Analysis of the Effect of Increased α2,3-Sialylation on RTK Activation in MKN45 Gastric Cancer Spheroids Treated with Crizotinib

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
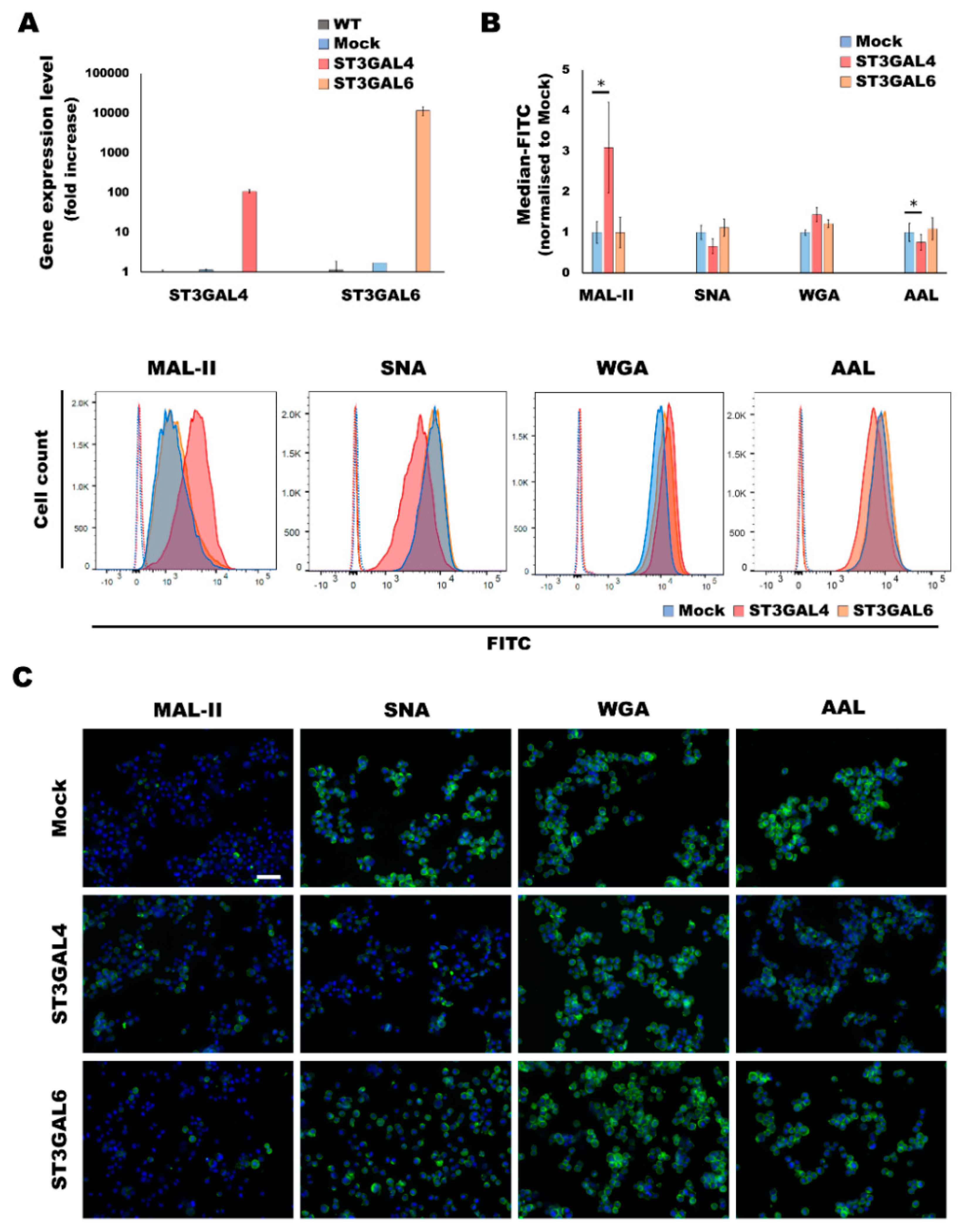
2.1. Overexpression of α2,3-Sialyltransferases Results in Different Cellular Glycocalyx
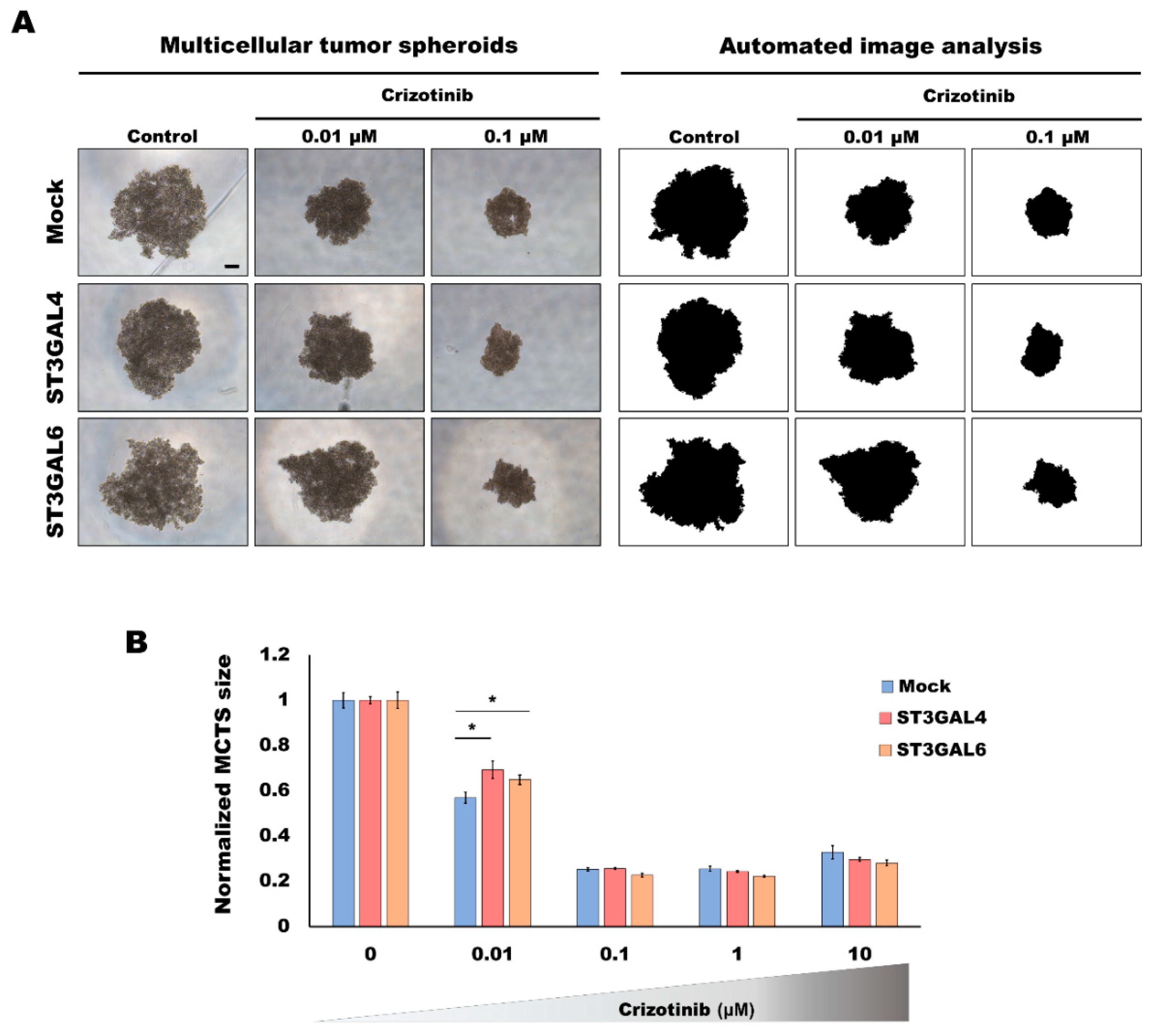
2.2. Overexpression of α2,3-Sialyltransferases Affects the Response to Crizotinib
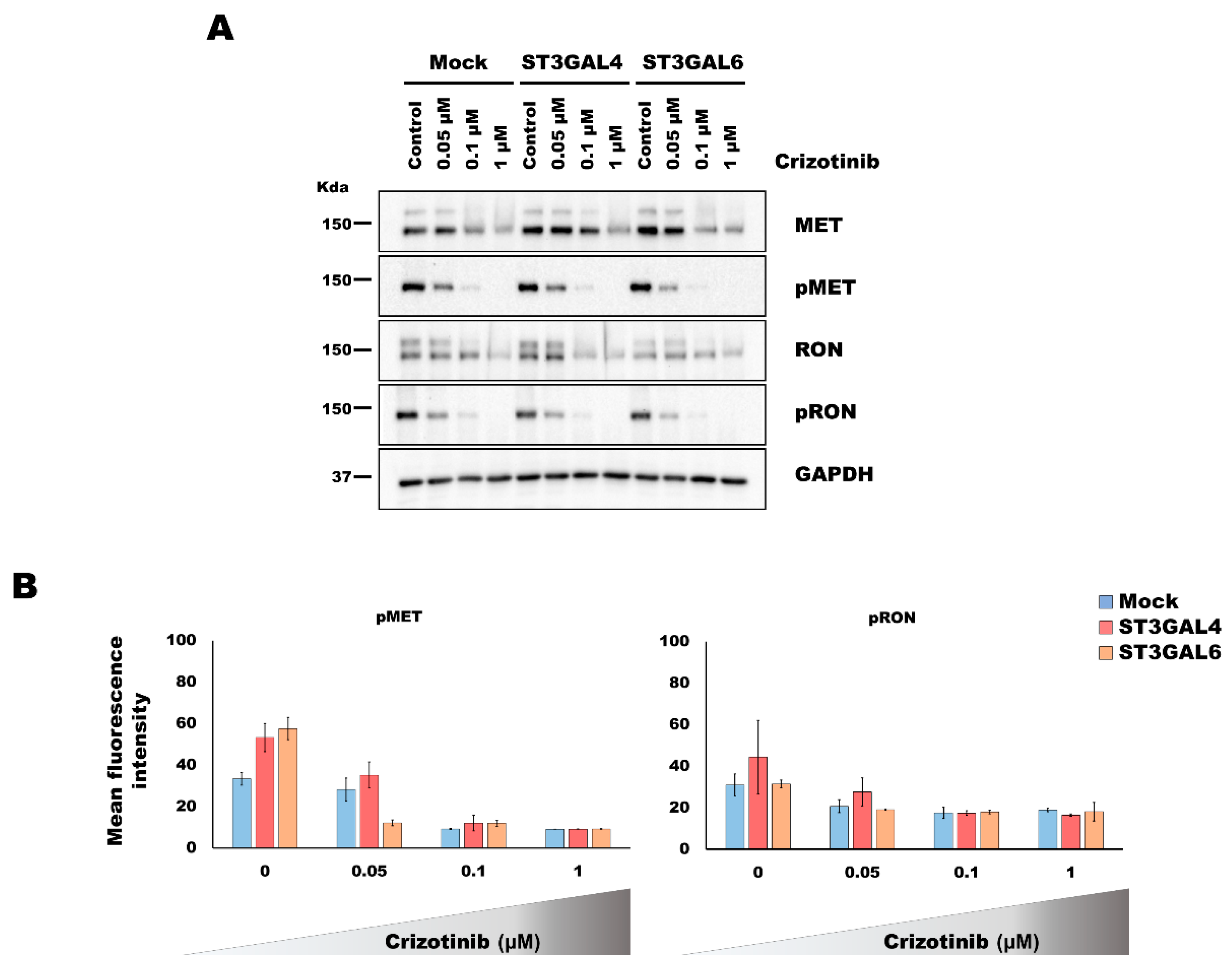
2.3. Analysis of the Activation of MET and RON by Overexpression of α2,3-Sialyltransferases
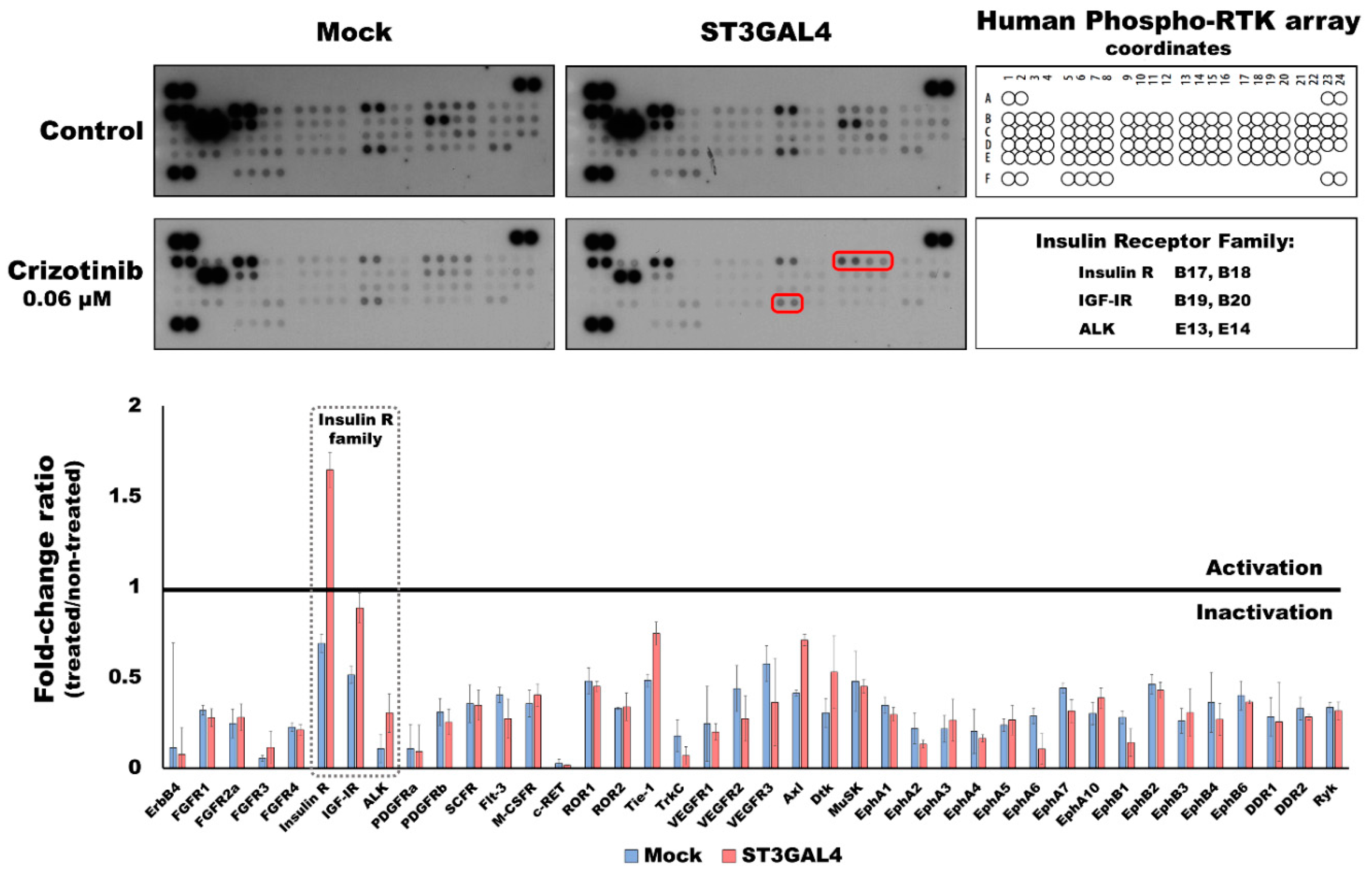
2.4. Overexpression of α2,3-Sialyltransferases Promotes Survival through the Activation of Insulin Receptors
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. RNA Extraction, cDNA Synthesis, and Real-Time Reverse Transcription PCR (RT-PCR)
4.3. Generation of Gastric Multicellular Tumor Spheroids
4.4. Spheroid Treatment
4.5. Image Analysis of Size of the Gastric Multicellular Tumor Spheroids
- Ratio of MCTS for each MCTS (x):
- Normalized ratio for each MCTS (x):
4.6. Flow Cytometry and Lectin Fluorescent Staining
4.7. Fluorescent Cell Staining of Gastric Spheroids
4.8. Image Analysis of Receptor Activation
4.9. Western Blot
4.10. Phosphoprotein Array
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AAL | Aleuria aurantia lectin |
| EGFR | Epidermal growth factor receptor 1 |
| ErbB2 | Epidermal growth factor receptor 2 |
| GC | Gastric cancer |
| HGFR | Hepatocyte growth factor receptor |
| IR | Insulin receptor |
| MAL-II | Maackia amurensis lectin II |
| MCTS | Multicellular tumor spheroids |
| MSI | Microsatellite instable |
| NSCLC | Non-small cell lung cancer |
| RON | Recepteur d’Origine Nantais (also known as macrophage-stimulating protein receptor (MSPR) or MST1R) |
| RT-qPCR | Real-time quantitative PCR |
| RTK | Receptor tyrosine kinase |
| SNA | Sambucus nigra agglutinin |
| TF | Thomsen-Friedenreich |
| TKI | Tyrosine kinase inhibitor |
| ULA | Ultra-low attachment |
| WGA | Wheat germ agglutinin |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Cutsem, E.; Sagaert, X.; Topal, B.; Haustermans, K.; Prenen, H. Gastric cancer. Lancet 2016, 388, 2654–2664. [Google Scholar] [CrossRef]
