Minimal Residual Disease Monitoring with Next-Generation Sequencing Methodologies in Hematological Malignancies



Abstract
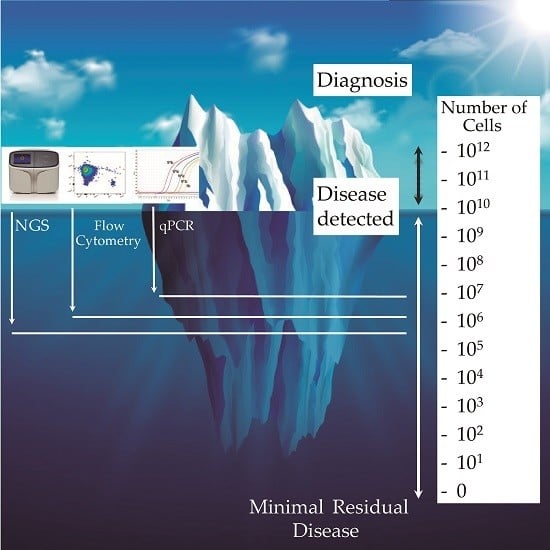
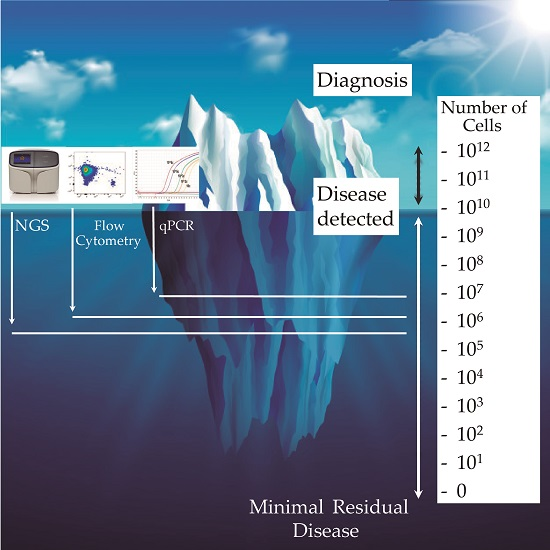
:
1. Introduction
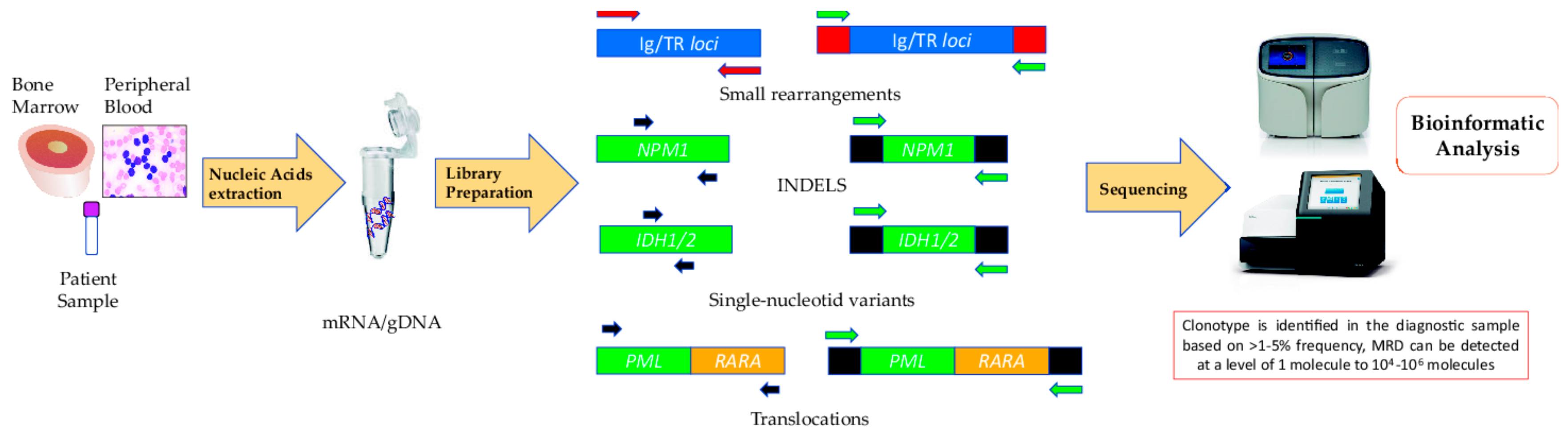
- Repertoire sequencing, including clonal rearrangements of immunoglobulin/T-cell antigen receptor genes: IGH (VDJ), IGH (DJ), IGK, TRG, TRD, and TRB [20]. In the case of lymphocytic disorders, several markers have been tested for their utility to monitor the disease. Rearrangement of the immunoglobulin heavy chain (IGH) gene occurs during B-cell differentiation, making it an ideal marker for MRD monitoring owing to the fact that each neoplastic cell has the identical sequence. Several NGS-based immunoglobulin clonality assays are commercially available—for example, ClonoSEQTM [27] and LymphoTrackTM [28] from Adaptive Biotechnologies® and Invivoscribe®, respectively; but other ‘in-house’ MRD tests with a more affordable and feasible design are also used [10].
