Cell Clearing Systems Bridging Neuro-Immunity and Synaptic Plasticity

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
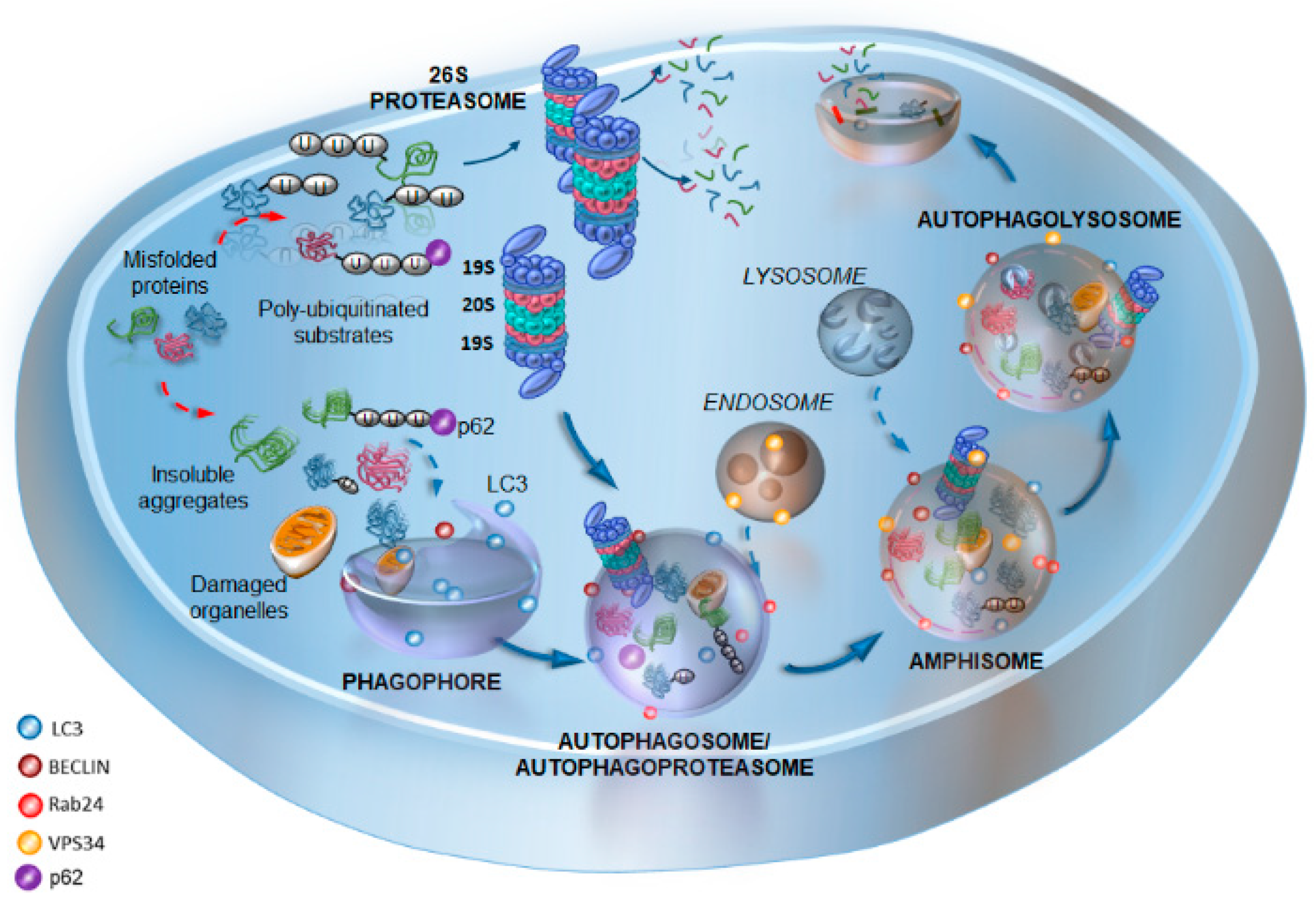
2. Cell Clearing Systems: Tracing the Path of the Interplay between Proteasome and Autophagy
3. Autophagy and Proteasome Tune Synaptic Plasticity by Modulating Neurotransmission and Immunity
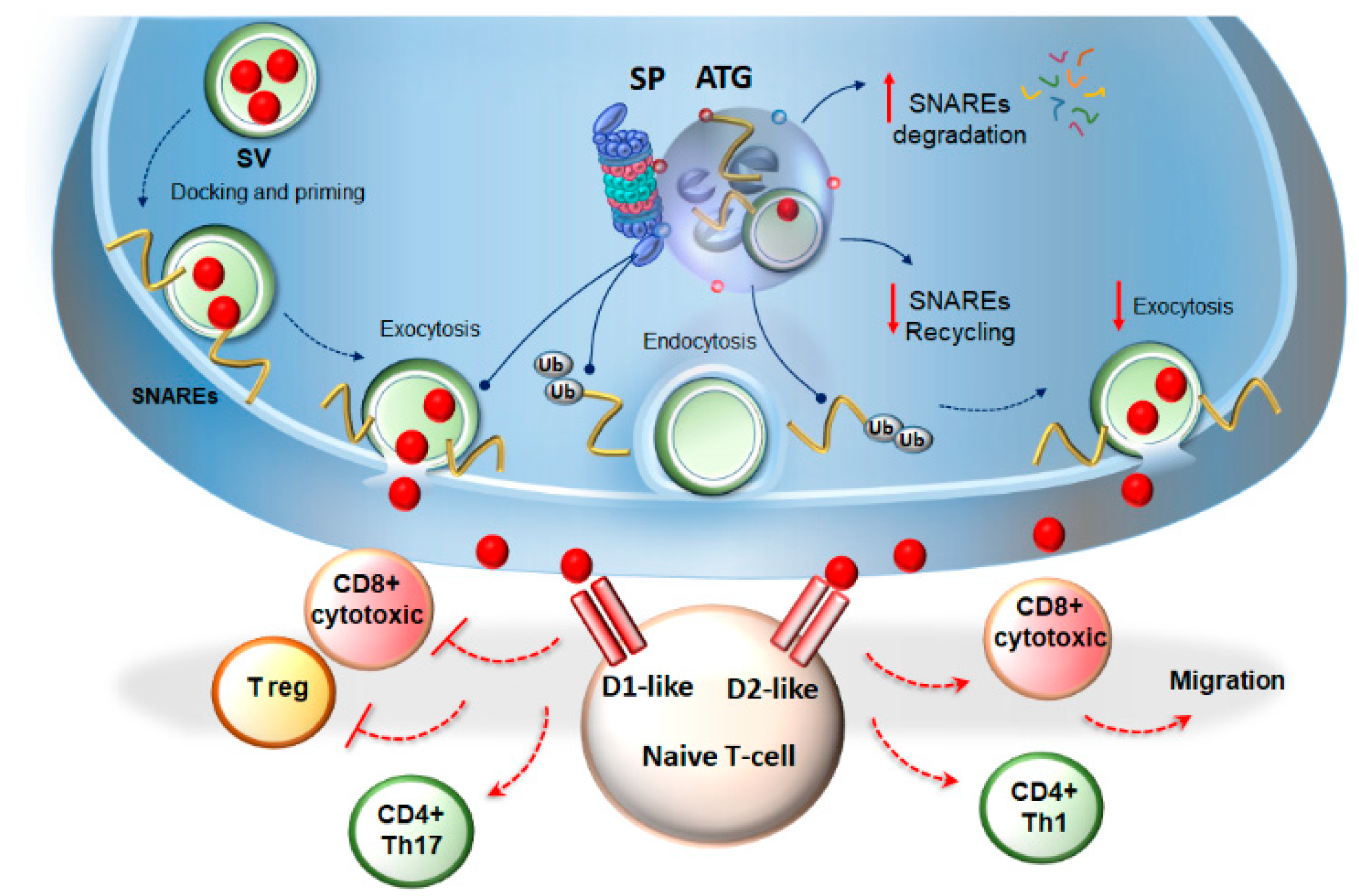
3.1. Autophagy- and Proteasome-Dependent Neurotransmission Linking Immune-Cells’ Activity and Synaptic Plasticity
3.2. Cell Clearing System in the Metabolism and Fate of Immune Cells
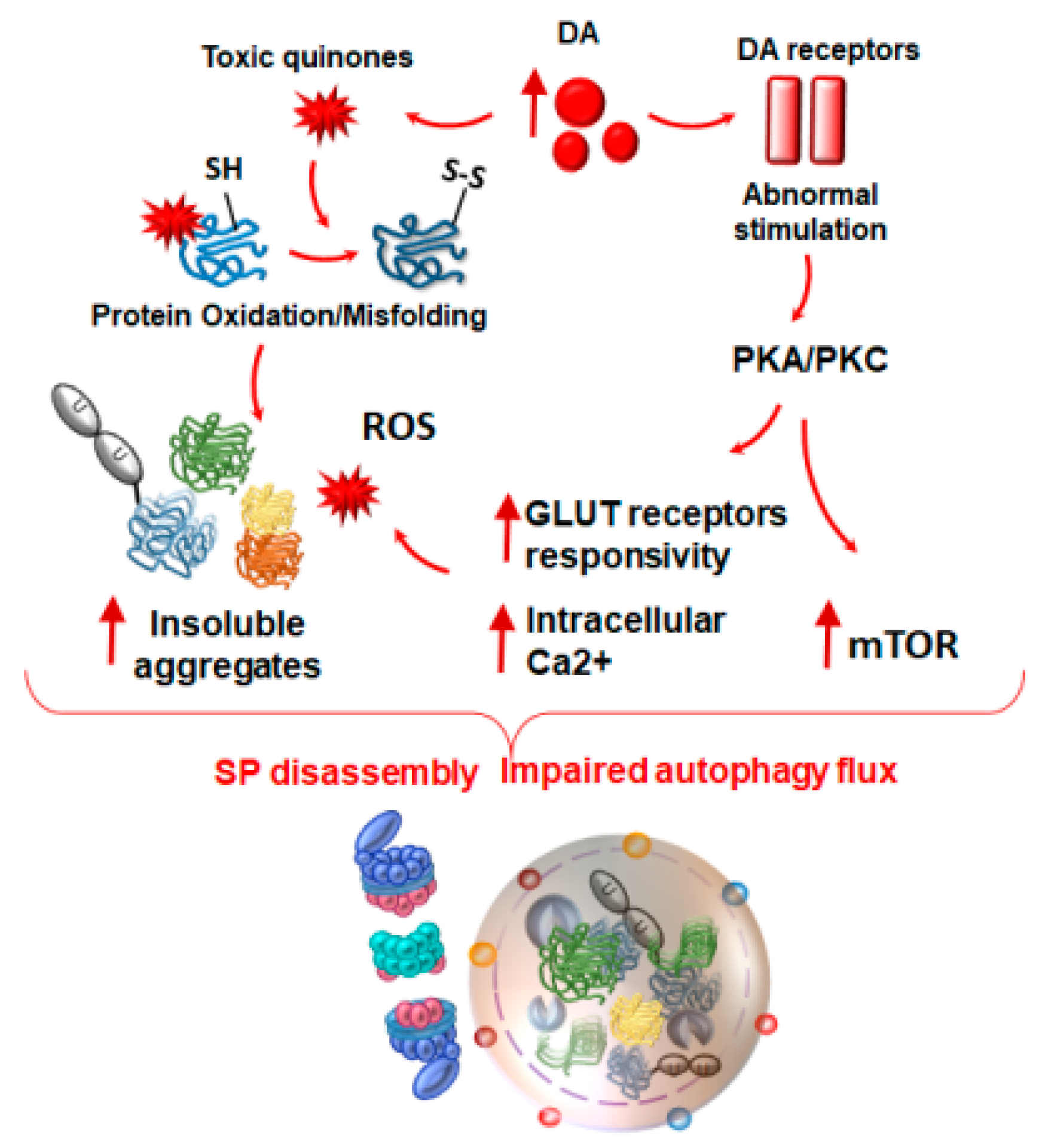
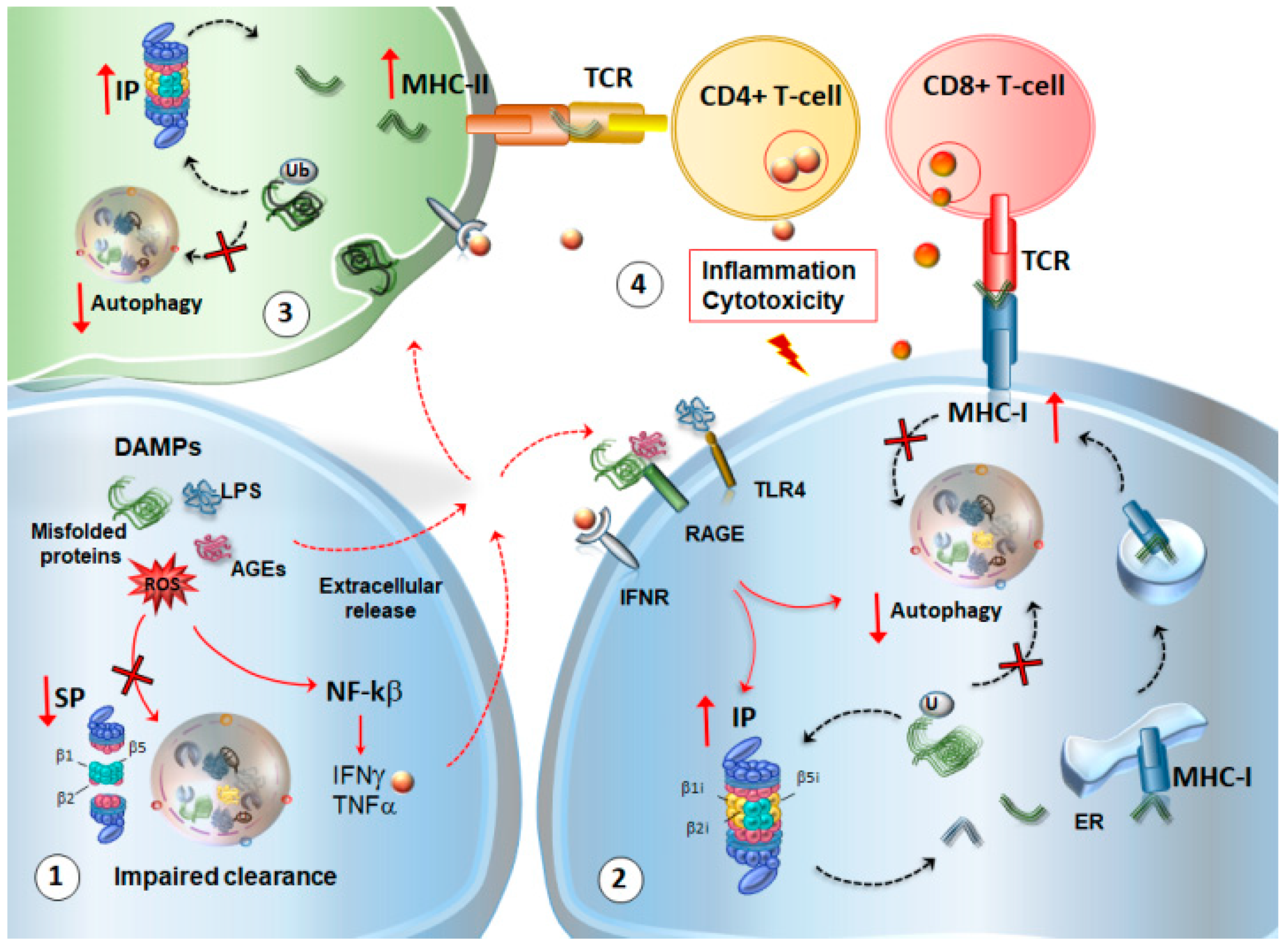
4. Autophagy and Proteasome Linking Altered Immunity and Synaptic Plasticity with Neurodegeneration
5. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| Ag | Antigen |
| APC | Antigen presenting cell |
| BDNF | Brain derived neurotrophic factor |
| CNS | Central nervous system |
| CTL | Cytotoxic T-lymphocyte |
| DA | Dopamine |
| DAMPs | Danger-associated molecular pattern molecules |
| DC | Dendritic cell |
| EAE | Experimental autoimmune encephalomyelitis |
| GLUT | Glutamate |
| HD | Huntingtin’s disease |
| HDAC6 | Histone deacetylase 6 |
| IFNγ | Interferon gamma |
| IL-1β | Interleukin 1 beta |
| IP | Immunoproteasome |
| Meth | Methamphetamine |
| MHC | Major histocompatibility complex |
| MS | Multiple sclerosis |
| mTOR | Mammalian target of rapamycin |
| NF-κB | Nuclear factor K beta |
| PD | Parkinson’s disease |
| Rab GTPase | Gtp bound ras proteins in brain |
| SNARE | Soluble Nsf attachment protein receptor |
| SP | Standard proteasome |
| SQSTM1 | Sequestosome-1 |
| SV | Synaptic vesicle |
| TCR | T-cells receptor |
| TLR4 | Toll-like receptor 4 |
| TNFα | Tumor necrosis factor alpha |
| UPS | Ubiquitin proteasome |
References
- Zipp, F.; Aktas, O. The brain as a target of inflammation: common pathways link inflammatory and neurodegenerative diseases. Trends Neurosci. 2006, 29, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Centonze, D.; Muzio, L.; Rossi, S.; Furlan, R.; Bernardi, G.; Martino, G. The link between inflammation, synaptic transmission and neurodegeneration in multiple sclerosis. Cell Death Differ. 2010, 17, 1083–1091. [Google Scholar] [CrossRef] [PubMed]
- Yirmiya, R.; Goshen, I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav. Immun. 2011, 25, 181–213. [Google Scholar] [CrossRef] [PubMed]
- Kerschensteiner, M.; Meinl, E.; Hohlfeld, R. Neuro-immune crosstalk in CNS diseases. Neuroscience 2009, 158, 1122–1132. [Google Scholar] [CrossRef] [PubMed]
- Kioussis, D.; Pachnis, V. Immune and nervous systems: more than just a superficial similarity? Immunity 2009, 31, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Limanaqi, F.; Biagioni, F.; Busceti, C.L.; Gaglione, A.; Fornai, F. A Sentinel in the Crosstalk Between the Nervous and Immune System: The (Immuno)-Proteasome. Front. Immunol. 2019, 10, 628. [Google Scholar] [CrossRef]
- Jovanova-Nesic, K.D.; Jankovic, B.D. The neuronal and immune memory systems as supervisors of neural plasticity and aging of the brain: from phenomenology to coding of information. Ann. N. Y. Acad. Sci. 2005, 1057, 279–295. [Google Scholar] [CrossRef]
- Schwartz, M.; Shechter, R. Protective autoimmunity functions by intracranial immunosurveillance to support the mind: The missing link between health and disease. Mol. Psychiatry. 2010, 15, 342–354. [Google Scholar] [CrossRef] [Green Version]
- Citri, A.; Malenka, R.C. Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology 2008, 33, 18–41. [Google Scholar] [CrossRef]
- Wang, J.; Hodes, G.E.; Zhang, H.; Zhang, S.; Zhao, W.; Golden, S.A.; Bi, W.; Menard, C.; Kana, V.; Leboeuf, M.; et al. Epigenetic modulation of inflammation and synaptic plasticity promotes resilience against stress in mice. Nat. Commun. 2018, 9, 477. [Google Scholar] [CrossRef]
- Limanaqi, F.; Gambardella, S.; Biagioni, F.; Busceti, C.L.; Fornai, F. Epigenetic Effects Induced by Methamphetamine and Methamphetamine-Dependent Oxidative Stress. Oxid. Med. Cell Longev. 2018, 22, 4982453. [Google Scholar] [CrossRef] [PubMed]
- Tournier, J.N.; Hellmann, A.Q. Neuro-immune connections: evidence for a neuro-immunological synapse. Trends Immunol. 2003, 24, 114–115. [Google Scholar] [CrossRef]
- Lorton, D.; Lubahn, C.L.; Estus, C.; Millar, B.A.; Carter, J.L.; Wood, C.A.; Bellinger, D.L. Bidirectional communication between the brain and the immune system: implications for physiological sleep and disorders with disrupted sleep. Neuroimmunomodulation 2006, 13, 357–374. [Google Scholar] [CrossRef] [PubMed]
- Lucin, K.M.; Wyss-Coray, T. Immune activation in brain aging and neurodegeneration: too much or too little? Neuron 2009, 64, 110–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, L.; Rauvala, H.; Gahmberg, C.G. Neuronal regulation of immune responses in the central nervous system. Trends Immunol. 2009, 30, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef] [PubMed]
- Plog, B.A.; Nedergaard, M. The Glymphatic System in Central Nervous System Health and Disease: Past, Present, and Future. Annu. Rev. Pathol. 2018, 24, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2016, 523, 337–341, Erratum in: Nature 2016, 533, 278. [Google Scholar] [CrossRef]
- Verheggen, I.C.M.; Van Boxtel, M.P.J.; Verhey, F.R.J.; Jansen, J.F.A.; Backes, W.H. Interaction between blood-brain barrier and glymphatic system in solute clearance. Neurosci. Biobehav. Rev. 2018, 90, 26–33. [Google Scholar] [CrossRef]
- Louveau, A.; Harris, T.H.; Kipnis, J. Revisiting the Mechanisms of CNS Immune Privilege. Trends Immunol. 2015, 36, 569–577. [Google Scholar] [CrossRef]
- Limanaqi, F.; Biagioni, F.; Gambardella, S.; Ryskalin, L.; Fornai, F. Interdependency Between Autophagy and Synaptic Vesicle Trafficking: Implications for Dopamine Release. Front. Mol. Neurosci. 2018, 11, 299. [Google Scholar] [CrossRef]
- Speese, S.D.; Trotta, N.; Rodesch, C.K.; Aravamudan, B.; Broadie, K. The ubiquitin proteasome system acutely regulates presynaptic protein turnover and synaptic efficacy. Curr. Biol. 2003, 13, 899–910. [Google Scholar] [CrossRef]
- Münz, C. Autophagy proteins in antigen processing for presentation on MHC molecules. Immunol. Rev. 2016, 272, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Palmowski, M.J.; Gileadi, U.; Salio, M.; Gallimore, A.; Millrain, M.; James, E.; Addey, C.; Scott, D.; Dyson, J.; Simpson, E.; et al. Role of immunoproteasomes in cross-presentation. J. Immunol. 2006, 177, 983–990. [Google Scholar] [CrossRef] [PubMed]
- Basler, M.; Kirk, C.J.; Groettrup, M. The immunoproteasome in antigen processing and other immunological functions. Curr. Opin. Immunol. 2013, 25, 74–80. [Google Scholar] [CrossRef] [Green Version]
- Hegde, A.N. The ubiquitin-proteasome pathway and synaptic plasticity. Learn. Mem. 2010, 17, 314–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegde, A.N. Proteolysis, synaptic plasticity and memory. Neurobiol. Learn. Mem. 2017, 138, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y. Emerging Concepts and Functions of Autophagy as a Regulator of Synaptic Components and Plasticity. Cells 2019, 8, 34. [Google Scholar] [CrossRef]
- Leung, C.S. Endogenous Antigen Presentation of MHC Class II Epitopes through Non-Autophagic Pathways. Front. Immunol. 2015, 6, 464. [Google Scholar] [CrossRef]
- Valečka, J.; Almeida, C.R.; Su, B.; Pierre, P.; Gatti, E. Autophagy and MHC-restricted antigen presentation. Mol. Immunol. 2018, 99, 163–170. [Google Scholar] [CrossRef]