- Sawyers, C. Targeted cancer therapy. Nature 2004, 432, 294–297. [Google Scholar] [CrossRef] [PubMed]
- Deng, N.; Goh, L.K.; Wang, H.; Das, K.; Tao, J.; Tan, I.B.; Zhang, S.; Lee, M.; Wu, J.; Lim, K.H.; et al. A comprehensive survey of genomic alterations in gastric cancer reveals systematic patterns of molecular exclusivity and co-occurrence among distinct therapeutic targets. Gut 2012, 61, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Lordick, F.; Allum, W.; Carneiro, F.; Mitry, E.; Tabernero, J.; Tan, P.; Van Cutsem, E.; van de Velde, C.; Cervantes, A. Unmet needs and challenges in gastric cancer: The way forward. Cancer Treat. Rev. 2014, 40, 692–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gherardi, E.; Birchmeier, W.; Birchmeier, C.; Vande Woude, G. Targeting met in cancer: Rationale and progress. Nat. Rev. Cancer 2012, 12, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef] [Green Version]
- Inokuchi, M.; Otsuki, S.; Fujimori, Y.; Sato, Y.; Nakagawa, M.; Kojima, K. Clinical significance of met in gastric cancer. World J. Gastrointest. Oncol. 2015, 7, 317–327. [Google Scholar] [CrossRef]
- Yao, H.P.; Zhou, Y.Q.; Zhang, R.; Wang, M.H. Msp-ron signalling in cancer: Pathogenesis and therapeutic potential. Nat. Rev. Cancer 2013, 13, 466–481. [Google Scholar] [CrossRef]
- Yang, S.Y.; Nguyen, T.T.; Ung, T.T.; Jung, Y.D. Role of recepteur d’origine nantais on gastric cancer development and progression. Chonnam. Med. J. 2017, 53, 178–186. [Google Scholar] [CrossRef] [Green Version]
- Gomes, C.; Osorio, H.; Pinto, M.T.; Campos, D.; Oliveira, M.J.; Reis, C.A. Expression of st3gal4 leads to sle(x) expression and induces c-met activation and an invasive phenotype in gastric carcinoma cells. PLoS ONE 2013, 8, e66737. [Google Scholar] [CrossRef] [PubMed]
- Mereiter, S.; Magalhaes, A.; Adamczyk, B.; Jin, C.; Almeida, A.; Drici, L.; Ibanez-Vea, M.; Gomes, C.; Ferreira, J.A.; Afonso, L.P.; et al. Glycomic analysis of gastric carcinoma cells discloses glycans as modulators of ron receptor tyrosine kinase activation in cancer. Biochim. Biophys. Acta 2016, 1860, 1795–1808. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.Y.; Shun, C.T.; Hung, K.Y.; Juan, H.F.; Hsu, C.L.; Huang, M.C.; Lai, I.R. Mucin glycosylating enzyme galnt2 suppresses malignancy in gastric adenocarcinoma by reducing met phosphorylation. Oncotarget 2016, 7, 11251–11262. [Google Scholar] [CrossRef] [PubMed]