2. MRD Monitoring in Acute Myeloid Leukemia
3. MRD Monitoring in Lymphoid Malignancies
3.1. Acute Lymphoblastic Leukemia
3.2. Chronic Lymphocytic Leukemia
3.3. Multiple Myeloma
4. Current Challenges
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ALL | Acute Lymphoblastic Leukemia |
| AML | Acute Myeloid Leukemia |
| ASO-PCR | Allele-Specific Oligonucleotide PCR |
| BM | Bone Marrow |
| CLL | Chronic Lymphocytic Leukemia |
| CNS | Central Nervous System |
| CR | Complete Remission |
| CSF | Cerebro-Spinal Fluid |
| DFS | Disease-Free Survival |
| ITD | Internal Tandem Repeats |
| MFC | Multiparametric Flow Cytometry |
| MM | Multiple Myeloma |
| MRD | Minimal Residual Disease |
| NGS | Next-Generation Sequencing |
| OS | Overall Survival |
| PB | Peripheral Blood |
| PFS | Progression-Free Survival |
| qPCR | Quantitative Polymerase Chain Reaction |
| RFS | Relapse-Free Survival |
| SNV | Single Nucleotide Variation |
| UMI | Unique Molecular Index |
| VAF | Variant Allele Frequency |
Appendix A

{kind=link}
{kind=link}
| Hematological Malignancy | Authors | NGS Methodology-Equipment | Target | Sensitivity | Year |
|---|---|---|---|---|---|
| AML | Thol et al. | Pyrosequencing 454 Junior | FLT3-ITD NPM1 | NE | 2012 |
| AML | Spencer et al. | Sure Select- Illumina-HiSeq | FLT3-ITD | NE | 2013 |
| AML | Klco et al. | Ion Torrent-PGM | xGen® AML Cancer Panel | NE | 2015 |
| AML | Jongen-Lavrencic et al. | Illumina-MiSeq | TruSight® Myeloid Sequencing Panel | NE | 2018 |
| AML | Levis et al. | Illumina-MiSeq | FLT3-ITD | 10−6 | 2018 |
| AML | Morita et al. | Illumina-HiSeq 2000 | NPM1 DNMT3A FLT3-ITD | NE | 2018 |
| AML | Thol. et al. | Illumina-MiSeq | IDH1 * DNMT3A | 5 × 10−5 | 2018 |
| AML | Onecha et al. | Ion Torrent-Proton PI | NPM1 * IDH1 IDH2 | 5 × 10−5 | 2019 |
| AML | Malmberg et al. | Illumina-MiSeq | TruSight Myeloid Sequencing Panel RUNX1/RUNX1T1 KMT2A/MLLT10 | NE | 2019 |
| CLL | Logan et al. | Pyrosequencing 454 Junior | IGH(VDJ) | 10−5 | 2011 |
| CLL | Logan et al. | Illumina | IGH(DJ) | 10−6 | 2013 |
| CLL | Rossi et al. | Pyrosequencing 454 Junior | TP53 | NE | 2014 |
| CLL | Stamatopoulos et al. | Illumina-MiSeq | IGH LEADER (VDJ) | NE | 2017 |
| ALL | Wu et al. | Illumina-HiSeq | TRB * TRG | 10−5 | 2011 |
| ALL | Gawad et al. | Illumina-MiSeq | IGH(VDJ) | NE | 2012 |
| ALL | Faham et al. | Illumina-MiSeq | IGH(VDJ) * IGH(DJ) * TRB TRD TRG | 10−6 | 2012 |
| ALL | Wu et al. | ClonoSEQ assay-Illumina | IGH(VDJ) IGH(DJ) | 10−6 | 2014 |
| ALL | Pulsipher et al. | Illumina-HiSeq | IGH(VDJ) | 10−7 | 2015 |
| ALL | Ferret et al. | Ion Torrent-PGM | IGH(VDJ) IGK TRG TRD | NE | 2016 |
| ALL | Sekiya et al. | Illumina-MiSeq | IGH(VDJ) * TRG | 10−6 | 2016 |
| ALL | Sala Torra et al. | ClonoSEQ assay-Illumina | IGH(VDJ) IGH(DJ) | NE | 2017 |
| ALL | Salson et al. | Ion Torrent- PGM | IGH(VDJ) * TRG | 2 × 10−5 | 2017 |
| ALL | Cheng et al. | Illumina-MiSeq | IGH(VDJ) | 10−6 | 2018 |
| ALL | Theunissen et al. | Illumina-MiSeq | IGH(VDJ) IGK TRG TRD TRB | NE | 2018 |
| ALL-CNS | Sanchez et al. | Ion Torrent-S5 XL | KD of BCR-ABL1 | NE | 2017 |
| ALL-CNS | Bartram et al. | Illumina-MiSeq | IGH(VDJ) | NE | 2018 |
| MM | Vij et al. | Illumina-MiSeq | IGH(VDJ) IGH(DJ) IGK | NE | 2014 |
| MM | Martinez-Lopez et al. | Illumina-MiSeq | IGH(VDJ) | 10−5 | 2014 |
| MM | Avet-Loiseau et al. | ClonoSEQ assay-Illumina | IGH(VDJ) IGH(DJ) | NE | 2016 |
| MM | Martinez-Lopez et al. | Ion Torrent-S5 XL Illumina-MiSeq | IGH(VDJ) * IGH(DJ) * IGK* | 10−5 | 2017 |
| MM | Perrot et al. | Illumina-MiSeq | IGH(VDJ) | 10−6 | 2018 |
References
- Brüggemann, M.; Kotrova, M. Minimal Residual Disease in Adult ALL: Technical Aspects and Implications for Correct Clinical Interpretation. Blood Adv. 2017, 2456–2466. [Google Scholar] [CrossRef] [PubMed]
- Van Dongen, J.J.; Seriu, T.; Panzer-Grümayer, E.R.; Biondi, A.; Pongers-Willemse, M.J.; Corral, L.; Stolz, F.; Schrappe, M.; Masera, G.; Kamps, W.A.; et al. Prognostic Value of Minimal Residual Disease in Acute Lymphoblastic Leukaemia in Childhood. Lancet 1998, 352, 1731–1738. [Google Scholar] [CrossRef]
- Brüggemann, M.; Schrauder, A.; Raff, T.; Pfeifer, H.; Dworzak, M.; Ottmann, O.G.; Asnafi, V.; Baruchel, A.; Bassan, R.; Benoit, Y.; et al. Standardized MRD Quantification in European ALL Trials: Proceedings of the Second International Symposium on MRD Assessment in Kiel, Germany, 18–20 September 2008. Leukemia 2010, 24, 521–535. [Google Scholar] [CrossRef]
