Calcium as a Key Player in Arrhythmogenic Cardiomyopathy: Adhesion Disorder or Intracellular Alteration?

 , ,
, ,
Abstract
:1. Introduction
2. Calcium Signaling and Cardiac Arrhythmias: A Tight Connection
3. Arrhythmogenic Cardiomyopathy
4. Arrhythmogenic Cardiomyopathy as Adhesion Disorder: Ca2+-Dependent Desmosomes’ Stability
5. Desmosomal Mutations and Arrhythmogenic Cardiomyopathy: Derangement of the Ca2+ Toolkit as a Consequence Rather than a Pathogenic Defect
PKP2
6. Non-Desmosomal Mutations and Altered Ca2+ Handling
6.1. RYR2
6.2. PLN
7. Conclusions
Funding
Conflicts of Interest
References
- Thiene, G.; Nava, A.; Corrado, D.; Rossi, L.; Pennelli, N. Right ventricular cardiomyopathy and sudden death in young people. N. Engl. J. Med. 1988, 318, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Thiene, G.; Basso, C. Arrhythmogenic right ventricular cardiomyopathy: An update. Cardiovasc. Pathol. 2001, 10, 109–117. [Google Scholar] [CrossRef]
- Corrado, D.; Basso, C.; Judge, D.P. Arrhythmogenic Cardiomyopathy. Circ. Res. 2017, 121, 784–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartoli, F.; Sabourin, J. Cardiac Remodeling and Disease: Current Understanding of STIM1/Orai1-Mediated Store-Operated Ca2+ Entry in Cardiac Function and Pathology. Adv. Exp. Med. Biol. 2017, 993, 523–534. [Google Scholar] [PubMed]
- Xie, W.; Santulli, G.; Guo, X.; Gao, M.; Chen, B.X.; Marks, A.R. Imaging atrial arrhythmic intracellular calcium in intact heart. J. Mol. Cell. Cardiol. 2013, 64, 120–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landstrom, A.P.; Dobrev, D.; Wehrens, X.H.T. Calcium Signaling and Cardiac Arrhythmias. Circ. Res. 2017, 120, 1969–1993. [Google Scholar] [CrossRef] [PubMed]
- Ter Keurs, H.E.; Boyden, P.A. Calcium and arrhythmogenesis. Physiol. Rev. 2007, 87, 457–506. [Google Scholar] [CrossRef] [PubMed]
- Venetucci, L.; Denegri, M.; Napolitano, C.; Priori, S.G. Inherited calcium channelopathies in the pathophysiology of arrhythmias. Nat. Rev. Cardiol. 2012, 9, 561–575. [Google Scholar] [CrossRef] [PubMed]
- Eisner, D.A.; Caldwell, J.L.; Kistamas, K.; Trafford, A.W. Calcium and Excitation-Contraction Coupling in the Heart. Circ. Res. 2017, 121, 181–195. [Google Scholar] [CrossRef]
- KC, S.; Nair, R.S.; Banerji, A.; Somasundaram, V.; Srinivas, P. Structure activity relationship of plumbagin in BRCA1 related cancer cells. Mol. Carcinog. 2013, 52, 392–403. [Google Scholar]
- Vangheluwe, P.; Sipido, K.R.; Raeymaekers, L.; Wuytack, F. New perspectives on the role of SERCA2’s Ca2+ affinity in cardiac function. Biochim. Et Biophys. Acta 2006, 1763, 1216–1228. [Google Scholar] [CrossRef] [PubMed]
- van Opbergen, C.J.; Delmar, M.; van Veen, T.A. Potential new mechanisms of pro-arrhythmia in arrhythmogenic cardiomyopathy: Focus on calcium sensitive pathways. Neth. Heart J. Mon. J. Neth. Soc. Cardiol. Neth. Heart Found. 2017, 25, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Bovo, E.; Huke, S.; Blatter, L.A.; Zima, A.V. The effect of PKA-mediated phosphorylation of ryanodine receptor on SR Ca2+ leak in ventricular myocytes. J. Mol. Cell. Cardiol. 2017, 104, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, S.; Terentyev, D. Altered Intracellular Calcium Homeostasis and Arrhythmogenesis in the Aged Heart. Int. J. Mol. Sci. 2019, 20, 2386. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Chen, S.R. Inherited dysfunction of sarcoplasmic reticulum Ca2+ handling and arrhythmogenesis. Circ. Res. 2011, 108, 871–883. [Google Scholar] [CrossRef] [PubMed]
- Rizzi, N.; Liu, N.; Napolitano, C.; Nori, A.; Turcato, F.; Colombi, B.; Bicciato, S.; Arcelli, D.; Spedito, A.; Scelsi, M.; et al. Unexpected structural and functional consequences of the R33Q homozygous mutation in cardiac calsequestrin: A complex arrhythmogenic cascade in a knock in mouse model. Circ. Res. 2008, 103, 298–306. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente, S.; Van Langen, I.M.; Postma, A.V.; Bikker, H.; Meijer, A. A case of catecholaminergic polymorphic ventricular tachycardia caused by two calsequestrin 2 mutations. Pacing Clin. Electrophysiol. 2008, 31, 916–919. [Google Scholar] [CrossRef]
- Faggioni, M.; Knollmann, B.C. Calsequestrin 2 and arrhythmias. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1250–H1260. [Google Scholar] [CrossRef] [Green Version]
- Wemhoner, K.; Friedrich, C.; Stallmeyer, B.; Coffey, A.J.; Grace, A.; Zumhagen, S.; Seebohm, G.; Ortiz-Bonnin, B.; Rinne, S.; Sachse, F.B.; et al. Gain-of-function mutations in the calcium channel CACNA1C (Cav1.2) cause non-syndromic long-QT but not Timothy syndrome. J. Mol. Cell. Cardiol. 2015, 80, 186–195. [Google Scholar] [CrossRef]