- Shechter, R.; London, A.; Schwartz, M. Orchestrated leukocyte recruitment to immune-privileged sites: absolute barriers versus educational gates. Nat. Rev. Immunol. 2013, 13, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Schwab, N.; Schneider-Hohendorf, T.; Wiendl, H. Trafficking of lymphocytes into the CNS. Oncotarget 2015, 6, 17863–17864. [Google Scholar] [CrossRef] [PubMed]
- Goverman, J. Autoimmune T cell responses in the central nervous system. Nat. Rev. Immunol. 2009, 9, 393–407. [Google Scholar] [CrossRef] [Green Version]
- Mignini, F.; Streccioni, V.; Amenta, F. Autonomic innervation of immune organs and neuroimmune modulation. Auton. Autacoid. Pharmacol. 2003, 23, 1–25. [Google Scholar] [CrossRef]
- Levite, M. Neurotransmitters activate T-cells and elicit crucial functions via neurotransmitter receptors. Curr. Opin. Pharmacol. 2008, 8, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, C.; Basu, B.; Chakroborty, D.; Dasgupta, P.S.; Basu, S. The immunoregulatory role of dopamine: an update. Brain Behav. Immun. 2009, 24, 525–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganor, Y.; Levite, M. The neurotransmitter glutamate and human T cells: glutamate receptors and glutamate-induced direct and potent effects on normal human T cells, cancerous human leukemia and lymphoma T cells, and autoimmune human T cells. J. Neural Transm. (Vienna) 2014, 121, 983–1006. [Google Scholar] [CrossRef]
- Brabb, T.; von Dassow, P.; Ordonez, N.; Schnabel, B.; Duke, B.; Goverman, J. In situ tolerance within the central nervous system as a mechanism for preventing autoimmunity. J. Exp. Med. 2000, 192, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Teige, I.; Birnir, B.; Issazadeh-Navikas, S. Neuron-mediated generation of regulatory T cells from encephalitogenic T cells suppresses EAE. Nat. Med. 2006, 12, 518–525. [Google Scholar] [CrossRef]
- Na, S.Y.; Cao, Y.; Toben, C.; Nitschke, L.; Stadelmann, C.; Gold, R.; Schimpl, A.; Hünig, T. Naive CD8 T-cells initiate spontaneous autoimmunity to a sequestered model antigen of the central nervous system. Brain 2008, 13, 2353–2365. [Google Scholar] [CrossRef] [PubMed]
- Sosa, R.A.; Forsthuber, T.G. The critical role of antigen-presentation-induced cytokine crosstalk in the central nervous system in multiple sclerosis and experimental autoimmune encephalomyelitis. J. Interferon Cytokine Res. 2011, 31, 753–768. [Google Scholar] [CrossRef] [PubMed]
- Jarry, U.; Jeannin, P.; Pineau, L.; Donnou, S.; Delneste, Y.; Couez, D. Efficiently stimulated adult microglia cross-prime naive CD8+ T cells injected in the brain. Eur. J. Immunol. 2013, 43, 1173–1184. [Google Scholar] [CrossRef] [PubMed]
- Krakowski, M.L.; Owens, T. Naive T lymphocytes traffic to inflamed central nervous system, but require antigen recognition for activation. Eur. J. Immunol. 2000, 30, 1002–1009. [Google Scholar] [CrossRef] [Green Version]
- McMahon, E.J.; Bailey, S.L.; Castenada, C.V.; Waldner, H.; Miller, S.D. Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nat. Med. 2005, 11, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Cose, S.; Brammer, C.; Khanna, K.M.; Masopust, D.; Lefrancois, L. Evidence that a significant number of naive T cells enter non-lymphoid organs as part of a normal migratory pathway. Eur. J. Immunol. 2006, 36, 1423–1433. [Google Scholar] [CrossRef] [Green Version]
- Herz, J.; Paterka, M.; Niesner, R.A.; Brandt, A.U.; Siffrin, V.; Leuenberger, T.; Birkenstock, J.; Mossakowski, A.; Glumm, R.; Zipp, F.; et al. In vivo imaging of lymphocytes in the CNS reveals different behaviour of naive T cells in health and autoimmunity. J. Neuroinflamm. 2011, 8, 131. [Google Scholar] [CrossRef] [PubMed]
- Chastain, E.M.; Duncan, D.S.; Rodgers, J.M.; Miller, S.D. The role of antigen presenting cells in multiple sclerosis. Biochim. Biophys. Acta. 2010, 1812, 265–274. [Google Scholar] [CrossRef]
- Cebrián, C.; Zucca, F.A.; Mauri, P.; Steinbeck, J.A.; Studer, L.; Scherzer, C.R.; Kanter, E.; Budhu, S.; Mandelbaum, J.; Vonsattel, J.P.; et al. MHC-I expression renders catecholaminergic neurons susceptible to T-cell-mediated degeneration. Nat. Commun. 2014, 5, 3633. [Google Scholar] [CrossRef]
- Schwartz, M.; Deczkowska, A. Neurological Disease as a Failure of Brain-Immune Crosstalk: The Multiple Faces of Neuroinflammation. Trends Immunol. 2016, 37, 668–679. [Google Scholar] [CrossRef]
- Ferretti, M.T.; Bruno, M.A.; Ducatenzeiler, A.; Klein, W.L.; Cuello, A.C. Intracellular Aβ-oligomers and early inflammation in a model of Alzheimer’s disease. Neurobiol. Aging 2012, 33, 1329–1342. [Google Scholar] [CrossRef]
- Alirezaei, M.; Kemball, C.C.; Whitton, J.L. Autophagy, inflammation and neurodegenerative disease. Eur. J. Neurosci. 2010, 33, 197–204. [Google Scholar] [CrossRef] [Green Version]
- Alirezaei, M.; Kiosses, W.B.; Flynn, C.T.; Brady, N.R.; Fox, H.S. Disruption of neuronal autophagy by infected microglia results in neurodegeneration. PLoS One. 2008, 3, e2906. [Google Scholar] [CrossRef]
- Plaza-Zabala, A.; Sierra-Torre, V.; Sierra, A. Autophagy and Microglia: Novel Partners in Neurodegeneration and Aging. Int. J. Mol. Sci. 2017, 18, 598. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, M.; Biagioni, F.; Ryskalin, L.; Limanaqi, F.; Gambardella, S.; Frati, A.; Fornai, F. Ambiguous Effects of Autophagy Activation Following Hypoperfusion/Ischemia. Int. J. Mol. Sci. 2018, 19, 2756. [Google Scholar] [CrossRef] [PubMed]
- Ryskalin, L.; Limanaqi, F.; Biagioni, F.; Frati, A.; Esposito, V.; Calierno, M.T.; Lenzi, P.; Fornai, F. The emerging role of m-TOR up-regulation in brain Astrocytoma. Histol. Histopathol. 2017, 32, 413–431. [Google Scholar] [CrossRef]
- Fabrizi, C.; Pompili, E.; De Vito, S.; Somma, F.; Catizone, A.; Ricci, G.; Lenzi, P.; Fornai, F.; Fumagalli, L. Impairment of the autophagic flux in astrocytes intoxicated by trimethyltin. Neurotoxicol. 2016, 52, 12–22. [Google Scholar] [CrossRef]
- Giorgi, F.S.; Biagioni, F.; Lenzi, P.; Frati, A.; Fornai, F. The role of autophagy in epileptogenesis and in epilepsy-induced neuronal alterations. J. Neural. Transm. (Vienna) 2015, 122, 849–862. [Google Scholar] [CrossRef]
- Lenzi, P.; Marongiu, R.; Falleni, A.; Gelmetti, V.; Busceti, C.L.; Michiorri, S.; Valente, E.M.; Fornai, F. A subcellular analysis of genetic modulation of PINK1 on mitochondrial alterations, autophagy and cell death. Arch. Ital. Biol. 2012, 150, 194–217. [Google Scholar] [CrossRef]
- Pasquali, L.; Ruggieri, S.; Murri, L.; Paparelli, A.; Fornai, F. Does autophagy worsen or improve the survival of dopaminergic neurons? Parkinsonism Relat. Disord. 2009, 15, S24–S27. [Google Scholar] [CrossRef]
- Ferrucci, M.; Pasquali, L.; Ruggieri, S.; Paparelli, A.; Fornai, F. Alpha-synuclein and autophagy as common steps in neurodegeneration. Parkinsonism Relat. Disord. 2008, 14, S180–S184. [Google Scholar] [CrossRef]
- Isidoro, C.; Biagioni, F.; Giorgi, F.S.; Fulceri, F.; Paparelli, A.; Fornai, F. The role of autophagy on the survival of dopamine neurons. Curr. Top. Med. Chem. 2009, 9, 869–879. [Google Scholar]
- Ryskalin, L.; Busceti, C.L.; Limanaqi, F.; Biagioni, F.; Gambardella, S.; Fornai, F. A Focus on the Beneficial Effects of Alpha Synuclein and a Re-Appraisal of Synucleinopathies. Curr. Protein Pept. Sci. 2018, 19, 598–611. [Google Scholar] [CrossRef]
- Thibaudeau, T.A.; Anderson, R.T.; Smith, D.M. A common mechanism of proteasome impairment by neurodegenerative disease-associated oligomers. Nat. Commun. 2018, 9, 1097. [Google Scholar] [CrossRef]
- Jansen, A.H.P.; Reits, E.A.J.; Hol, E.M. The ubiquitin proteasome system in glia and its role in neurodegenerative diseases. Front. Mol. Neurosci. 2014, 7, 73. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, C.E.; Orr, A.; Tydlacka, S.; Li, S.H.; Li, X.J. Impaired ubiquitin-proteasome system activity in the synapses of Huntington’s disease mice. J. Cell Biol. 2008, 180, 1177–1189. [Google Scholar] [CrossRef] [PubMed]