- Dogliotti, I.; Drandi, D.; Genuardi, E.; Ferrero, S. New Molecular Technologies for Minimal Residual Disease Evaluation in B-Cell Lymphoid Malignancies. J. Clin. Med. 2018, 7, 288. [Google Scholar] [CrossRef] [PubMed]
- Flores-Montero, J.; Sanoja-Flores, L.; Paiva, B.; Puig, N.; García-Sánchez, O.; Böttcher, S.; van der Velden, V.H.J.; Pérez-Morán, J.-J.; Vidriales, M.-B.; García-Sanz, R.; et al. Next Generation Flow for Highly Sensitive and Standardized Detection of Minimal Residual Disease in Multiple Myeloma. Leukemia 2017, 31, 2094–2103. [Google Scholar] [CrossRef] [PubMed]
- Theunissen, P.; Mejstrikova, E.; Sedek, L.; van der Sluijs-Gelling, A.J.; Gaipa, G.; Bartels, M.; Sobral da Costa, E.; Kotrová, M.; Novakova, M.; Sonneveld, E.; et al. Standardized Flow Cytometry for Highly Sensitive MRD Measurements in B-Cell Acute Lymphoblastic Leukemia. Blood 2017, 129, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Rawstron, A.C.; Fazi, C.; Agathangelidis, A.; Villamor, N.; Letestu, R.; Nomdedeu, J.; Palacio, C.; Stehlikova, O.; Kreuzer, K.-A.; Liptrot, S.; et al. A Complementary Role of Multiparameter Flow Cytometry and High-Throughput Sequencing for Minimal Residual Disease Detection in Chronic Lymphocytic Leukemia: An European Research Initiative on CLL Study. Leukemia 2016, 30, 929–936. [Google Scholar] [CrossRef]
- Cheng, S.; Inghirami, G.; Cheng, S.; Tam, W. Simple Deep Sequencing-Based Post-Remission MRD Surveillance Predicts Clinical Relapse in B-ALL. J. Hematol. Oncol. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Van der Velden, V.; Jacobs, D.; Wijkhuijs, A.; Comans-Bitter, W.; Willemse, M.; Hählen, K.; Kamps, W.; van Wering, E.; van Dongen, J. Minimal Residual Disease Levels in Bone Marrow and Peripheral Blood Are Comparable in Children with T Cell Acute Lymphoblastic Leukemia (ALL), but Not in Precursor-B-ALL. Leukemia 2002, 16, 1432–1436. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lopez, J.; Sanchez-Vega, B.; Barrio, S.; Cuenca, I.; Ruiz-Heredia, Y.; Alonso, R.; Rapado, I.; Marin, C.; Cedena, M.-T.; Paiva, B.; et al. Analytical and Clinical Validation of a Novel In-House Deep-Sequencing Method for Minimal Residual Disease Monitoring in a Phase II Trial for Multiple Myeloma. Leukemia 2017, 31, 1446–1449. [Google Scholar] [CrossRef]
- Kumar, S.; Paiva, B.; Anderson, K.C.; Durie, B.; Landgren, O.; Moreau, P.; Munshi, N.; Lonial, S.; Bladé, J.; Mateos, M.-V.; et al. International Myeloma Working Group Consensus Criteria for Response and Minimal Residual Disease Assessment in Multiple Myeloma. Lancet Oncol. 2016, 17, e328–e346. [Google Scholar] [CrossRef]
- Schuurhuis, G.J.; Heuser, M.; Freeman, S.; Béné, M.-C.; Buccisano, F.; Cloos, J.; Grimwade, D.; Haferlach, T.; Hills, R.K.; Hourigan, C.S.; et al. Minimal/Measurable Residual Disease in AML: A Consensus Document from the European LeukemiaNet MRD Working Party. Blood 2018, 131, 1275–1291. [Google Scholar] [CrossRef] [PubMed]
- Ivey, A.; Hills, R.K.; Simpson, M.A.; Jovanovic, J.V.; Gilkes, A.; Grech, A.; Patel, Y.; Bhudia, N.; Farah, H.; Mason, J.; et al. Assessment of Minimal Residual Disease in Standard-Risk AML. N. Engl. J. Med. 2016, 374, 422–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thol, F.; Gabdoulline, R.; Liebich, A.; Klement, P.; Schiller, J.; Kandziora, C.; Hambach, L.; Stadler, M.; Koenecke, C.; Flintrop, M.; et al. Measurable Residual Disease Monitoring by NGS before Allogeneic Hematopoietic Cell Transplantation in AML. Blood 2018, 132, 1703–1713. [Google Scholar] [CrossRef]
- Berry, D.A.; Zhou, S.; Higley, H.; Mukundan, L.; Fu, S.; Reaman, G.H.; Wood, B.L.; Kelloff, G.J.; Jessup, J.M.; Radich, J.P. Association of Minimal Residual Disease with Clinical Outcome in Pediatric and Adult Acute Lymphoblastic Leukemia: A Meta-Analysis. JAMA Oncol. 2017, 3, e170580. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.; Robrecht, S.; Fink, A.M.; Bahlo, J.; Cramer, P.; von Tresckow, J.; Maurer, C.; Langerbeins, P.; Fingerle-Rowson, G.; Ritgen, M.; et al. Minimal Residual Disease Assessment Improves Prediction of Outcome in Patients With Chronic Lymphocytic Leukemia (CLL) Who Achieve Partial Response: Comprehensive Analysis of Two Phase III Studies of the German CLL Study Group. J. Clin. Oncol. 2016, 34, 3758–3765. [Google Scholar] [CrossRef]