- Mereiter, S.; Balmana, M.; Campos, D.; Gomes, J.; Reis, C.A. Glycosylation in the era of cancer-targeted therapy: Where are we heading? Cancer Cell 2019, 36, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Varki, A.; Kannagi, R.; Toole, B.; Stanley, P. Glycosylation changes in cancer. In Essentials of Glycobiology, 3rd ed.; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor: Suffolk, NY, USA, 2015; pp. 597–609. [Google Scholar]
- Pinho, S.S.; Reis, C.A. Glycosylation in cancer: Mechanisms and clinical implications. Nat. Rev. Cancer 2015, 15, 540–555. [Google Scholar] [CrossRef] [PubMed]
- Pearce, O.M.; Laubli, H. Sialic acids in cancer biology and immunity. Glycobiology 2016, 26, 111–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dall’Olio, F.; Malagolini, N.; Trinchera, M.; Chiricolo, M. Sialosignaling: Sialyltransferases as engines of self-fueling loops in cancer progression. Biochim. Biophys. Acta 2014, 1840, 2752–2764. [Google Scholar] [CrossRef] [Green Version]
- Marcos, N.T.; Bennett, E.P.; Gomes, J.; Magalhaes, A.; Gomes, C.; David, L.; Dar, I.; Jeanneau, C.; DeFrees, S.; Krustrup, D.; et al. St6galnac-i controls expression of sialyl-tn antigen in gastrointestinal tissues. Front. Biosci. (Elite Ed.) 2011, 3, 1443–1455. [Google Scholar] [CrossRef]
- Harduin-Lepers, A.; Krzewinski-Recchi, M.A.; Colomb, F.; Foulquier, F.; Groux-Degroote, S.; Delannoy, P. Sialyltransferases functions in cancers. Front. Biosci. (Elite Ed.) 2012, 4, 499–515. [Google Scholar] [CrossRef]
- Harduin-Lepers, A.; Vallejo-Ruiz, V.; Krzewinski-Recchi, M.A.; Samyn-Petit, B.; Julien, S.; Delannoy, P. The human sialyltransferase family. Biochimie 2001, 83, 727–737. [Google Scholar] [CrossRef]
- Lordick, F. Targeting the hgf/met pathway in gastric cancer. Lancet Oncol. 2014, 15, 914–916. [Google Scholar] [CrossRef]
