Evolutionary Toxicogenomics of the Striped Killifish (Fundulus majalis) in the New Bedford Harbor (Massachusetts, USA)


Abstract
:1. Introduction
2. Results
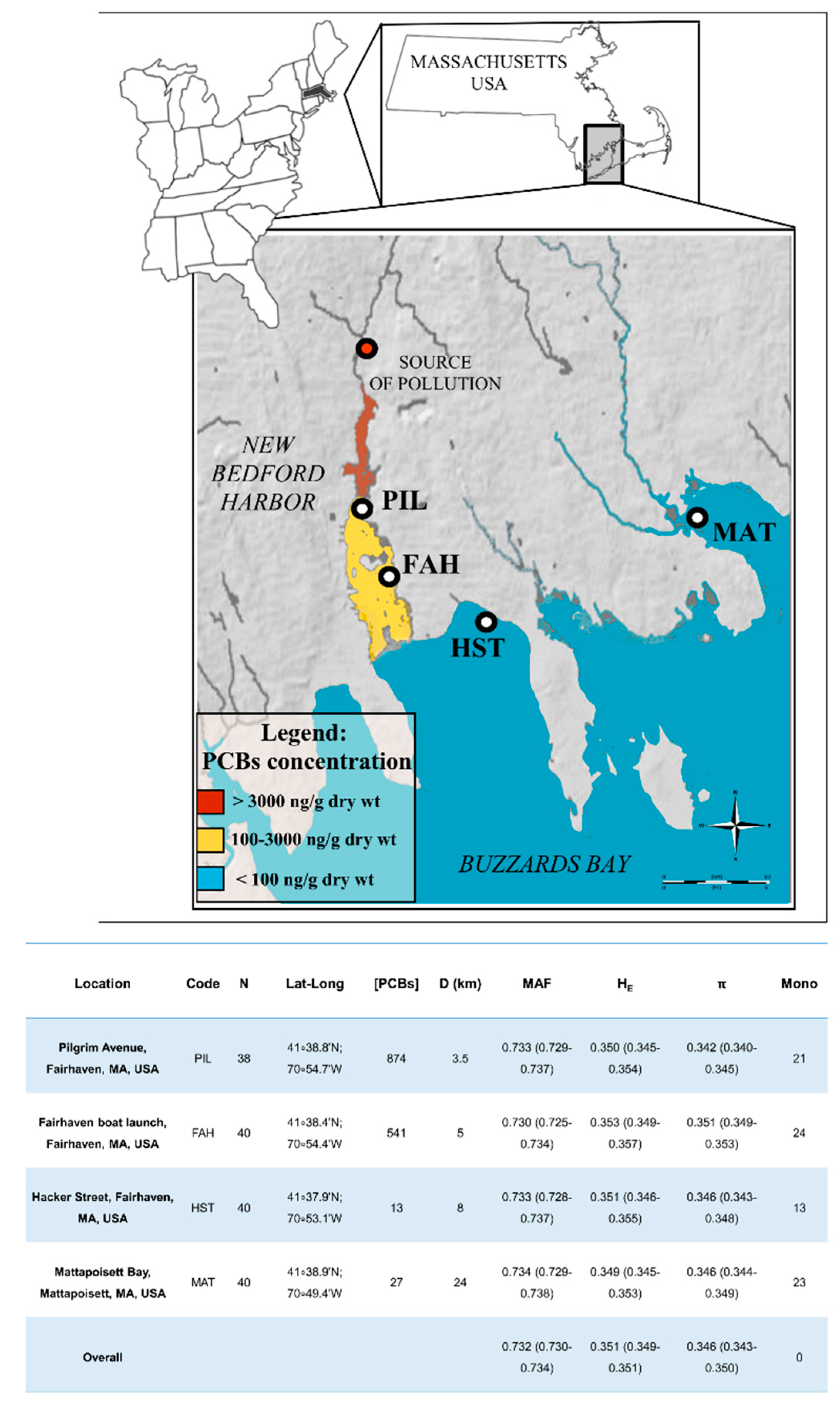
2.1. NGS Sequencing
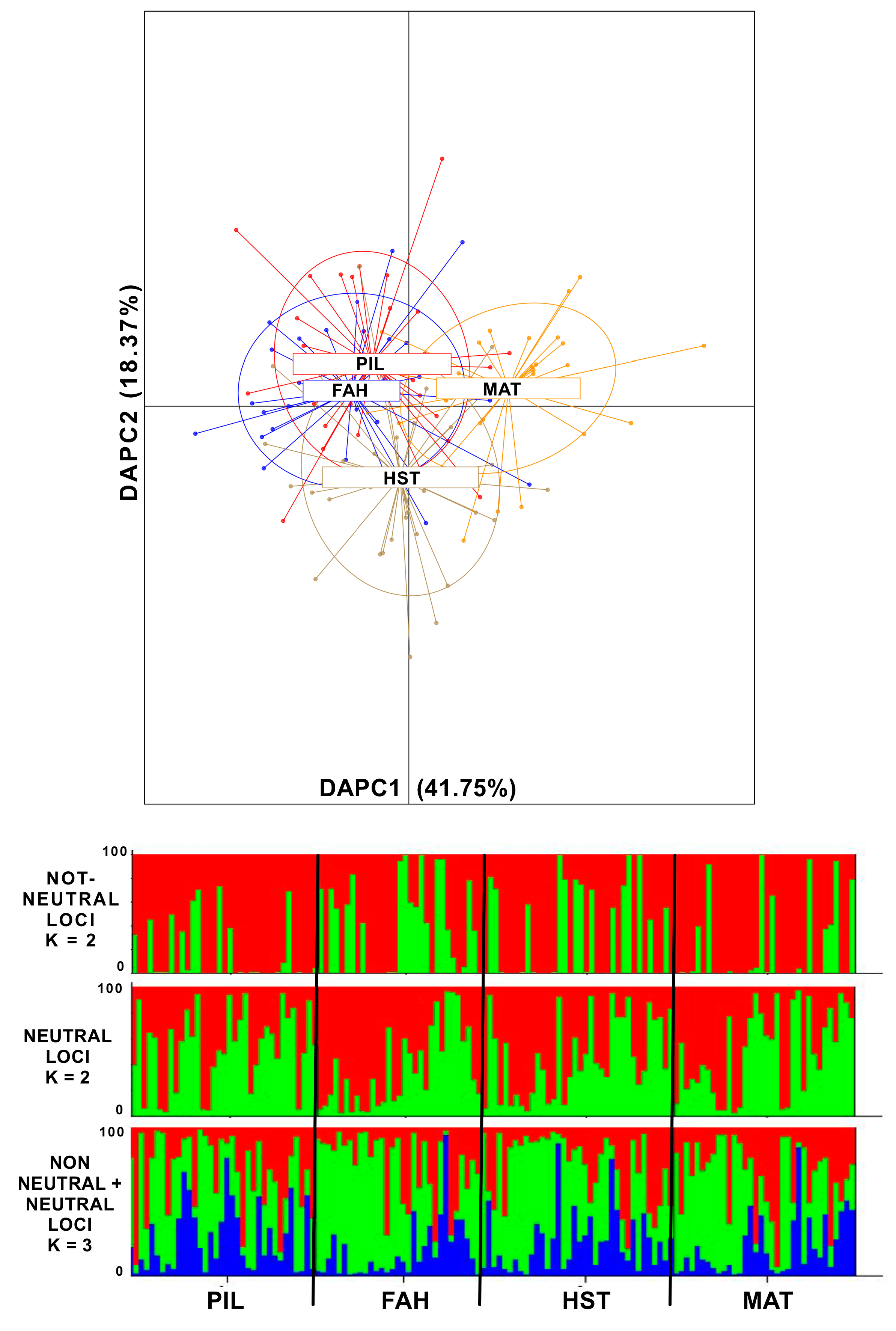
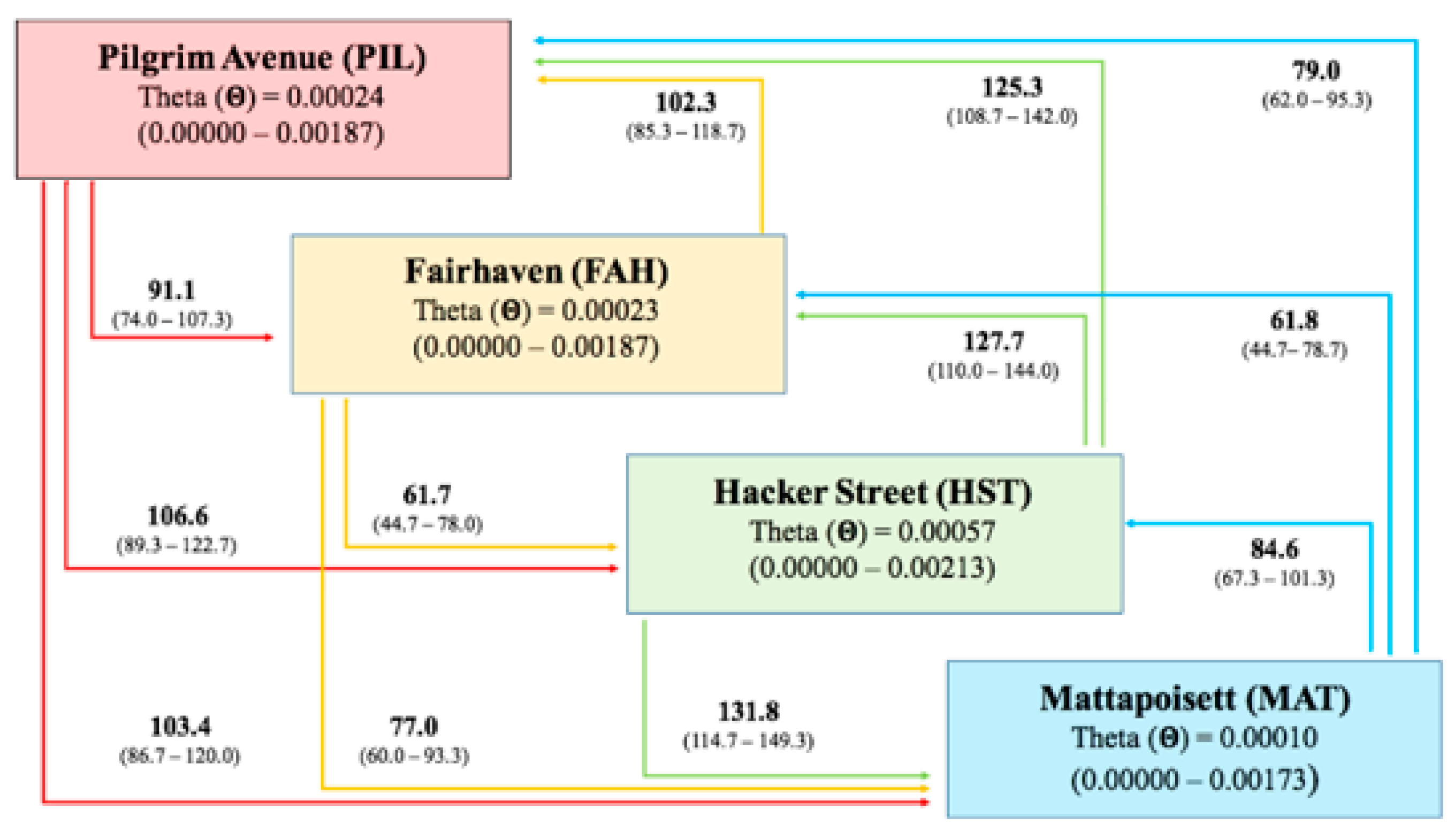
2.2. Genetic Differentiation Between Populations and Gene Flow
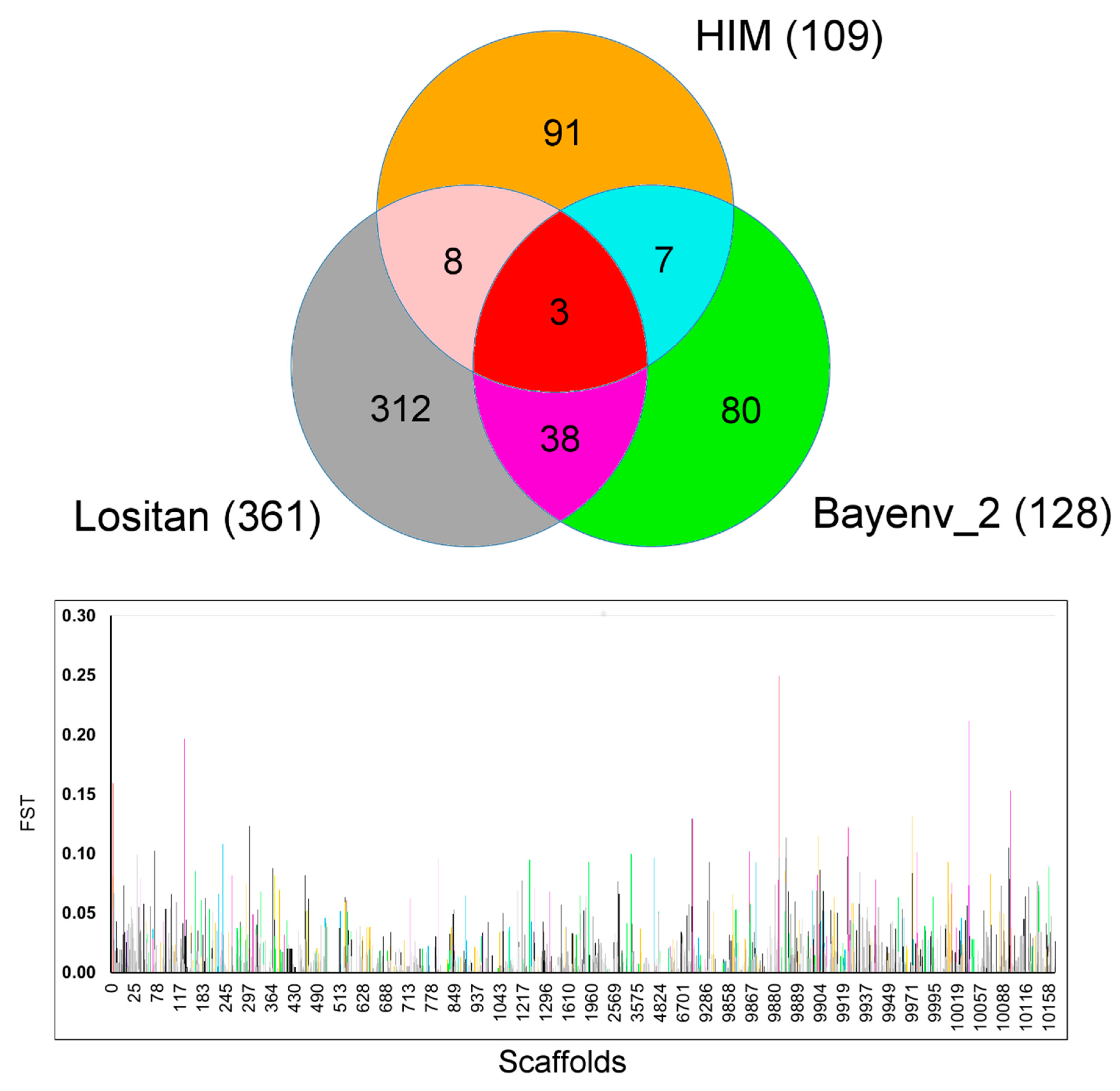
2.3. Tests for Detecting Signatures of Selection
2.4. Tests for Functional Annotation of Candidate Outliers
3. Discussion
3.1. Genetic Variability and Population Differentiation for F. majalis in the New Bedford Harbor
3.2. What Tests for Candidate Outliers can Tell Us about Selective Response to Pollutants: A Matter of Relative Performance
3.3. Genome-Environment Interaction in F. majalis from NBH
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cope, W.G.; Leidy, R.B.; Hodgson, E. Classes of toxicants: use classes. In A Textbook of Modern Toxicology; Hodgson, E., Ed.; John Wiley and Sons Inc.: New York, NY, USA, 2004; pp. 49–74. [Google Scholar]