- McNaught, K.S.; Belizaire, R.; Isacson, O.; Jenner, P.; Olanow, C.W. Altered proteasomal function in sporadic Parkinson’s disease. Exp. Neurol. 2003, 179, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Bizzozero, O.A. Decreased activity of the 20S proteasome in the brain white matter and gray matter of patients with multiple sclerosis. J. Neurochem. 2011, 117, 143–153. [Google Scholar] [CrossRef] [Green Version]
- van Scheppingen, J.; Broekaart, D.W.; Scholl, T.; Zuidberg, M.R.; Anink, J.J.; Spliet, W.G.; van Rijen, P.C.; Czech, T.; Hainfellner, J.A.; Feucht, M.; et al. Dysregulation of the (immuno)proteasome pathway in malformations of cortical development. J. Neuroinflammation 2016, 13, 202. [Google Scholar] [CrossRef] [Green Version]
- Graham, S.H.; Liu, H. Life and death in the trash heap: The ubiquitin proteasome pathway and UCHL1 in brain aging, neurodegenerative disease and cerebral Ischemia. Ageing Res. Rev. 2017, 34, 30–38. [Google Scholar] [CrossRef] [Green Version]
- Castino, R.; Lazzeri, G.; Lenzi, P.; Bellio, N.; Follo, C.; Ferrucci, M.; Fornai, F.; Isidoro, C. Suppression of autophagy precipitates neuronal cell death following low doses of methamphetamine. J. Neurochem. 2008, 106, 1426–1439. [Google Scholar] [CrossRef] [Green Version]
- Moszczynska, A.; Yamamoto, B.K. Methamphetamine oxidatively damages parkin and decreases the activity of 26S proteasome in vivo. J. Neurochem. 2011, 116, 1005–1017. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.; Chandramani-Shivalingappa, P.; Jin, H.; Ghosh, A.; Anantharam, V.; Ali, S.; Kanthasamy, A.G.; Kanthasamy, A. Methamphetamine-induced neurotoxicity linked to ubiquitin-proteasome system dysfunction and autophagy-related changes that can be modulated by protein kinase C delta in dopaminergic neuronal cells. Neuroscience 2012, 210, 308–332. [Google Scholar] [CrossRef]
- Lazzeri, G.; Biagioni, F.; Fulceri, F.; Busceti, C.L.; Scavuzzo, M.C.; Ippolito, C.; Salvetti, A.; Lenzi, P.; Fornai, F. mTOR Modulates Methamphetamine-Induced Toxicity through Cell Clearing Systems. Oxid. Med. Cell. Longev. 6124. [Google Scholar] [CrossRef] [PubMed]
- Pla, A.; Pascual, M.; Renau-Piqueras, J.; Guerri, C. TLR4 mediates the impairment of ubiquitin-proteasome and autophagy-lysosome pathways induced by ethanol treatment in brain. Cell Death Dis. 2014, 5, e1066. [Google Scholar] [CrossRef] [PubMed]
- Ryskalin, L.; Limanaqi, F.; Frati, A.; Busceti, C.L.; Fornai, F. mTOR-Related Brain Dysfunctions in Neuropsychiatric Disorders. Int. J. Mol. Sci. 2018, 19, 2226. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Cho, M.H.; Shim, W.H.; Kim, J.K.; Jeon, E.Y.; Kim, D.H.; Yoon, S.Y. Deficient autophagy in microglia impairs synaptic pruning and causes social behavioral defects. Mol. Psychiatry 2017, 22, 1576–1584. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Du, X.; Jiang, Y.; Botchway, B.O.A.; Hu, Z.; Fang, M. Inhibition of Autophagy in Microglia Alters Depressive-Like Behavior via BDNF Pathway in Postpartum Depression. Front. Psychiatry 2018, 9, 434. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Bento, C.F.; Deretic, V. Therapeutic targeting of autophagy in neurodegenerative and infectious diseases. J. Exp. Med. 2015, 212, 979–990. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef] [Green Version]
- Cadwell, K. Crosstalk between autophagy and inflammatory signalling pathways: balancing defence and homeostasis. Nat. Rev. Immunol. 2016, 16, 661–675. [Google Scholar] [CrossRef] [Green Version]
- Nikoletopoulou, V.; Sidiropoulou, K.; Kallergi, E.; Dalezios, Y.; Tavernarakis, N. Modulation of Autophagy by BDNF Underlies Synaptic Plasticity. Cell Metab. 2017, 26, 230–242. [Google Scholar] [CrossRef] [PubMed]
- Shehata, M.; Inokuchi, K. Does autophagy work in synaptic plasticity and memory? Rev. Neurosci. 2014, 25, 543–557. [Google Scholar] [CrossRef]
- Niedermann, G.; Grimm, R.; Geier, E.; Maurer, M.; Realini, C.; Gartmann, C.; Soll, J.; Omura, S.; Rechsteiner, M.C.; Baumeister, W.; et al. Potential Immunocompetence of Proteolytic Fragments Produced by Proteasomes before Evolution of the Vertebrate Immune System. J. Exp. Med. 1997, 186, 209–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrington, D.A.; Gregerson, D.S. Immunoproteasomes: structure, function, and antigen presentation. Prog. Mol. Biol. Transl. Sci. 2012, 109, 75–112. [Google Scholar] [PubMed]
- Johnston-Carey, H.K.; Pomatto, L.C.D.; Davies, K.J.A. The Immunoproteasome in Oxidative Stress, Aging, and Disease. Crit. Rev. Biochem. Mol. Biol. 2015, 51, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Hakim, V.; Cohen, L.D.; Zuchman, R.; Ziv, T.; Ziv, N.E. The effects of proteasomal inhibition on synaptic proteostasis. EMBO J. 2016, 35, 2238–2262. [Google Scholar] [CrossRef] [PubMed]
- Bingol, B.; Schuman, E.M. Synaptic protein degradation by the ubiquitin proteasome system. Curr. Opin. Neurobiol. 2005, 15, 536–541. [Google Scholar] [CrossRef] [PubMed]
- Lenzi, P.; Lazzeri, G.; Biagioni, F.; Busceti, C.L.; Gambardella, S.; Salvetti, A.; Fornai, F. The autophagoproteasome a novel cell clearing organelle in baseline and stimulated conditions. Front. Neuroanat. 2016, 10, 78. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhai, B.; Gygi, S.P.; Goldberg, A.L. mTOR inhibition activates overall protein degradation by the ubiquitin proteasome system as well as by autophagy. Proc. Natl. Acad. Sci. USA 2015, 112, 15790–15797. [Google Scholar] [CrossRef] [Green Version]
- Cohen-Kaplan, V.; Ciechanover, A.; Livneh, I. p62 at the crossroad of the ubiquitin-proteasome system and autophagy. Oncotarget 2016, 7, 83833–83834. [Google Scholar] [CrossRef] [Green Version]
- Ji, C.H.; Kwon, Y.T. Crosstalk and Interplay between the Ubiquitin-Proteasome System and Autophagy. Mol. Cells 2017, 40, 441–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korolchuk, V.I.; Menzies, F.M.; Rubinsztein, D.C. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 2010, 584, 1393–1398. [Google Scholar] [CrossRef] [PubMed]
- Ugun-Klusek, A.; Tatham, M.H.; Elkharaz, J.; Constantin-Teodosiu, D.; Lawler, K.; Mohamed, H.; Paine, S.M.; Anderson, G.; John Mayer, R.; Lowe, J.; et al. Continued 26S proteasome dysfunction in mouse brain cortical neurons impairs autophagy and the Keap1-Nrf2 oxidative defence pathway. Cell Death Dis. 2017, 8, e2531. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo-Pereira, M.E.; Rockwell, P.; Schmidt-Glenewinkel, T.; Serrano, P. Neuroinflammation and J2 prostaglandins: linking impairment of the ubiquitin-proteasome pathway and mitochondria to neurodegeneration. Front. Mol. Neurosci. 2015, 7, 104. [Google Scholar] [CrossRef]
- Kulbe, J.R.; Mulcahy Levy, J.M.; Coultrap, S.J.; Thorburn, A.; Bayer, K.U. Excitotoxic glutamate insults block autophagic flux in hippocampal neurons. Brain Res. 2014, 1542, 12–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Ji, X.; Liu, J.; Li, Z.; Zhang, X. Dopamine Receptor Subtypes Differentially Regulate Autophagy. Int. J. Mol. Sci. 2018, 19, 1540. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Yuan, W.; Li, Z.; Hou, Y.; Liu, F.; Feng, J. 6-Hydroxydopamine induces autophagic flux dysfunction by impairing transcription factor EB activation and lysosomal function in dopaminergic neurons and SH-SY5Y cells. Toxicol. Lett. 2018, 283, 58–68. [Google Scholar] [CrossRef]
- Du, D.; Hu, L.; Wu, J.; Wu, Q.; Cheng, W.; Guo, Y.; Guan, R.; Wang, Y.; Chen, X.; Yan, X.; et al. Neuroinflammation contributes to autophagy flux blockage in the neurons of rostral ventrolateral medulla in stress-induced hypertension rats. J. Neuroinflammation 2017, 14. [Google Scholar] [CrossRef]
