Identification of Novel Pathways Associated with Patterned Cerebellar Purkinje Neuron Degeneration in Niemann-Pick Disease, Type C1

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Immunohistochemistry and RNA-Sequencing
2.2. Differential Gene Expression
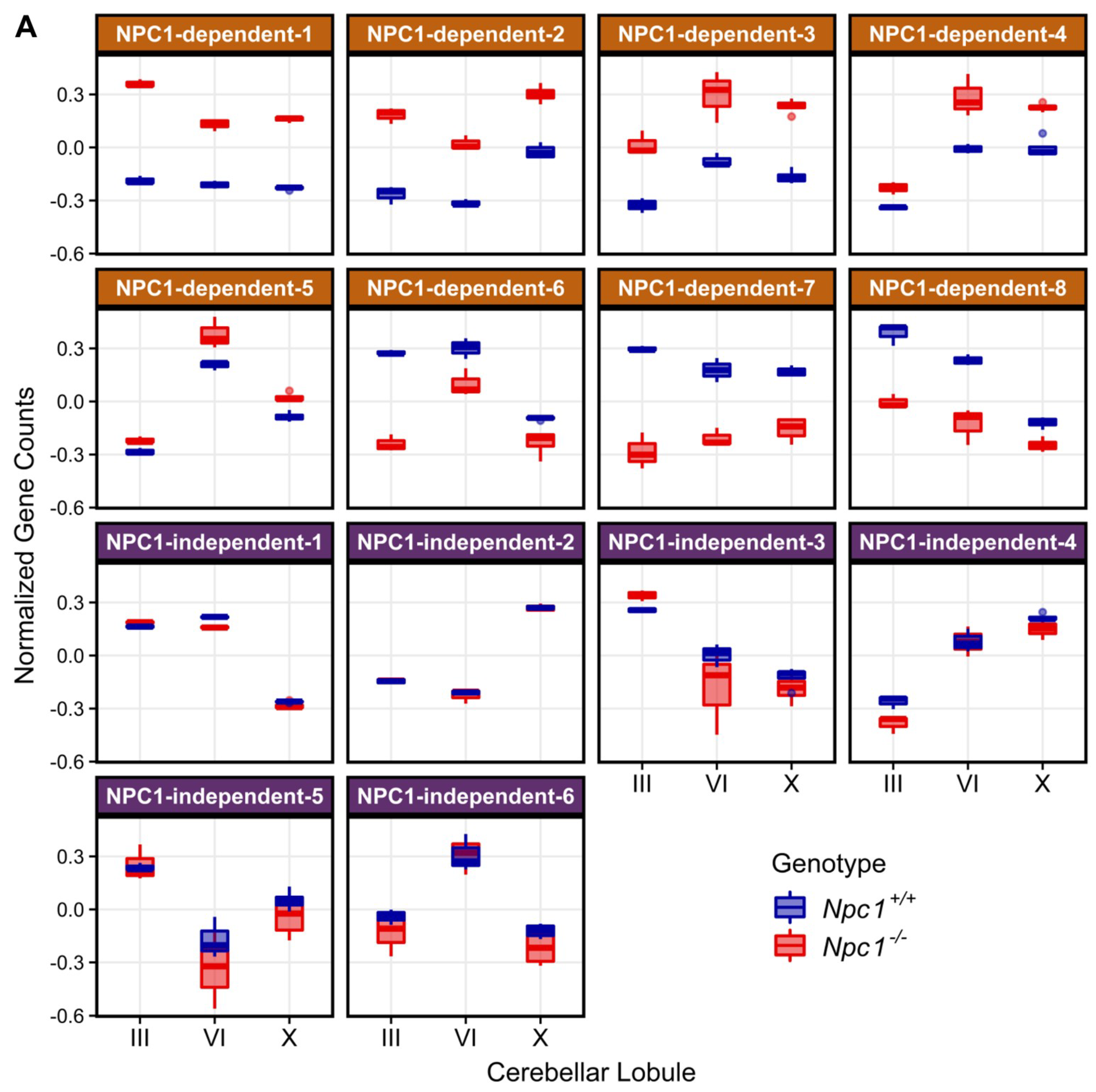
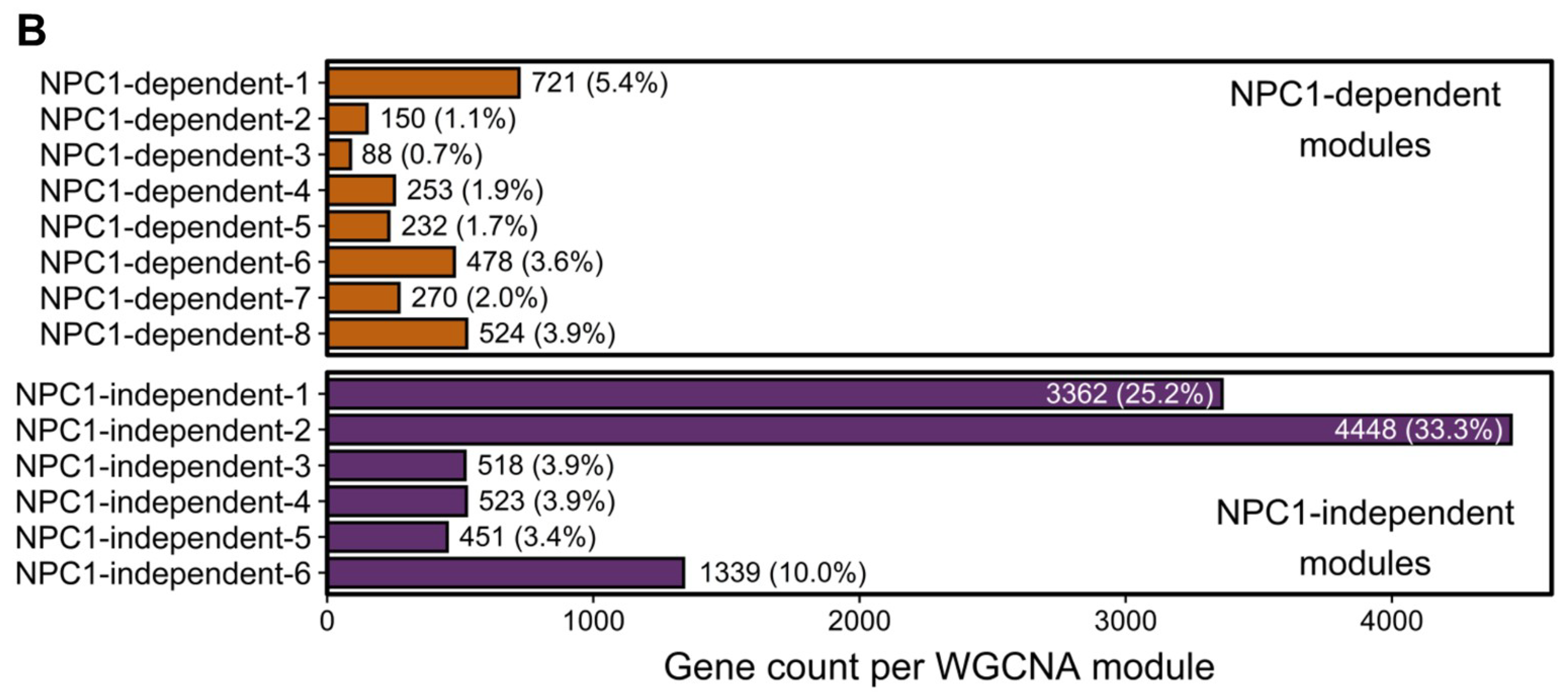
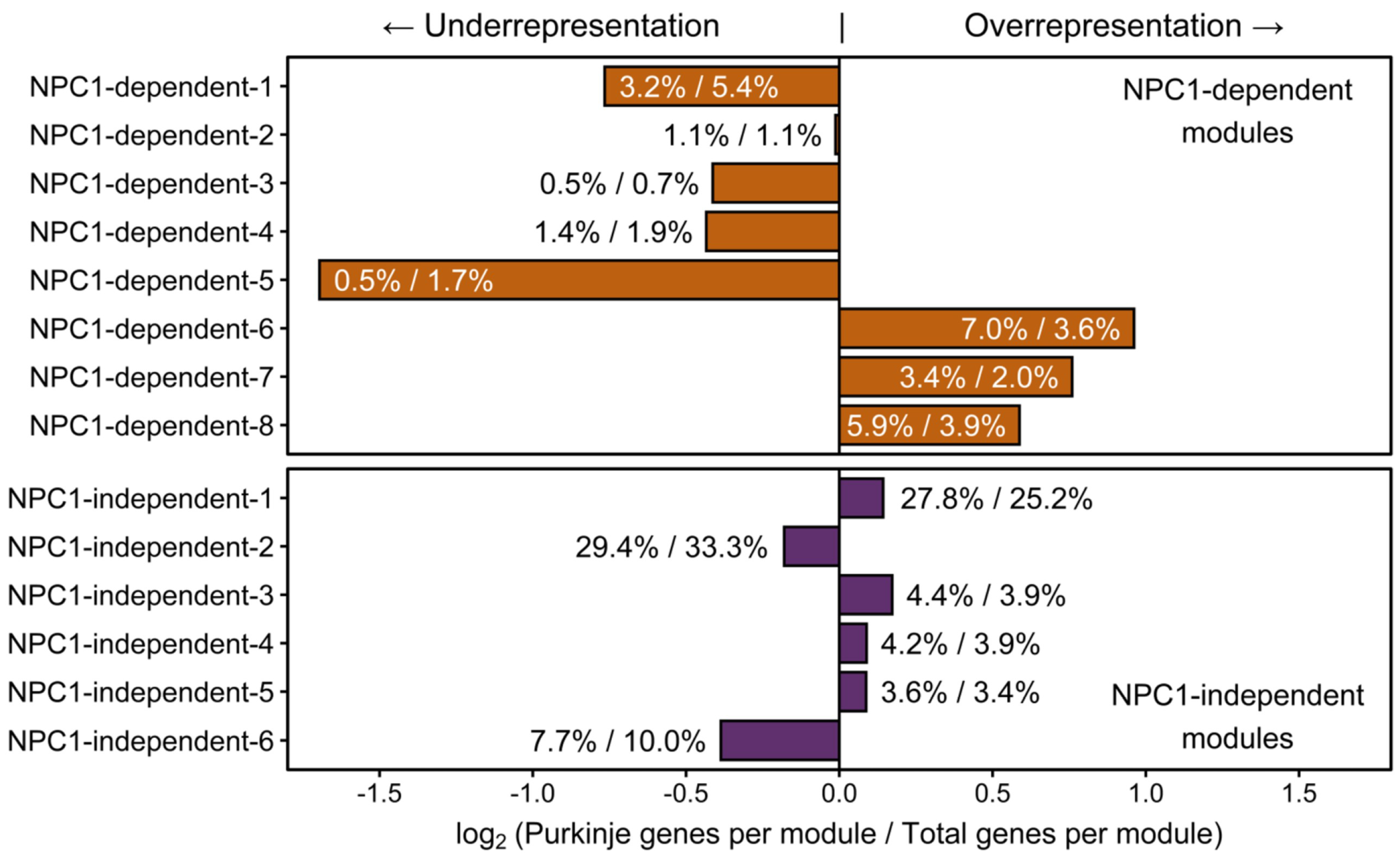
2.3. Weighted Gene Co-expression Network Analysis
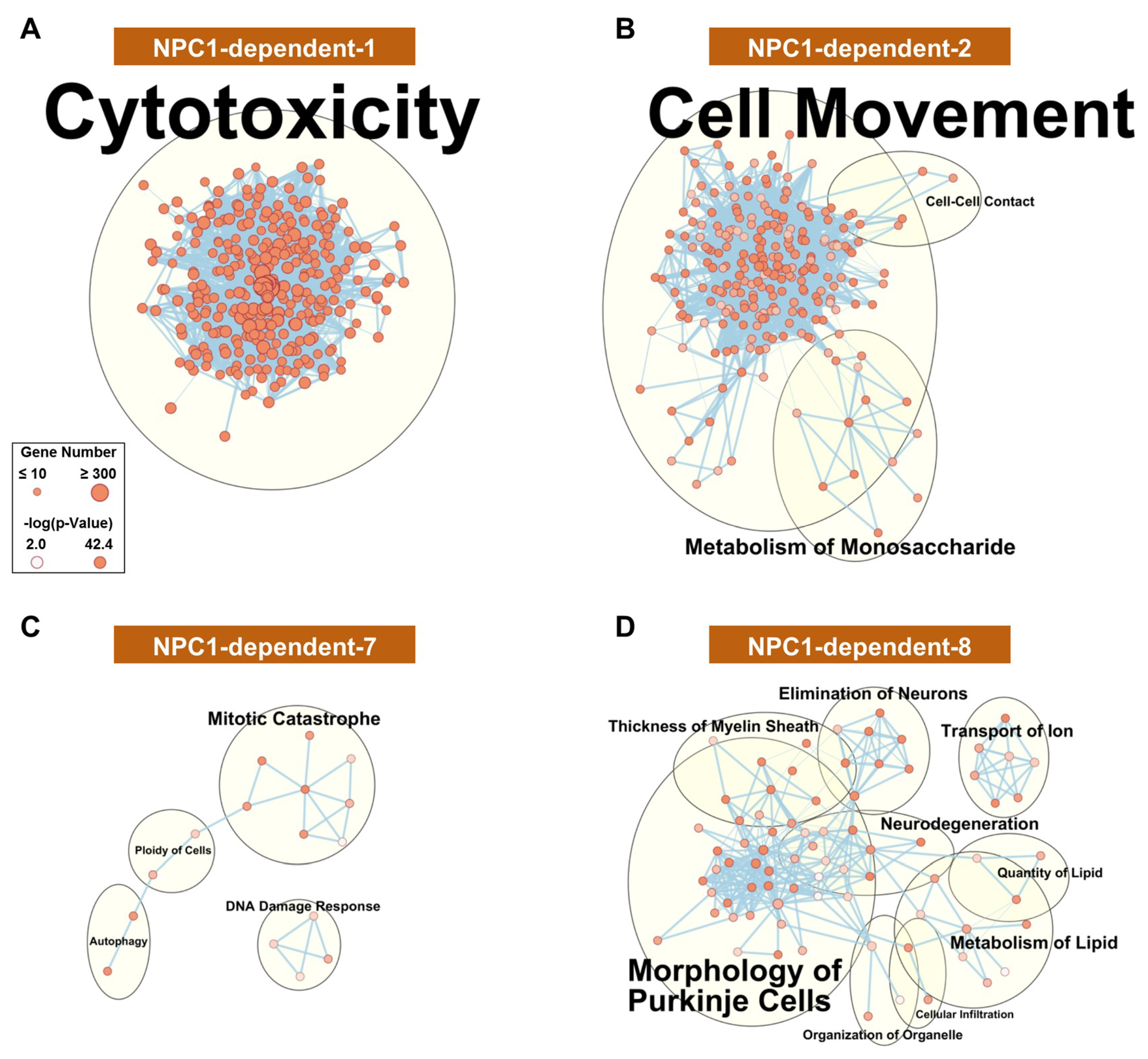
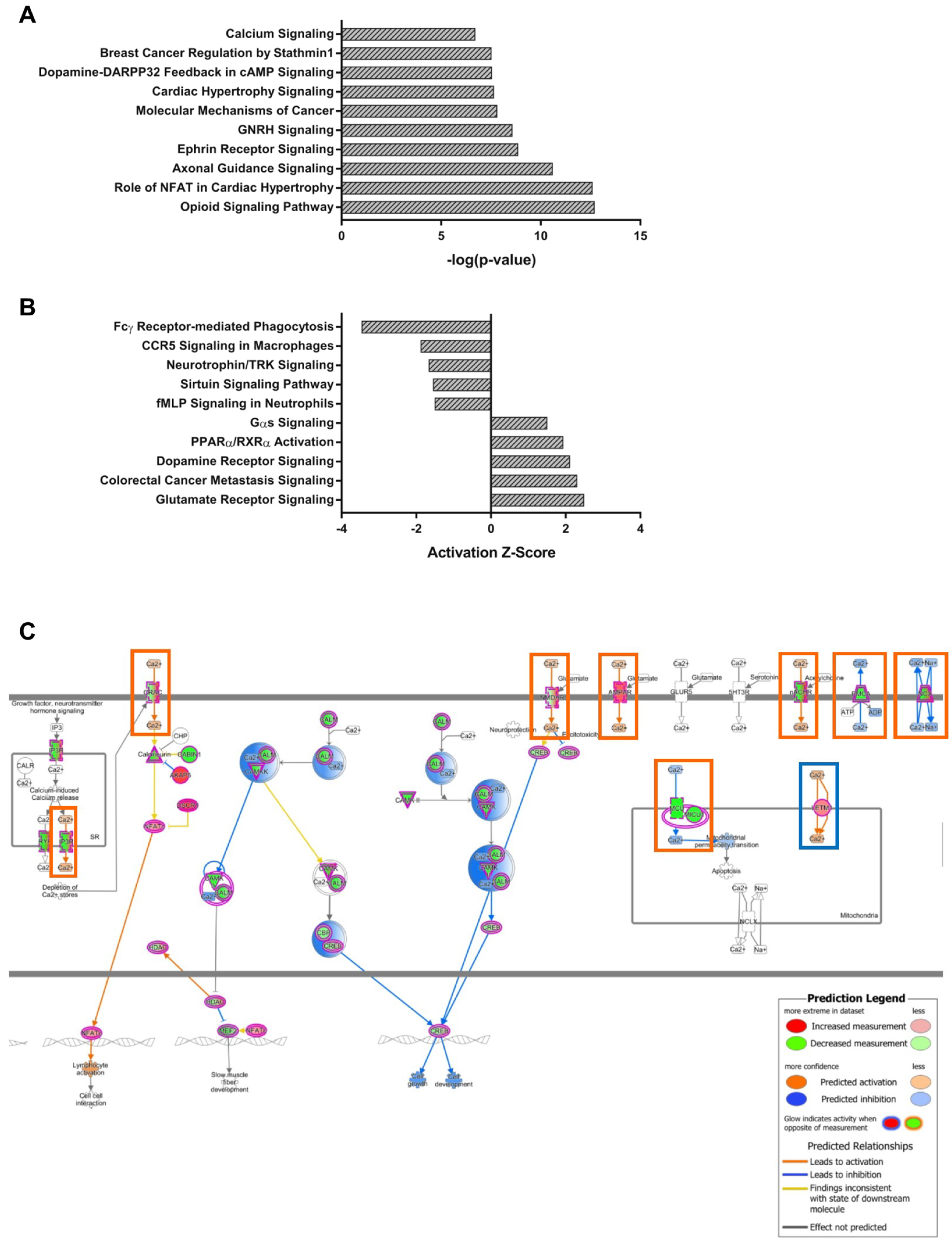
2.3.1. NPC1-dependent Modules
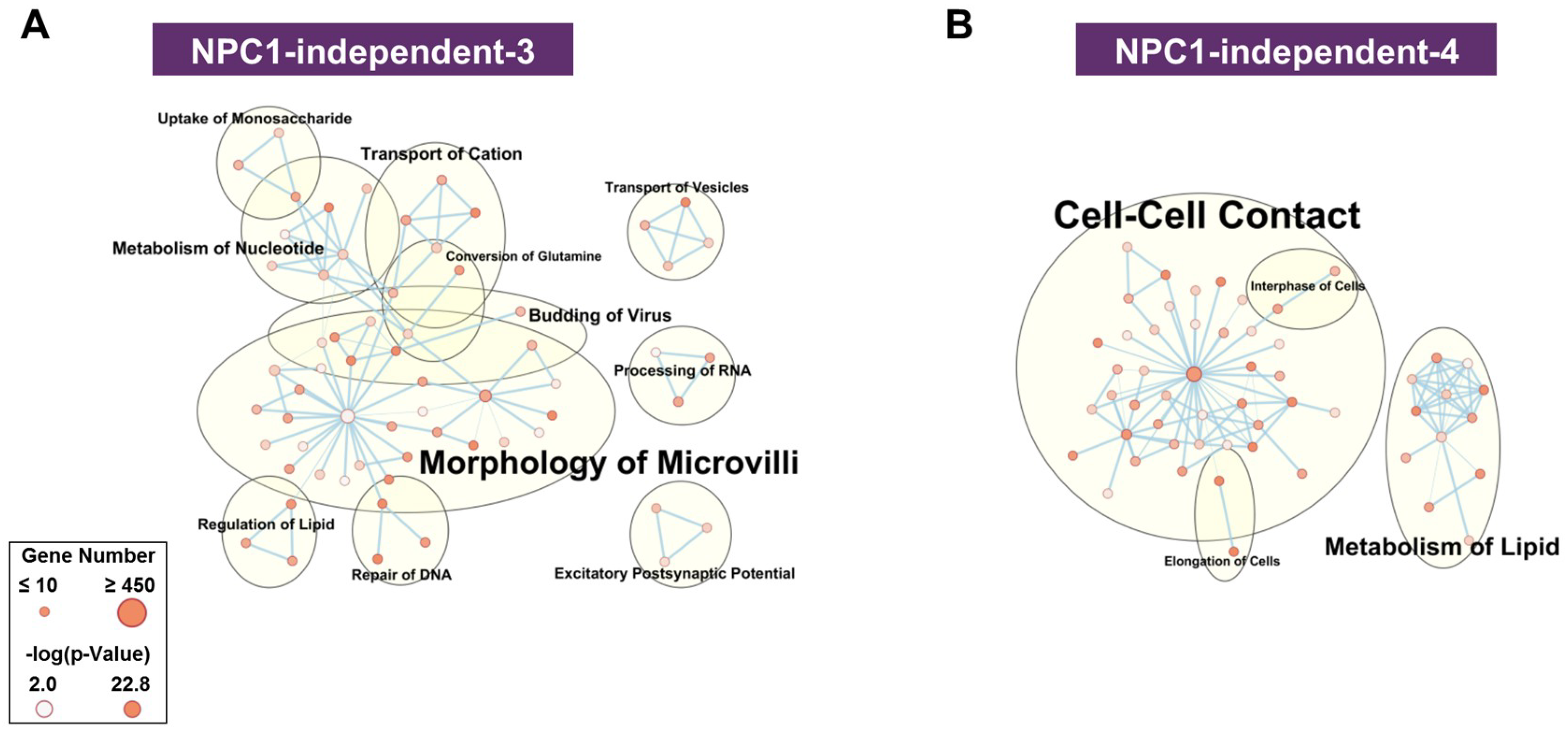
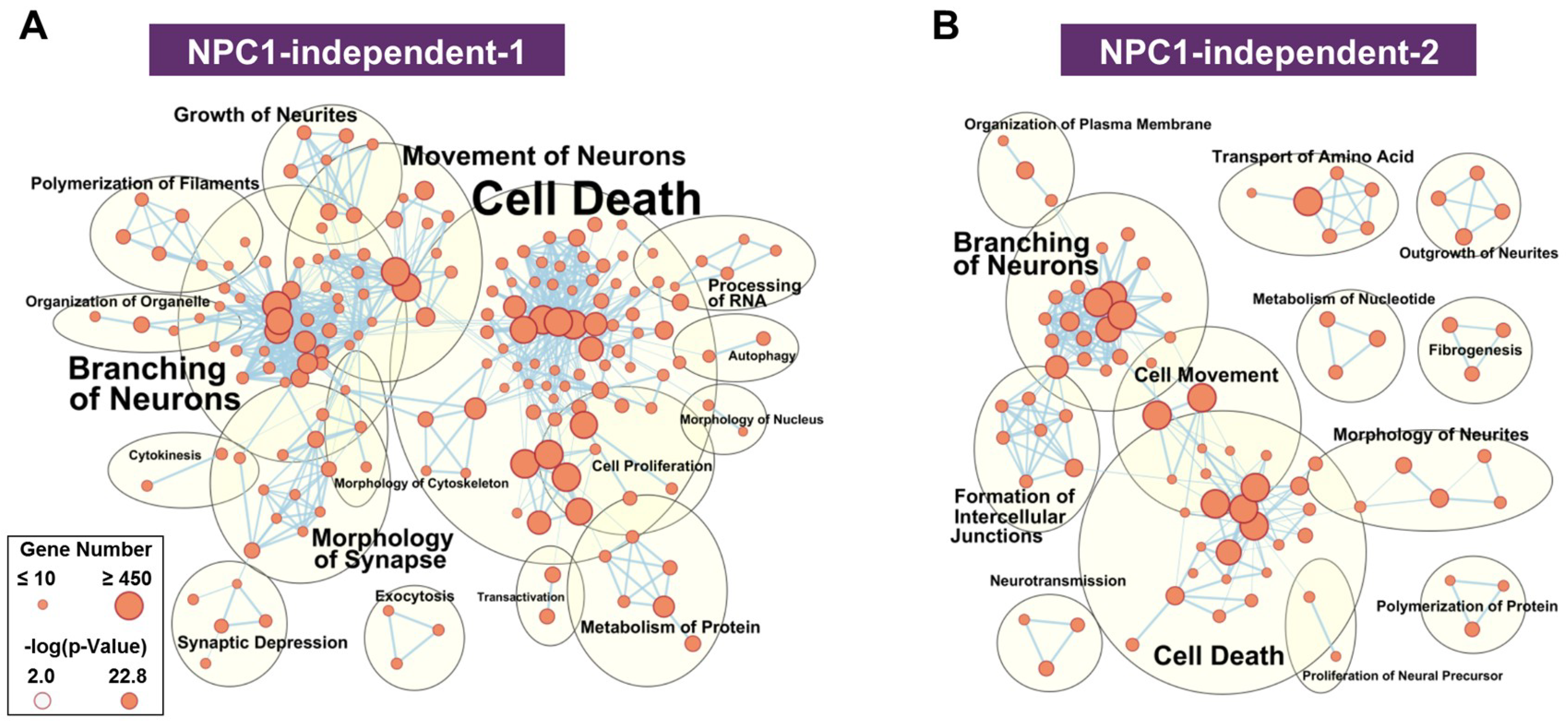
2.3.2. NPC1-independent Expression Modules
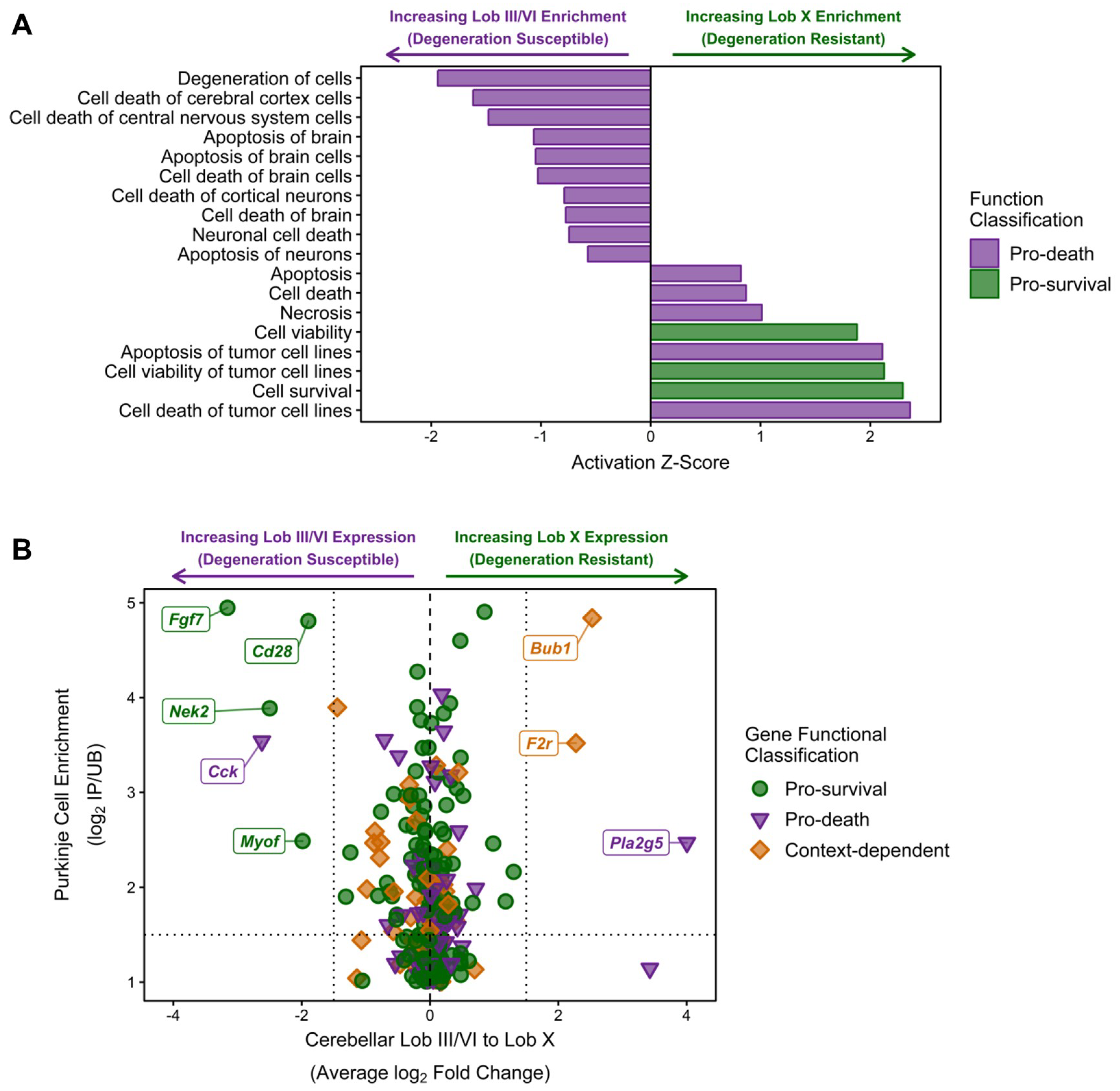
2.4. Analysis of Purkinje Neuron Specific Transcripts
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. RNA-Sequencing
4.3. Gene Expression Analyses
4.4. Weighted Gene Co-expression Network Analysis
4.5. Pathway Analysis
4.6. Immunohistochemistry
4.7. β-secretase Assay
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| NPC | Niemann-Pick disease, type C |
| NPC1 | Niemann-Pick disease, type C1 |
| WGCNA | Weighted gene co-expression network analysis |
| RNA-seq | RNA-sequencing |
| PCA | Principal component analysis |
| IPA | Ingenuity Pathway Analysis |
| Aβ | Amyloid-β |
| SHH | Sonic Hedgehog |
| CCK | Cholecystokinin |
| ISH | In situ hybridization |
| PBS | Phosphate buffered saline |
References
- Patterson, M.C.; Mengel, E.; Wijburg, F.A.; Muller, A.; Schwierin, B.; Drevon, H.; Vanier, M.T.; Pineda, M. Disease and patient characteristics in NP-C patients: Findings from an international disease registry. Orphanet J. Rare Dis. 2013, 8, 12. [Google Scholar] [CrossRef]
- Vanier, M.T. Niemann-Pick disease type C. Orphanet J. Rare Dis. 2010, 5, 16. [Google Scholar] [CrossRef] [Green Version]
- Naureckiene, S.; Sleat, D.E.; Lackland, H.; Fensom, A.; Vanier, M.T.; Wattiaux, R.; Jadot, M.; Lobel, P. Identification of HE1 as the second gene of Niemann-Pick C disease. Science 2000, 290, 2298–2301. [Google Scholar] [CrossRef]
- Walkley, S.U.; Suzuki, K. Consequences of NPC1 and NPC2 loss of function in mammalian neurons. Biochim. Biophys. Acta 2004, 1685, 48–62. [Google Scholar] [CrossRef]
- Lloyd-Evans, E.; Morgan, A.J.; He, X.; Smith, D.A.; Elliot-Smith, E.; Sillence, D.J.; Churchill, G.C.; Schuchman, E.H.; Galione, A.; Platt, F.M. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med. 2008, 14, 1247–1255. [Google Scholar] [CrossRef]
- Sevin, M.; Lesca, G.; Baumann, N.; Millat, G.; Lyon-Caen, O.; Vanier, M.T.; Sedel, F. The adult form of Niemann-Pick disease type C. Brain A J. Neurol. 2007, 130, 120–133. [Google Scholar] [CrossRef] [Green Version]
- Apps, R.; Hawkes, R. Cerebellar cortical organization: A one-map hypothesis. Nat. Rev. Neurosci. 2009, 10, 670–681. [Google Scholar] [CrossRef]
- Brochu, G.; Maler, L.; Hawkes, R. Zebrin II: A polypeptide antigen expressed selectively by Purkinje cells reveals compartments in rat and fish cerebellum. J. Comp. Neurol. 1990, 291, 538–552. [Google Scholar] [CrossRef] [PubMed]
- Sillitoe, R.V.; Hawkes, R. Whole-mount immunohistochemistry: A high-throughput screen for patterning defects in the mouse cerebellum. J. Histochem. Cytochem. Off. J. Histochem. Soc. 2002, 50, 235–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, A.H.; Dziennis, S.; Hawkes, R.; Herrup, K. The cloning of zebrin II reveals its identity with aldolase C. Development 1994, 120, 2081–2090. [Google Scholar] [PubMed]
