Adenosine Induces EBV Lytic Reactivation through ADORA1 in EBV-Associated Gastric Carcinoma

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
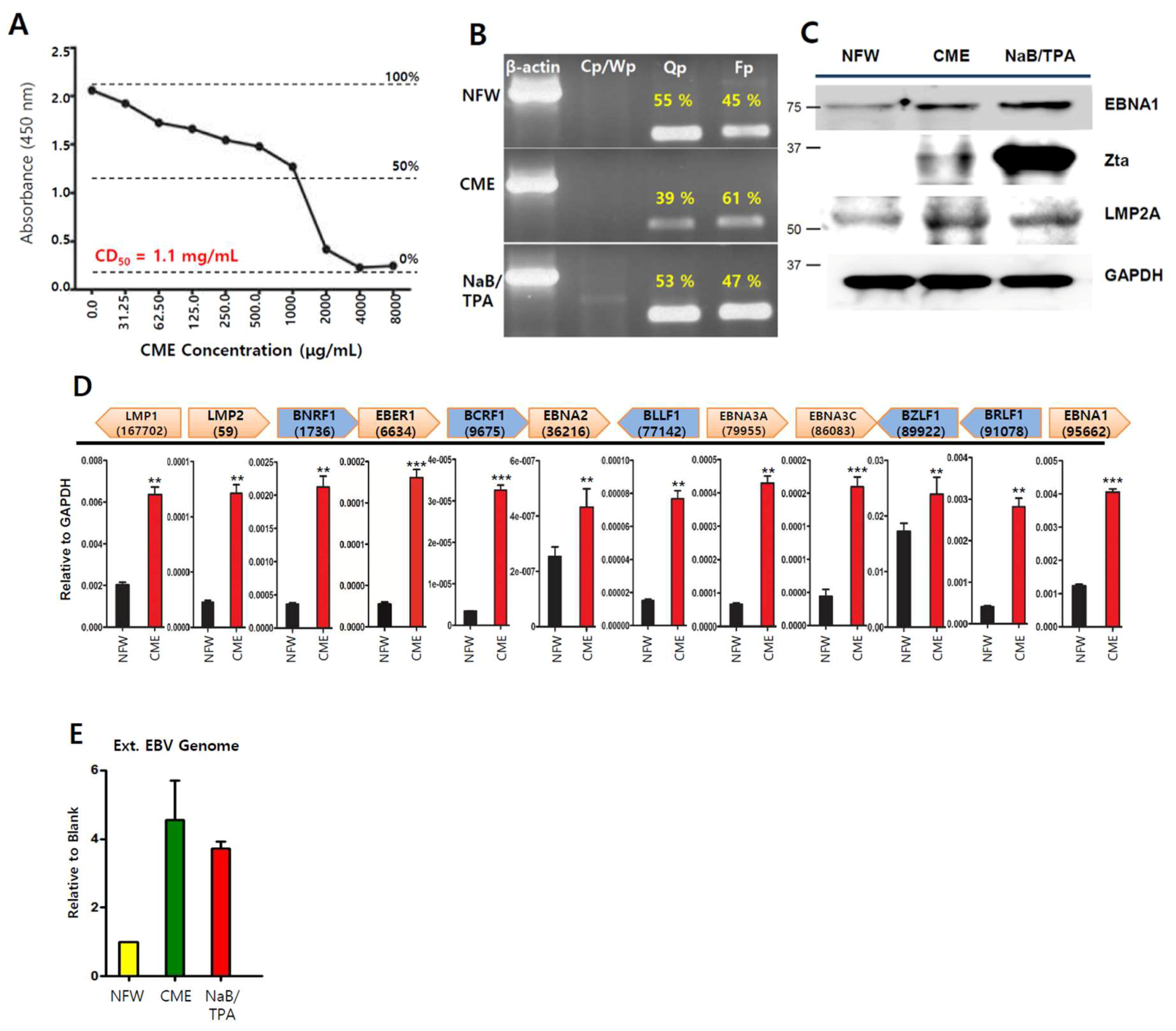
2.1. Cordyceps Extracts Induce EBV Lytic Infection in EBVaGC Cells
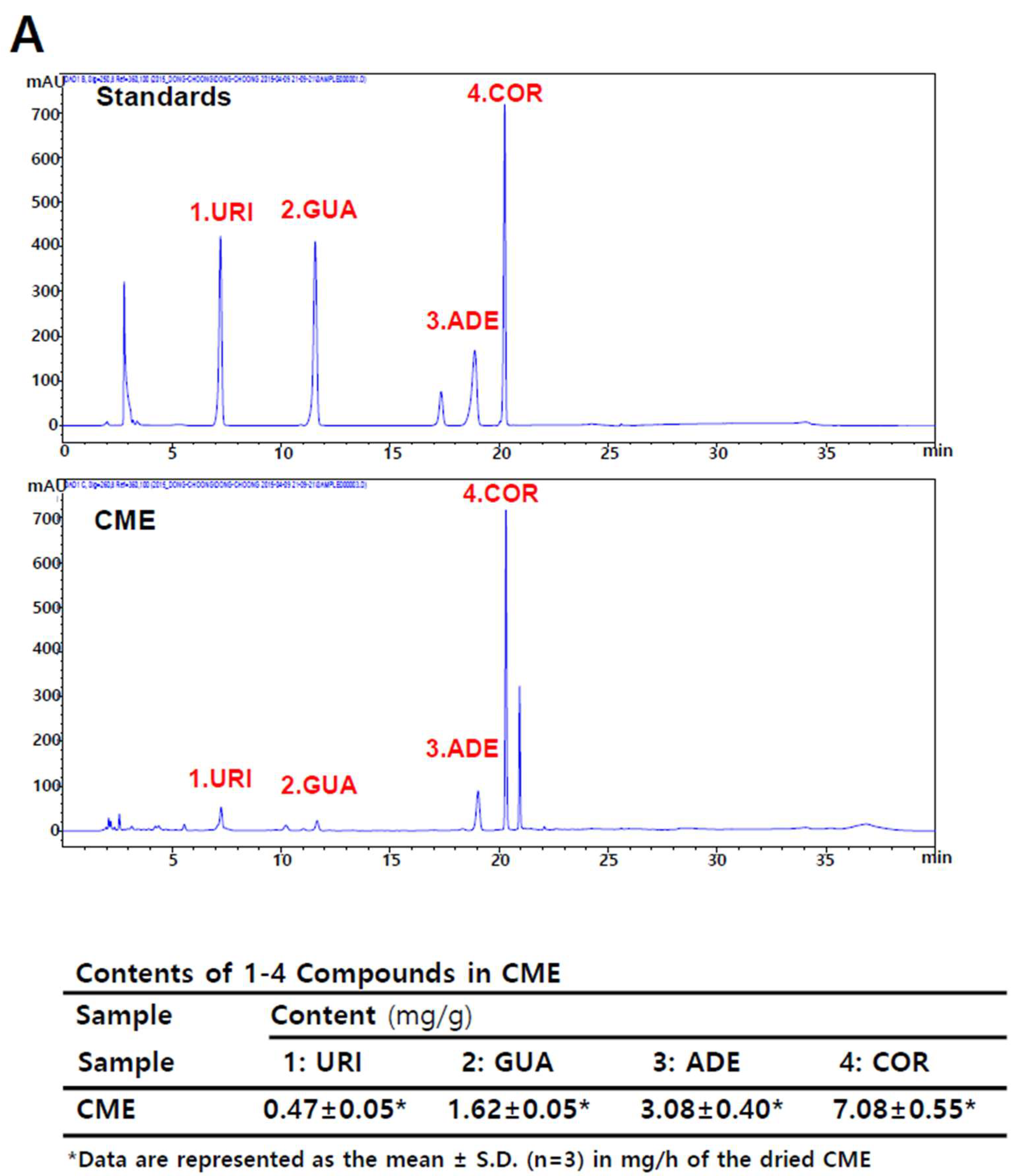
2.2. Adenosine is Present in Cordyceps militaris (CME)
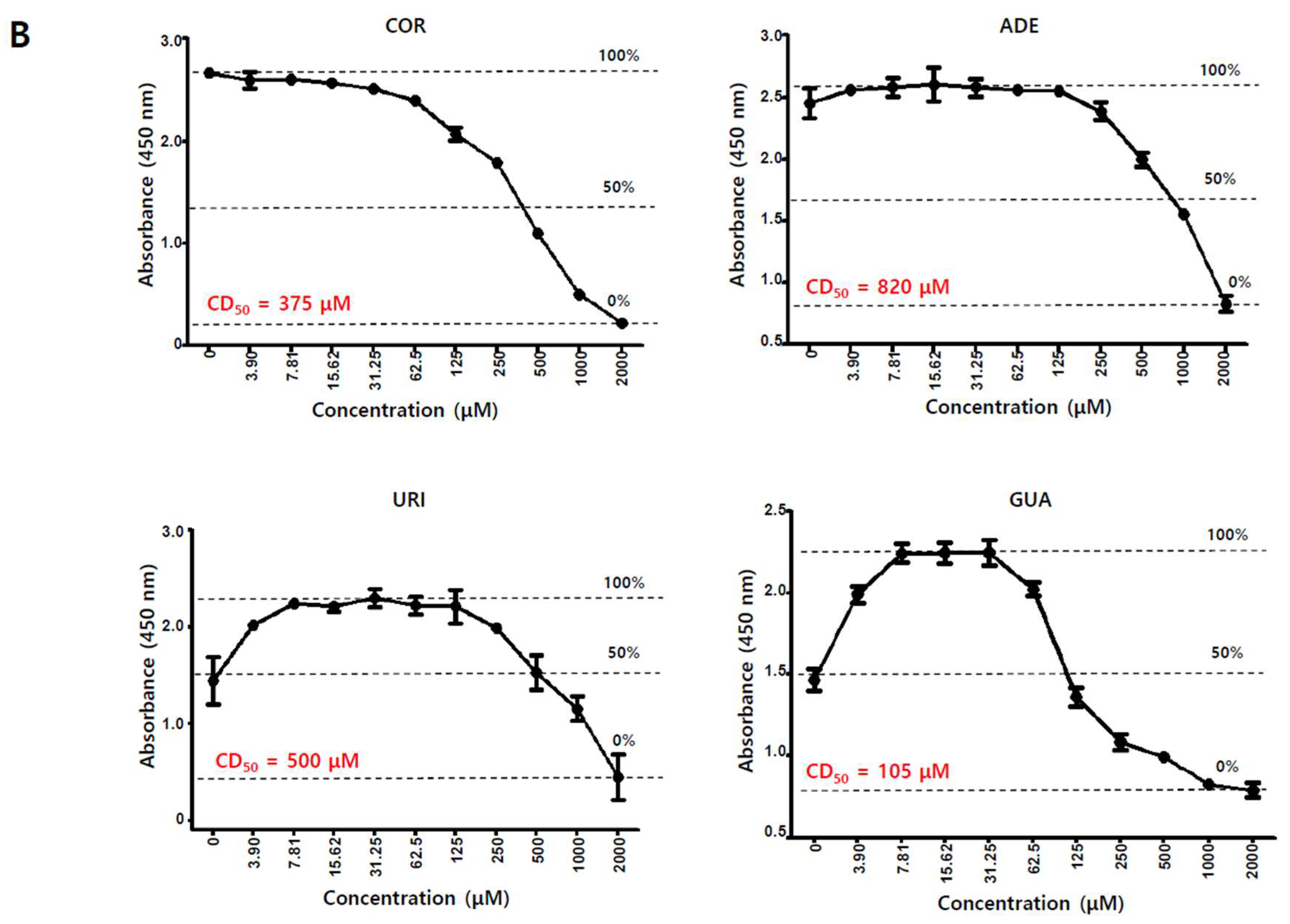
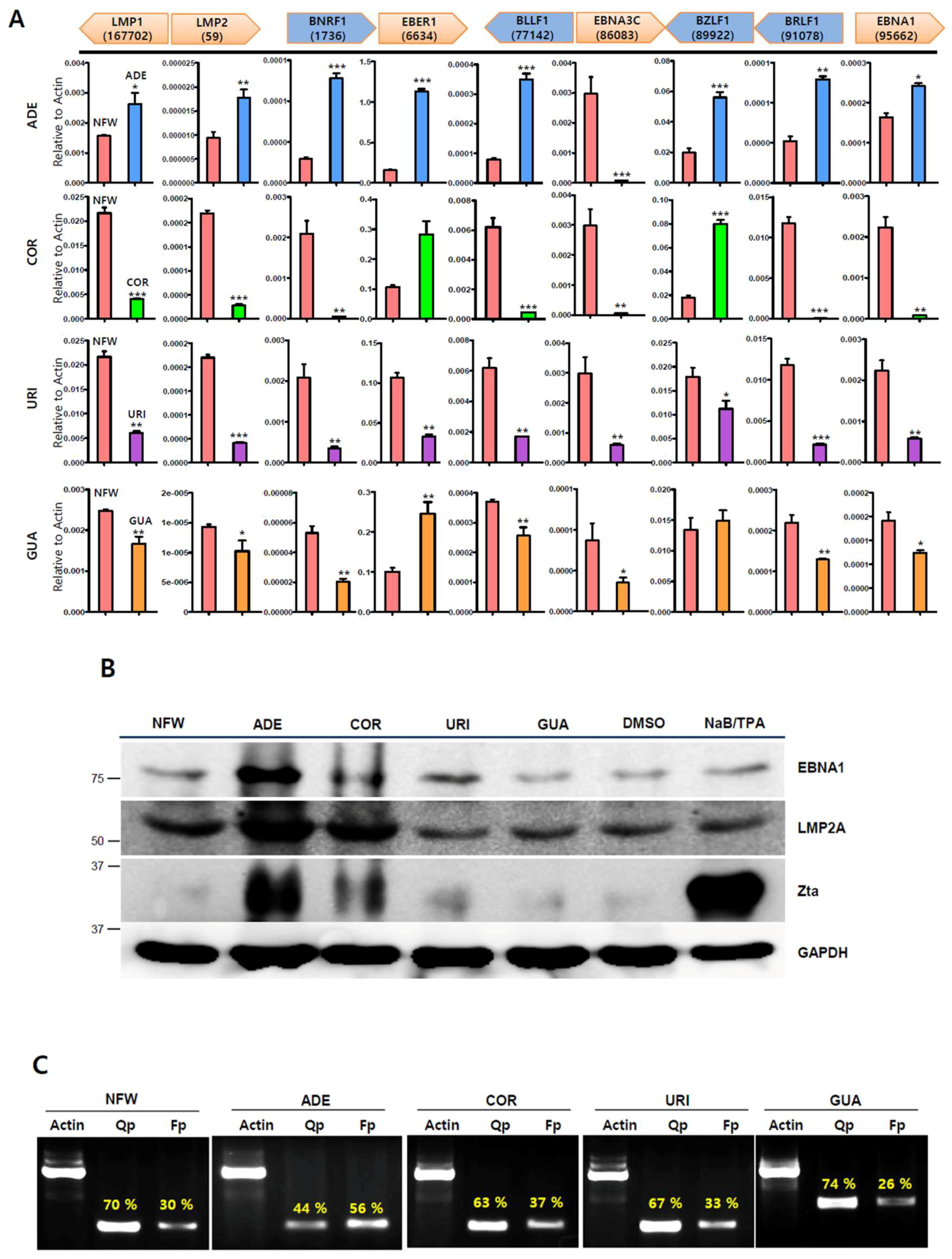
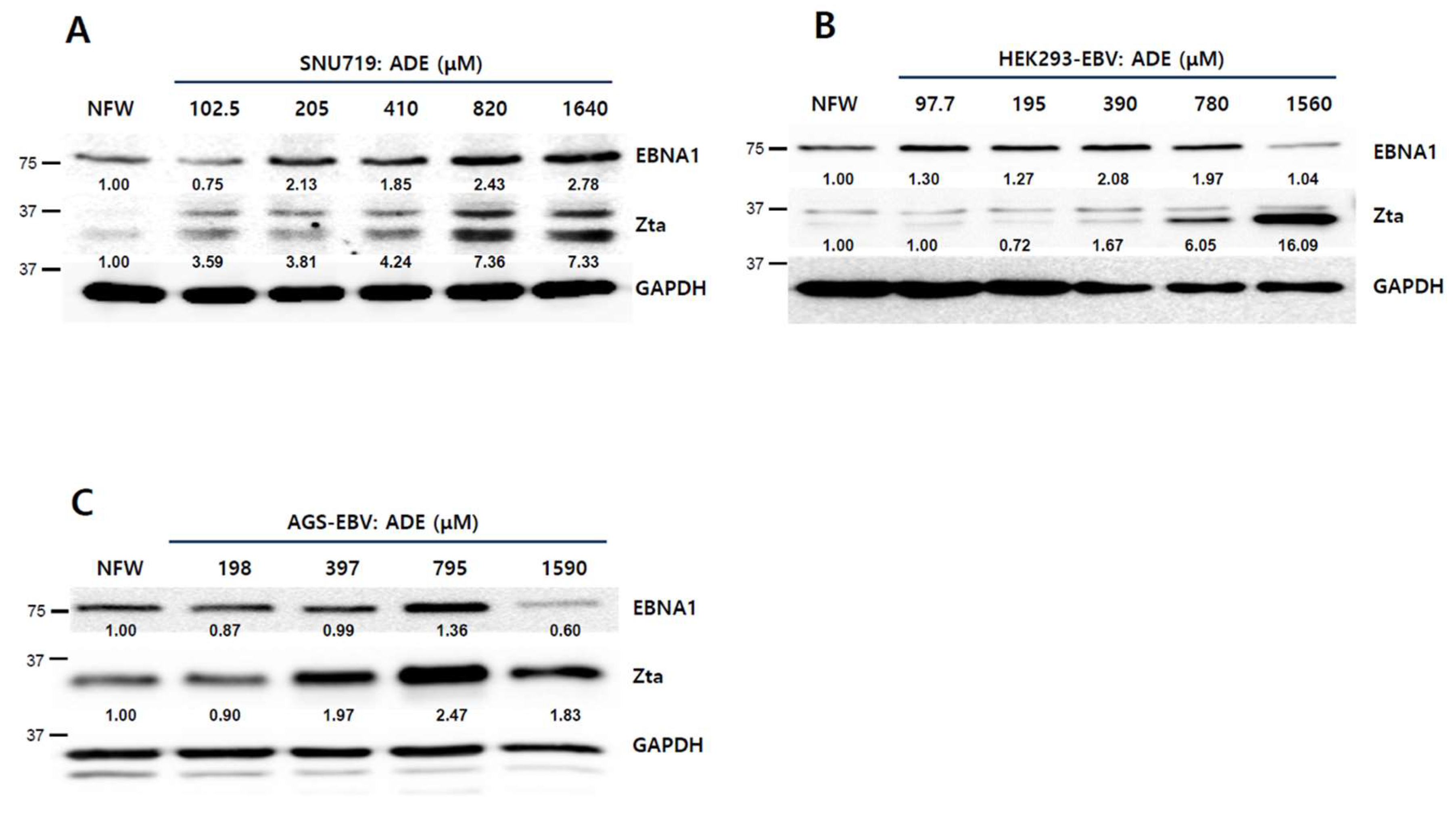
2.3. Adenosine Induces EBV Gene Expression
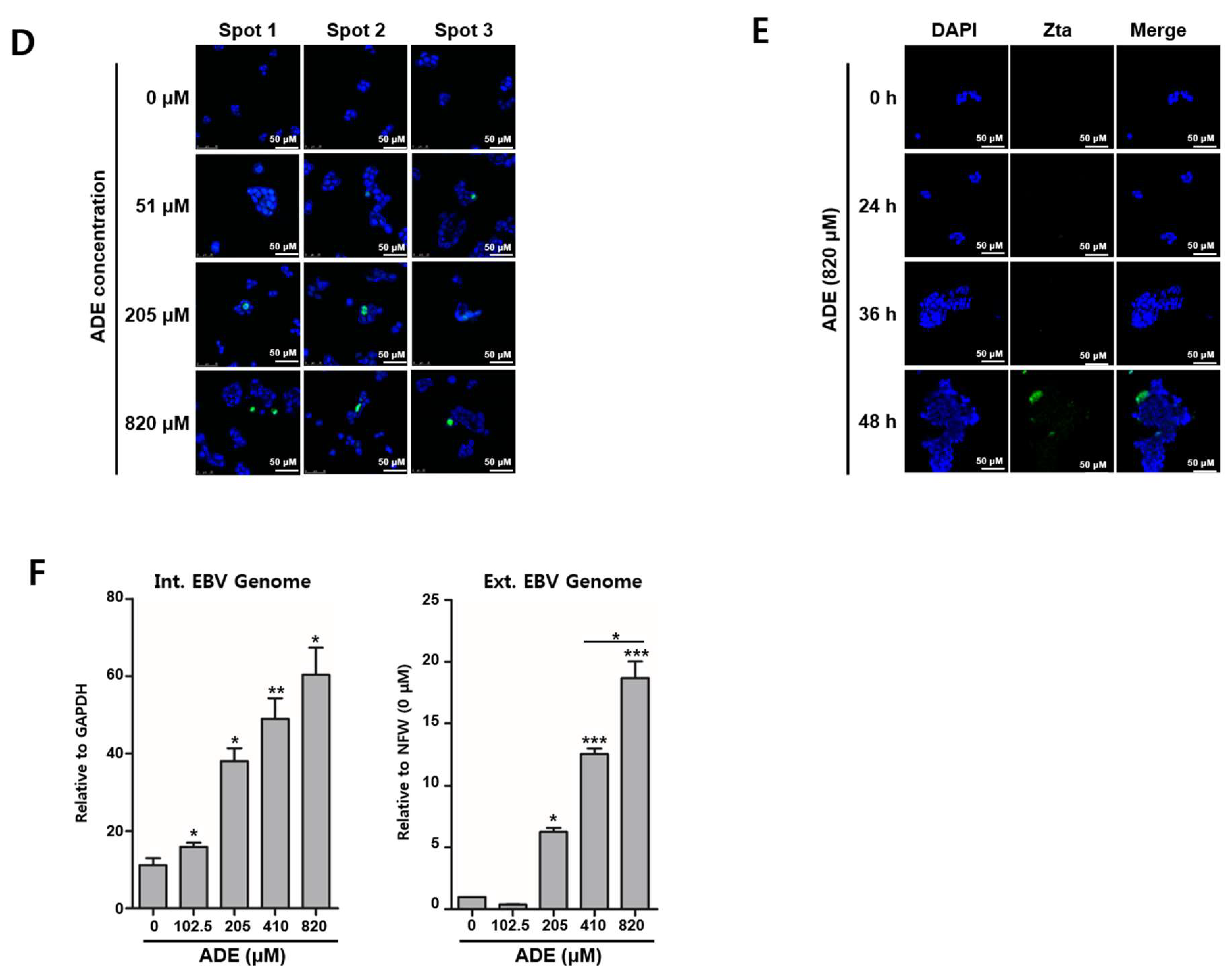
2.4. Adenosine Induces EBV Lytic Reactivation
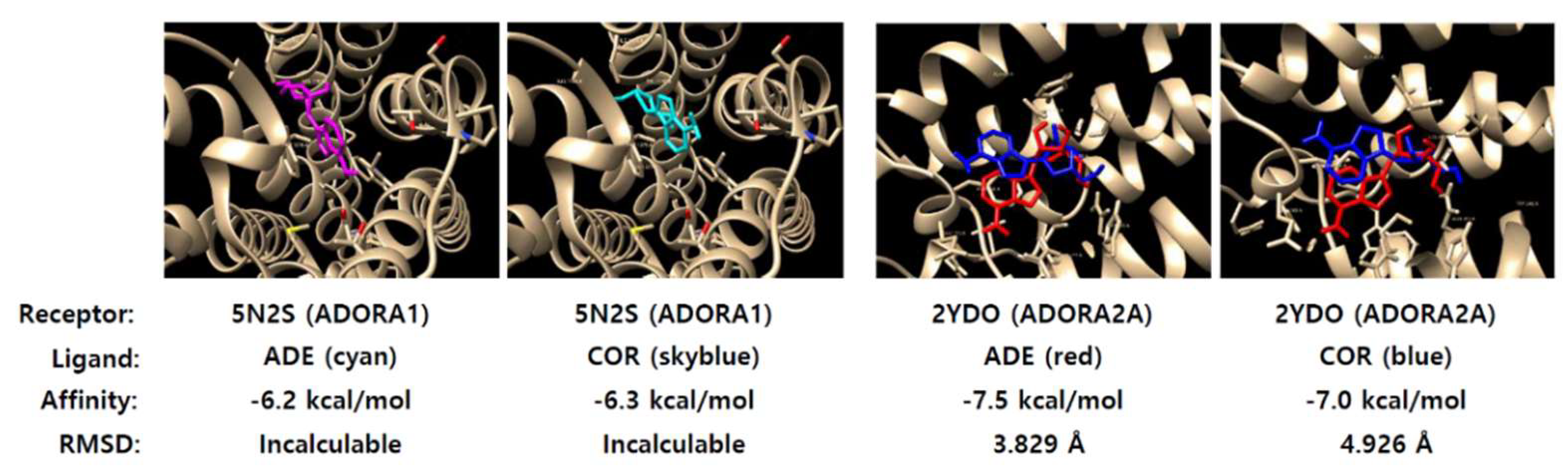
2.5. Adenosine Has Strong Affinity to Adenosine A1 Receptor (ADORA1)
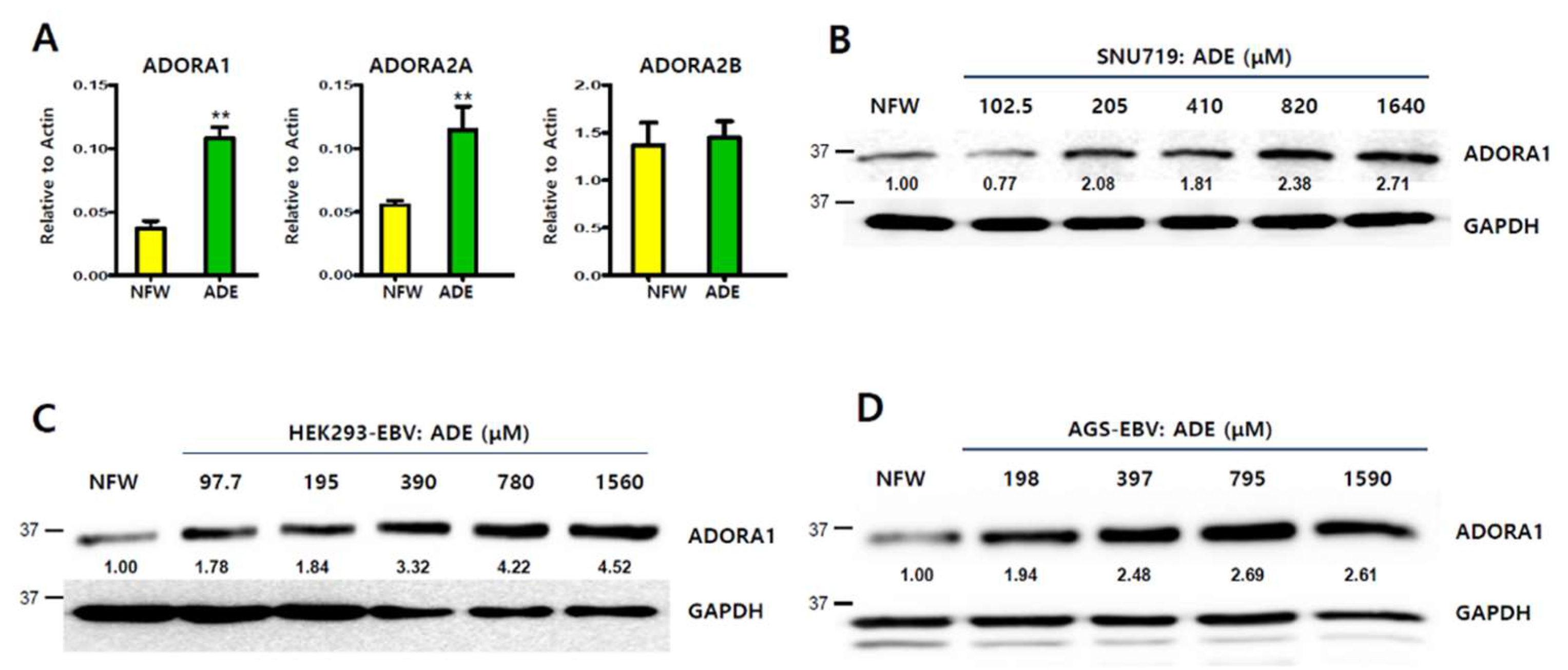
2.6. Induction of Adenosine A1 Receptor by Adenosine
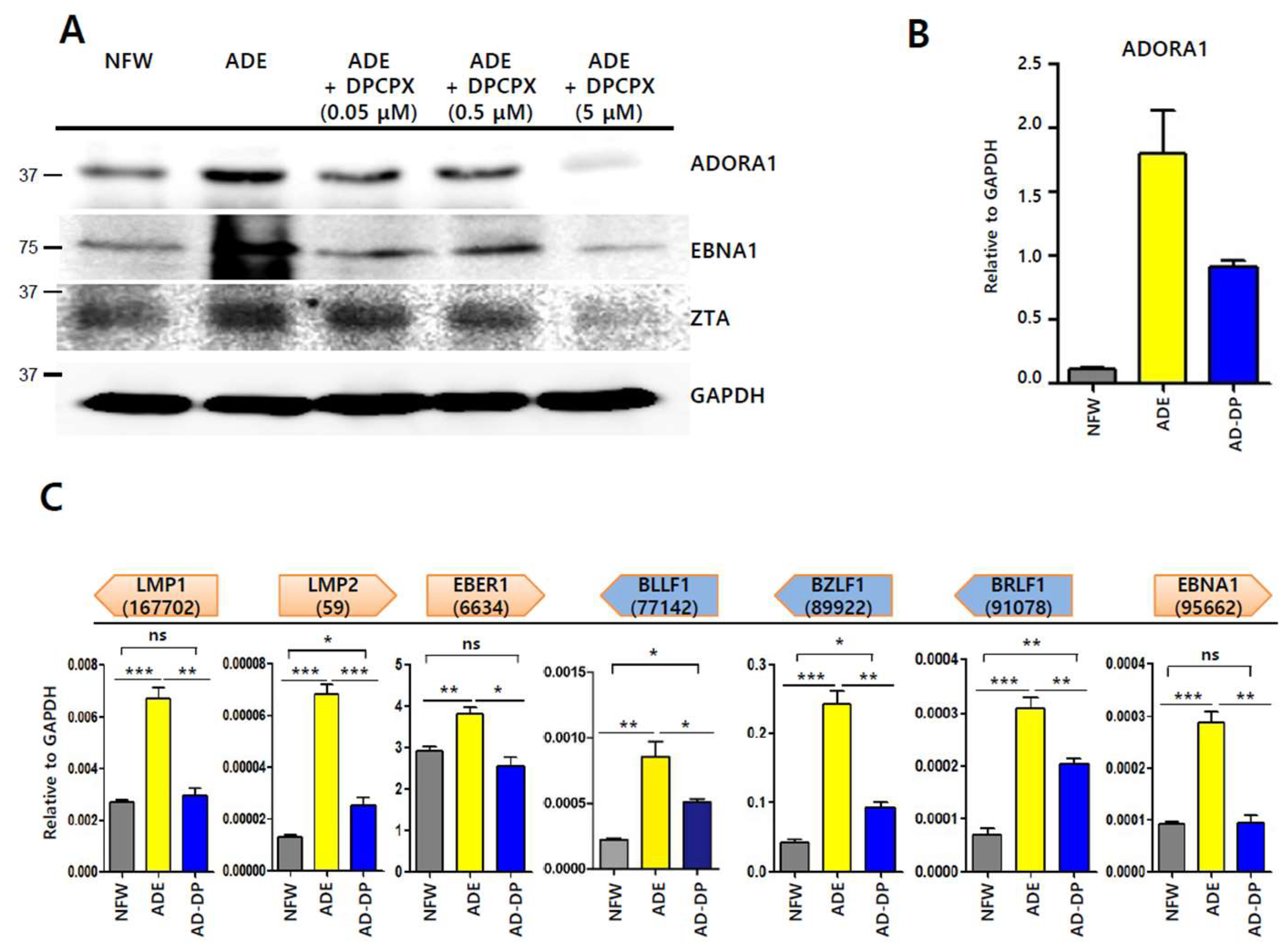
2.7. Inhibitor of Adenosine A1 Receptor Suppresses EBV Gene Induction
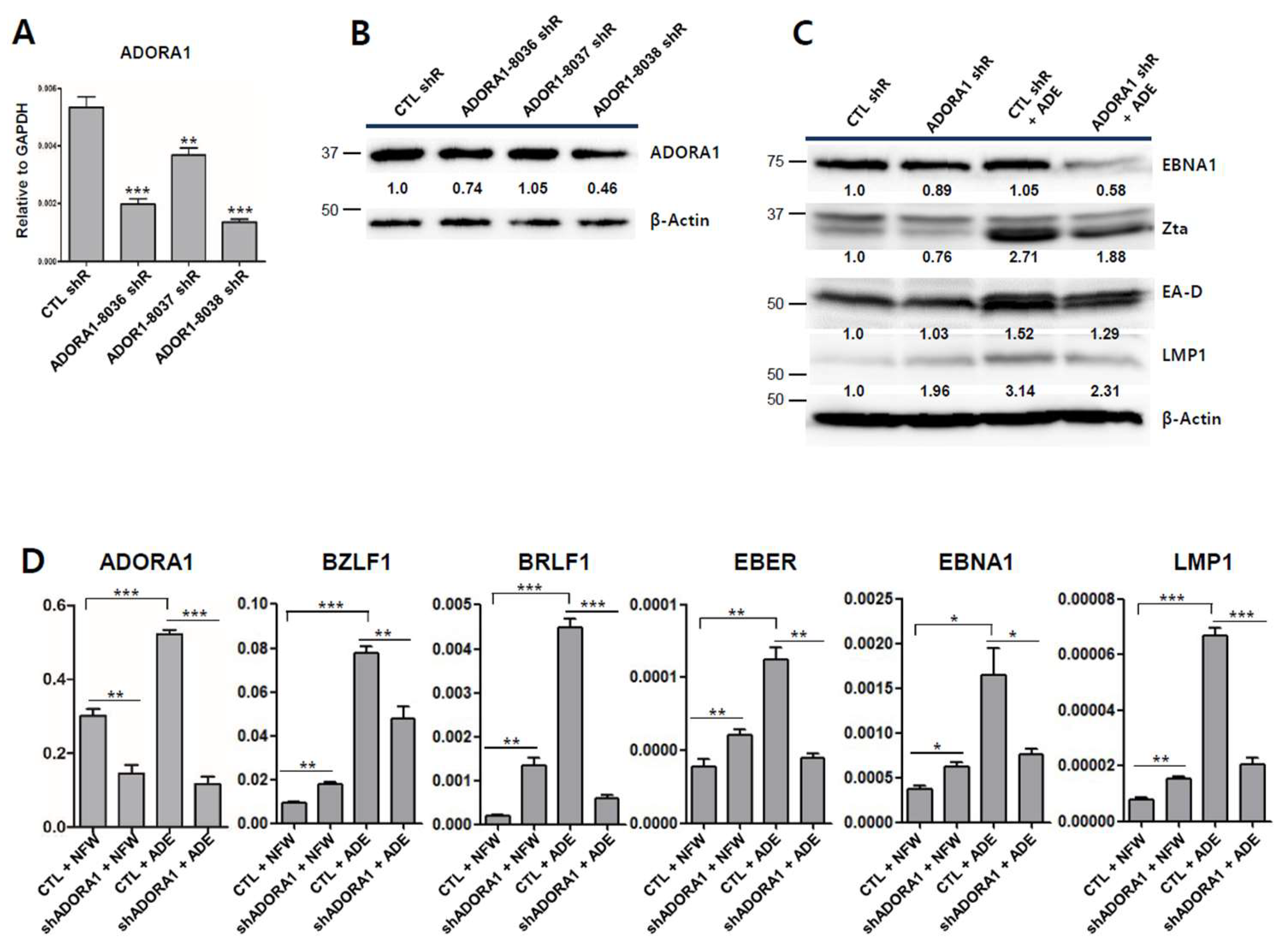
2.8. Partial Depletion of ADORA1 Suppresses BZLF1 Induction by Adenosine
2.9. Adenosine Increases ATP but Decreases Cyclic Adenosine Monophosphate
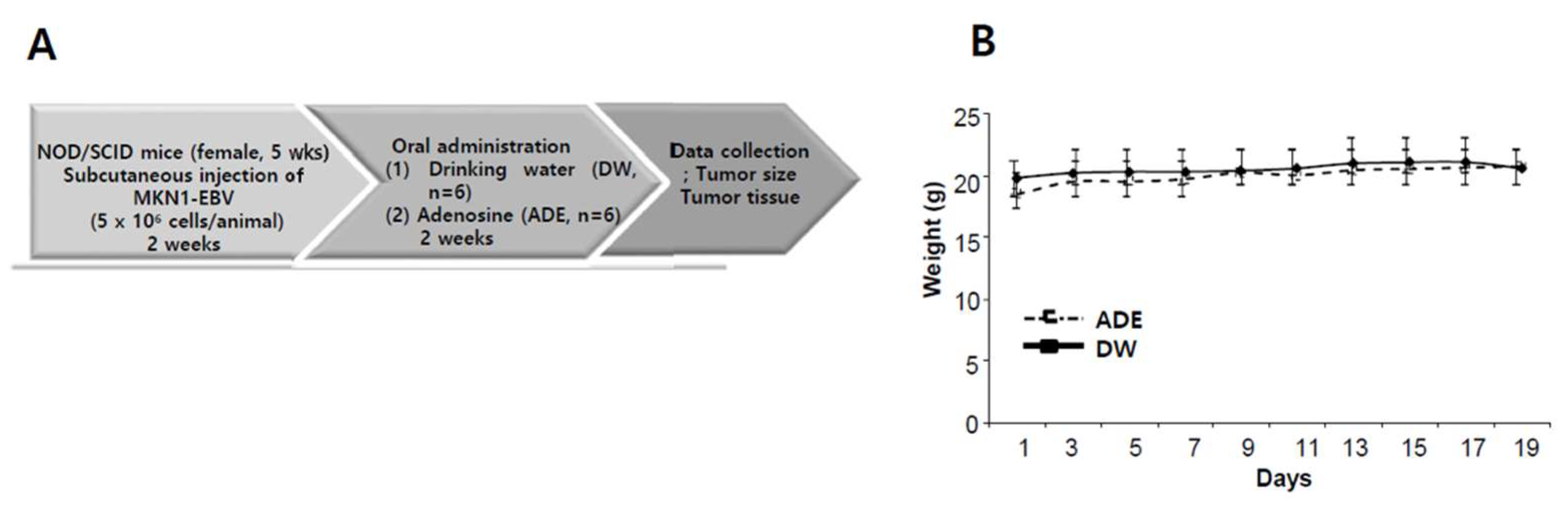
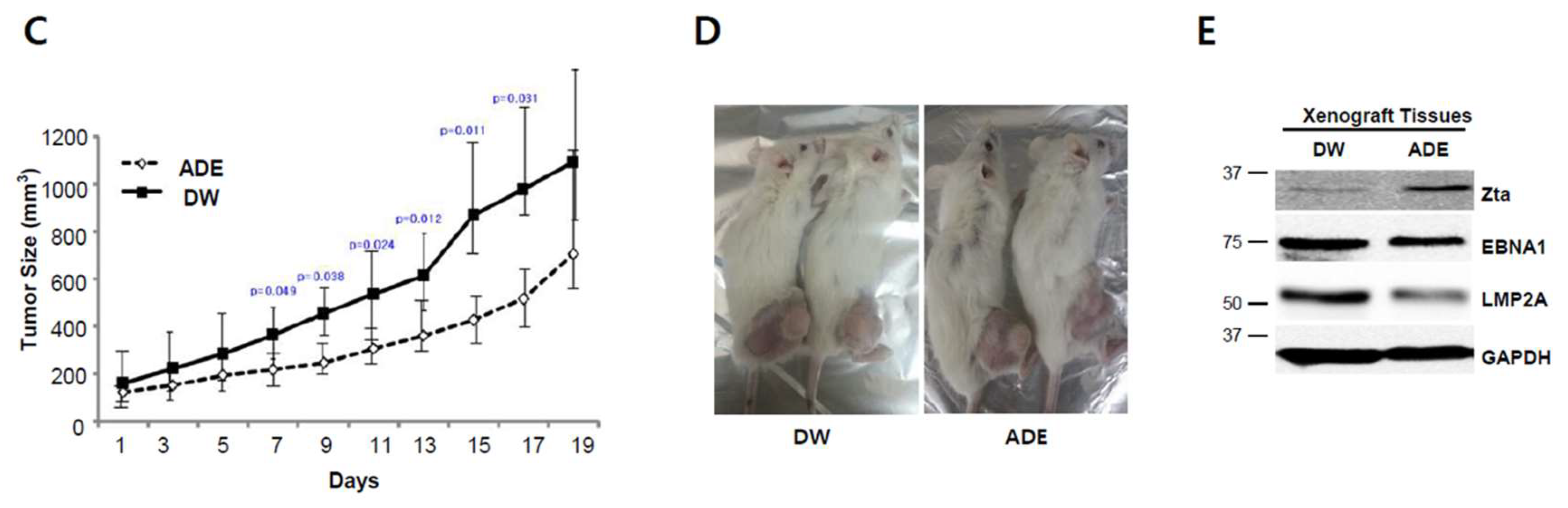
2.10. Adenosine Suppresses Tumor Development in a Xenograft Mouse Model
3. Discussion
4. Materials and Methods
4.1. Preparation of CMEs
4.2. Cell lines and Reagents
4.3. Cytotoxicity Assay
4.4. Western Blot Assay
4.5. Reverse-Transcriptional Quantitative PCR (RT-qPCR) Assay
4.6. Promoter Usage Assay
4.7. EBV Genome Measurement Assay
4.8. High-Performance Liquid Chromatography (HPLC) Assay
4.9. Immunofluorescence (IFA) and Immunocytochemistry (ICA) Assays