- Burashnikov, E.; Pfeiffer, R.; Barajas-Martinez, H.; Delpon, E.; Hu, D.; Desai, M.; Borggrefe, M.; Haissaguerre, M.; Kanter, R.; Pollevick, G.D.; et al. Mutations in the cardiac L-type calcium channel associated with inherited J-wave syndromes and sudden cardiac death. Heart Rhythm 2010, 7, 1872–1882. [Google Scholar] [CrossRef] [Green Version]
- Templin, C.; Ghadri, J.R.; Rougier, J.S.; Baumer, A.; Kaplan, V.; Albesa, M.; Sticht, H.; Rauch, A.; Puleo, C.; Hu, D.; et al. Identification of a novel loss-of-function calcium channel gene mutation in short QT syndrome (SQTS6). Eur. Heart J. 2011, 32, 1077–1088. [Google Scholar] [CrossRef] [PubMed]
- Devalla, H.D.; Gelinas, R.; Aburawi, E.H.; Beqqali, A.; Goyette, P.; Freund, C.; Chaix, M.A.; Tadros, R.; Jiang, H.; Le Bechec, A.; et al. TECRL, a new life-threatening inherited arrhythmia gene associated with overlapping clinical features of both LQTS and CPVT. Embo Mol. Med. 2016, 8, 1390–1408. [Google Scholar] [CrossRef] [PubMed]
- Nyegaard, M.; Overgaard, M.T.; Sondergaard, M.T.; Vranas, M.; Behr, E.R.; Hildebrandt, L.L.; Lund, J.; Hedley, P.L.; Camm, A.J.; Wettrell, G.; et al. Mutations in calmodulin cause ventricular tachycardia and sudden cardiac death. Am. J. Hum. Genet. 2012, 91, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Chopra, N.; Yang, T.; Asghari, P.; Moore, E.D.; Huke, S.; Akin, B.; Cattolica, R.A.; Perez, C.F.; Hlaing, T.; Knollmann-Ritschel, B.E.; et al. Ablation of triadin causes loss of cardiac Ca2+ release units, impaired excitation-contraction coupling, and cardiac arrhythmias. Proc. Natl. Acad. Sci. USA 2009, 106, 7636–7641. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Basso, C.; Thiene, G.; McKenna, W.J.; Davies, M.J.; Fontaliran, F.; Nava, A.; Silvestri, F.; Blomstrom-Lundqvist, C.; Wlodarska, E.K.; et al. Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: A multicenter study. J. Am. Coll. Cardiol. 1997, 30, 1512–1520. [Google Scholar] [CrossRef]
- Dalal, D.; Nasir, K.; Bomma, C.; Prakasa, K.; Tandri, H.; Piccini, J.; Roguin, A.; Tichnell, C.; James, C.; Russell, S.D.; et al. Arrhythmogenic right ventricular dysplasia: A United States experience. Circulation 2005, 112, 3823–3832. [Google Scholar] [CrossRef]
- Marcus, F.I.; Fontaine, G.H.; Guiraudon, G.; Frank, R.; Laurenceau, J.L.; Malergue, C.; Grosgogeat, Y. Right ventricular dysplasia: A report of 24 adult cases. Circulation 1982, 65, 384–398. [Google Scholar] [CrossRef]
- Cheedipudi, S.M.; Hu, J.; Fan, S.; Yuan, P.; Karmouch, J.; Czernuszewicz, G.; Robertson, M.J.; Coarfa, C.; Hong, K.; Yao, Y.; et al. Exercise Restores Dysregulated Gene Expression in a Mouse Model of Arrhythmogenic Cardiomyopathy. Cardiovasc. Res. 2019. [Google Scholar] [CrossRef]
- Agullo-Pascual, E.; Cerrone, M.; Delmar, M. Arrhythmogenic cardiomyopathy and Brugada syndrome: Diseases of the connexome. Febs Lett. 2014, 588, 1322–1330. [Google Scholar] [CrossRef]
- Marcus, F.I.; McKenna, W.J.; Sherrill, D.; Basso, C.; Bauce, B.; Bluemke, D.A.; Calkins, H.; Corrado, D.; Cox, M.G.; Daubert, J.P.; et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: Proposed modification of the Task Force Criteria. Eur. Heart J. 2010, 31, 806–814. [Google Scholar] [CrossRef]
- Dalal, D.; Tandri, H.; Judge, D.P.; Amat, N.; Macedo, R.; Jain, R.; Tichnell, C.; Daly, A.; James, C.; Russell, S.D.; et al. Morphologic variants of familial arrhythmogenic right ventricular dysplasia/cardiomyopathy a genetics-magnetic resonance imaging correlation study. J. Am. Coll. Cardiol. 2009, 53, 1289–1299. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Link, M.S.; Calkins, H. Arrhythmogenic Right Ventricular Cardiomyopathy. N. Engl. J. Med. 2017, 376, 1489–1490. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ortega, M.; Rodriguez-Vita, J.; Sanchez-Lopez, E.; Carvajal, G.; Egido, J. TGF-beta signaling in vascular fibrosis. Cardiovasc. Res. 2007, 74, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Akdis, D.; Brunckhorst, C.; Duru, F.; Saguner, A.M. Arrhythmogenic Cardiomyopathy: Electrical and Structural Phenotypes. Arrhythmia Electrophysiol. Rev. 2016, 5, 90–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asimaki, A.; Syrris, P.; Wichter, T.; Matthias, P.; Saffitz, J.E.; McKenna, W.J. A novel dominant mutation in plakoglobin causes arrhythmogenic right ventricular cardiomyopathy. Am. J. Hum. Genet. 2007, 81, 964–973. [Google Scholar] [CrossRef] [PubMed]
- Rampazzo, A.; Nava, A.; Malacrida, S.; Beffagna, G.; Bauce, B.; Rossi, V.; Zimbello, R.; Simionati, B.; Basso, C.; Thiene, G.; et al. Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am. J. Hum. Genet. 2002, 71, 1200–1206. [Google Scholar] [CrossRef] [PubMed]