- Armstrong, C.L.; Hawkes, R. Pattern formation in the cerebellar cortex. Biochem. Cell Biol. 2000, 78, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Ozol, K.; Hayden, J.M.; Oberdick, J.; Hawkes, R. Transverse zones in the vermis of the mouse cerebellum. J. Comp. Neurol. 1999, 412, 95–111. [Google Scholar] [CrossRef]
- Sarna, J.R.; Hawkes, R. Patterned Purkinje cell death in the cerebellum. Prog. Neurobiol. 2003, 70, 473–507. [Google Scholar] [CrossRef]
- Wang, T.; Morgan, J.I. The Purkinje cell degeneration (pcd) mouse: An unexpected molecular link between neuronal degeneration and regeneration. Brain Res. 2007, 1140, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, C.F.; Lutz, C.M.; O’Sullivan, T.N.; Shaughnessy, J.D., Jr.; Hawkes, R.; Frankel, W.N.; Copeland, N.G.; Jenkins, N.A. Absence epilepsy in tottering mutant mice is associated with calcium channel defects. Cell 1996, 87, 607–617. [Google Scholar] [CrossRef] [Green Version]
- Duffin, C.A.; McFarland, R.; Sarna, J.R.; Vogel, M.W.; Armstrong, C.L. Heat shock protein 25 expression and preferential Purkinje cell survival in the lurcher mutant mouse cerebellum. J. Comp. Neurol. 2010, 518, 1892–1907. [Google Scholar] [CrossRef] [PubMed]
- O’Hearn, E.; Molliver, M.E. Degeneration of Purkinje cells in parasagittal zones of the cerebellar vermis after treatment with ibogaine or harmaline. Neuroscience 1993, 55, 303–310. [Google Scholar] [CrossRef]
- Winkelman, M.D.; Hines, J.D. Cerebellar degeneration caused by high-dose cytosine arabinoside: A clinicopathological study. Ann. Neurol. 1983, 14, 520–527. [Google Scholar] [CrossRef]
- Ciesielski, K.T.; Yanofsky, R.; Ludwig, R.N.; Hill, D.E.; Hart, B.L.; Astur, R.S.; Snyder, T. Hypoplasia of the cerebellar vermis and cognitive deficits in survivors of childhood leukemia. Arch. Neurol. 1994, 51, 985–993. [Google Scholar] [CrossRef]
- Torvik, A.; Torp, S. The prevalence of alcoholic cerebellar atrophy. A morphometric and histological study of an autopsy material. J. Neurol. Sci. 1986, 75, 43–51. [Google Scholar] [CrossRef]
- Yokota, O.; Tsuchiya, K.; Terada, S.; Oshima, K.; Ishizu, H.; Matsushita, M.; Kuroda, S.; Akiyama, H. Alcoholic cerebellar degeneration: A clinicopathological study of six Japanese autopsy cases and proposed potential progression pattern in the cerebellar lesion. Neuropathol. Off. J. Jpn. Soc. Neuropathol. 2007, 27, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I.; Fabregues, I.; Pineda, M.; Gracia, I.; Ribalta, T. A Golgi study of cerebellar atrophy in human chronic alcoholism. Neuropathol. Appl. Neurobiol. 1984, 10, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Robitaille, Y.; Schut, L.; Kish, S.J. Structural and immunocytochemical features of olivopontocerebellar atrophy caused by the spinocerebellar ataxia type 1 (SCA-1) mutation define a unique phenotype. Acta Neuropathol. 1995, 90, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, C.F.; Tottene, A.; Lennon, V.A.; Wilson, S.M.; Dubel, S.J.; Paylor, R.; Hosford, D.A.; Tessarollo, L.; McEnery, M.W.; Pietrobon, D.; et al. Dystonia and cerebellar atrophy in Cacna1a null mice lacking P/Q calcium channel activity. FASEB J. 2001, 15, 1288–1290. [Google Scholar] [CrossRef] [PubMed]
- Gilman, S. The spinocerebellar ataxias. Clin. Neuropharmacol. 2000, 23, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Iwata, M.; Hirano, A.; French, J.H. Degeneration of the cerebellar system in X-chromosome-linked copper malabsorption. Ann. Neurol. 1979, 5, 542–549. [Google Scholar] [CrossRef]