4.10. Lentiviral Transduction
4.11. ATP Assay
4.12. cAMP Assay
4.13. Docking Assay and Binding Score
4.14. Ethics Statement
4.15. Xenograft model Antitumor Assay
4.16. Statistical Analysis
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Young, L.S.; Yap, L.F.; Murray, P.G. Epstein-Barr virus: More than 50 years old and still providing surprises. Nat. Rev. Cancer 2016, 16, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Maeda, E.; Akahane, M.; Kiryu, S.; Kato, N.; Yoshikawa, T.; Hayashi, N.; Aoki, S.; Minami, M.; Uozaki, H.; Fukayama, M.; et al. Spectrum of Epstein-Barr virus-related diseases: A pictorial review. Jpn. J. Radiol. 2009, 27, 4–19. [Google Scholar] [CrossRef] [PubMed]
- Khan, G.; Hashim, M.J. Global burden of deaths from Epstein-Barr virus attributable malignancies 1990–2010. Infect. Agents Cancer 2014, 9, 38. [Google Scholar] [CrossRef] [PubMed]
- Jacome, A.A.; Lima, E.M.; Kazzi, A.I.; Chaves, G.F.; Mendonca, D.C.; Maciel, M.M.; Santos, J.S. Epstein-Barr virus-positive gastric cancer: A distinct molecular subtype of the disease? Rev. Soc. Bras. Med. Trop. 2016, 49, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki-Ushiku, A.; Kunita, A.; Fukayama, M. Update on Epstein-Barr virus and gastric cancer (review). Int. J. Oncol. 2015, 46, 1421–1434. [Google Scholar] [CrossRef]
- Ma, S.D.; Hegde, S.; Young, K.H.; Sullivan, R.; Rajesh, D.; Zhou, Y.; Jankowska-Gan, E.; Burlingham, W.J.; Sun, X.; Gulley, M.L.; et al. A new model of Epstein-Barr virus infection reveals an important role for early lytic viral protein expression in the development of lymphomas. J. Virol. 