhibition of miR-590-3p promotes interleukin-18 expression and angiogenesis of endothelial progenitor cells. Int. J. Mol. Sci. 2017, 18, 920. [Google Scholar] [CrossRef] [PubMed]
- Müller-Ladner, U.; Bosserhoff, A.K.; Dreher, K.; Hein, R.; Neidhart, M.; Gay, S.; Schölmerich, J.; Buettner, R.; Lang, B. MIA (melanoma inhibitory activity): A potential serum marker for rheumatoid arthritis. Rheumatology 1999, 38, 148–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niki, Y.; Takeuchi, T.; Nakayama, M.; Nagasawa, H.; Kurasawa, T.; Yamada, H.; Toyama, Y.; Miyamoto, T. Clinical significance of cartilage biomarkers for monitoring structural joint damage in rheumatoid arthritis patients treated with anti-TNF therapy. PLoS ONE 2012, 7, e37447. [Google Scholar] [CrossRef] [PubMed]
- Landewé, R.; Geusens, P.; Boers, M.; van der Heijde, D.; Lems, W.; te Koppele, J.; van der Linden, S.; Garnero, P. Markers for type II collagen breakdown predict the effect of disease-modifying treatment on long-term radiographic progression in patients with rheumatoid arthritis. Arthritis Rheum. 2004, 50, 1390–1399. [Google Scholar] [CrossRef]
- Aletaha, D.; Funovits, J.; Smolen, J.S. Physical disability in rheumatoid arthritis is associated with cartilage damage rather than bone destruction. Ann. Rheum. Dis. 2011, 70, 733–739. [Google Scholar] [CrossRef] [Green Version]
- Hayer, S.; Bauer, G.; Willburger, M.; Sinn, K.; Alasti, F.; Plasenzotti, R.; Shvets, T.; Niederreiter, B.; Aschauer, C.; Steiner, G.; et al. Cartilage damage and bone erosion are more prominent determinants of functional impairment in longstanding experimental arthritis than synovial inflammation. Dis. Model. Mech. 2016, 9, 1329–1338. [Google Scholar] [CrossRef] [Green Version]
- Krivoruchko, N.; Tuganbekova, S.; Rakhimbekova, G.; Kuzembaeva, K.; Zaripova, L. The results of fetal chondrocytes transplantation in patients with rheumatoid arthritis. Cent. Asian J. Glob. Health 2014, 3, 164. [Google Scholar] [CrossRef] [Green Version]
- Aletaha, D.; Smolen, J.S. Diagnosis and management of rheumatoid arthritis: A review. JAMA 2018, 320, 1360–1372. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tseng, C.-C.; Chen, Y.-J.; Chang, W.-A.; Tsai, W.-C.; Ou, T.-T.; Wu, C.-C.; Sung, W.-Y.; Yen, J.-H.; Kuo, P.-L. Dual Role of Chondrocytes in Rheumatoid Arthritis: The Chicken and the Egg. Int. J. Mol. Sci. 2020, 21, 1071. https://doi.org/10.3390/ijms21031071
Tseng C-C, Chen Y-J, Chang W-A, Tsai W-C, Ou T-T, Wu C-C, Sung W-Y, Yen J-H, Kuo P-L. Dual Role of Chondrocytes in Rheumatoid Arthritis: The Chicken and the Egg. International Journal of Molecular Sciences. 2020; 21(3):1071. https://doi.org/10.3390/ijms21031071
Chicago/Turabian StyleTseng, Chia-Chun, Yi-Jen Chen, Wei-An Chang, Wen-Chan Tsai, Tsan-Teng Ou, Cheng-Chin Wu, Wan-Yu Sung, Jeng-Hsien Yen, and Po-Lin Kuo. 2020. "Dual Role of Chondrocytes in Rheumatoid Arthritis: The Chicken and the Egg" International Journal of Molecular Sciences 21, no. 3: 1071. https://doi.org/10.3390/ijms21031071