- Aynaci, F.M.; Mocan, H.; Bahadir, S.; Sari, A.; Aksoy, A. A case of Menkes’ syndrome associated with deafness and inferior cerebellar vermian hypoplasia. Acta Paediatr. 1997, 86, 121–123. [Google Scholar] [CrossRef]
- Sarna, J.; Miranda, S.R.; Schuchman, E.H.; Hawkes, R. Patterned cerebellar Purkinje cell death in a transgenic mouse model of Niemann Pick type A/B disease. Eur. J. Neurosci. 2001, 13, 1873–1880. [Google Scholar] [CrossRef]
- Higashi, Y.; Murayama, S.; Pentchev, P.G.; Suzuki, K. Cerebellar degeneration in the Niemann-Pick type C mouse. Acta Neuropathol. 1993, 85, 175–184. [Google Scholar] [CrossRef]
- Tanaka, J.; Nakamura, H.; Miyawaki, S. Cerebellar involvement in murine sphingomyelinosis: A new model of Niemann-Pick disease. J. Neuropathol. Exp. Neurol. 1988, 47, 291–300. [Google Scholar] [CrossRef]
- Sarna, J.R.; Larouche, M.; Marzban, H.; Sillitoe, R.V.; Rancourt, D.E.; Hawkes, R. Patterned Purkinje cell degeneration in mouse models of Niemann-Pick type C disease. J. Comp. Neurol. 2003, 456, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Baudry, M.; Yao, Y.; Simmons, D.; Liu, J.; Bi, X. Postnatal development of inflammation in a murine model of Niemann-Pick type C disease: Immunohistochemical observations of microglia and astroglia. Exp. Neurol. 2003, 184, 887–903. [Google Scholar] [CrossRef]
- Williams, I.M.; Wallom, K.L.; Smith, D.A.; Al Eisa, N.; Smith, C.; Platt, F.M. Improved neuroprotection using miglustat, curcumin and ibuprofen as a triple combination therapy in Niemann-Pick disease type C1 mice. Neurobiol. Dis. 2014, 67, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Cluzeau, C.V.; Watkins-Chow, D.E.; Fu, R.; Borate, B.; Yanjanin, N.; Dail, M.K.; Davidson, C.D.; Walkley, S.U.; Ory, D.S.; Wassif, C.A.; et al. Microarray expression analysis and identification of serum biomarkers for Niemann-Pick disease, type C1. Hum. Mol. Genet. 2012, 21, 3632–3646. [Google Scholar] [CrossRef] [Green Version]
- Liao, G.; Wen, Z.; Irizarry, K.; Huang, Y.; Mitsouras, K.; Darmani, M.; Leon, T.; Shi, L.; Bi, X. Abnormal gene expression in cerebellum of Npc1−/− mice during postnatal development. Brain Res. 2010, 1325, 128–140. [Google Scholar] [CrossRef] [Green Version]
- Alam, M.S.; Getz, M.; Safeukui, I.; Yi, S.; Tamez, P.; Shin, J.; Velazquez, P.; Haldar, K. Genomic expression analyses reveal lysosomal, innate immunity proteins, as disease correlates in murine models of a lysosomal storage disorder. PLoS ONE 2012, 7, e48273. [Google Scholar] [CrossRef]
- Cougnoux, A.; Drummond, R.A.; Collar, A.L.; Iben, J.R.; Salman, A.; Westgarth, H.; Wassif, C.A.; Cawley, N.X.; Farhat, N.Y.; Ozato, K.; et al. Microglia activation in Niemann-Pick disease, type C1 is amendable to therapeutic intervention. Hum. Mol. Genet. 2018, 27, 2076–2089. [Google Scholar] [CrossRef] [Green Version]
- Mancarci, B.O.; Toker, L.; Tripathy, S.J.; Li, B.; Rocco, B.; Sibille, E.; Pavlidis, P. Cross-Laboratory Analysis of Brain Cell Type Transcriptomes with Applications to Interpretation of Bulk Tissue Data. eNeuro 2017, 4. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Antalffy, B.; Kang, D.; Orr, H.T.; Zoghbi, H.Y. Polyglutamine expansion down-regulates specific neuronal genes before pathologic changes in SCA1. Nat. Neurosci. 2000, 3, 157–163. [Google Scholar] [CrossRef]