- Gerull, B.; Heuser, A.; Wichter, T.; Paul, M.; Basson, C.T.; McDermott, D.A.; Lerman, B.B.; Markowitz, S.M.; Ellinor, P.T.; MacRae, C.A.; et al. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat. Genet. 2004, 36, 1162–1164. [Google Scholar] [CrossRef]
- Awad, M.M.; Dalal, D.; Cho, E.; Amat-Alarcon, N.; James, C.; Tichnell, C.; Tucker, A.; Russell, S.D.; Bluemke, D.A.; Dietz, H.C.; et al. DSG2 mutations contribute to arrhythmogenic right ventricular dysplasia/cardiomyopathy. Am. J. Hum. Genet. 2006, 79, 136–142. [Google Scholar] [CrossRef]
- Pilichou, K.; Nava, A.; Basso, C.; Beffagna, G.; Bauce, B.; Lorenzon, A.; Frigo, G.; Vettori, A.; Valente, M.; Towbin, J.; et al. Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation 2006, 113, 1171–1179. [Google Scholar] [CrossRef]
- Syrris, P.; Ward, D.; Evans, A.; Asimaki, A.; Gandjbakhch, E.; Sen-Chowdhry, S.; McKenna, W.J. Arrhythmogenic right ventricular dysplasia/cardiomyopathy associated with mutations in the desmosomal gene desmocollin-2. Am. J. Hum. Genet. 2006, 79, 978–984. [Google Scholar] [CrossRef]
- Merner, N.D.; Hodgkinson, K.A.; Haywood, A.F.; Connors, S.; French, V.M.; Drenckhahn, J.D.; Kupprion, C.; Ramadanova, K.; Thierfelder, L.; McKenna, W.; et al. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am. J. Hum. Genet. 2008, 82, 809–821. [Google Scholar] [CrossRef] [PubMed]
- Quarta, G.; Syrris, P.; Ashworth, M.; Jenkins, S.; Zuborne Alapi, K.; Morgan, J.; Muir, A.; Pantazis, A.; McKenna, W.J.; Elliott, P.M. Mutations in the Lamin A/C gene mimic arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2012, 33, 1128–1136. [Google Scholar] [CrossRef] [PubMed]
- Klauke, B.; Kossmann, S.; Gaertner, A.; Brand, K.; Stork, I.; Brodehl, A.; Dieding, M.; Walhorn, V.; Anselmetti, D.; Gerdes, D.; et al. De novo desmin-mutation N116S is associated with arrhythmogenic right ventricular cardiomyopathy. Hum. Mol. Genet. 2010, 19, 4595–4607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Hengel, J.; Calore, M.; Bauce, B.; Dazzo, E.; Mazzotti, E.; De Bortoli, M.; Lorenzon, A.; Li Mura, I.E.; Beffagna, G.; Rigato, I.; et al. Mutations in the area composita protein alphaT-catenin are associated with arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2013, 34, 201–210. [Google Scholar] [CrossRef] [PubMed]
- van der Zwaag, P.A.; van Rijsingen, I.A.; Asimaki, A.; Jongbloed, J.D.; van Veldhuisen, D.J.; Wiesfeld, A.C.; Cox, M.G.; van Lochem, L.T.; de Boer, R.A.; Hofstra, R.M.; et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: Evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur. J. Heart Fail. 2012, 14, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Rampazzo, A.; Nava, A.; Danieli, G.A.; Buja, G.; Daliento, L.; Fasoli, G.; Scognamiglio, R.; Corrado, D.; Thiene, G. The gene for arrhythmogenic right ventricular cardiomyopathy maps to chromosome 14q23-q24. Hum. Mol. Genet. 1994, 3, 959–962. [Google Scholar] [CrossRef]
- Taylor, M.; Graw, S.; Sinagra, G.; Barnes, C.; Slavov, D.; Brun, F.; Pinamonti, B.; Salcedo, E.E.; Sauer, W.; Pyxaras, S.; et al. Genetic variation in titin in arrhythmogenic right ventricular cardiomyopathy-overlap syndromes. Circulation 2011, 124, 876–885. [Google Scholar] [CrossRef] [PubMed]
- Erkapic, D.; Neumann, T.; Schmitt, J.; Sperzel, J.; Berkowitsch, A.; Kuniss, M.; Hamm, C.W.; Pitschner, H.F. Electrical storm in a patient with arrhythmogenic right ventricular cardiomyopathy and SCN5A mutation. Europace 2008, 10, 884–887. [Google Scholar] [CrossRef]
- Mayosi, B.M.; Fish, M.; Shaboodien, G.; Mastantuono, E.; Kraus, S.; Wieland, T.; Kotta, M.C.; Chin, A.; Laing, N.; Ntusi, N.B.; et al. Identification of Cadherin 2 (CDH2) Mutations in Arrhythmogenic Right Ventricular Cardiomyopathy. Circ. Cardiovasc. Genet. 2017, 10, e001605. [Google Scholar] [CrossRef]
- Rampazzo, A.; Nava, A.; Erne, P.; Eberhard, M.; Vian, E.; Slomp, P.; Tiso, N.; Thiene, G.; Danieli, G.A. A new locus for arrhythmogenic right ventricular cardiomyopathy (ARVD2) maps to chromosome 1q42-q43. Hum. Mol. Genet. 1995, 4, 2151–2154. [Google Scholar] [CrossRef] [Green Version]
- Tiso, N.; Stephan, D.A.; Nava, A.; Bagattin, A.; Devaney, J.M.; Stanchi, F.; Larderet, G.; Brahmbhatt, B.; Brown, K.; Bauce, B.; et al. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum. Mol. Genet. 2001, 10, 189–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buikema, J.W.; Mady, A.S.; Mittal, N.V.; Atmanli, A.; Caron, L.; Doevendans, P.A.; Sluijter, J.P.; Domian, I.J. Wnt/beta-catenin signaling directs the regional expansion of first and second heart field-derived ventricular cardiomyocytes. Development 2013, 140, 4165–4176. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gras, E.; Lombardi, R.; Giocondo, M.J.; Willerson, J.T.; Schneider, M.D.; Khoury, D.S.; Marian, A.J. Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J. Clin. Investig. 2006, 116, 2012–2021. [Google Scholar] [CrossRef] [PubMed]