- Eder, J.P.; Vande Woude, G.F.; Boerner, S.A.; LoRusso, P.M. Novel therapeutic inhibitors of the c-met signaling pathway in cancer. Clin. Cancer Res. 2009, 15, 2207–2214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahu, A.; Prabhash, K.; Noronha, V.; Joshi, A.; Desai, S. Crizotinib: A comprehensive review. South Asian J. Cancer 2013, 2, 91–97. [Google Scholar]
- Pampaloni, F.; Reynaud, E.G.; Stelzer, E.H. The third dimension bridges the gap between cell culture and live tissue. Nat. Rev. Mol. Cell Biol. 2007, 8, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Abbott, A. Cell culture: Biology’s new dimension. Nature 2003, 424, 870–872. [Google Scholar] [CrossRef] [PubMed]
- Breslin, S.; O’Driscoll, L. Three-dimensional cell culture: The missing link in drug discovery. Drug Discov. Today 2013, 18, 240–249. [Google Scholar] [CrossRef]
- Weigelt, B.; Ghajar, C.M.; Bissell, M.J. The need for complex 3d culture models to unravel novel pathways and identify accurate biomarkers in breast cancer. Adv. Drug Deliv. Rev. 2014, 69–70, 42–51. [Google Scholar] [CrossRef] [Green Version]
- Vinci, M.; Gowan, S.; Boxall, F.; Patterson, L.; Zimmermann, M.; Court, W.; Lomas, C.; Mendiola, M.; Hardisson, D.; Eccles, S.A. Advances in establishment and analysis of three-dimensional tumor spheroid-based functional assays for target validation and drug evaluation. BMC Biol. 2012, 10, 29. [Google Scholar] [CrossRef] [Green Version]
- Balmana, M.; Mereiter, S.; Diniz, F.; Feijao, T.; Barrias, C.C.; Reis, C.A. Multicellular human gastric-cancer spheroids mimic the glycosylation phenotype of gastric carcinomas. Molecules 2018, 23, 2815. [Google Scholar] [CrossRef] [Green Version]
- Nath, S.; Devi, G.R. Three-dimensional culture systems in cancer research: Focus on tumor spheroid model. Pharmacol. Ther. 2016, 163, 94–108. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, A.S.; Harduin-Lepers, A.; Magalhaes, A.; Machado, E.; Mendes, N.; Costa, L.T.; Matthiesen, R.; Almeida, R.; Costa, J.; Reis, C.A. Differential expression of alpha-2,3-sialyltransferases and alpha-1,3/4-fucosyltransferases regulates the levels of sialyl lewis a and sialyl lewis x in gastrointestinal carcinoma cells. Int. J. Biochem. Cell Biol. 2010, 42, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Itakura, Y.; Nakamura-Tsuruta, S.; Kominami, J.; Tateno, H.; Hirabayashi, J. Sugar-binding profiles of chitin-binding lectins from the hevein family: A comprehensive study. Int. J. Mol. Sci. 2017, 18, 1160. [Google Scholar] [CrossRef] [PubMed]