2011, 85, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Haan, K.M.; Aiyar, A.; Longnecker, R. Establishment of latent Epstein-Barr virus infection and stable episomal maintenance in murine B-cell lines. J. Virol. 2001, 75, 3016–3020. [Google Scholar] [CrossRef]
- Kenney, S.C.; Mertz, J.E. Regulation of the latent-lytic switch in Epstein-Barr virus. Semin. Cancer Biol. 2014, 26, 60–68. [Google Scholar] [CrossRef]
- Field, H.J.; Vere Hodge, R.A. Recent developments in anti-herpesvirus drugs. Br. Med. Bull. 2013, 106, 213–249. [Google Scholar] [CrossRef] [Green Version]
- Elion, G.B.; Furman, P.A.; Fyfe, J.A.; de Miranda, P.; Beauchamp, L.; Schaeffer, H.J. Selectivity of action of an antiherpetic agent, 9-(2-hydroxyethoxymethyl) guanine. Proc. Natl. Acad. Sci. USA 1977, 74, 5716–5720. [Google Scholar] [CrossRef]
- Gustafson, E.A.; Chillemi, A.C.; Sage, D.R.; Fingeroth, J.D. The Epstein-Barr virus thymidine kinase does not phosphorylate ganciclovir or acyclovir and demonstrates a narrow substrate specificity compared to the herpes simplex virus type 1 thymidine kinase. Antimicrob. Agents Chemother. 1998, 42, 2923–2931. [Google Scholar] [CrossRef] [PubMed]
- Hellden, A.; Odar-Cederlof, I.; Diener, P.; Barkholt, L.; Medin, C.; Svensson, J.O.; Sawe, J.; Stahle, L. High serum concentrations of the acyclovir main metabolite 9-carboxymethoxymethylguanine in renal failure patients with acyclovir-related neuropsychiatric side effects: An observational study. Nephrol. Dial. Transplant. 2003, 18, 1135–1141. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.B.; Wang, H.X. Pharmacological actions of Cordyceps, a prized folk medicine. J. Pharm. Pharmacol. 2005, 57, 1509–1519. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Yu, J.; Du, L.; Tang, J.; Feng, W.H. Cordycepin enhances Epstein-Barr virus lytic infection and Epstein-Barr virus-positive tumor treatment efficacy by doxorubicin. Cancer Lett. 2016, 376, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Ryu, E.; Son, M.; Lee, M.; Lee, K.; Cho, J.Y.; Cho, S.; Lee, S.K.; Lee, Y.M.; Cho, H.; Sung, G.H.; et al. Cordycepin is a novel chemical suppressor of Epstein-Barr virus replication. Oncoscience 2014, 1, 866–881. [Google Scholar] [CrossRef] [PubMed]
- Cristalli, G.; Costanzi, S.; Lambertucci, C.; Lupidi, G.; Vittori, S.; Volpini, R.; Camaioni, E. Adenosine deaminase: Functional implications and different classes of inhibitors. Med. Res. Rev. 2001, 21, 105–128. [Google Scholar] [CrossRef]
- Spiers, A.S.; Parekh, S.J.; Bishop, M.B. Hairy-cell leukemia: Induction of complete remission with pentostatin (2′-deoxycoformycin). J. Clin. Oncol. 1984, 2, 1336–1342. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Nakagome, I.; Hirono, S.; Itoh, T.; Fujiwara, R. Inhibition of adenosine deaminase (ADA)-mediated metabolism of cordycepin by natural substances. Pharmacol. Res. Perspect. 2015, 3, e00121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, R.P.; Tachibana, K.; Hegen, M.; Munakata, Y.; Cho, D.; Schlossman, S.F.; Morimoto, C. Determination of adenosine deaminase binding domain on CD26 and its immunoregulatory effect on T cell activation. J. Immunol. 1997, 159, 6070–6076. [Google Scholar]