- Sommariva, E.; Brambilla, S.; Carbucicchio, C.; Gambini, E.; Meraviglia, V.; Dello Russo, A.; Farina, F.M.; Casella, M.; Catto, V.; Pontone, G.; et al. Cardiac mesenchymal stromal cells are a source of adipocytes in arrhythmogenic cardiomyopathy. Eur. Heart J. 2016, 37, 1835–1846. [Google Scholar] [CrossRef]
- Chen, S.N.; Gurha, P.; Lombardi, R.; Ruggiero, A.; Willerson, J.T.; Marian, A.J. The hippo pathway is activated and is a causal mechanism for adipogenesis in arrhythmogenic cardiomyopathy. Circ. Res. 2014, 114, 454–468. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Leach, J.; Wang, J.; Martin, J.F. Hippo/Yap Signaling in Cardiac Development and Regeneration. Curr. Treat. Options Cardiovasc. Med. 2016, 18, 38. [Google Scholar] [CrossRef]
- Piersma, B.; Bank, R.A.; Boersema, M. Signaling in Fibrosis: TGF-beta, WNT, and YAP/TAZ Converge. Front. Med. 2015, 2, 59. [Google Scholar] [CrossRef]
- Azzolin, L.; Panciera, T.; Soligo, S.; Enzo, E.; Bicciato, S.; Dupont, S.; Bresolin, S.; Frasson, C.; Basso, G.; Guzzardo, V.; et al. YAP/TAZ incorporation in the beta-catenin destruction complex orchestrates the Wnt response. Cell 2014, 158, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Oxford, E.M.; Musa, H.; Maass, K.; Coombs, W.; Taffet, S.M.; Delmar, M. Connexin43 remodeling caused by inhibition of plakophilin-2 expression in cardiac cells. Circ. Res. 2007, 101, 703–711. [Google Scholar] [CrossRef]
- Jansen, J.A.; Noorman, M.; Musa, H.; Stein, M.; de Jong, S.; van der Nagel, R.; Hund, T.J.; Mohler, P.J.; Vos, M.A.; van Veen, T.A.; et al. Reduced heterogeneous expression of Cx43 results in decreased Nav1.5 expression and reduced sodium current that accounts for arrhythmia vulnerability in conditional Cx43 knockout mice. Heart Rhythm 2012, 9, 600–607. [Google Scholar] [CrossRef] [Green Version]
- Cerrone, M.; Delmar, M. Desmosomes and the sodium channel complex: Implications for arrhythmogenic cardiomyopathy and Brugada syndrome. Trends Cardiovasc. Med. 2014, 24, 184–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agullo-Pascual, E.; Lin, X.; Leo-Macias, A.; Zhang, M.; Liang, F.X.; Li, Z.; Pfenniger, A.; Lubkemeier, I.; Keegan, S.; Fenyo, D.; et al. Super-resolution imaging reveals that loss of the C-terminus of connexin43 limits microtubule plus-end capture and NaV1.5 localization at the intercalated disc. Cardiovasc. Res. 2014, 104, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Sato, P.Y.; Musa, H.; Coombs, W.; Guerrero-Serna, G.; Patino, G.A.; Taffet, S.M.; Isom, L.L.; Delmar, M. Loss of plakophilin-2 expression leads to decreased sodium current and slower conduction velocity in cultured cardiac myocytes. Circ. Res. 2009, 105, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Bonne, S.; Hatzfeld, M.; van Roy, F.; Green, K.J. Protein binding and functional characterization of plakophilin 2. Evidence for its diverse roles in desmosomes and beta -catenin signaling. J. Biol. Chem. 2002, 277, 10512–10522. [Google Scholar] [CrossRef] [PubMed]
- Godsel, L.M.; Dubash, A.D.; Bass-Zubek, A.E.; Amargo, E.V.; Klessner, J.L.; Hobbs, R.P.; Chen, X.; Green, K.J. Plakophilin 2 couples actomyosin remodeling to desmosomal plaque assembly via RhoA. Mol. Biol. Cell 2010, 21, 2844–2859. [Google Scholar] [CrossRef] [PubMed]
- Dorn, T.; Kornherr, J.; Parrotta, E.I.; Zawada, D.; Ayetey, H.; Santamaria, G.; Iop, L.; Mastantuono, E.; Sinnecker, D.; Goedel, A.; et al. Interplay of cell-cell contacts and RhoA/MRTF-A signaling regulates cardiomyocyte identity. EMBO J. 2018, 37. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, S.; Zhang, H.; Hu, S.; Wei, Y. RhoA activity increased in myocardium of arrhythmogenic cardiomyopathy patients and affected connexin 43 protein expression in HL-1 cells. Int. J. Clin. Exp. Med. 2015, 8, 12906–12913. [Google Scholar]
- Xu, H.F.; Ding, Y.J.; Zhang, Z.X.; Wang, Z.F.; Luo, C.L.; Li, B.X.; Shen, Y.W.; Tao, L.Y.; Zhao, Z.Q. MicroRNA21 regulation of the progression of viral myocarditis to dilated cardiomyopathy. Mol. Med. Rep. 2014, 10, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Vogel, B.; Keller, A.; Frese, K.S.; Leidinger, P.; Sedaghat-Hamedani, F.; Kayvanpour, E.; Kloos, W.; Backe, C.; Thanaraj, A.; Brefort, T.; et al. Multivariate miRNA signatures as biomarkers for non-ischaemic systolic heart failure. Eur. Heart J. 2013, 34, 2812–2822. [Google Scholar] [CrossRef] [PubMed]
- Thum, T.; Gross, C.; Fiedler, J.; Fischer, T.; Kissler, S.; Bussen, M.; Galuppo, P.; Just, S.; Rottbauer, W.; Frantz, S.; et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 2008, 456, 980–984. [Google Scholar] [CrossRef] [PubMed]