- Thompson, P.A.; Peterson, C.B.; Strati, P.; Jorgensen, J.; Keating, M.J.; O’Brien, S.M.; Ferrajoli, A.; Burger, J.A.; Estrov, Z.; Jain, N.; et al. Serial Minimal Residual Disease (MRD) Monitoring during First-Line FCR Treatment for CLL May Direct Individualized Therapeutic Strategies. Leukemia 2018, 32, 2388–2398. [Google Scholar] [CrossRef]
- Ho, C.; Arcila, M.E. Minimal Residual Disease Detection of Myeloma Using Sequencing of Immunoglobulin Heavy Chain Gene VDJ Regions. Semin. Hematol. 2018, 55, 13–18. [Google Scholar] [CrossRef]
- Bai, Y.; Orfao, A.; Chim, C.S. Molecular Detection of Minimal Residual Disease in Multiple Myeloma. Br. J. Haematol. 2018, 181, 11–26. [Google Scholar] [CrossRef]
- Theunissen, P.M.J.; de Bie, M.; van Zessen, D.; de Haas, V.; Stubbs, A.P.; van der Velden, V.H.J. Next-Generation Antigen Receptor Sequencing of Paired Diagnosis and Relapse Samples of B-Cell Acute Lymphoblastic Leukemia: Clonal Evolution and Implications for Minimal Residual Disease Target Selection. Leuk. Res. 2019, 76, 98–104. [Google Scholar] [CrossRef]
- Logan, A.C.; Zhang, B.; Narasimhan, B.; Carlton, V.; Zheng, J.; Moorhead, M.; Krampf, M.R.; Jones, C.D.; Waqar, A.N.; Faham, M.; et al. Minimal Residual Disease Quantification Using Consensus Primers and High-Throughput IGH Sequencing Predicts Post-Transplant Relapse in Chronic Lymphocytic Leukemia. Leukemia 2013, 27, 1659–1665. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Vicente, A.E.; Bikos, V.; Hernández-Sánchez, M.; Hernández-Rivas, J.-M.; Pospisilova, S. Next-Generation Sequencing in Chronic Lymphocytic Leukemia. Oncotarget 2017, 8, 71234–71248. [Google Scholar] [CrossRef] [PubMed]
- Ferrero, S.; Drandi, D.; Mantoan, B.; Ghione, P.; Omedè, P.; Ladetto, M. Minimal Residual Disease Detection in Lymphoma and Multiple Myeloma: Impact on Therapeutic Paradigms: MRD Detection in Lymphoma and Multiple Myeloma. Hematol. Oncol. 2011, 29, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Onecha, E.; Linares, M.; Rapado, I.; Ruiz-Heredia, Y.; Martinez-Sanchez, P.; Cedena, T.; Pratcorona, M.; Oteyza, J.P.; Herrera, P.; Barragan, E.; et al. A Novel Deep Targeted Sequencing Method for Minimal Residual Disease Monitoring in Acute Myeloid Leukemia. Haematologica 2019, 104, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Jongen-Lavrencic, M.; Grob, T.; Hanekamp, D.; Kavelaars, F.G.; al Hinai, A.; Zeilemaker, A.; Erpelinck-Verschueren, C.A.J.; Gradowska, P.L.; Meijer, R.; Cloos, J.; et al. Molecular Minimal Residual Disease in Acute Myeloid Leukemia. N. Engl. J. Med. 2018, 378, 1189–1199. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, R.M.; Kim, D.D.H. Next-Generation Sequencing-Based Minimal Residual Disease Monitoring in Patients Receiving Allogeneic Hematopoietic Stem Cell Transplantation for Acute Myeloid Leukemia or Myelodysplastic Syndrome. Curr. Opin. Hematol. 2018, 25, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Faham, M.; Zheng, J.; Moorhead, M.; Carlton, V.E.H.; Stow, P.; Coustan-Smith, E.; Pui, C.-H.; Campana, D. Deep-Sequencing Approach for Minimal Residual Disease Detection in Acute Lymphoblastic Leukemia. Blood 2012, 120, 5173–5180. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Hwang, I.S.; Kim, J.; Lee, K.-A.; Lee, S.-T.; Choi, J.R. Detection of Immunoglobulin Heavy Chain Gene Clonality by Next-Generation Sequencing for Minimal Residual Disease Monitoring in B-Lymphoblastic Leukemia. Ann. Lab. Med. 2017, 37, 331. [Google Scholar] [CrossRef] [PubMed]
- Delsing Malmberg, E.; Rehammar, A.; Pereira, M.B.; Abrahamsson, J.; Samuelsson, T.; Ståhlman, S.; Asp, J.; Tierens, A.; Palmqvist, L.; Kristiansson, E.; et al. Accurate and Sensitive Analysis of Minimal Residual Disease in Acute Myeloid Leukemia Using Deep Sequencing of Single Nucleotide Variations. J. Mol. Diagn. 2019, 21, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Dillon, L.W.; Hayati, S.; Roloff, G.W.; Tunc, I.; Pirooznia, M.; Mitrofanova, A.; Hourigan, C.S. Targeted RNA-Sequencing for the Quantification of Measurable Residual Disease in Acute Myeloid Leukemia. Haematologica 2019, 104, 297–304. [Google Scholar] [CrossRef]