- Liu, Y.; Wang, J.; Wang, W.; Zhang, H.; Zhang, X.; Han, C. The Chemical Constituents and Pharmacological Actions of Cordyceps sinensis. Evid. Based Complement. Altern. Med. 2015, 2015, 575063. [Google Scholar]
- Lee, H.H.; Lee, S.; Lee, K.; Shin, Y.S.; Kang, H.; Cho, H. Anti-cancer effect of Cordyceps militaris in human colorectal carcinoma RKO cells via cell cycle arrest and mitochondrial apoptosis. DARU J. Pharm. Sci. 2015, 23, 35. [Google Scholar] [CrossRef] [PubMed]
- Paterson, R.R. Cordyceps: A traditional Chinese medicine and another fungal therapeutic biofactory? Phytochemistry 2008, 69, 1469–1495. [Google Scholar] [CrossRef]
- Zheng, P.; Xia, Y.; Xiao, G.; Xiong, C.; Hu, X.; Zhang, S.; Zheng, H.; Huang, Y.; Zhou, Y.; Wang, S.; et al. Genome sequence of the insect pathogenic fungus Cordyceps militaris, a valued traditional Chinese medicine. Genome Biol. 2011, 12, R116. [Google Scholar] [CrossRef] [PubMed]
- Suh, W.; Nam, G.; Yang, W.S.; Sung, G.H.; Shim, S.H.; Cho, J.Y. Chemical Constituents Identified from Fruit Body of Cordyceps bassiana and Their Anti-Inflammatory Activity. Biomol. Ther. (Seoul) 2017, 25, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Mediero, A.; Cronstein, B.N. Adenosine and bone metabolism. Trends Endocrinol. Metab. 2013, 24, 290–300. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinson, E.A.; Johnson, R.A.; Wells, J.N. Potent adenosine receptor antagonists that are selective for the A1 receptor subtype. Mol. Pharmacol. 1987, 31, 247–252. [Google Scholar] [PubMed]