- Wisdom, H.L. Contamination of the marine environment from land–based sources. Mar. Pollut. Bull. 1992, 25, 32–36. [Google Scholar]
- GESAMP. Reducing environmental impacts of coastal aquaculture. Rep. Stud. 1991, 47. [Google Scholar]
- Johnston, E.L.; Mayer–Pinto, M.; Crowe, T.P.; Frid, C. Chemical contaminant effects on marine ecosystem functioning. J. Appl. Ecol. 2015, 52, 140–149. [Google Scholar] [CrossRef]
- Hamilton, P.B.; Cowx, I.G.; Oleksiak, M.F.; Grahn, M.; Stevens, J.R.; Carvalho, G.R.; Nicol, E.; Tyler, C.R. Population-level consequences for wild fish exposed to sublethal concentrations of chemicals–a critical review. Fish Fish. 2015, 17, 545–566. [Google Scholar] [CrossRef]
- Oziolor, E.M.; Matson, C.W. Evolutionary toxicology: population adaptation in response to anthropogenic pollution. In Extremophile Fishes: Ecology, Evolution, and Physiology of Teleosts in Extreme Environments; Riesch, R., Tobler, M., Plath, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2015; pp. 247–277. [Google Scholar]
- Ribeiro, R.; Baird, D.J.; Soares, A.M.; Lopes, I. Contaminant driven genetic erosion: a case study with Daphnia longispina. Environ. Toxicol. Chem. 2012, 31, 977–982. [Google Scholar] [CrossRef] [PubMed]
- Dallinger, R.; Hockner, M. Evolutionary concepts in ecotoxicology: tracing the genetic background of differential cadmium sensitivities in invertebrate lineages. Ecotoxicology 2013, 22, 767–778. [Google Scholar] [CrossRef] [PubMed]
- Williams, L.M.; Oleksiak, M.F. Signatures of selection in natural populations adapted to chronic pollution. BMC Evol. Biol. 2008, 8, 282. [Google Scholar] [CrossRef] [PubMed]
- Bozinovic, G.; Oleksiak, M.F. Genomic approaches with natural fish populations from polluted environments. Environ. Toxicol. Chem. 2011, 30, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, A.; Galvez, F.; Zhang, S.; Williams, L.M.; Oleksiak, M.F. Functional genomics of physiological plasticity and local adaptation in killifish. J. Hered. 