- Kawakita, A.; Yanamoto, S.; Yamada, S.; Naruse, T.; Takahashi, H.; Kawasaki, G.; Umeda, M. MicroRNA-21 promotes oral cancer invasion via the Wnt/beta-catenin pathway by targeting DKK2. Pathol. Oncol. Res. Por. 2014, 20, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.W.; Chang, Y.L.; Chang, Y.C.; Lin, J.C.; Chen, C.C.; Pan, S.H.; Wu, C.T.; Chen, H.Y.; Yang, S.C.; Hong, T.M.; et al. MicroRNA-135b promotes lung cancer metastasis by regulating multiple targets in the Hippo pathway and LZTS1. Nat. Commun. 2013, 4, 1877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skillington, J.; Choy, L.; Derynck, R. Bone morphogenetic protein and retinoic acid signaling cooperate to induce osteoblast differentiation of preadipocytes. J. Cell Biol. 2002, 159, 135–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurha, P.; Chen, X.; Lombardi, R.; Willerson, J.T.; Marian, A.J. Knockdown of Plakophilin 2 Downregulates miR-184 Through CpG Hypermethylation and Suppression of the E2F1 Pathway and Leads to Enhanced Adipogenesis In Vitro. Circ. Res. 2016, 119, 731–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rainer, J.; Meraviglia, V.; Blankenburg, H.; Piubelli, C.; Pramstaller, P.P.; Paolin, A.; Cogliati, E.; Pompilio, G.; Sommariva, E.; Domingues, F.S.; et al. The arrhythmogenic cardiomyopathy-specific coding and non-coding transcriptome in human cardiac stromal cells. BMC Genom. 2018, 19, 491. [Google Scholar] [CrossRef]
- van Rooij, E.; Sutherland, L.B.; Thatcher, J.E.; DiMaio, J.M.; Naseem, R.H.; Marshall, W.S.; Hill, J.A.; Olson, E.N. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13027–13032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurinna, S.; Schafer, M.; Ostano, P.; Karouzakis, E.; Chiorino, G.; Bloch, W.; Bachmann, A.; Gay, S.; Garrod, D.; Lefort, K.; et al. A novel Nrf2-miR-29-desmocollin-2 axis regulates desmosome function in keratinocytes. Nat. Commun. 2014, 5, 5099. [Google Scholar] [CrossRef] [Green Version]
- Kostetskii, I.; Li, J.; Xiong, Y.; Zhou, R.; Ferrari, V.A.; Patel, V.V.; Molkentin, J.D.; Radice, G.L. Induced deletion of the N-cadherin gene in the heart leads to dissolution of the intercalated disc structure. Circ. Res. 2005, 96, 346–354. [Google Scholar] [CrossRef]
- Li, J.; Levin, M.D.; Xiong, Y.; Petrenko, N.; Patel, V.V.; Radice, G.L. N-cadherin haploinsufficiency affects cardiac gap junctions and arrhythmic susceptibility. J. Mol. Cell. Cardiol. 2008, 44, 597–606. [Google Scholar] [CrossRef] [Green Version]
- Sheikh, F.; Ross, R.S.; Chen, J. Cell-cell connection to cardiac disease. Trends Cardiovasc. Med. 2009, 19, 182–190. [Google Scholar] [CrossRef]
- Garrod, D.R.; Berika, M.Y.; Bardsley, W.F.; Holmes, D.; Tabernero, L. Hyper-adhesion in desmosomes: Its regulation in wound healing and possible relationship to cadherin crystal structure. J. Cell Sci. 2005, 118, 5743–5754. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.E.; Merritt, A.J.; Garrod, D.R. Calcium-independent desmosomes of keratinocytes are hyper-adhesive. J. Investig. Dermatol. 2007, 127, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Wallis, S.; Lloyd, S.; Wise, I.; Ireland, G.; Fleming, T.P.; Garrod, D. The alpha isoform of protein kinase C is involved in signaling the response of desmosomes to wounding in cultured epithelial cells. Mol. Biol. Cell 2000, 11, 1077–1092. [Google Scholar] [CrossRef] [PubMed]
- Hennings, H.; Holbrook, K.A. Calcium regulation of cell-cell contact and differentiation of epidermal cells in culture. An ultrastructural study. Exp. Cell Res. 1983, 143, 127–142. [Google Scholar] [CrossRef]
- Kartenbeck, J.; Schmid, E.; Franke, W.W.; Geiger, B. Different modes of internalization of proteins associated with adhaerens junctions and desmosomes: Experimental separation of lateral contacts induces endocytosis of desmosomal plaque material. Embo J. 1982, 1, 725–732. [Google Scholar] [CrossRef]
- Mattey, D.L.; Garrod, D.R. Splitting and internalization of the desmosomes of cultured kidney epithelial cells by reduction in calcium concentration. J. Cell Sci. 1986, 85, 113–124. [Google Scholar]
- Nollet, F.; Kools, P.; van Roy, F. Phylogenetic analysis of the cadherin superfamily allows identification of six major subfamilies besides several solitary members. J. Mol. Biol. 2000, 299, 551–572. [Google Scholar] [CrossRef]
- Allen, T.D.; Potten, C.S. Desmosomal form, fate, and function in mammalian epidermis. J. Ultrastruct. Res. 1975, 51, 94–105. [Google Scholar] [CrossRef]