- Xiao, J.H.; Sun, Z.H.; Pan, W.D.; Lu, Y.H.; Chen, D.X.; Zhong, J.J. Jiangxienone, a new compound with potent cytotoxicity against tumor cells from traditional Chinese medicinal mushroom Cordyceps jiangxiensis. Chem. Biodivers. 2012, 9, 1349–1355. [Google Scholar] [CrossRef] [PubMed]
- Park, J.G.; Yang, H.K.; Kim, W.H.; Chung, J.K.; Kang, M.S.; Lee, J.H.; Oh, J.H.; Park, H.S.; Yeo, K.S.; Kang, S.H.; et al. Establishment and characterization of human gastric carcinoma cell lines. Int. J. Cancer 1997, 70, 443–449. [Google Scholar] [CrossRef] [Green Version]
- Yoshiyama, H.; Imai, S.; Shimizu, N.; Takada, K. Epstein-Barr virus infection of human gastric carcinoma cells: Implication of the existence of a new virus receptor different from CD21. J. Virol. 1997, 71, 5688–5691. [Google Scholar] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, K.F.; Yeung, P.L.; Tam, K.P.; Chiang, A.K.S. Counteracting survival functions of EBNA3C in Epstein-Barr virus (EBV)-driven lymphoproliferative diseases by combination of SAHA and bortezomib. Oncotarget 2018, 9, 25101–25114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, K.F.; Cheung, A.K.; Choi, C.K.; Yeung, P.L.; Middeldorp, J.M.; Lung, M.L.; Tsao, S.W.; Chiang, A.K. Inhibition of class I histone deacetylases by romidepsin potently induces Epstein-Barr virus lytic cycle and mediates enhanced cell death with ganciclovir. Int. J. Cancer 2016, 138, 125–136. [Google Scholar] [CrossRef]
- Hui, K.F.; Ho, D.N.; Tsang, C.M.; Middeldorp, J.M.; Tsao, G.S.; Chiang, A.K. Activation of lytic cycle of Epstein-Barr virus by suberoylanilide hydroxamic acid leads to apoptosis and tumor growth suppression of nasopharyngeal carcinoma. Int. J. Cancer 2012, 131, 1930–1940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klinger, M.; Freissmuth, M.; Nanoff, C. Adenosine receptors: G protein-mediated signalling and the role of accessory proteins. Cell. Signal. 