- Barroso-Chinea, P.; Thiolat, M.L.; Bido, S.; Martinez, A.; Doudnikoff, E.; Baufreton, J.; Bourdenx, M.; Bloch, B.; Bezard, E.; Martin-Negrier, M.L. D1 dopamine receptor stimulation impairs striatal proteasome activity in Parkinsonism through 26S proteasome disassembly. Neurobiol. Dis. 2015, 78, 77–87. [Google Scholar] [CrossRef]
- Caldeira, M.V.; Curcio, M.; Leal, G.; Salazar, I.L.; Mele, M.; Santos, A.R.; Melo, C.V.; Pereira, P.; Canzoniero, L.M.; Duarte, C.B. Excitotoxic stimulation downregulates the ubiquitin-proteasome system through activation of NMDA receptors in cultured hippocampal neurons. Biochim. Biophys. Acta. 2013, 1832, 263–274. [Google Scholar] [CrossRef]
- Davies, K.J. Protein modification by oxidants and the role of proteolytic enzymes. Biochem. Soc. Trans. 1993, 21, 346–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coux, O.; Tanaka, K.; Goldberg, A.L. Structure and functions of the 20S and 26S proteasomes. Annu. Rev. Biochem. 1996, 65, 801–847. [Google Scholar] [CrossRef] [PubMed]
- Humbard, M.A.; Maupin-Furlow, J.A. Prokaryotic proteasomes: nanocompartments of degradation. J. Mol. Microbiol. Biotechnol. 2013, 23, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A.; Kwon, Y.T. Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp. Mol. Med. 2015, 47, e147. [Google Scholar] [CrossRef] [PubMed]
- Webb, J.L.; Ravikumar, B.; Atkins, J.; Skepper, J.N.; Rubinsztein, D.C. Alpha-Synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 2003, 278, 25009–25013. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Lee, J.H.; Rubinsztein, D.C. Tau degradation: the ubiquitin-proteasome system versus the autophagy-lysosome system. Prog. Neurobiol. 2013, 105, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Rudolf, R.; Khan, M.M.; Wild, F.; Hashemolhosseini, S. The impact of autophagy on peripheral synapses in health and disease. Front. Biosci. (Landmark Ed) 2016, 21, 1474–1487. [Google Scholar] [CrossRef] [PubMed]
- Akwa, Y.; Gondard, E.; Mann, A.; Capetillo-Zarate, E.; Alberdi, E.; Matute, C.; Marty, S.; Vaccari, T.; Lozano, A.M.; Baulieu, E.E.; et al. Synaptic activity protects against AD and FTD-like pathology via autophagic-lysosomal degradation. Mol. Psychiatry 2018, 23, 1530–1540. [Google Scholar] [CrossRef]
- Shen, D.N.; Zhang, L.H.; Wei, E.Q.; Yang, Y. Autophagy in synaptic development, function, and pathology. Neurosci. Bull. 2015, 31, 416–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vijayan, V.; Verstreken, P. Autophagy in the presynaptic compartment in health and disease. J. Cell Biol. 2017, 216, 1895–1906. [Google Scholar] [CrossRef]
- Münz, C. The Macroautophagy Machinery in Endo- and Exocytosis. J. Mol. Biol. 2017, 429, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Meijer, A.J.; Codogno, P. Signalling and autophagy regulation in health, aging and disease. Mol. Asp. Med. 2006, 27, 411–425. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Zientara-Rytter, K.; Subramani, S. The Roles of Ubiquitin-Binding Protein Shuttles in the Degradative Fate of Ubiquitinated Proteins in the Ubiquitin-Proteasome System and Autophagy. Cells 2019, 8, 40. [Google Scholar] [CrossRef] [PubMed]
- Chin, L.S.; Olzmann, J.A.; Li, L. Parkin-mediated ubiquitin signalling in aggresome formation and autophagy. Biochem. Soc. Trans. 2010, 38, 144–149. [Google Scholar] [CrossRef] [Green Version]
- Cakir, Z.; Funk, K.; Lauterwasser, J.; Todt, F.; Zerbes, R.M.; Oelgeklaus, A.; Tanaka, A.; van der Laan, M.; Edlich, F. Parkin promotes proteasomal degradation of misregulated BAX. J. Cell Sci. 2017, 130, 2903–2913. [Google Scholar] [CrossRef]
- Hook, S.S.; Orian, A.; Cowley, S.M.; Eisenman, R.N. Histone deacetylase 6 binds polyubiquitin through its zinc finger (PAZ domain) and copurifies with deubiquitinating enzymes. Proc. Natl. Acad. Sci. USA 2002, 99, 13425–13430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olzmann, J.A.; Li, L.; Chudaev, M.V.; Chen, J.; Perez, F.A.; Palmiter, R.D.; Chin, L.S. Parkin-mediated K63-linked polyubiquitination targets misfolded DJ-1 to aggresomes via binding to HDAC6. J. Cell Biol. 2007, 178, 1025–1038. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, H.; Ali, Y.O.; Ravichandran, M.; Dong, A.; Qiu, W.; MacKenzie, F.; Dhe-Paganon, S.; Arrowsmith, C.H.; Zhai, R.G. Protein aggregates are recruited to aggresome by histone deacetylase 6 via unanchored ubiquitin C termini. J. Biol. Chem. 2012, 287, 2317–2327. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, Y.; Kovacs, J.J.; McLaurin, A.; Vance, J.M.; Ito, A.; Yao, T.P. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003, 115, 727–738. [Google Scholar] [CrossRef]
- Iwata, A.; Riley, B.E.; Johnston, J.A.; Kopito, R.R. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J. Biol. Chem. 2005, 280, 40282–40292. [Google Scholar] [CrossRef]
- Into, T.; Inomata, M.; Takayama, E.; Takigawa, T. Autophagy in regulation of Toll-like receptor signaling. Cell Signal. 2012, 24, 1150–1162. [Google Scholar] [CrossRef]
- Lee, J.Y.; Koga, H.; Kawaguchi, Y.; Tang, W.; Wong, E.; Gao, Y.S.; Pandey, U.B.; Kaushik, S.; Tresse, E.; Lu, J.; et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010, 29, 969–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, U.B.; Nie, Z.; Batlevi, Y.; McCray, B.A.; Ritson, G.P.; Nedelsky, N.B.; Schwartz, S.L.; DiProspero, N.A.; Knight, M.A.; Schuldiner, O.; et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 2007, 447, 859–863. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell. Mol. Biol. Lett. 2016, 21, 29. [Google Scholar] [CrossRef] [PubMed]
- Bjørkøy, G.; Lamark, T.; Pankiv, S.; Øvervatn, A.; Brech, A.; Johansen, T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009, 452, 181–197. [Google Scholar] [CrossRef] [PubMed]
- Livneh, I.; Cohen-Kaplan, V.; Cohen-Rosenzweig, C.; Avni, N.; Ciechanover, A. The life cycle of the 26S proteasome: from birth, through regulation and function, and onto its death. Cell Res. 2016, 26, 869–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otero, M.G.; Alloatti, M.; Cromberg, L.E.; Almenar-Queralt, A.; Encalada, S.E.; Pozo Devoto, V.M.; Bruno, L.; Goldstein, L.S.; Falzone, T.L. Fast axonal transport of the proteasome complex depends on membrane interaction and molecular motor function. J. Cell. Sci. 2014, 127, 1537–1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryskalin, L.; Lazzeri, G.; Flaibani, M.; Biagioni, F.; Gambardella, S.; Frati, A.; Fornai, F. mTOR-Dependent Cell Proliferation in the Brain. Biomed. Res. Int. 2017, 2017, 7082696. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.G.; Zhang, H. ULK1 cycling: The ups and downs of the autophagy response. J. Cell Biol. 2016, 215, 757–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papinski, D.; Kraft, C. Regulation of autophagy by signaling through the Atg1/ULK1 complex. J. Mol. Biol. 2016, 428, 1725–1741. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Nazio, F.; Carinci, M.; Valacca, C.; Bielli, P.; Strappazzon, F.; Antonioli, M.; Ciccosanti, F.; Rodolfo, C.; Campello, S.; Fimia, G.M.; et al. Fine-tuning of ULK1 mRNA and protein levels is required for autophagy oscillation. J. Cell Biol. 2016, 215, 841–856. [Google Scholar] [CrossRef]
- Liu, C.C.; Lin, Y.C.; Chen, Y.H.; Chen, C.M.; Pang, L.Y.; Chen, H.A.; Wu, P.R.; Lin, M.Y.; Jiang, S.T.; Tsai, T.F.; et al. Cul3-KLHL20 ubiquitin ligase governs the turnover of ULK1 and VPS34 complexes to control autophagy termination. Mol. Cell. 2016, 61, 84–97. [Google Scholar] [CrossRef]