- Knollmann, B.C.; Chopra, N.; Hlaing, T.; Akin, B.; Yang, T.; Ettensohn, K.; Knollmann, B.E.; Horton, K.D.; Weissman, N.J.; Holinstat, I.; et al. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J. Clin. Investig. 2006, 116, 2510–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neidert, N.; von Ehr, A.; Zoller, T.; Spittau, B. Microglia-Specific Expression of Olfml3 Is Directly Regulated by Transforming Growth Factor beta1-Induced Smad2 Signaling. Front. Immunol. 2018, 9, 1728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majer, A.; Medina, S.J.; Sorensen, D.; Martin, M.J.; Frost, K.L.; Phillipson, C.; Manguiat, K.; Booth, S.A. The cell type resolved mouse transcriptome in neuron-enriched brain tissues from the hippocampus and cerebellum during prion disease. Sci. Rep. 2019, 9, 1099. [Google Scholar] [CrossRef] [Green Version]
- Mathys, H.; Adaikkan, C.; Gao, F.; Young, J.Z.; Manet, E.; Hemberg, M.; De Jager, P.L.; Ransohoff, R.M.; Regev, A.; Tsai, L.H. Temporal Tracking of Microglia Activation in Neurodegeneration at Single-Cell Resolution. Cell Rep. 2017, 21, 366–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- German, D.C.; Liang, C.L.; Song, T.; Yazdani, U.; Xie, C.; Dietschy, J.M. Neurodegeneration in the Niemann-Pick C mouse: Glial involvement. Neuroscience 2002, 109, 437–450. [Google Scholar] [CrossRef]
- Hess, E.J.; Wilson, M.C. Tottering and leaner mutations perturb transient developmental expression of tyrosine hydroxylase in embryologically distinct Purkinje cells. Neuron 1991, 6, 123–132. [Google Scholar] [CrossRef]
- Sawada, K.; Komatsu, S.; Haga, H.; Sun, X.Z.; Hisano, S.; Fukui, Y. Abnormal expression of tyrosine hydroxylase immunoreactivity in cerebellar cortex of ataxic mutant mice. Brain Res. 1999, 829, 107–112. [Google Scholar] [CrossRef]
- Jeong, Y.G.; Kim, M.K.; Hawkes, R. Ectopic expression of tyrosine hydroxylase in Zebrin II immunoreactive Purkinje cells in the cerebellum of the ataxic mutant mouse, pogo. Dev. Brain Res. 2001, 129, 201–209. [Google Scholar] [CrossRef]
- Yadid, G.; Sotnik-Barkai, I.; Tornatore, C.; Baker-Cairns, B.; Harvey-White, J.; Pentchev, P.G.; Goldin, E. Neurochemical alterations in the cerebellum of a murine model of Niemann-Pick type C disease. Brain Res. 1998, 799, 250–256. [Google Scholar] [CrossRef]
- Abbott, L.C.; Isaacs, K.R.; Heckroth, J.A. Co-localization of tyrosine hydroxylase and zebrin II immunoreactivities in Purkinje cells of the mutant mice, tottering and tottering/leaner. Neuroscience 1996, 71, 461–475. [Google Scholar] [CrossRef]
- Sawada, K.; Sakata-Haga, H.; Fukui, Y. Alternating array of tyrosine hydroxylase and heat shock protein 25 immunopositive Purkinje cell stripes in zebrin II-defined transverse zone of the cerebellum of rolling mouse Nagoya. Brain Res. 2010, 1343, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Kilbourne, E.J.; Nankova, B.B.; Lewis, E.J.; McMahon, A.; Osaka, H.; Sabban, D.B.; Sabban, E.L. Regulated expression of the tyrosine hydroxylase gene by membrane depolarization. Identification of the responsive element and possible second messengers. J. Biol. Chem. 1992, 267, 7563–7569. [Google Scholar] [PubMed]
- Nankova, B.; Hiremagalur, B.; Menezes, A.; Zeman, R.; Sabban, E. Promoter elements and second messenger pathways involved in transcriptional activation of tyrosine hydroxylase by ionomycin. Brain Res. Mol. Brain Res. 1996, 35, 164–172. [Google Scholar] [CrossRef]