- Mereiter, S.; Magalhaes, A.; Adamczyk, B.; Jin, C.; Almeida, A.; Drici, L.; Ibanez-Vea, M.; Larsen, M.R.; Kolarich, D.; Karlsson, N.G.; et al. Glycomic and sialoproteomic data of gastric carcinoma cells overexpressing st3gal4. Data Brief 2016, 7, 814–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, A.M.; Huang, P.H. Receptor tyrosine kinase coactivation networks in cancer. Cancer Res. 2010, 70, 3857–3860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58. [Google Scholar] [CrossRef] [PubMed]
- Bhullar, K.S.; Lagaron, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef]
- Alexander, P.B.; Wang, X.F. Resistance to receptor tyrosine kinase inhibition in cancer: Molecular mechanisms and therapeutic strategies. Front. Med. 2015, 9, 134–138. [Google Scholar] [CrossRef] [Green Version]
- Niederst, M.J.; Engelman, J.A. Bypass mechanisms of resistance to receptor tyrosine kinase inhibition in lung cancer. Sci. Signal 2013, 6, re6. [Google Scholar] [CrossRef] [Green Version]
- Mereiter, S.; Polom, K.; Williams, C.; Polonia, A.; Guergova-Kuras, M.; Karlsson, N.G.; Roviello, F.; Magalhaes, A.; Reis, C.A. The thomsen-friedenreich antigen: A highly sensitive and specific predictor of microsatellite instability in gastric cancer. J. Clin. Med. 2018, 7, 256. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.C.; Yen, H.Y.; Chen, C.Y.; Chen, C.H.; Cheng, P.F.; Juan, Y.H.; Chen, C.H.; Khoo, K.H.; Yu, C.J.; Yang, P.C.; et al. Sialylation and fucosylation of epidermal growth factor receptor suppress its dimerization and activation in lung cancer cells. Proc. Natl. Acad. Sci. USA 2011, 108, 11332–11337. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Zhu, M.; Linhai, Y.; Song, Y.; Gui, X.; Tan, G.; Li, J.; Liu, Y.; Deng, Z.; Chen, X.; et al. Increasing her2 alpha2,6 sialylation facilitates gastric cancer progression and resistance via the akt and erk pathways. Oncol. Rep. 2018, 40, 2997–3005. [Google Scholar] [PubMed]