- McHarg, S.; Hopkins, G.; Lim, L.; Garrod, D. Down-regulation of desmosomes in cultured cells: The roles of PKC, microtubules and lysosomal/proteasomal degradation. PLoS ONE 2014, 9, e108570. [Google Scholar] [CrossRef]
- He, W.; Cowin, P.; Stokes, D.L. Untangling desmosomal knots with electron tomography. Science 2003, 302, 109–113. [Google Scholar] [CrossRef]
- Thomason, H.A.; Cooper, N.H.; Ansell, D.M.; Chiu, M.; Merrit, A.J.; Hardman, M.J.; Garrod, D.R. Direct evidence that PKCalpha positively regulates wound re-epithelialization: Correlation with changes in desmosomal adhesiveness. J. Pathol. 2012, 227, 346–356. [Google Scholar] [CrossRef]
- Ohno, S. The genetic background of arrhythmogenic right ventricular cardiomyopathy. J. Arrhythmia 2016, 32, 398–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Austin, K.M.; Trembley, M.A.; Chandler, S.F.; Sanders, S.P.; Saffitz, J.E.; Abrams, D.J.; Pu, W.T. Molecular mechanisms of arrhythmogenic cardiomyopathy. Nat. Rev. Cardiol. 2019. [Google Scholar] [CrossRef]
- Norgett, E.E.; Hatsell, S.J.; Carvajal-Huerta, L.; Cabezas, J.C.; Common, J.; Purkis, P.E.; Whittock, N.; Leigh, I.M.; Stevens, H.P.; Kelsell, D.P. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum. Mol. Genet. 2000, 9, 2761–2766. [Google Scholar] [CrossRef] [Green Version]
- Cerrone, M.; Montnach, J.; Lin, X.; Zhao, Y.T.; Zhang, M.; Agullo-Pascual, E.; Leo-Macias, A.; Alvarado, F.J.; Dolgalev, I.; Karathanos, T.V.; et al. Plakophilin-2 is required for transcription of genes that control calcium cycling and cardiac rhythm. Nat. Commun. 2017, 8, 106. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, O.; Fernandez-Falgueras, A.; Iglesias, A.; Brugada, R. Brugada Syndrome and PKP2: Evidences and uncertainties. Int. J. Cardiol. 2016, 214, 403–405. [Google Scholar] [CrossRef] [PubMed]
- Novelli, V.; Malkani, K.; Cerrone, M. Pleiotropic Phenotypes Associated With PKP2 Variants. Front. Cardiovasc. Med. 2018, 5, 184. [Google Scholar] [CrossRef] [PubMed]
- Agullo-Pascual, E.; Reid, D.A.; Keegan, S.; Sidhu, M.; Fenyo, D.; Rothenberg, E.; Delmar, M. Super-resolution fluorescence microscopy of the cardiac connexome reveals plakophilin-2 inside the connexin43 plaque. Cardiovasc. Res. 2013, 100, 231–240. [Google Scholar] [CrossRef]
- Camors, E.; Mohler, P.J.; Bers, D.M.; Despa, S. Ankyrin-B reduction enhances Ca spark-mediated SR Ca release promoting cardiac myocyte arrhythmic activity. J. Mol. Cell. Cardiol. 2012, 52, 1240–1248. [Google Scholar] [CrossRef] [Green Version]
- Chopra, N.; Knollmann, B.C. Triadin regulates cardiac muscle couplon structure and microdomain Ca2+ signalling: A path towards ventricular arrhythmias. Cardiovasc. Res. 2013, 98, 187–191. [Google Scholar] [CrossRef]
- Kim, J.C.; Perez-Hernandez Duran, M.; Alvarado, F.J.; Maurya, S.R.; Montnach, J.; Yin, Y.; Zhang, M.; Lin, X.; Vasquez, C.; Heguy, A.; et al. Disruption of Ca2+i Homeostasis and Cx43 Hemichannel Function in the Right Ventricle Precedes Overt Arrhythmogenic Cardiomyopathy in PKP2-Deficient Mice. Circulation 2019. [Google Scholar] [CrossRef] [PubMed]
- De Bock, M.; Wang, N.; Bol, M.; Decrock, E.; Ponsaerts, R.; Bultynck, G.; Dupont, G.; Leybaert, L. Connexin 43 hemichannels contribute to cytoplasmic Ca2+ oscillations by providing a bimodal Ca2+-dependent Ca2+ entry pathway. J. Biol. Chem. 2012, 287, 12250–12266. [Google Scholar] [CrossRef] [PubMed]
- Berra-Romani, R.; Raqeeb, A.; Avelino-Cruz, J.E.; Moccia, F.; Oldani, A.; Speroni, F.; Taglietti, V.; Tanzi, F. Ca2+ signaling in injured in situ endothelium of rat aorta. Cell Calcium 2008, 44, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Tanzi, F.; Munaron, L. Endothelial remodelling and intracellular calcium machinery. Curr. Mol. Med. 2014, 14, 457–480. [Google Scholar] [CrossRef] [PubMed]
- Montnach, J.; Agullo-Pascual, E.; Tadros, R.; Bezzina, C.R.; Delmar, M. Bioinformatic analysis of a plakophilin-2-dependent transcription network: Implications for the mechanisms of arrhythmogenic right ventricular cardiomyopathy in humans and in boxer dogs. EP. Eur. 2018, 20, iii125–iii132. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Wong, J.; Wen, J.; Wang, S.; Wang, C.; Spiering, S.; Kan, N.G.; Forcales, S.; Puri, P.L.; Leone, T.C.; et al. Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature 2013, 494, 105–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Battrawy, I.; Zhao, Z.; Lan, H.; Cyganek, L.; Tombers, C.; Li, X.; Buljubasic, F.; Lang, S.; Tiburcy, M.; Zimmermann, W.H.; et al. Electrical dysfunctions in human-induced pluripotent stem cell-derived cardiomyocytes from a patient with an arrhythmogenic right ventricular cardiomyopathy. EP Eur. 2018, 20, f46–f56. [Google Scholar] [CrossRef]