- Nagamoto-Combs, K.; Piech, K.M.; Best, J.A.; Sun, B.; Tank, A.W. Tyrosine hydroxylase gene promoter activity is regulated by both cyclic AMP-responsive element and AP1 sites following calcium influx. Evidence for cyclic amp-responsive element binding protein-independent regulation. J. Biol. Chem. 1997, 272, 6051–6058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.H.; Brkanac, Z.; Verlinde, C.L.; Tan, X.J.; Bylenok, L.; Nochlin, D.; Matsushita, M.; Lipe, H.; Wolff, J.; Fernandez, M.; et al. Missense mutations in the regulatory domain of PKC gamma: A new mechanism for dominant nonepisodic cerebellar ataxia. Am. J. Hum. Genet. 2003, 72, 839–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chopra, R.; Wasserman, A.H.; Pulst, S.M.; De Zeeuw, C.I.; Shakkottai, V.G. Protein kinase C activity is a protective modifier of Purkinje neuron degeneration in cerebellar ataxia. Hum. Mol. Genet. 2018, 27, 1396–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, N.M.; Shekhar, K.; Whitney, I.E.; Jacobi, A.; Benhar, I.; Hong, G.; Yan, W.; Adiconis, X.; Arnold, M.E.; Lee, J.M.; et al. Single-Cell Profiles of Retinal Ganglion Cells Differing in Resilience to Injury Reveal Neuroprotective Genes. Neuron 2019, 104, 1039–1055.e1012. [Google Scholar] [CrossRef]
- Boisvert, M.M.; Erikson, G.A.; Shokhirev, M.N.; Allen, N.J. The Aging Astrocyte Transcriptome from Multiple Regions of the Mouse Brain. Cell Rep. 2018, 22, 269–285. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.; Lau, S.K.; Doering, L.C. Astrocyte-secreted thrombospondin-1 modulates synapse and spine defects in the fragile X mouse model. Mol. Brain 2016, 9, 74. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Boil. 2005, 4. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. Fast R Functions for Robust Correlations and Hierarchical Clustering. J. Stat. Softw. 2012, 46, 46. [Google Scholar] [CrossRef] [Green Version]
- Lopez, M.E.; Klein, A.D.; Scott, M.P. Complement is dispensable for neurodegeneration in Niemann-Pick disease type C. J. Neuroinflamm. 2012, 9, 216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulinski, A.; Vance, J.E. Lipid homeostasis and lipoprotein secretion in Niemann-Pick C1-deficient hepatocytes. J. Biol. Chem. 2007, 282, 1627–1637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uronen, R.L.; Lundmark, P.; Orho-Melander, M.; Jauhiainen, M.; Larsson, K.; Siegbahn, A.; Wallentin, L.; Zethelius, B.; Melander, O.; Syvanen, A.C.; et al. Niemann-Pick C1 modulates hepatic triglyceride metabolism and its genetic variation contributes to serum triglyceride levels. Arter. Thromb. Vasc. Biol. 2010, 30, 1614–1620. [Google Scholar] [CrossRef]
- Mattsson, N.; Zetterberg, H.; Bianconi, S.; Yanjanin, N.M.; Fu, R.; Mansson, J.E.; Porter, F.D.; Blennow, K. Gamma-secretase-dependent amyloid-beta is increased in Niemann-Pick type C: A cross-sectional study. Neurology 2011, 76, 366–372. [Google Scholar] [CrossRef] [Green Version]
- Kodam, A.; Maulik, M.; Peake, K.; Amritraj, A.; Vetrivel, K.S.; Thinakaran, G.; Vance, J.E.; Kar, S. Altered levels and distribution of amyloid precursor protein and its processing enzymes in Niemann-Pick type C1-deficient mouse brains. Glia 2010, 58, 1267–1281. [Google Scholar] [CrossRef] [Green Version]
- Lewis, P.M.; Gritli-Linde, A.; Smeyne, R.; Kottmann, A.; McMahon, A.P. Sonic hedgehog signaling is required for expansion of granule neuron precursors and patterning of the mouse cerebellum. Dev. Biol. 2004, 270, 393–410. [Google Scholar] [CrossRef] [Green Version]