- Thiede, C. Analysis of FLT3-Activating Mutations in 979 Patients with Acute Myelogenous Leukemia: Association with FAB Subtypes and Identification of Subgroups with Poor Prognosis. Blood 2002, 99, 4326–4335. [Google Scholar] [CrossRef] [PubMed]
- Kottaridis, P.D. The Presence of a FLT3 Internal Tandem Duplication in Patients with Acute Myeloid Leukemia (AML) Adds Important Prognostic Information to Cytogenetic Risk Group and Response to the First Cycle of Chemotherapy: Analysis of 854 Patients from the United Kingdom Medical Research Council AML 10 and 12 Trials. Blood 2001, 98, 1752–1759. [Google Scholar] [CrossRef] [PubMed]
- Small, D. FLT3 Mutations: Biology and Treatment. Hematology 2006, 2006, 178–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhaak, R.G.W. Mutations in Nucleophosmin (NPM1) in Acute Myeloid Leukemia (AML): Association with Other Gene Abnormalities and Previously Established Gene Expression Signatures and Their Favorable Prognostic Significance. Blood 2005, 106, 3747–3754. [Google Scholar] [CrossRef] [PubMed]
- Thol, F.; Kölking, B.; Damm, F.; Reinhardt, K.; Klusmann, J.-H.; Reinhardt, D.; von Neuhoff, N.; Brugman, M.H.; Schlegelberger, B.; Suerbaum, S.; et al. Next-Generation Sequencing for Minimal Residual Disease Monitoring in Acute Myeloid Leukemia Patients with FLT3-ITD or NPM1 Mutations. Genes. Chromosomes Cancer 2012, 51, 689–695. [Google Scholar] [CrossRef]
- Spencer, D.H.; Abel, H.J.; Lockwood, C.M.; Payton, J.E.; Szankasi, P.; Kelley, T.W.; Kulkarni, S.; Pfeifer, J.D.; Duncavage, E.J. Detection of FLT3 Internal Tandem Duplication in Targeted, Short-Read-Length, Next-Generation Sequencing Data. J. Mol. Diagn. 2013, 15, 81–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, K.; Schulz, M.H.; Long, Q.; Apweiler, R.; Ning, Z. Pindel: A Pattern Growth Approach to Detect Break Points of Large Deletions and Medium Sized Insertions from Paired-End Short Reads. Bioinformatics 2009, 25, 2865–2871. [Google Scholar] [CrossRef]
- Levis, M.J.; Perl, A.E.; Altman, J.K.; Gocke, C.D.; Bahceci, E.; Hill, J.; Liu, C.; Xie, Z.; Carson, A.R.; McClain, V.; et al. A Next-Generation Sequencing–Based Assay for Minimal Residual Disease Assessment in AML Patients with FLT3 -ITD Mutations. Blood Adv. 2018, 2, 825–831. [Google Scholar] [CrossRef]
- Morita, K.; Kantarjian, H.M.; Wang, F.; Yan, Y.; Bueso-Ramos, C.; Sasaki, K.; Issa, G.C.; Wang, S.; Jorgensen, J.; Song, X.; et al. Clearance of Somatic Mutations at Remission and the Risk of Relapse in Acute Myeloid Leukemia. J. Clin. Oncol. 2018, 36, 1788–1797. [Google Scholar] [CrossRef]
- Klco, J.M.; Miller, C.A.; Griffith, M.; Petti, A.; Spencer, D.H.; Ketkar-Kulkarni, S.; Wartman, L.D.; Christopher, M.; Lamprecht, T.L.; Helton, N.M.; et al. Association Between Mutation Clearance After Induction Therapy and Outcomes in Acute Myeloid Leukemia. JAMA 2015, 314, 811. [Google Scholar] [CrossRef]
- Gawad, C.; Pepin, F.; Carlton, V.E.H.; Klinger, M.; Logan, A.C.; Miklos, D.B.; Faham, M.; Dahl, G.; Lacayo, N. Massive Evolution of the Immunoglobulin Heavy Chain Locus in Children with B Precursor Acute Lymphoblastic Leukemia. Blood 2012, 120, 4407–4417. [Google Scholar] [CrossRef] [PubMed]
- Salson, M.; Giraud, M.; Caillault, A.; Grardel, N.; Duployez, N.; Ferret, Y.; Duez, M.; Herbert, R.; Rocher, T.; Sebda, S.; et al. High-Throughput Sequencing in Acute Lymphoblastic Leukemia: Follow-up of Minimal Residual Disease and Emergence of New Clones. Leuk. Res. 2017, 53, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ferret, Y.; Caillault, A.; Sebda, S.; Duez, M.; Grardel, N.; Duployez, N.; Villenet, C.; Figeac, M.; Preudhomme, C.; Salson, M.; et al. Multi-Loci Diagnosis of Acute Lymphoblastic Leukaemia with High-Throughput Sequencing and Bioinformatics Analysis. Br. J. Haematol. 2016, 173, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Duez, M.; Giraud, M.; Herbert, R.; Rocher, T.; Salson, M.; Thonier, F. Vidjil: A Web Platform for Analysis of High-Throughput Repertoire Sequencing. PLoS ONE 2016, 11, e0166126. [Google Scholar] [CrossRef] [PubMed]