- Lombardi, R.; Marian, A.J. Arrhythmogenic right ventricular cardiomyopathy is a disease of cardiac stem cells. Curr. Opin. Cardiol. 2010, 25, 222–228. [Google Scholar] [CrossRef] [Green Version]
- Stadiotti, I.; Pompilio, G.; Maione, A.S.; Pilato, C.A.; D’Alessandra, Y.; Sommariva, E. Arrhythmogenic cardiomyopathy: What blood can reveal? Heart Rhythm 2019, 16, 470–477. [Google Scholar] [CrossRef]
- Laitinen, P.J.; Brown, K.M.; Piippo, K.; Swan, H.; Devaney, J.M.; Brahmbhatt, B.; Donarum, E.A.; Marino, M.; Tiso, N.; Viitasalo, M.; et al. Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation 2001, 103, 485–490. [Google Scholar] [CrossRef]
- Priori, S.G.; Napolitano, C.; Tiso, N.; Memmi, M.; Vignati, G.; Bloise, R.; Sorrentino, V.; Danieli, G.A. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation 2001, 103, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Nava, A.; Canciani, B.; Daliento, L.; Miraglia, G.; Buja, G.; Fasoli, G.; Martini, B.; Scognamiglio, R.; Thiene, G. Juvenile sudden death and effort ventricular tachycardias in a family with right ventricular cardiomyopathy. Int. J. Cardiol. 1988, 21, 111–126. [Google Scholar] [CrossRef]
- McCarthy, T.V.; Quane, K.A.; Lynch, P.J. Ryanodine receptor mutations in malignant hyperthermia and central core disease. Hum. Mutat. 2000, 15, 410–417. [Google Scholar] [CrossRef]
- Marx, S.O.; Reiken, S.; Hisamatsu, Y.; Jayaraman, T.; Burkhoff, D.; Rosemblit, N.; Marks, A.R. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): Defective regulation in failing hearts. Cell 2000, 101, 365–376. [Google Scholar] [CrossRef]
- Censier, K.; Urwyler, A.; Zorzato, F.; Treves, S. Intracellular calcium homeostasis in human primary muscle cells from malignant hyperthermia-susceptible and normal individuals. Effect Of overexpression of recombinant wild-type and Arg163Cys mutated ryanodine receptors. J. Clin. Investig. 1998, 101, 1233–1242. [Google Scholar] [CrossRef] [PubMed]
- Kannankeril, P.J.; Mitchell, B.M.; Goonasekera, S.A.; Chelu, M.G.; Zhang, W.; Sood, S.; Kearney, D.L.; Danila, C.I.; De Biasi, M.; Wehrens, X.H.; et al. Mice with the R176Q cardiac ryanodine receptor mutation exhibit catecholamine-induced ventricular tachycardia and cardiomyopathy. Proc. Natl. Acad. Sci. USA 2006, 103, 12179–12184. [Google Scholar] [CrossRef] [Green Version]
- Amador, F.J.; Stathopulos, P.B.; Enomoto, M.; Ikura, M. Ryanodine receptor calcium release channels: Lessons from structure-function studies. Febs J. 2013, 280, 5456–5470. [Google Scholar] [CrossRef]
- Mallat, Z.; Tedgui, A.; Fontaliran, F.; Frank, R.; Durigon, M.; Fontaine, G. Evidence of apoptosis in arrhythmogenic right ventricular dysplasia. N. Engl. J. Med. 1996, 335, 1190–1196. [Google Scholar] [CrossRef]
- Roux-Buisson, N.; Gandjbakhch, E.; Donal, E.; Probst, V.; Deharo, J.C.; Chevalier, P.; Klug, D.; Mansencal, N.; Delacretaz, E.; Cosnay, P.; et al. Prevalence and significance of rare RYR2 variants in arrhythmogenic right ventricular cardiomyopathy/dysplasia: Results of a systematic screening. Heart Rhythm 2014, 11, 1999–2009. [Google Scholar] [CrossRef] [Green Version]
- Larsen, M.K.; Berge, K.E.; Leren, T.P.; Nissen, P.H.; Hansen, J.; Kristensen, I.B.; Banner, J.; Jensen, H.K. Postmortem genetic testing of the ryanodine receptor 2 (RYR2) gene in a cohort of sudden unexplained death cases. Int. J. Leg. Med. 2013, 127, 139–144. [Google Scholar] [CrossRef]
- Thomas, N.L.; George, C.H.; Williams, A.J.; Lai, F.A. Ryanodine receptor mutations in arrhythmias: Advances in understanding the mechanisms of channel dysfunction. Biochem. Soc. Trans. 2007, 35, 946–951. [Google Scholar] [CrossRef] [PubMed]
- Okudaira, N.; Kuwahara, M.; Hirata, Y.; Oku, Y.; Nishio, H. A knock-in mouse model of N-terminal R420W mutation of cardiac ryanodine receptor exhibits arrhythmogenesis with abnormal calcium dynamics in cardiomyocytes. Biochem. Biophys. Res. Commun. 2014, 452, 665–668. [Google Scholar] [CrossRef] [PubMed]
- Milting, H.; Lukas, N.; Klauke, B.; Korfer, R.; Perrot, A.; Osterziel, K.J.; Vogt, J.; Peters, S.; Thieleczek, R.; Varsanyi, M. Composite polymorphisms in the ryanodine receptor 2 gene associated with arrhythmogenic right ventricular cardiomyopathy. Cardiovasc. Res. 2006, 71, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Braz, J.C.; Gregory, K.; Pathak, A.; Zhao, W.; Sahin, B.; Klevitsky, R.; Kimball, T.F.; Lorenz, J.N.; Nairn, A.C.; Liggett, S.B.; et al. PKC-alpha regulates cardiac contractility and propensity toward heart failure. Nat. Med. 2004, 10, 248–254. [Google Scholar] [CrossRef] [PubMed]
- del Monte, F.; Harding, S.E.; Dec, G.W.; Gwathmey, J.K.; Hajjar, R.J. Targeting phospholamban by gene transfer in human heart failure. Circulation 2002, 105, 904–907. [Google Scholar] [CrossRef] [PubMed]