- Canterini, S.; Dragotto, J.; Dardis, A.; Zampieri, S.; De Stefano, M.E.; Mangia, F.; Erickson, R.P.; Fiorenza, M.T. Shortened primary cilium length and dysregulated Sonic hedgehog signaling in Niemann-Pick C1 disease. Hum. Mol. Genet. 2017, 26, 2277–2289. [Google Scholar] [CrossRef]
- Nusca, S.; Canterini, S.; Palladino, G.; Bruno, F.; Mangia, F.; Erickson, R.P.; Fiorenza, M.T. A marked paucity of granule cells in the developing cerebellum of the Npc1(−/−) mouse is corrected by a single injection of hydroxypropyl-beta-cyclodextrin. Neurobiol. Dis. 2014, 70, 117–126. [Google Scholar] [CrossRef] [Green Version]
- D’Angelo, E.; Mazzarello, P.; Prestori, F.; Mapelli, J.; Solinas, S.; Lombardo, P.; Cesana, E.; Gandolfi, D.; Congi, L. The cerebellar network: From structure to function and dynamics. Brain Res. Rev. 2011, 66, 5–15. [Google Scholar] [CrossRef]
- Efthymiou, A.G.; Steiner, J.; Pavan, W.J.; Wincovitch, S.; Larson, D.M.; Porter, F.D.; Rao, M.S.; Malik, N. Rescue of an in vitro neuron phenotype identified in Niemann-Pick disease, type C1 induced pluripotent stem cell-derived neurons by modulating the WNT pathway and calcium signaling. Stem Cells Transl. Med. 2015, 4, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Hurley, M.J.; Mash, D.C.; Jenner, P. Markers for dopaminergic neurotransmission in the cerebellum in normal individuals and patients with Parkinson’s disease examined by RT-PCR. Eur. J. Neurosci. 2003, 18, 2668–2672. [Google Scholar] [CrossRef] [PubMed]
- Takayasu, Y.; Iino, M.; Takatsuru, Y.; Tanaka, K.; Ozawa, S. Functions of glutamate transporters in cerebellar Purkinje cell synapses. Acta Physiol. 2009, 197, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, F.A.; Lehre, K.P.; van Lookeren Campagne, M.; Ottersen, O.P.; Danbolt, N.C.; Storm-Mathisen, J. Glutamate transporters in glial plasma membranes: Highly differentiated localizations revealed by quantitative ultrastructural immunocytochemistry. Neuron 1995, 15, 711–720. [Google Scholar] [CrossRef] [Green Version]
- Lehre, K.P.; Danbolt, N.C. The number of glutamate transporter subtype molecules at glutamatergic synapses: Chemical and stereological quantification in young adult rat brain. J. Neurosci. Off. J. Soc. Neurosci. 1998, 18, 8751–8757. [Google Scholar] [CrossRef] [Green Version]
- Dehnes, Y.; Chaudhry, F.A.; Ullensvang, K.; Lehre, K.P.; Storm-Mathisen, J.; Danbolt, N.C. The glutamate transporter EAAT4 in rat cerebellar Purkinje cells: A glutamate-gated chloride channel concentrated near the synapse in parts of the dendritic membrane facing astroglia. J. Neurosci. Off. J. Soc. Neurosci. 1998, 18, 3606–3619. [Google Scholar] [CrossRef] [Green Version]
- Elrick, M.J.; Pacheco, C.D.; Yu, T.; Dadgar, N.; Shakkottai, V.G.; Ware, C.; Paulson, H.L.; Lieberman, A.P. Conditional Niemann-Pick C mice demonstrate cell autonomous Purkinje cell neurodegeneration. Hum. Mol. Genet. 2010, 19, 837–847. [Google Scholar] [CrossRef] [Green Version]
- Ko, D.C.; Milenkovic, L.; Beier, S.M.; Manuel, H.; Buchanan, J.; Scott, M.P. Cell-autonomous death of cerebellar purkinje neurons with autophagy in Niemann-Pick type C disease. PLoS Genet. 2005, 1, 81–95. [Google Scholar] [CrossRef]
- Doyle, J.P.; Dougherty, J.D.; Heiman, M.; Schmidt, E.F.; Stevens, T.R.; Ma, G.; Bupp, S.; Shrestha, P.; Shah, R.D.; Doughty, M.L.; et al. Application of a translational profiling approach for the comparative analysis of CNS cell types. Cell 2008, 135, 749–762. [Google Scholar] [CrossRef] [Green Version]