- Marshall, R.S.; Vierstra, R.D. To save or degrade: balancing proteasome homeostasis to maximize cell survival. Autophagy 2018, 14, 2029–2031. [Google Scholar] [CrossRef] [PubMed]
- Marshall, R.S.; Vierstra, R.D. Eat or be eaten: The autophagic plight of inactive 26S proteasomes. Autophagy 2015, 11, 1927–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen-Kaplan, V.; Livneh, I.; Avni, N.; Fabre, B.; Ziv, T.; Kwon, Y.T.; Ciechanover, A. p62-and ubiquitin-dependent stress-induced autophagy of the mammalian 26S proteasome. Proc. Natl. Acad. Sci. USA 2016, 113, E7490–E7499. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef]
- Sato, S.; Uchihara, T.; Fukuda, T.; Noda, S.; Kondo, H.; Saiki, S.; Komatsu, M.; Uchiyama, Y.; Tanaka, K.; Hattori, N. Loss of autophagy in dopaminergic neurons causes Lewy pathology and motor dysfunction in aged mice. Sci. Rep. 2018, 8, 2813. [Google Scholar] [CrossRef] [Green Version]
- Romero-Granados, R.; Fontán-Lozano, Á.; Aguilar-Montilla, F.J.; Carrión, Á.M. Postnatal proteasome inhibition induces neurodegeneration and cognitive deficiencies in adult mice: a new model of neurodevelopment syndrome. PLoS ONE 2011, 6, e28927. [Google Scholar] [CrossRef] [PubMed]
- Fornai, F.; Lenzi, P.; Gesi, M.; Ferrucci, M.; Lazzeri, G.; Busceti, C.L.; Ruffoli, R.; Soldani, P.; Ruggieri, S.; Alessandri, M.G.; et al. Fine structure and biochemical mechanisms underlying nigrostriatal inclusions and cell death after proteasome inhibition. J. Neurosci. 2003, 23, 8955–8966. [Google Scholar] [CrossRef]
- Fornai, F.; Lenzi, P.; Gesi, M.; Ferrucci, M.; Lazzeri, G.; Capobianco, L.; de Blasi, A.; Battaglia, G.; Nicoletti, F.; Ruggieri, S.; et al. Similarities between methamphetamine toxicity and proteasome inhibition. Ann. N. Y. Acad. Sci. 2004, 1025, 162–170. [Google Scholar] [CrossRef]
- Korolchuk, V.I.; Mansilla, A.; Menzies, F.M.; Rubinsztein, D.C. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol. Cell 2009, 33, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Yang, Y.P.; Mao, C.J.; Liu, L.; Zheng, H.F.; Hu, L.F.; Liu, C.F. Crosstalk between the proteasome system and autophagy in the clearance of α-synuclein. Acta Pharmacol. Sin. 2013, 34, 674–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, K.; Dunner, K.; McConkey, D.J. Proteasome inhibitors activate autophagy as a cytoprotective response in human prostate cancer cells. Oncogene 2009, 29, 451–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, D.; Wang, W.; Zhuang, J.; Zhao, Z. Proteasome inhibitor-induced autophagy in PC12 cells overexpressing A53T mutant α-synuclein. Mol. Med. Rep. 2014, 11, 1655–1660. [Google Scholar] [CrossRef] [Green Version]
- Bao, W.; Gu, Y.; Ta, L.; Wang, K.; Xu, Z. Induction of autophagy by the MG 132 proteasome inhibitor is associated with endoplasmic reticulum stress in MCF 7 cells. Mol. Med. Rep. 2016, 13, 796–804. [Google Scholar] [CrossRef]
- Van Kerkhof, P.; dos Santos, C.M.A.; Sachse, M.; Klumperman, J.; Bu, G.; Strous, G.J. Proteasome Inhibitors Block a Late Step in Lysosomal Transport of Selected Membrane but not Soluble Proteins. Pfeffer SR, ed. Mol. Biol. Cell 2001, 12, 2556–2566. [Google Scholar] [CrossRef] [PubMed]
- Kleijnen, M.F.; Kirkpatrick, D.S.; Gygi, S.P. The ubiquitin–proteasome system regulates membrane fusion of yeast vacuoles. EMBO J. 2007, 26, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Villarroel-Campos, D.; Henríquez, D.R.; Bodaleo, F.J.; Oguchi, M.E.; Bronfman, F.C.; Fukuda, M.; Gonzalez-Billault, C. Rab35 Functions in Axon Elongation Are Regulated by P53-Related Protein Kinase in a Mechanism That Involves Rab35 Protein Degradation and the Microtubule-Associated Protein 1B. J. Neurosci. 2016, 36, 7298–7313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheehan, P.; Zhu, M.; Beskow, A.; Vollmer, C.; Waites, C.L. Activity-dependent degradation of synaptic vesicle proteins requires Rab35 and the ESCRT pathway. J. Neurosci. 2016, 36, 8668–8686. [Google Scholar] [CrossRef]
- Shin, D.; Na, W.; Lee, J.H.; Kim, G.; Baek, J.; Park, S.H.; Choi, C.Y.; Lee, S. Site-specific monoubiquitination downregulates Rab5 by disrupting effector binding and guanine nucleotide conversion. Elife 2017, 6, e29154. [Google Scholar] [CrossRef] [PubMed]
- Yun, Y.S.; Kim, K.H.; Tschida, B.; Chen, L.; Largaespada, D.; Kim, D-H. mTORC1 Coordinates Protein Synthesis and Immunoproteasome Formation via PRAS40 to Prevent Accumulation of Protein Stress. Mol. Cell 2016, 61, 625–639. [Google Scholar] [CrossRef]
- Zhang, H.M.; Fu, J.; Hamilton, R.; Diaz, V.; Zhang, Y. The mammalian target of rapamycin modulates the immunoproteasome system in the heart. J. Mol. Cell Cardiol. 2015, 86, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Lelegren, M.; Liu, Y.; Ross, C.; Tardif, S.; Salmon, A.B. Pharmaceutical inhibition of mTOR in the common marmoset: effect of rapamycin on regulators of proteostasis in a non-human primate. Pathobiol. Aging Age Relat. Dis. 2016, 6, 31793. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zou, L.X.; Lin, Q.Y.; Yan, X.; Bi, H.L.; Xie, X.; Wang, S.; Wang, Q.S.; Zhang, Y.L.; Li, H.H. Resveratrol as a new inhibitor of immunoproteasome prevents PTEN degradation and attenuates cardiac hypertrophy after pressure overload. Redox Biol. 2019, 20, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Krüger, E.; Kloetzel, P.M. Immunoproteasomes at the interface of innate and adaptive immune responses: two faces of one enzyme. Curr. Opin. Immunol. 2012, 24, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Dodson, M.; Redmann, M.; Rajasekaran, N.S.; Darley-Usmar, V.; Zhang, J. KEAP1-NRF2 signalling and autophagy in protection against oxidative and reductive proteotoxicity. Biochem. J. 2015, 469, 347–355. [Google Scholar] [CrossRef]
- Simon, H.U.; Friis, R.; Tait, S.W.; Ryan, K.M. Retrograde signaling from autophagy modulates stress responses. Sci. Signal. 2017, 10, eaag2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez, D.; Torres, C.A.; Setlik, W.; Cebrián, C.; Mosharov, E.V.; Tang, G.; Cheng, H.C.; Kholodilov, N.; Yarygina, O.; Burke, R.E.; et al. Regulation of presynaptic neurotransmission by macroautophagy. Neuron 2012, 74, 277–284. [Google Scholar] [CrossRef] [Green Version]
- Torres, C.A.; Sulzer, D. Macroautophagy can press a brake on presynaptic neurotransmission. Autophagy 2012, 8, 1540–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, M.; Shen, K. The role of the ubiquitin proteasome system in synapse remodeling and neurodegenerative diseases. Bioessays 2008, 30, 1075–1083. [Google Scholar] [CrossRef] [Green Version]
- Bingol, B.; Sheng, M. Deconstruction for reconstruction: the role of proteolysis in neural plasticity and disease. Neuron 2011, 69, 22–32. [Google Scholar] [CrossRef]
- Durairaj, G.; Kaiser, P. The 26S proteasome and initiation of gene transcription. Biomolecules 2014, 4, 827–847. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Hegde, A.N.; Martin, K.C. The ubiquitin proteasome system functions as an inhibitory constraint on synaptic strengthening. Curr. Biol. 2003, 13, 887–898. [Google Scholar] [CrossRef]
- Tang, S.J.; Reis, G.; Kang, H.; Gingras, A.-C.; Sonenberg, N.; Schuman, E.M. A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc. Natl. Acad. Sci. USA 2002, 99, 467–472. [Google Scholar] [CrossRef]