- Haghighi, K.; Kolokathis, F.; Gramolini, A.O.; Waggoner, J.R.; Pater, L.; Lynch, R.A.; Fan, G.C.; Tsiapras, D.; Parekh, R.R.; Dorn, G.W., 2nd; et al. A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy. Proc. Natl. Acad. Sci. USA 2006, 103, 1388–1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akdis, D.; Medeiros-Domingo, A.; Gaertner-Rommel, A.; Kast, J.I.; Enseleit, F.; Bode, P.; Klingel, K.; Kandolf, R.; Renois, F.; Andreoletti, L.; et al. Myocardial expression profiles of candidate molecules in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia compared to those with dilated cardiomyopathy and healthy controls. Heart Rhythm 2016, 13, 731–741. [Google Scholar] [CrossRef]
- Karakikes, I.; Stillitano, F.; Nonnenmacher, M.; Tzimas, C.; Sanoudou, D.; Termglinchan, V.; Kong, C.W.; Rushing, S.; Hansen, J.; Ceholski, D.; et al. Correction of human phospholamban R14del mutation associated with cardiomyopathy using targeted nucleases and combination therapy. Nat. Commun. 2015, 6, 6955. [Google Scholar] [CrossRef]
- Raja, S.B.; Rajendiran, V.; Kasinathan, N.K.; Amrithalakshmi, P.; Venkatabalasubramanian, S.; Murali, M.R.; Devaraj, H.; Devaraj, S.N. Differential cytotoxic activity of Quercetin on colonic cancer cells depends on ROS generation through COX-2 expression. Food Chem. Toxicol. 2017, 106, 92–106. [Google Scholar] [CrossRef]
- Liu, G.S.; Morales, A.; Vafiadaki, E.; Lam, C.K.; Cai, W.F.; Haghighi, K.; Adly, G.; Hershberger, R.E.; Kranias, E.G. A novel human R25C-phospholamban mutation is associated with super-inhibition of calcium cycling and ventricular arrhythmia. Cardiovasc. Res. 2015, 107, 164–174. [Google Scholar] [CrossRef] [Green Version]
- Groeneweg, J.A.; van der Zwaag, P.A.; Olde Nordkamp, L.R.; Bikker, H.; Jongbloed, J.D.; Jongbloed, R.; Wiesfeld, A.C.; Cox, M.G.; van der Heijden, J.F.; Atsma, D.E.; et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy according to revised 2010 task force criteria with inclusion of non-desmosomal phospholamban mutation carriers. Am. J. Cardiol. 2013, 112, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Te Rijdt, W.P.; Asimaki, A.; Jongbloed, J.D.H.; Hoorntje, E.T.; Lazzarini, E.; van der Zwaag, P.A.; de Boer, R.A.; van Tintelen, J.P.; Saffitz, J.E.; van den Berg, M.P.; et al. Distinct molecular signature of phospholamban p.Arg14del arrhythmogenic cardiomyopathy. Cardiovasc. Pathol. Off. J. Soc. Cardiovasc. Pathol. 2019, 40, 2–6. [Google Scholar] [CrossRef] [PubMed]
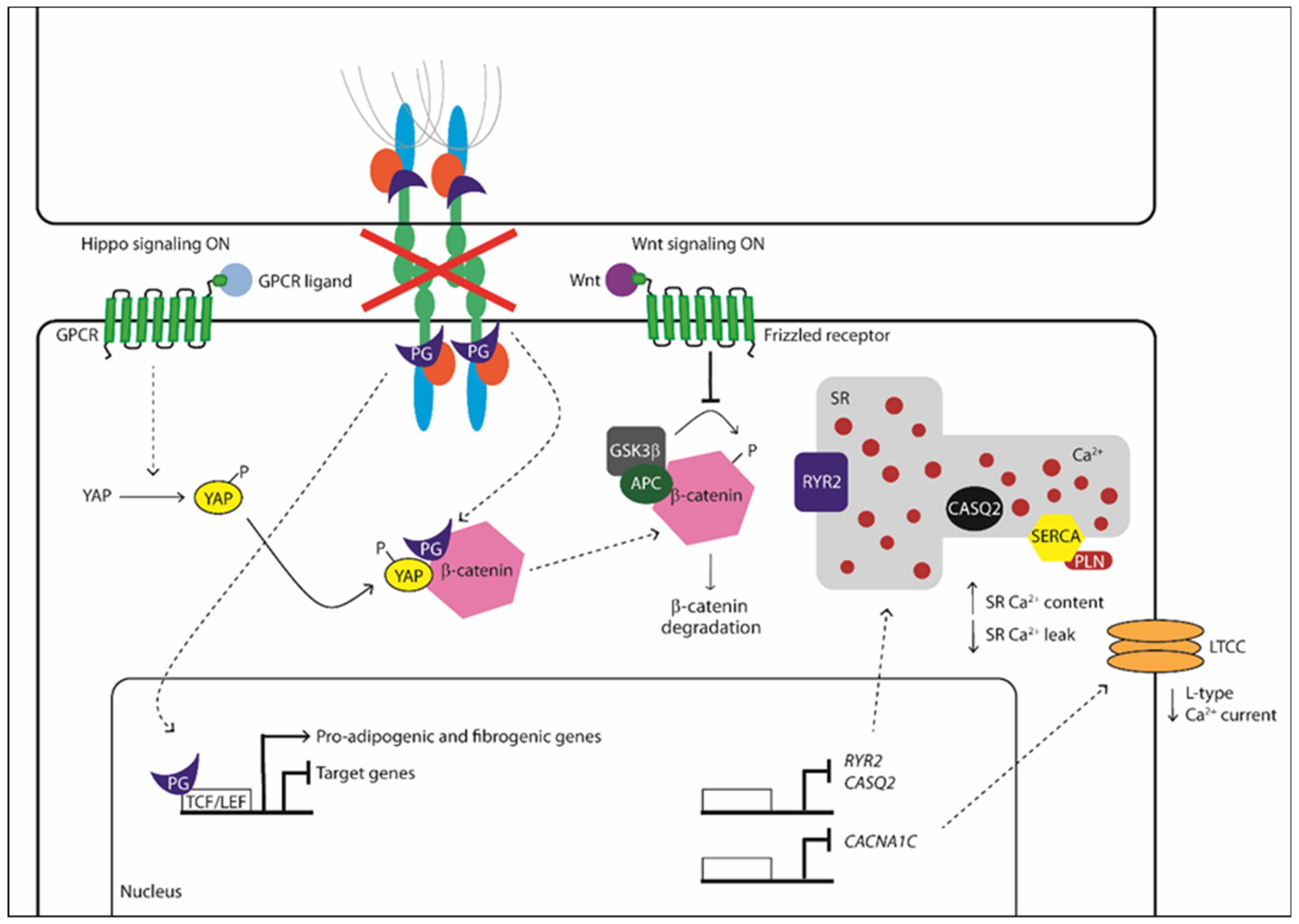
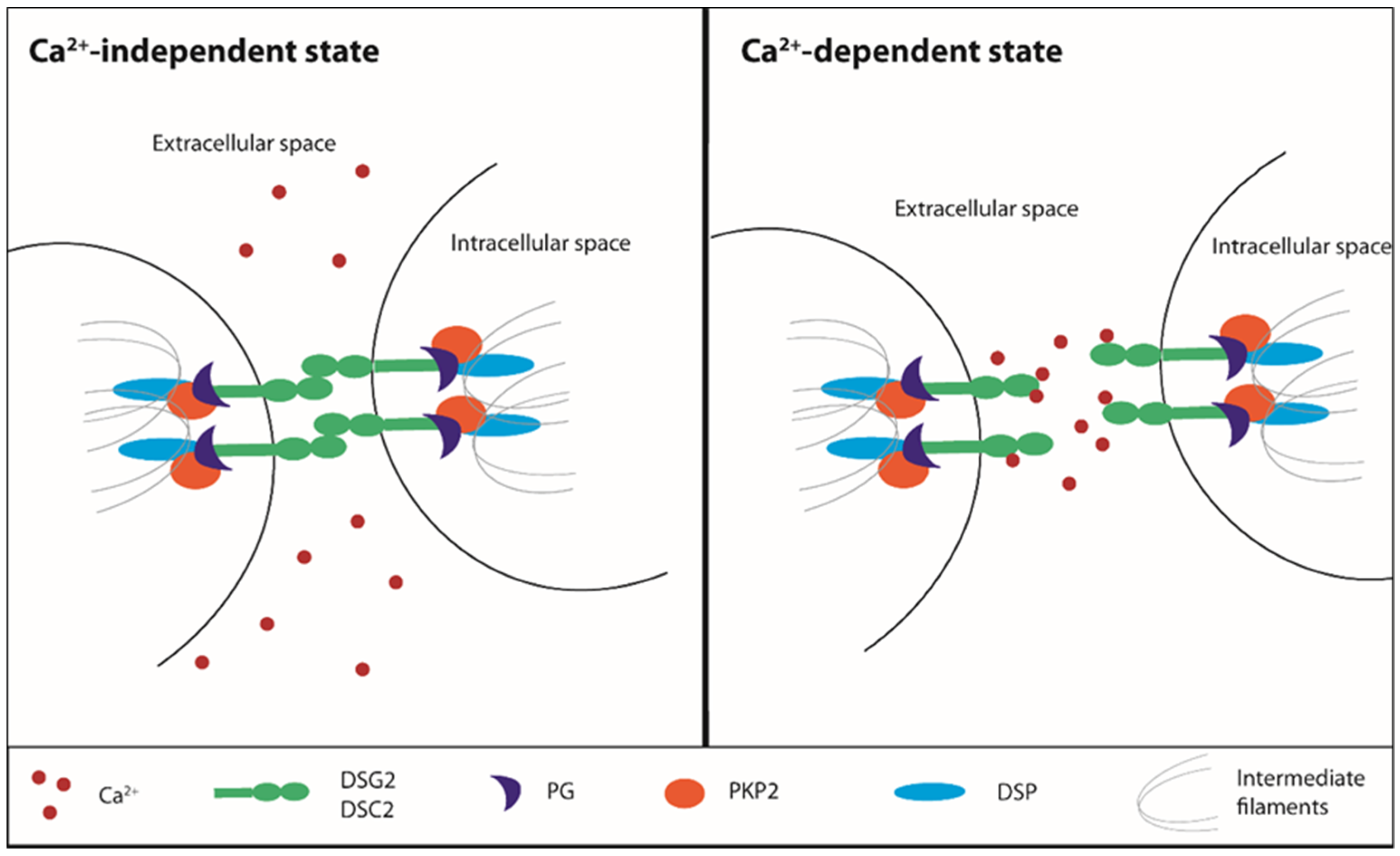
 Activation/Phosphorylation,
Activation/Phosphorylation,  Translocation,
Translocation,  Inhibition).
Activation/Phosphorylation, Translocation, Inhibition).
Inhibition).
Activation/Phosphorylation, Translocation, Inhibition).


{kind=link}
{kind=link}
| Gene | Coding Protein | Locus | Reference | |
|---|---|---|---|---|
| desmosomal mutations | JUP | Junction Plakoglobin | 17q21.2 | [35] |
| DSP | Desmoplakin | 6p24.3 | [36] | |
| PKP2 | Plakophilin 2 | 12p11.21 | [37] | |
| DSG2 | Desmoglein 2 | 18q12.1 | [38,39] | |
| DSC2 | Desmocollin 2 | 18q12.1 | [40] | |
| non-desmosomal mutations | TMEM43 | Transmembrane protein 43 | 3p25.1 | [41] |
| LMNA | Lamin A/C | 1q22 | [42] | |
| DES | Desmin | 2q35 | [43] | |
| CTNNA3 | Alpha-T-catenin | 10q21.3 | [44] | |
| PLN | Phospholamban | 6q22.31 | [45] | |
| TGFB3 | Transforming growth factor-3 | 14q24.3 | [27,46] | |
| TTN | Titin | 2q31.2 | [47] | |
| SCN5A | Sodium voltage gated channel alpha subunit 5 (NaV 1.5) | 3p22.2 | [48] | |
| CDH2 | Cadherin C | 18q12.1 | [49] | |
| RYR2 | Type 2 ryanodine receptor | 1q42-q43 | [50,51] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moccia, F.; Lodola, F.; Stadiotti, I.; Pilato, C.A.; Bellin, M.; Carugo, S.; Pompilio, G.; Sommariva, E.; Maione, A.S. Calcium as a Key Player in Arrhythmogenic Cardiomyopathy: Adhesion Disorder or Intracellular Alteration? Int. J. Mol. Sci. 2019, 20, 3986. https://doi.org/10.3390/ijms20163986
Moccia F, Lodola F, Stadiotti I, Pilato CA, Bellin M, Carugo S, Pompilio G, Sommariva E, Maione AS. Calcium as a Key Player in Arrhythmogenic Cardiomyopathy: Adhesion Disorder or Intracellular Alteration? International Journal of Molecular Sciences. 2019; 20(16):3986. https://doi.org/10.3390/ijms20163986
Chicago/Turabian StyleMoccia, Francesco, Francesco Lodola, Ilaria Stadiotti, Chiara Assunta Pilato, Milena Bellin, Stefano Carugo, Giulio Pompilio, Elena Sommariva, and Angela Serena Maione. 2019. "Calcium as a Key Player in Arrhythmogenic Cardiomyopathy: Adhesion Disorder or Intracellular Alteration?" International Journal of Molecular Sciences 20, no. 16: 3986. https://doi.org/10.3390/ijms20163986