- Gukovskaya, A.S.; Gukovsky, I.; Jung, Y.; Mouria, M.; Pandol, S.J. Cholecystokinin induces caspase activation and mitochondrial dysfunction in pancreatic acinar cells. Roles in cell injury processes of pancreatitis. J. Boil. Chem. 2002, 277, 22595–22604. [Google Scholar] [CrossRef] [Green Version]
- Niederau, C.; Luthen, R.; Heintges, T. Effects of CCK on pancreatic function and morphology. Ann. N. Y. Acad. Sci. 1994, 713, 180–198. [Google Scholar] [CrossRef] [PubMed]
- Dockray, G.J.; Gregory, R.A.; Hutchison, J.B.; Harris, J.I.; Runswick, M.J. Isolation, structure and biological activity of two cholecystokinin octapeptides from sheep brain. Nature 1978, 274, 711–713. [Google Scholar] [CrossRef] [PubMed]
- Emson, P.C.; Lee, C.M.; Rehfeld, J.F. Cholecystokinin octapeptide: Vesicular localization and calcium dependent release from rat brain in vitro. Life Sci. 1980, 26, 2157–2163. [Google Scholar] [CrossRef]
- Lee, S.Y.; Soltesz, I. Cholecystokinin: A multi-functional molecular switch of neuronal circuits. Dev. Neurobiol. 2011, 71, 83–91. [Google Scholar] [CrossRef] [Green Version]
- Katona, I.; Sperlagh, B.; Sik, A.; Kafalvi, A.; Vizi, E.S.; Mackie, K.; Freund, T.F. Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. J. Neurosci. Off. J. Soc. Neurosci. 1999, 19, 4544–4558. [Google Scholar] [CrossRef]
- Nyiri, G.; Cserep, C.; Szabadits, E.; Mackie, K.; Freund, T.F. CB1 cannabinoid receptors are enriched in the perisynaptic annulus and on preterminal segments of hippocampal GABAergic axons. Neuroscience 2005, 136, 811–822. [Google Scholar] [CrossRef]
- Migaud, M.; Roques, B.P.; Durieux, C. Effects of cholecystokinin octapeptide and BC 264, a potent and selective CCK-B agonist on aspartate and glutamate release from rat hippocampal slices. Neuropharmacology 1994, 33, 737–743. [Google Scholar] [CrossRef]
- Breukel, A.I.; Lopes da Silva, F.H.; Ghijsen, W.E. Cholecystokinin (CCK-8) modulates vesicular release of excitatory amino acids in rat hippocampal nerve endings. Neurosci. Lett. 1997, 234, 67–70. [Google Scholar] [CrossRef]
- Chung, C.; Elrick, M.J.; Dell’Orco, J.M.; Qin, Z.S.; Kalyana-Sundaram, S.; Chinnaiyan, A.M.; Shakkottai, V.G.; Lieberman, A.P. Heat Shock Protein Beta-1 Modifies Anterior to Posterior Purkinje Cell Vulnerability in a Mouse Model of Niemann-Pick Type C Disease. PLoS Genet. 2016, 12, e1006042. [Google Scholar] [CrossRef]
- Lein, E.S.; Hawrylycz, M.J.; Ao, N.; Ayres, M.; Bensinger, A.; Bernard, A.; Boe, A.F.; Boguski, M.S.; Brockway, K.S.; Byrnes, E.J.; et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature 2007, 445, 168–176. [Google Scholar] [CrossRef]
- Nagao, S.; Kwak, S.; Kanazawa, I. EAAT4, a glutamate transporter with properties of a chloride channel, is predominantly localized in Purkinje cell dendrites, and forms parasagittal compartments in rat cerebellum. Neuroscience 1997, 78, 929–933. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, R.; Sakata, S.; Naito, A.; Hirashima, N.; Tanaka, M. Dendritic differentiation of cerebellar Purkinje cells is promoted by ryanodine receptors expressed by Purkinje and granule cells. Dev. Neurobiol. 2014, 74, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Kwan, K.M.; Mailloux, C.M.; Lee, W.K.; Grinberg, A.; Wurst, W.; Behringer, R.R.; Westphal, H. LIM-homeodomain proteins Lhx1 and Lhx5, and their cofactor Ldb1, control Purkinje cell differentiation in the developing cerebellum. Proc. Natl. Acad. Sci. USA 2007, 104, 13182–13186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cologna, S.M.; Cluzeau, C.V.; Yanjanin, N.M.; Blank, P.S.; Dail, M.K.; Siebel, S.; Toth, C.L.; Wassif, C.A.; Lieberman, A.P.; Porter, F.D. Human and mouse neuroinflammation markers in Niemann-Pick disease, type C1. J. Inherit. Metab. Dis. 2014, 37, 83–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, M.E.; Klein, A.D.; Hong, J.; Dimbil, U.J.; Scott, M.P. Neuronal and epithelial cell rescue resolves chronic systemic inflammation in the lipid storage disorder Niemann-Pick C. Hum. Mol. Genet. 