- Britain, C.M.; Holdbrooks, A.T.; Anderson, J.C.; Willey, C.D.; Bellis, S.L. Sialylation of egfr by the st6gal-i sialyltransferase promotes egfr activation and resistance to gefitinib-mediated cell death. J. Ovarian Res. 2018, 11, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yen, H.Y.; Liu, Y.C.; Chen, N.Y.; Tsai, C.F.; Wang, Y.T.; Chen, Y.J.; Hsu, T.L.; Yang, P.C.; Wong, C.H. Effect of sialylation on egfr phosphorylation and resistance to tyrosine kinase inhibition. Proc. Natl. Acad. Sci. USA 2015, 112, 6955–6960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Wu, N.; Shen, J.; Teixido, C.; Sun, X.; Lin, Z.; Qian, X.; Zou, Z.; Guan, W.; Yu, L.; et al. Met overexpression and amplification define a distinct molecular subgroup for targeted therapies in gastric cancer. Gastric Cancer 2016, 19, 778–788. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, W.; Okamoto, I.; Arao, T.; Kuwata, K.; Hatashita, E.; Yamaguchi, H.; Sakai, K.; Yanagihara, K.; Nishio, K.; Nakagawa, K. Antitumor action of the met tyrosine kinase inhibitor crizotinib (pf-02341066) in gastric cancer positive for met amplification. Mol. Cancer Ther. 2012, 11, 1557–1564. [Google Scholar] [CrossRef] [Green Version]
- Okajima, T.; Fukumoto, S.; Miyazaki, H.; Ishida, H.; Kiso, M.; Furukawa, K.; Urano, T.; Furukawa, K. Molecular cloning of a novel alpha2,3-sialyltransferase (st3gal vi) that sialylates type ii lactosamine structures on glycoproteins and glycolipids. J. Biol. Chem. 1999, 274, 11479–11486. [Google Scholar] [CrossRef] [Green Version]
- Groux-Degroote, S.; Rodriguez-Walker, M.; Dewald, J.H.; Daniotti, J.L.; Delannoy, P. Gangliosides in cancer cell signaling. Prog. Mol. Biol. Transl. Sci. 2018, 156, 197–227. [Google Scholar]
- Shintani, Y.; Takashima, S.; Asano, Y.; Kato, H.; Liao, Y.; Yamazaki, S.; Tsukamoto, O.; Seguchi, O.; Yamamoto, H.; Fukushima, T.; et al. Glycosaminoglycan modification of neuropilin-1 modulates vegfr2 signaling. EMBO J. 2006, 25, 3045–3055. [Google Scholar] [CrossRef] [Green Version]