- Sekiya, Y.; Xu, Y.; Muramatsu, H.; Okuno, Y.; Narita, A.; Suzuki, K.; Wang, X.; Kawashima, N.; Sakaguchi, H.; Yoshida, N.; et al. Clinical Utility of Next-Generation Sequencing-Based Minimal Residual Disease in Paediatric B-Cell Acute Lymphoblastic Leukaemia. Br. J. Haematol. 2017, 176, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Sala Torra, O.; Othus, M.; Williamson, D.W.; Wood, B.; Kirsch, I.; Robins, H.; Beppu, L.; O’Donnell, M.R.; Forman, S.J.; Appelbaum, F.R.; et al. Next-Generation Sequencing in Adult B Cell Acute Lymphoblastic Leukemia Patients. Biol. Blood Marrow Transplant. 2017, 23, 691–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulsipher, M.A.; Carlson, C.; Langholz, B.; Wall, D.A.; Schultz, K.R.; Bunin, N.; Kirsch, I.; Gastier-Foster, J.M.; Borowitz, M.; Desmarais, C.; et al. IgH-V(D)J NGS-MRD Measurement Pre- and Early Post-Allotransplant Defines Very Low- and Very High-Risk ALL Patients. Blood 2015, 125, 3501–3508. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Sherwood, A.; Fromm, J.R.; Winter, S.S.; Dunsmore, K.P.; Loh, M.L.; Greisman, H.A.; Sabath, D.E.; Wood, B.L.; Robins, H. High-Throughput Sequencing Detects Minimal Residual Disease in Acute T Lymphoblastic Leukemia. Sci. Transl. Med. 2012, 4, 134ra63. [Google Scholar] [CrossRef]
- Bartram, J.; Goulden, N.; Wright, G.; Adams, S.; Brooks, T.; Edwards, D.; Inglott, S.; Yousafzai, Y.; Hubank, M.; Halsey, C. High Throughput Sequencing in Acute Lymphoblastic Leukemia Reveals Clonal Architecture of Central Nervous System and Bone Marrow Compartments. Haematologica 2018, 103, e110–e114. [Google Scholar] [CrossRef]
- Sanchez, R.; Ayala, R.; Alonso, R.A.; Martínez, M.P.; Ribera, J.; García, O.; Sanchez-Pina, J.; Mercadal, S.; Montesinos, P.; Martino, R.; et al. Clinical Characteristics of Patients with Central Nervous System Relapse in BCR-ABL1-Positive Acute Lymphoblastic Leukemia: The Importance of Characterizing ABL1 Mutations in Cerebrospinal Fluid. Ann. Hematol. 2017, 96, 1069–1075. [Google Scholar] [CrossRef]
- Logan, A.C.; Gao, H.; Wang, C.; Sahaf, B.; Jones, C.D.; Marshall, E.L.; Buno, I.; Armstrong, R.; Fire, A.Z.; Weinberg, K.I.; et al. High-Throughput VDJ Sequencing for Quantification of Minimal Residual Disease in Chronic Lymphocytic Leukemia and Immune Reconstitution Assessment. Proc. Natl. Acad. Sci. USA 2011, 108, 21194–21199. [Google Scholar] [CrossRef] [PubMed]
- Van Dongen, J.J.M.; Langerak, A.W.; Brüggemann, M.; Evans, P.A.S.; Hummel, M.; Lavender, F.L.; Delabesse, E.; Davi, F.; Schuuring, E.; García-Sanz, R.; et al. Design and Standardization of PCR Primers and Protocols for Detection of Clonal Immunoglobulin and T-Cell Receptor Gene Recombinations in Suspect Lymphoproliferations: Report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia 2003, 17, 2257–2317. [Google Scholar] [CrossRef] [PubMed]
- Stamatopoulos, B.; Timbs, A.; Bruce, D.; Smith, T.; Clifford, R.; Robbe, P.; Burns, A.; Vavoulis, D.V.; Lopez, L.; Antoniou, P.; et al. Targeted Deep Sequencing Reveals Clinically Relevant Subclonal IgHV Rearrangements in Chronic Lymphocytic Leukemia. Leukemia 2017, 31, 837–845. [Google Scholar] [CrossRef] [PubMed]
- Nadeu, F.; Delgado, J.; Royo, C.; Baumann, T.; Stankovic, T.; Pinyol, M.; Jares, P.; Navarro, A.; Martin-Garcia, D.; Bea, S.; et al. Clinical Impact of Clonal and Subclonal TP53, SF3B1, BIRC3, NOTCH1, and ATM Mutations in Chronic Lymphocytic Leukemia. Blood 2016, 127, 2122–2130. [Google Scholar] [CrossRef] [PubMed]
- Rigolin, G.M.; Saccenti, E.; Bassi, C.; Lupini, L.; Quaglia, F.M.; Cavallari, M.; Martinelli, S.; Formigaro, L.; Lista, E.; Bardi, M.A.; et al. Extensive Next-Generation Sequencing Analysis in Chronic Lymphocytic Leukemia at Diagnosis: Clinical and Biological Correlations. J. Hematol. Oncol. 2016, 9, 88. [Google Scholar] [CrossRef] [PubMed]
- Pospisilova, S.; Gonzalez, D.; Malcikova, J.; Trbusek, M.; Rossi, D.; Kater, A.P.; Cymbalista, F.; Eichhorst, B.; Hallek, M.; Hillmen, P.; et al. ERIC Recommendations on TP53 Mutation Analysis in Chronic Lymphocytic Leukemia. Leukemia 2012, 26, 1458–1461. [Google Scholar] [CrossRef]
- Dreger, P.; Ritgen, M.; Böttcher, S.; Schmitz, N.; Kneba, M. The Prognostic Impact of Minimal Residual Disease Assessment after Stem Cell Transplantation for Chronic Lymphocytic Leukemia: Is Achievement of Molecular Remission Worthwhile? Leukemia 2005, 19, 1135–1138. [Google Scholar] [CrossRef]