- Upadhya, S.C.; Smith, T.K.; Hegde, A.N. Ubiquitin-proteasome-mediated CREB repressor degradation during induction of long-term facilitation. J. Neurochem. 2004, 91, 210–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinetti, G.V.; Schweizer, F.E. Ubiquitination acutely regulates presynaptic neurotransmitter release in mammalian neurons. J. Neurosci. 2010, 30, 3157–3166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, C.; Bach, S.V.; Haynes, K.A.; Hegde, A.N. Proteasome Modulates Positive and Negative Translational Regulators in Long-Term Synaptic Plasticity. J. Neurosci. 2014, 34, 3171–3182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aravamudan, B.; Broadie, K. Synaptic Drosophila UNC-13 is regulated by antagonistic G-protein pathways via a proteasome-dependent degradation mechanism. J. Neurobiol. 2003, 54, 417–438. [Google Scholar] [CrossRef] [PubMed]
- Wentzel, C.; Delvendahl, I.; Sydlik, S.; Georgiev, O.; Müller, M. Dysbindin links presynaptic proteasome function to homeostatic recruitment of low release probability vesicles. Nat. Commun. 2018, 9, 267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willeumier, K.; Pulst, S.M.; Schweizer, F.E. Proteasome Inhibition Triggers Activity-Dependent Increase in the Size of the Recycling Vesicle Pool in Cultured Hippocampal Neurons. J. Neurosci. 2006, 26, 11333–11341. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.K.; Kashyap, M.P.; Tripathi, V.K.; Singh, S.; Garg, G.; Rizvi, S.I. Neuroprotection Through Rapamycin-Induced Activation of Autophagy and PI3K/Akt1/mTOR/CREB Signaling Against Amyloid-β-Induced Oxidative Stress, Synaptic/Neurotransmission Dysfunction, and Neurodegeneration in Adult Rats. Mol. Neurobiol. 2017, 54, 5815–5828. [Google Scholar] [CrossRef] [PubMed]
- Sunkaria, A.; Yadav, A.; Bhardwaj, S.; Sandhir, R. Postnatal Proteasome Inhibition Promotes Amyloid-β Aggregation in Hippocampus and Impairs Spatial Learning in Adult Mice. Neuroscience 2017, 367, 47–59. [Google Scholar] [CrossRef]
- Laurén, J.; Gimbel, D.A.; Nygaard, H.B.; Gilbert, J.W.; Strittmatter, S.M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 2009, 457, 1128–1132. [Google Scholar] [CrossRef] [PubMed]
- Ghiglieri, V.; Calabrese, V.; Calabresi, P. Alpha-Synuclein: From Early Synaptic Dysfunction to Neurodegeneration. Front. Neurol. 2018, 9, 295. [Google Scholar] [CrossRef] [PubMed]
- Phan, J.A.; Stokholm, K.; Zareba-Paslawska, J.; Jakobsen, S.; Vang, K.; Gjedde, A.; Landau, A.M.; Romero-Ramos, M. Early synaptic dysfunction induced by α-synuclein in a rat model of Parkinson’s disease. Sci. Rep. 2017, 7, 6363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, K.; Wei, Q.; Liu, F.F.; Hu, F.; Xie, A.J.; Zhu, L.Q.; Liu, D. Synaptic Dysfunction in Alzheimer’s Disease: Aβ, Tau, and Epigenetic Alterations. Mol. Neurobiol. 2018, 55, 3021–3032. [Google Scholar] [CrossRef]
- Chin, L.S.; Vavalle, J.P.; Li, L. Staring, a novel E3 ubiquitin-protein ligase that targets syntaxin 1 for degradation. J. Biol. Chem. 2002, 277, 35071–35079. [Google Scholar] [CrossRef]
- Subramaniam, M.; Kern, B.; Vogel, S.; Klose, V.; Schneider, G.; Roeper, J. Selective increase of in vivo firing frequencies in DA SN neurons after proteasome inhibition in the ventral midbrain. Eur. J. Neurosci. 2014, 40, 2898–2909. [Google Scholar] [CrossRef] [PubMed]
- Konieczny, J.; Lenda, T.; Czarnecka, A. Early increase in dopamine release in the ipsilateral striatum after unilateral intranigral administration of lactacystin produces spontaneous contralateral rotations in rats. Neuroscience 2016, 324, 92–106. [Google Scholar] [CrossRef]
- Lillethorup, T.P.; Glud, A.N.; Alstrup, A.K.O.; Mikkelsen, T.W.; Nielsen, E.H.; Zaer, H.; Doudet, D.J.; Brooks, D.J.; Sørensen, J.C.H.; Orlowski, D.; et al. Nigrostriatal proteasome inhibition impairs dopamine neurotransmission and motor function in minipigs. Exp. Neurol. 2018, 303, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Kim, O.J. A single mutation at lysine 241 alters expression and trafficking of the D2 dopamine receptor. J. Recept. Signal Transduct. Res. 2008, 28, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Rondou, P.; Haegeman, G.; Vanhoenacker, P.; Van Craenenbroeck, K. BTB protein KLHL12 targets the dopamine D4 receptor for ubiquitination by a Cul3-based E3 ligase. J. Biol. Chem. 2008, 283, 11083–11096. [Google Scholar] [CrossRef]
- Alonso, V.; Friedman, P.A. Minireview: ubiquitination-regulated G protein-coupled receptor signaling and trafficking. Mol. Endocrinol. 2013, 27, 558–572. [Google Scholar] [CrossRef] [PubMed]
- Peeler, J.C.; Schedin-Weiss, S.; Soula, M.; Kazmi, M.A.; Sakmar, T.P. Isopeptide and ester bond ubiquitination both regulate degradation of the human dopamine receptor 4. J. Biol. Chem. 2017, 292, 21623–21630. [Google Scholar] [CrossRef] [Green Version]
- Milnerwood, A.J.; Raymond, L.A. Early synaptic pathophysiology in neurodegeneration: insights from Huntington’s disease. Trends Neurosci. 2010, 33, 513–523. [Google Scholar] [CrossRef]
- Schirinzi, T.; Madeo, G.; Martella, G.; Maltese, M.; Picconi, B.; Calabresi, P.; Pisani, A. Early synaptic dysfunction in Parkinson’s disease: Insights from animal models. Mov. Disord. 2016, 31, 802–813. [Google Scholar] [CrossRef]
- Krasnova, I.N.; Justinova, Z.; Cadet, J.L. Methamphetamine addiction: involvement of CREB and neuroinflammatory signaling pathways. Psychopharmacology (Berl) 2016, 233, 1945–1962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krasnova, I.N.; Cadet, J.L. Methamphetamine toxicity and messengers of death. Brain Res. Rev. 2009, 60, 379–407. [Google Scholar] [CrossRef] [PubMed]
- Moratalla, R.; Khairnar, A.; Simola, N.; Granado, N.; García-Montes, J.R.; Porceddu, P.F.; Tizabi, Y.; Costa, G.; Morelli, M. Amphetamine-related drugs neurotoxicity in humans and in experimental animals: Main mechanisms. Prog. Neurobiol. 2017, 155, 149–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tata, D.A.; Yamamoto, B.K. Interactions between methamphetamine and environmental stress: role of oxidative stress, glutamate and mitochondrial dysfunction. Addiction 2007, 102, 49–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.H.; Wu, W.R.; Lee, L.M.; Huang, P.R.; Chen, J.C. mTOR signaling in the nucleus accumbens mediates behavioral sensitization to methamphetamine. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 86, 331–339. [Google Scholar] [CrossRef]
- Bentea, E.; Verbruggen, L.; Massie, A. The Proteasome Inhibition Model of Parkinson’s Disease. J. Parkinsons Dis. 2017, 7, 31–63. [Google Scholar] [CrossRef] [Green Version]
- Keller, J.N.; Hanni, K.B.; Marksberry, W.R. Impaired proteasome function in Alzheimer’s disease. J. Neurochem. 2000, 75, 436–439. [Google Scholar] [CrossRef]
- Seo, H.; Sonntag, K.C.; Isacson, O. Generalized brain and skin proteasome inhibition in Huntington’s disease. Ann. Neurol. 2004, 56, 319–328. [Google Scholar] [CrossRef]
- Rubio, M.D.; Wood, K.; Haroutunian, V.; Meador-Woodruff, J.H. Dysfunction of the ubiquitin proteasome and ubiquitin-like systems in schizophrenia. Neuropsychopharmacology 2013, 38, 1910–1920. [Google Scholar] [CrossRef]
- Siman, R.; Cocca, R.; Dong, Y. The mTOR Inhibitor Rapamycin Mitigates Perforant Pathway Neurodegeneration and Synapse Loss in a Mouse Model of Early-Stage Alzheimer-Type Tauopathy. PLoS ONE 2015, 10, e0142340. [Google Scholar] [CrossRef]
- Schneider, M.; de Vries, P.J.; Schönig, K.; Rößner, V.; Waltereit, R. mTOR inhibitor reverses autistic-like social deficit behaviours in adult rats with both Tsc2 haploinsufficiency and developmental status epilepticus. Eur. Arch. Psychiatry Clin. Neurosci. 2017, 267, 455–463. [Google Scholar] [CrossRef]
- Masini, D.; Bonito-Oliva, A.; Bertho, M.; Fisone, G. Inhibition of mTORC1 Signaling Reverts Cognitive and Affective Deficits in a Mouse Model of Parkinson’s Disease. Front. Neurol. 2018, 9, 208. [Google Scholar] [CrossRef] [PubMed]
- Kara, N.Z.; Flaisher-Grinberg, S.; Anderson, G.W.; Agam, G.; Einat, H. Mood-stabilizing effects of rapamycin and its analog temsirolimus: relevance to autophagy. Behav. Pharmacol. 2018, 29, 379–384. [Google Scholar] [CrossRef]