2012, 21, 2946–2960. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Sugimoto, Y.; Ohsaki, Y.; Ueno, M.; Kato, S.; Kitamura, Y.; Hosokawa, H.; Davies, J.P.; Ioannou, Y.A.; Vanier, M.T.; et al. Endosomal accumulation of Toll-like receptor 4 causes constitutive secretion of cytokines and activation of signal transducers and activators of transcription in Niemann-Pick disease type C (NPC) fibroblasts: A potential basis for glial cell activation in the NPC brain. J. Neurosci. Off. J. Soc. Neurosci. 2007, 27, 1879–1891. [Google Scholar] [CrossRef] [Green Version]
- Loftus, S.K.; Morris, J.A.; Carstea, E.D.; Gu, J.Z.; Cummings, C.; Brown, A.; Ellison, J.; Ohno, K.; Rosenfeld, M.A.; Tagle, D.A.; et al. Murine model of Niemann-Pick C disease: Mutation in a cholesterol homeostasis gene. Science 1997, 277, 232–235. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Zhu, A.; Ibrahim, J.G.; Love, M.I. Heavy-tailed prior distributions for sequence count data: Removing the noise and preserving large differences. Bioinformatics 2019, 35, 2084–2092. [Google Scholar] [CrossRef]
- Leek, J.T. svaseq: Removing batch effects and other unwanted noise from sequencing data. Nucleic Acids Res. 2014, 42. [Google Scholar] [CrossRef]
- Merico, D.; Isserlin, R.; Stueker, O.; Emili, A.; Bader, G.D. Enrichment map: A network-based method for gene-set enrichment visualization and interpretation. PLoS ONE 2010, 5, e13984. [Google Scholar] [CrossRef] [PubMed]
- Kucera, M.; Isserlin, R.; Arkhangorodsky, A.; Bader, G.D. AutoAnnotate: A Cytoscape app for summarizing networks with semantic annotations. F1000Research 2016, 5, 1717. [Google Scholar] [CrossRef] [PubMed]
- Williams, I.M.; Carletti, B.; Leto, K.; Magrassi, L.; Rossi, F. Cerebellar granule cells transplanted in vivo can follow physiological and unusual migratory routes to integrate into the recipient cortex. Neurobiol. Dis. 2008, 30, 139–149. [Google Scholar] [CrossRef] [PubMed]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martin, K.B.; Williams, I.M.; Cluzeau, C.V.; Cougnoux, A.; Dale, R.K.; Iben, J.R.; Cawley, N.X.; Wassif, C.A.; Porter, F.D. Identification of Novel Pathways Associated with Patterned Cerebellar Purkinje Neuron Degeneration in Niemann-Pick Disease, Type C1. Int. J. Mol. Sci. 2020, 21, 292. https://doi.org/10.3390/ijms21010292
Martin KB, Williams IM, Cluzeau CV, Cougnoux A, Dale RK, Iben JR, Cawley NX, Wassif CA, Porter FD. Identification of Novel Pathways Associated with Patterned Cerebellar Purkinje Neuron Degeneration in Niemann-Pick Disease, Type C1. International Journal of Molecular Sciences. 2020; 21(1):292. https://doi.org/10.3390/ijms21010292
Chicago/Turabian StyleMartin, Kyle B., Ian M. Williams, Celine V. Cluzeau, Antony Cougnoux, Ryan K. Dale, James R. Iben, Niamh X. Cawley, Christopher A. Wassif, and Forbes D. Porter. 2020. "Identification of Novel Pathways Associated with Patterned Cerebellar Purkinje Neuron Degeneration in Niemann-Pick Disease, Type C1" International Journal of Molecular Sciences 21, no. 1: 292. https://doi.org/10.3390/ijms21010292