- Varghese, A.M.; Rawstron, A.C.; Hillmen, P. Eradicating Minimal Residual Disease in Chronic Lymphocytic Leukemia: Should This Be the Goal of Treatment? Curr. Hematol. Malig. Rep. 2010, 5, 35–44. [Google Scholar] [CrossRef]
- Farina, L.; Carniti, C.; Dodero, A.; Vendramin, A.; Raganato, A.; Spina, F.; Patriarca, F.; Narni, F.; Benedetti, F.; Olivieri, A.; et al. Qualitative and Quantitative Polymerase Chain Reaction Monitoring of Minimal Residual Disease in Relapsed Chronic Lymphocytic Leukemia: Early Assessment Can Predict Long-Term Outcome after Reduced Intensity Allogeneic Transplantation. Haematologica 2009, 94, 654–662. [Google Scholar] [CrossRef]
- Nabhan, C.; Coutré, S.; Hillmen, P. Minimal Residual Disease in Chronic Lymphocytic Leukaemia: Is It Ready for Primetime? Br. J. Haematol. 2007, 136, 379–392. [Google Scholar] [CrossRef]
- Moreno, C. Clinical Significance of Minimal Residual Disease, as Assessed by Different Techniques, after Stem Cell Transplantation for Chronic Lymphocytic Leukemia. Blood 2006, 107, 4563–4569. [Google Scholar] [CrossRef] [PubMed]
- Rawstron, A.C.; Böttcher, S.; Letestu, R.; Villamor, N.; Fazi, C.; Kartsios, H.; de Tute, R.M.; Shingles, J.; Ritgen, M.; Moreno, C.; et al. Improving Efficiency and Sensitivity: European Research Initiative in CLL (ERIC) Update on the International Harmonised Approach for Flow Cytometric Residual Disease Monitoring in CLL. Leukemia 2013, 27, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Ladetto, M.; Pagliano, G.; Ferrero, S.; Cavallo, F.; Drandi, D.; Santo, L.; Crippa, C.; De Rosa, L.; Pregno, P.; Grasso, M.; et al. Major Tumor Shrinking and Persistent Molecular Remissions after Consolidation With Bortezomib, Thalidomide, and Dexamethasone in Patients With Autografted Myeloma. J. Clin. Oncol. 2010, 28, 2077–2084. [Google Scholar] [CrossRef] [PubMed]
- Galimberti, S.; Benedetti, E.; Morabito, F.; Papineschi, F.; Callea, V.; Fazzi, R.; Stelitano, C.; Andreazzoli, F.; Guerrini, F.; Ciabatti, E.; et al. Prognostic Role of Minimal Residual Disease in Multiple Myeloma Patients after Non-Myeloablative Allogeneic Transplantation. Leuk. Res. 2005, 29, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Putkonen, M.; Kairisto, V.; Juvonen, V.; Pelliniemi, T.-T.; Rauhala, A.; Itälä-Remes, M.; Remes, K. Depth of Response Assessed by Quantitative ASO-PCR Predicts the Outcome after Stem Cell Transplantation in Multiple Myeloma: Post-Transplant MRD in MM. Eur. J. Haematol. 2010, 85, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lopez, J.; Lahuerta, J.J.; Pepin, F.; Gonzalez, M.; Barrio, S.; Ayala, R.; Puig, N.; Montalban, M.A.; Paiva, B.; Weng, L.; et al. Prognostic Value of Deep Sequencing Method for Minimal Residual Disease Detection in Multiple Myeloma. Blood 2014, 123, 3073–3079. [Google Scholar] [CrossRef] [PubMed]
- Vij, R.; Mazumder, A.; Klinger, M.; O’Dea, D.; Paasch, J.; Martin, T.; Weng, L.; Park, J.; Fiala, M.; Faham, M.; et al. Deep Sequencing Reveals Myeloma Cells in Peripheral Blood in Majority of Multiple Myeloma Patients. Clin. Lymphoma Myeloma Leuk. 2014, 14, 131–139. [Google Scholar] [CrossRef]
- Avet-Loiseau, H.; Casneuf, T.; Chiu, C.; Laubach, J.P.; Lee, J.-J.; Moreau, P.; Plesner, T.; Nahi, H.; Khokhar, N.Z.; Qi, M.; et al. Evaluation of Minimal Residual Disease (MRD) in Relapsed/Refractory Multiple Myeloma (RRMM) Patients Treated with Daratumumab in Combination with Lenalidomide Plus Dexamethasone or Bortezomib Plus Dexamethasone. Blood 2016, 128, 246. [Google Scholar]
- Perrot, A.; Lauwers-Cances, V.; Corre, J.; Robillard, N.; Hulin, C.; Chretien, L.; Dejoie, T.; Maheo, S.; Stoppa, A.-M.; Karlin, L.; et al. Minimal Residual Disease Negativity Using Deep Sequencing Is a Major Prognostic Factor in Multiple Myeloma. Blood 2018, 132, 2456–2464. [Google Scholar] [CrossRef]
- Kotrova, M.; Trka, J.; Kneba, M.; Brüggemann, M. Is Next-Generation Sequencing the Way to Go for Residual Disease Monitoring in Acute Lymphoblastic Leukemia? Mol. Diagn. Ther. 2017, 21, 481–492. [Google Scholar] [CrossRef]
- Coustan-Smith, E. Use of Peripheral Blood Instead of Bone Marrow to Monitor Residual Disease in Children with Acute Lymphoblastic Leukemia. Blood 2002, 100, 2399–2402. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Emerson, R.O.; Sherwood, A.; Loh, M.L.; Angiolillo, A.; Howie, B.; Vogt, J.; Rieder, M.; Kirsch, I.; Carlson, C.; et al. Detection of Minimal Residual Disease in B Lymphoblastic Leukemia by High-Throughput Sequencing of IGH. Clin. Cancer Res. 2014, 20, 4540–4548. [Google Scholar] [CrossRef] [PubMed]