2002, 14, 99–108. [Google Scholar] [CrossRef]
- Blue, W.T.; Winland, R.D.; Stobbs, D.G.; Kirksey, D.F.; Savage, R.E. Effects of adenosine monophosphate on the reactivation of latent herpes simplex virus type 1 infections of mice. Antimicrob. Agents Chemother. 1981, 20, 547–548. [Google Scholar] [CrossRef] [PubMed]
- Colgin, M.A.; Smith, R.L.; Wilcox, C.L. Inducible cyclic AMP early repressor produces reactivation of latent herpes simplex virus type 1 in neurons in vitro. J. Virol. 2001, 75, 2912–2920. [Google Scholar] [CrossRef] [PubMed]
- Wildeman, M.A.; Novalic, Z.; Verkuijlen, S.A.; Juwana, H.; Huitema, A.D.; Tan, I.B.; Middeldorp, J.M.; de Boer, J.P.; Greijer, A.E. Cytolytic virus activation therapy for Epstein-Barr virus-driven tumors. Clin. Cancer Res. 2012, 18, 5061–5070. [Google Scholar] [CrossRef] [PubMed]
- Novalic, Z.; van Rossen, T.; Greijer, A.; Middeldorp, J. Agents and approaches for lytic induction therapy of Epstein-Barr Virus associated malignancies. Med. Chem. (Los Angel.) 2016, 6, 449–466. [Google Scholar]
- Lee, M.; Son, M.; Ryu, E.; Shin, Y.S.; Kim, J.G.; Kang, B.W.; Cho, H.; Kang, H. Quercetin-induced apoptosis prevents EBV infection. Oncotarget 2015, 6, 12603–12624. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Zhang, Y.; Yang, M.; Zhang, Y.; Xie, Q.; Li, Z.; Dong, Z.; Yang, Y.; Deng, B.; Feng, A.; et al. Hypoxia induces T-cell apoptosis by inhibiting chemokine C receptor 7 expression: The role of adenosine receptor A(2). Cell. Mol. Immunol. 