- Sillitoe, R.V.; Vogel, M.W. Desire, disease, and the origins of the dopaminergic system. Schizophr. Bull. 2008, 34, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Levite, M.; Chowers, Y.; Ganor, Y.; Besser, M.; Hershkovits, R.; Cahalon, L. Dopamine interacts directly with its D3 and D2 receptors on normal human T-cells, and activates beta1 integrin function. Eur. J. Immunol. 2001, 31, 3504–3512. [Google Scholar] [CrossRef]
- Fiserová, A.; Starec, M.; Kuldová, M.; Kovárů, H.; Páv, M.; Vannucci, L.; Pospísil, M. Effects of D2-dopamine and alpha-adrenoceptor antagonists in stress induced changes on immune responsiveness of mice. J. Neuroimmunol. 2002, 130, 55–65. [Google Scholar]
- Buttarelli, F.R.; Fanciulli, A.; Pellicano, C.; Pontieri, F.E. The dopaminergic system in peripheral blood lymphocytes: from physiology to pharmacology and potential applications to neuropsychiatric disorders. Curr. Neuropharmacol. 2011, 9, 278–288. [Google Scholar] [PubMed]
- Tanabe, S.; Yamashita, T. The role of immune cells in brain development and neurodevelopmental diseases. Int. Immunol. 2018, 30, 437–444. [Google Scholar] [CrossRef]
- Smolders, J.; Heutinck, K.M.; Fransen, N.L.; Remmerswaal, E.B.M.; Hombrink, P.; Ten Berge, I.J.M.; van Lier, R.A.W.; Huitinga, I.; Hamann, J. Tissue-resident memory T-cells populate the human brain. Nat. Commun. 2018, 9, 4593. [Google Scholar] [CrossRef]
- Zarif, H.; Hosseiny, S.; Paquet, A.; Lebrigand, K.; Arguel, M.J.; Cazareth, J.; Lazzari, A.; Heurteaux, C.; Glaichenhaus, N.; Chabry, J.; et al. CD4(+) T Cells Have a Permissive Effect on Enriched Environment-Induced Hippocampus Synaptic Plasticity. Front. Synaptic Neurosci. 2018, 10, 14. [Google Scholar] [CrossRef]
- Zarif, H.; Nicolas, S.; Guyot, M.; Hosseiny, S.; Lazzari, A.; Canali, M.M.; Cazareth, J.; Brau, F.; Golzné, V.; Dourneau, E.; et al. CD8(+) T cells are essential for the effects of enriched environment on hippocampus-dependent behavior, hippocampal neurogenesis and synaptic plasticity. Brain Behav. Immun. 2018, 69, 235–254. [Google Scholar] [CrossRef]
- Haas, K.F.; Miller, S.L.; Friedman, D.B.; Broadie, K. The ubiquitin-proteasome system postsynaptically regulates glutamatergic synaptic function. Mol. Cell. Neurosci. 2007, 35, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Nikoletopoulou, V.; Tavernarakis, N. Regulation and Roles of Autophagy at Synapses. Trends Cell. Biol. 2018, 28, 646–661. [Google Scholar] [CrossRef]
- Hogins, J.; Crawford, D.C.; Jiang, X.; Mennerick, S. Presynaptic silencing is an endogenous neuroprotectant during excitotoxic insults. Neurobiol. Dis. 2011, 43, 516–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schori, H.; Yoles, E.; Schwartz, M. T-cell-based immunity counteracts the potential toxicity of glutamate in the central nervous system. J. Neuroimmunol. 2001, 119, 199–204. [Google Scholar] [CrossRef]
- Lombardi, G.; Miglio, G.; Canonico, P.L.; Naldi, P.; Comi, C.; Monaco, F. Abnormal response to glutamate of T lymphocytes from multiple sclerosis patients. Neurosci. Lett. 2003, 340, 5–8. [Google Scholar] [CrossRef]
- Sommer, A.; Winner, B.; Prots, I. The Trojan horse - neuroinflammatory impact of T-cells in neurodegenerative diseases. Mol. Neurodegener. 2017, 12, 78. [Google Scholar] [CrossRef] [PubMed]
- Levite, M.; Marino, F.; Cosentino, M. Dopamine, T-cells and multiple sclerosis (MS). J. Neural. Transm. (Vienna) 2017, 124, 525–542. [Google Scholar] [CrossRef]
- Saunders, J.A.; Estes, K.A.; Kosloski, L.M.; Allen, H.E.; Dempsey, K.M.; Torres-Russotto, D.R.; Meza, J.L.; Santamaria, P.M.; Bertoni, J.M.; Murman, D.L.; et al. CD4+ regulatory and effector/memory T cell subsets profile motor dysfunction in Parkinson’s disease. J. Neuroimmune Pharmacol. 2012, 7, 927–938. [Google Scholar] [CrossRef]
- González, H.; Contreras, F.; Prado, C.; Elgueta, D.; Franz, D.; Bernales, S.; Pacheco, R. Dopamine receptor D3 expressed on CD4+ T-cells favors neurodegeneration of dopaminergic neurons during Parkinson’s disease. J. Immunol. 2013, 190, 5048–5056. [Google Scholar] [CrossRef]
- Talhada, D.; Rabenstein, M.; Ruscher, K. The role of dopaminergic immune cell signalling in poststroke inflammation. Ther. Adv. Neurol. Disord. 2018, 11, 1756286418774225. [Google Scholar] [CrossRef]
- Lee, M. Neurotransmitters and microglial-mediated neuroinflammation. Curr. Protein Pept. Sci. 2013, 14, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Mastroeni, D.; Grover, A.; Leonard, B.; Joyce, J.N.; Coleman, P.D.; Kozik, B.; Bellinger, D.L.; Rogers, J. Microglial responses to dopamine in a cell culture model of Parkinson’s disease. Neurobiol. Aging 2008, 30, 1805–1817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fornai, F.; Carrizzo, A.; Ferrucci, M.; Damato, A.; Biagioni, F.; Gaglione, A.; Puca, A.A.; Vecchione, C. Brain diseases and tumorigenesis: The good and bad cops of pentraxin3. Int. J. Biochem. Cell Biol. 2015, 69, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.C.; Chang, J.H.; Jin, J. Regulation of nuclear factor-κB in autoimmunity. Trends Immunol. 2013, 34, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; You, M.; Shi, H.; Hou, Y. Ubiquitin-mediated NFκB degradation pathway. Cell. Mol. Immunol. 2015, 12, 653–655. [Google Scholar] [CrossRef] [PubMed]
- Arbogast, F.; Gros, F. Lymphocyte Autophagy in Homeostasis, Activation, and Inflammatory Diseases. Front. Immunol. 2018, 9, 1801, Erratum in: Front. Immunol. 2018, 9, 2627. [Google Scholar] [CrossRef]
- Bronietzki, A.W.; Schuster, M.; Schmitz, I. Autophagy in T-cell development, activation and differentiation. Immunol. Cell Biol. 2015, 93, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Gaczynska, M.; Rock, K.L.; Spies, T.; Goldberg, A.L. Peptidase activities of proteasomes are differentially regulated by the major histocompatibility complex-encoded genes for LMP2 and LMP7. Proc. Natl. Acad. Sci. USA 1994, 91, 9213–9217. [Google Scholar] [CrossRef]
- Huber, E.M.; Basler, M.; Schwab, R.; Heinemeyer, W.; Kirk, C.J.; Groettrup, M. Immuno- and constitutive proteasome crystal structures reveal differences in substrate and inhibitor specificity. Cell 2012, 148, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Driscoll, J.; Brown, M.G.; Finley, D.; Monaco, J.J. MHC-linked LMP gene products specifically alter peptidase activities of the proteasome. Nature 1993, 365, 262–264. [Google Scholar] [CrossRef]
- Chapiro, J.; Claverol, S.; Piette, F.; Ma, W.; Stroobant, V.; Guillaume, B.; Gairin, J.E.; Morel, S.; Burlet-Schiltz, O.; Monsarrat, B.; et al. Destructive cleavage of antigenic peptides either by the immunoproteasome or by the standard proteasome results in differential antigen presentation. J. Immunol. 2006, 176, 1053–1061. [Google Scholar] [CrossRef] [PubMed]
- Lei, B.; Abdul Hameed, M.D.; Hamza, A.; Wehenkel, M.; Muzyka, J.L.; Yao, X.J.; Kim, K.B.; Zhan, C.G. Molecular basis of the selectivity of the immunoproteasome catalytic subunit LMP2-specific inhibitor revealed by molecular modeling and dynamics simulations. J. Phys. Chem. B. 2010, 114, 12333–12339. [Google Scholar] [CrossRef]
- Loi, M.; Müller, A.; Steinbach, K.; Niven, J.; Barreira da Silva, R.; Paul, P.; Ligeon, L.A.; Caruso, A.; Albrecht, R.A.; Becker, A.C.; et al. Macroautophagy Proteins Control MHC Class I Levels on Dendritic Cells and Shape Anti-viral CD8(+) T Cell Responses. Cell Rep. 2016, 15, 1076–1087. [Google Scholar] [CrossRef]