- Julien, S.; Bobowski, M.; Steenackers, A.; Le Bourhis, X.; Delannoy, P. How do gangliosides regulate rtks signaling? Cells 2013, 2, 751–767. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, J.G.; Balmana, M.; Macedo, J.A.; Pocas, J.; Fernandes, A.; de-Freitas-Junior, J.C.M.; Pinho, S.S.; Gomes, J.; Magalhaes, A.; Gomes, C.; et al. Glycosylation in cancer: Selected roles in tumour progression, immune modulation and metastasis. Cell. Immunol. 2018, 333, 46–57. [Google Scholar] [CrossRef] [Green Version]
- Raiber, E.A.; Wilkinson, J.A.; Manetti, F.; Botta, M.; Deakin, J.; Gallagher, J.; Lyon, M.; Ducki, S.W. Novel heparin/heparan sulfate mimics as inhibitors of hgf/sf-induced met activation. Bioorg. Med. Chem. Lett. 2007, 17, 6321–6325. [Google Scholar] [CrossRef] [PubMed]
- Sakata, H.; Stahl, S.J.; Taylor, W.G.; Rosenberg, J.M.; Sakaguchi, K.; Wingfield, P.T.; Rubin, J.S. Heparin binding and oligomerization of hepatocyte growth factor/scatter factor isoforms. Heparan sulfate glycosaminoglycan requirement for met binding and signaling. J. Biol. Chem. 1997, 272, 9457–9463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, K.L.; Hunter, L.A.; Ethier, S.P.; Boerner, J.L. Met and c-src cooperate to compensate for loss of epidermal growth factor receptor kinase activity in breast cancer cells. Cancer Res. 2008, 68, 3314–3322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shattuck, D.L.; Miller, J.K.; Carraway, K.L., 3rd; Sweeney, C. Met receptor contributes to trastuzumab resistance of her2-overexpressing breast cancer cells. Cancer Res. 2008, 68, 1471–1477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. Met amplification leads to gefitinib resistance in lung cancer by activating erbb3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Zerillo, C.; Kolmakova, J.; Christensen, J.G.; Harris, L.N.; Rimm, D.L.; Digiovanna, M.P.; Stern, D.F. Association of constitutively activated hepatocyte growth factor receptor (met) with resistance to a dual egfr/her2 inhibitor in non-small-cell lung cancer cells. Br. J. Cancer 2009, 100, 941–949. [Google Scholar] [CrossRef]