2010, 7, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Wang, Z.; Stong, N.; Plasschaert, R.; Moczan, A.; Chen, H.S.; Hu, S.; Wikramasinghe, P.; Davuluri, R.V.; Bartolomei, M.S.; et al. A role for CTCF and cohesin in subtelomere chromatin organization, TERRA transcription, and telomere end protection. EMBO J. 2012, 31, 4165–4178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.H.; Lee, S.; Shin, Y.S.; Cho, M.; Kang, H.; Cho, H. Anti-Cancer Effect of Quercetin in Xenograft Models with EBV-Associated Human Gastric Carcinoma. Molecules 2016, 21, 1286. [Google Scholar] [CrossRef] [PubMed]














© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, S.J.; Ryu, E.; Lee, S.; Huh, S.; Shin, Y.S.; Kang, B.W.; Kim, J.G.; Cho, H.; Kang, H. Adenosine Induces EBV Lytic Reactivation through ADORA1 in EBV-Associated Gastric Carcinoma. Int. J. Mol. Sci. 2019, 20, 1286. https://doi.org/10.3390/ijms20061286
Choi SJ, Ryu E, Lee S, Huh S, Shin YS, Kang BW, Kim JG, Cho H, Kang H. Adenosine Induces EBV Lytic Reactivation through ADORA1 in EBV-Associated Gastric Carcinoma. International Journal of Molecular Sciences. 2019; 20(6):1286. https://doi.org/10.3390/ijms20061286
Chicago/Turabian StyleChoi, Su Jin, Eunhyun Ryu, Seulki Lee, Sora Huh, Yu Su Shin, Byung Woog Kang, Jong Gwang Kim, Hyosun Cho, and Hyojeung Kang. 2019. "Adenosine Induces EBV Lytic Reactivation through ADORA1 in EBV-Associated Gastric Carcinoma" International Journal of Molecular Sciences 20, no. 6: 1286. https://doi.org/10.3390/ijms20061286