- Loi, M.; Ligeon, L.A.; Münz, C. MHC Class I Internalization via Autophagy Proteins. Methods Mol. Biol. 2019, 1880, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Loi, M.; Gannagé, M.; Münz, C. ATGs help MHC class II, but inhibit MHC class I antigen presentation. Autophagy 2016, 12, 1681–1682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tey, S.K.; Khanna, R. Autophagy mediates transporter associated with antigen processing-independent presentation of viral epitopes through MHC class I pathway. Blood 2012, 120, 994–1004. [Google Scholar] [CrossRef] [Green Version]
- Nil, A.; Firat, E.; Sobek, V.; Eichmann, K.; Niedermann, G. Expression of housekeeping and immunoproteasome subunit genes is differentially regulated in positively and negatively selecting thymic stroma subsets. Eur. J. Immunol. 2004, 34, 2681–2689. [Google Scholar] [CrossRef] [Green Version]
- Ugras, S.; Daniels, M.J.; Fazelinia, H.; Gould, N.S.; Yocum, A.K.; Luk, K.C.; Luna, E.; Ding, H.; McKennan, C.; Seeholzer, S.; et al. Induction of the immunoproteasome subunit Lmp7 links proteostasis and immunity in α-synuclein aggregation disorders. EBioMedicine 2018, 31, 307–319. [Google Scholar] [CrossRef]
- Moritz, K.E.; McCormack, N.M.; Abera, M.B.; Viollet, C.; Yauger, Y.J.; Sukumar, G.; Dalgard, C.L.; Burnett, B.G. The role of the immunoproteasome in interferon-γ-mediated microglial activation. Sci. Rep. 2017, 7, 9365. [Google Scholar] [CrossRef] [Green Version]
- Seifert, U.; Bialy, L.P.; Ebstein, F.; Bech-Otschir, D.; Voigt, A.; Schröter, F.; Prozorovski, T.; Lange, N.; Steffen, J.; Rieger, M.; et al. Immunoproteasomes preserve protein homeostasis upon interferon-induced oxidative stress. Cell 2010, 142, 613–624. [Google Scholar] [CrossRef]
- Nathan, J.A.; Spinnenhirn, V.; Schmidtke, G.; Basler, M.; Groettrup, M.; Goldberg, A.L. Immuno- and constitutive proteasomes do not differ in their abilities to degrade ubiquitinated proteins. Cell 2013, 152, 1184–1194. [Google Scholar] [CrossRef]
- Piccinini, M.; Mostert, M.; Croce, S.; Baldovino, S.; Papotti, M.; Rinaudo, M.T. Interferon-gamma-inducible subunits are incorporated in human brain 20S proteasome. J. Neuroimmunol. 2003, 135, 135–140. [Google Scholar] [CrossRef]
- Cebrián, C.; Loike, J.D.; Sulzer, D. Neuronal MHC-I expression and its implications in synaptic function, axonal regeneration and Parkinson’s and other brain diseases. Front. Neuroanat. 2014, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cullheim, S.; Thams, S. Classic major histocompatibility complex class I molecules: new actors at the neuromuscular junction. Neuroscientist. 2010, 16, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Lazarczyk, M.J.; Kemmler, J.E.; Eyford, B.A.; Short, J.A.; Varghese, M.; Sowa, A.; Dickstein, D.R.; Yuk, F.J.; Puri, R.; Biron, K.E.; et al. Major Histocompatibility Complex class I proteins are critical for maintaining neuronal structural complexity in the aging brain. Sci. Rep 2016, 6, 26199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edamura, M.; Murakami, G.; Meng, H.; Itakura, M.; Shigemoto, R.; Fukuda, A.; Nakahara, D. Functional deficiency of MHC class I enhances LTP and abolishes LTD in the nucleus accumbens of mice. PLoS ONE 2014, 9, e107099. [Google Scholar] [CrossRef]
- Heink, S.; Ludwig, D.; Kloetzel, P.M.; Krüger, E. IFN-gamma-induced immune adaptation of the proteasome system is an accelerated and transient response. Proc. Natl. Acad. Sci. USA 2005, 102, 9241–9246. [Google Scholar] [CrossRef]
- Mo, M.S.; Li, G.H.; Sun, C.C.; Huang, S.X.; Wei, L.; Zhang, L.M.; Zhou, M.M.; Wu, Z.H.; Guo, W.Y.; Yang, X.L.; et al. Dopaminergic neurons show increased low-molecular-mass protein 7 activity induced by 6-hydroxydopamine in vitro and in vivo. Transl. Neurodegener. 2018, 7, 19. [Google Scholar] [CrossRef]
- Basler, M.; Mundt, S.; Muchamuel, T.; Moll, C.; Jiang, J.; Groettrup, M.; Kirk, C.J. Inhibition of the immunoproteasome ameliorates experimental autoimmune encephalomyelitis. EMBO Mol. Med. 2014, 6, 226–238. [Google Scholar] [CrossRef] [Green Version]
- Fissolo, N.; Kraus, M.; Reich, M.; Ayturan, M.; Overkleeft, H.; Driessen, C.; Weissert, R. Dual inhibition of proteasomal and lysosomal proteolysis ameliorates autoimmune central nervous system inflammation. Eur. J. Immunol. 2008, 38, 2401–2411. [Google Scholar] [CrossRef]
- Díaz-Hernández, M.; Hernández, F.; Martín-Aparicio, E.; Gómez-Ramos, P.; Morán, M.A.; Castaño, J.G.; Ferrer, I.; Avila, J.; Lucas, J.J. Neuronal induction of the immunoproteasome in Huntington’s disease. J. Neurosci. 2003, 23, 11653–11661. [Google Scholar] [CrossRef] [PubMed]
- Schmeisser, H.; Bekisz, J.; Zoon, K.C. New function of type I IFN: induction of autophagy. J. Interferon Cytokine Res. 2014, 34, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Mocholi, E.; Dowling, S.D.; Botbol, Y.; Gruber, R.C.; Ray, A.K.; Vastert, S.; Shafit-Zagardo, B.; Coffer, P.J.; Macian, F. Autophagy Is a Tolerance-Avoidance Mechanism that Modulates TCR-Mediated Signaling and Cell Metabolism to Prevent Induction of T Cell Anergy. Cell Rep. 2018, 24, 1136–1150. [Google Scholar] [CrossRef] [PubMed]
- Paunovic, V.; Petrovic, I.V.; Milenkovic, M.; Janjetovic, K.; Pravica, V.; Dujmovic, I.; Milosevic, E.; Martinovic, V.; Mesaros, S.; Drulovic, J.; et al. Autophagy-independent increase of ATG5 expression in T cells of multiplesclerosis patients. J. Neuroimmunol. 2018, 319, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Zhang, C.; Jiang, N.; He, D.; Bai, Y.; Xin, Y. Rapamycin combined with MCC950 to treat multiple sclerosis in experimental autoimmune encephalomyelitis. J. Cell Biochem. 2019, 120, 5160–5168. [Google Scholar] [CrossRef]
- Su, P.; Zhang, J.; Wang, D.; Zhao, F.; Cao, Z.; Aschner, M.; Luo, W. The role of autophagy in modulation of neuroinflammation in microglia. Neuroscience 2016, 319, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.M.; Wang, F.; Qi, D.; Liu, W.W.; Gu, C.; Mao, C.J.; Yang, Y.P.; Zhao, Z.; Hu, L.F.; Liu, C.F. A Critical Role of Autophagy in Regulating Microglia Polarization in Neurodegeneration. Front. Aging Neurosci. 2018, 10, 378. [Google Scholar] [CrossRef]
- Cho, M.H.; Cho, K.; Kang, H.J.; Jeon, E.Y.; Kim, H.S.; Kwon, H.J.; Kim, H.M.; Kim, D.H.; Yoon, S.Y. Autophagy in microglia degrades extracellular β-amyloid fibrils and regulates theNLRP3 inflammasome. Autophagy 2014, 10, 1761–1775. [Google Scholar] [CrossRef]
- Berthet, A.; Bezard, E.; Porras, G.; Fasano, S.; Barroso-Chinea, P.; Dehay, B.; Martinez, A.; Thiolat, M.L.; Nosten-Bertrand, M.; Giros, B.; et al. L-DOPA impairs proteasome activity in parkinsonism through D1 dopamine receptor. J. Neurosci. 2012, 32, 681–691. [Google Scholar] [CrossRef]
- Lazzeri, G.; Lenzi, P.; Busceti, C.L.; Ferrucci, M.; Falleni, A.; Bruno, V.; Paparelli, A.; Fornai, F. Mechanisms involved in the formation of dopamine-induced intracellular bodies within striatal neurons. J. Neurochem. 2007, 101, 1414–1427. [Google Scholar] [CrossRef] [Green Version]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Limanaqi, F.; Biagioni, F.; Busceti, C.L.; Ryskalin, L.; Soldani, P.; Frati, A.; Fornai, F. Cell Clearing Systems Bridging Neuro-Immunity and Synaptic Plasticity. Int. J. Mol. Sci. 2019, 20, 2197. https://doi.org/10.3390/ijms20092197
Limanaqi F, Biagioni F, Busceti CL, Ryskalin L, Soldani P, Frati A, Fornai F. Cell Clearing Systems Bridging Neuro-Immunity and Synaptic Plasticity. International Journal of Molecular Sciences. 2019; 20(9):2197. https://doi.org/10.3390/ijms20092197
Chicago/Turabian StyleLimanaqi, Fiona, Francesca Biagioni, Carla Letizia Busceti, Larisa Ryskalin, Paola Soldani, Alessandro Frati, and Francesco Fornai. 2019. "Cell Clearing Systems Bridging Neuro-Immunity and Synaptic Plasticity" International Journal of Molecular Sciences 20, no. 9: 2197. https://doi.org/10.3390/ijms20092197