- Liska, D.; Chen, C.T.; Bachleitner-Hofmann, T.; Christensen, J.G.; Weiser, M.R. Hgf rescues colorectal cancer cells from egfr inhibition via met activation. Clin. Cancer Res. 2011, 17, 472–482. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.T.; Kim, H.; Liska, D.; Gao, S.; Christensen, J.G.; Weiser, M.R. Met activation mediates resistance to lapatinib inhibition of her2-amplified gastric cancer cells. Mol. Cancer Ther. 2012, 11, 660–669. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, J. Combining targeted drugs to stop resistant tumors. Science 2011, 331, 1542–1545. [Google Scholar] [CrossRef]
- Ferguson, F.M.; Gray, N.S. Kinase inhibitors: The road ahead. Nat. Rev. Drug Discov. 2018, 17, 353–377. [Google Scholar] [CrossRef]
- Lopez Sambrooks, C.; Baro, M.; Quijano, A.; Narayan, A.; Cui, W.; Greninger, P.; Egan, R.; Patel, A.; Benes, C.H.; Saltzman, W.M.; et al. Oligosaccharyltransferase inhibition overcomes therapeutic resistance to egfr tyrosine kinase inhibitors. Cancer Res. 2018, 78, 5094–5106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative pcr and the 2(-delta delta c(t)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Bauman, E.; Feijao, T.; Carvalho, D.T.O.; Granja, P.L.; Barrias, C.C. Xeno-free pre-vascularized spheroids for therapeutic applications. Sci. Rep. 2018, 8, 230. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balmaña, M.; Diniz, F.; Feijão, T.; Barrias, C.C.; Mereiter, S.; Reis, C.A. Analysis of the Effect of Increased α2,3-Sialylation on RTK Activation in MKN45 Gastric Cancer Spheroids Treated with Crizotinib. Int. J. Mol. Sci. 2020, 21, 722. https://doi.org/10.3390/ijms21030722
Balmaña M, Diniz F, Feijão T, Barrias CC, Mereiter S, Reis CA. Analysis of the Effect of Increased α2,3-Sialylation on RTK Activation in MKN45 Gastric Cancer Spheroids Treated with Crizotinib. International Journal of Molecular Sciences. 2020; 21(3):722. https://doi.org/10.3390/ijms21030722
Chicago/Turabian StyleBalmaña, Meritxell, Francisca Diniz, Tália Feijão, Cristina C. Barrias, Stefan Mereiter, and Celso A. Reis. 2020. "Analysis of the Effect of Increased α2,3-Sialylation on RTK Activation in MKN45 Gastric Cancer Spheroids Treated with Crizotinib" International Journal of Molecular Sciences 21, no. 3: 722. https://doi.org/10.3390/ijms21030722