- Kotrova, M.; van der Velden, V.H.J.; van Dongen, J.J.M.; Formankova, R.; Sedlacek, P.; Brüggemann, M.; Zuna, J.; Stary, J.; Trka, J.; Fronkova, E. Next-Generation Sequencing Indicates False-Positive MRD Results and Better Predicts Prognosis after SCT in Patients with Childhood ALL. Bone Marrow Transplant. 2017, 52, 962–968. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, N.; Wesemann, D.R. Analyzing Immunoglobulin Repertoires. Front. Immunol. 2018, 9, 462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, W.H.; Tong, R.S.; Young, A.L.; Druley, T.E. Rare Event Detection Using Error-Corrected DNA and RNA Sequencing. J. Vis. Exp. 2018, 138, 57509. [Google Scholar] [CrossRef]
- Young, A.L.; Wong, T.N.; Hughes, A.E.O.; Heath, S.E.; Ley, T.J.; Link, D.C.; Druley, T.E. Quantifying Ultra-Rare Pre-Leukemic Clones via Targeted Error-Corrected Sequencing. Leukemia 2015, 29, 1608–1611. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, S.R.; Schmitt, M.W.; Fox, E.J.; Kohrn, B.F.; Salk, J.J.; Ahn, E.H.; Prindle, M.J.; Kuong, K.J.; Shen, J.-C.; Risques, R.-A.; et al. Detecting Ultralow-Frequency Mutations by Duplex Sequencing. Nat. Protoc. 2014, 9, 2586–2606. [Google Scholar] [CrossRef]
- The EuroClonality-NGS Working Group; Knecht, H.; Reigl, T.; Kotrová, M.; Appelt, F.; Stewart, P.; Vojtech, B.; Krejci, A.; Grioni, A.; Pal, K.; et al. Quality Control and Quantification in IG/TR next-Generation Sequencing Marker Identification: Protocols and Bioinformatic Functionalities by EuroClonality-NGS. Leukemia 2019, in press. [Google Scholar]
- The EuroClonality-NGS Working Group; Brüggemann, M.; Kotrová, M.; Knecht, H.; Bartram, J.; Boudjoghra, M.; Bystry, V.; Fazio, G.; Fronkova, E.; Giraud, M.; et al. Standardized Next-Generation Sequencing of Immunoglobulin and T-Cell Receptor Gene Recombinations for MRD Marker Identification in Acute Lymphoblastic Leukemia. A EuroClonality-NGS Validation Study. Leukemia 2019, in press. [Google Scholar]
- Langerak, A.W.; Brüggemann, M.; Davi, F.; Darzentas, N.; van Dongen, J.J.M.; Gonzalez, D.; Cazzaniga, G.; Giudicelli, V.; Lefranc, M.-P.; Giraud, M.; et al. High-Throughput Immunogenetics for Clinical and Research Applications in Immunohematology: Potential and Challenges. J. Immunol. 2017, 198, 3765–3774. [Google Scholar] [CrossRef] [Green Version]

| Methodology | Strengths | Weaknesses |
|---|---|---|
| Multiparameter Flow Cytometry | Fast; Absolute Quantification; Information at a cellular level; Wide availability | Variable antigen expression could lead to false negative results; High grade of expertise needed; Medium sensitivity with less than 8-colours |
| Allele-Specific Oligonucleotide PCR | High sensitivity | Time-consuming in the design of patient-specific primers; Requirement for optimal DNA quality and quantity |
| Digital PCR | Absolute quantification; High sensitivity; Avoids PCR inhibitors due to compartmentalization of target sequences | Lack of standardization No possibility to find new variants Allele-specific design |
| Next-Generation Sequencing or High-Throughput Sequencing | High Sensitivity (>10−6); Patient-specific primers not necessary; Versatility | Lack of standardization; High degree of bioinformatics expertise; Expensive |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez, R.; Ayala, R.; Martínez-López, J. Minimal Residual Disease Monitoring with Next-Generation Sequencing Methodologies in Hematological Malignancies. Int. J. Mol. Sci. 2019, 20, 2832. https://doi.org/10.3390/ijms20112832
Sánchez R, Ayala R, Martínez-López J. Minimal Residual Disease Monitoring with Next-Generation Sequencing Methodologies in Hematological Malignancies. International Journal of Molecular Sciences. 2019; 20(11):2832. https://doi.org/10.3390/ijms20112832
Chicago/Turabian StyleSánchez, Ricardo, Rosa Ayala, and Joaquín Martínez-López. 2019. "Minimal Residual Disease Monitoring with Next-Generation Sequencing Methodologies in Hematological Malignancies" International Journal of Molecular Sciences 20, no. 11: 2832. https://doi.org/10.3390/ijms20112832