2011, 102, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Laporte, M.; Pavey, S.A.; Rougeux, C.; Pierron, F.; Lauzent, M.; Budzinki, H.; Labadie, P.; Geneste, E.; Couture, P.; Baudrimont, M.; Bernatchez, L. RAD sequencing reveals within–generation polygenic selection in response to anthropogenic organic and metal contamination in North Atlantic Eels. Mol. Ecol. 2016, 25, 219–237. [Google Scholar] [CrossRef] [PubMed]
- Narum, S.R.; Buerkle, C.A.; Davey, J.W.; Miller, M.R.; Hohenlohe, P.A. Genotyping–by–sequencing in ecological and conservation genomics. Mol. Ecol. 2013, 22, 2841–2847. [Google Scholar] [CrossRef] [PubMed]
- Savolainen, O.; Lascoux, M.; Merila, J. Ecological genomics of local adaptation. Nat. Rev. Genet. 2013, 14, 807–820. [Google Scholar] [CrossRef] [PubMed]
- Ellegren, H. Genome sequencing and population genomics in non–model organisms. Trends Ecol. Evol. 2014, 29, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Reid, N.M.; Whitehead, A. Functional genomics to assess biological responses to marine pollution at physiological and evolutionary timescales: toward a vision of predictive ecotoxicology. Brief Funct. Genom. 2015, 15, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Svingen, T.; Vinggaard, A.M. The risk of chemical cocktail effects and how to deal with the issue. J. Epidemiol. Community Health 2016, 70, 322–323. [Google Scholar] [CrossRef] [PubMed]
- Tiffin, P.; Ross–Ibarra, J. Advances and limits of using population genetics to understand local adaptation. Trends Ecol. Evol. 2014, 29, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping–by–sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed]
- Pruell, R.J.; Norwood, C.B.; Bowen, R.D.; Boothman, W.S.; Rogerson, P.F.; Hackett, M.; Butterworth, B.C. Geochemical study of sediment contamination in New Bedford Harbor, Massachusetts. Mar. Environ. Res. 1990, 29, 77–101. [Google Scholar] [CrossRef]
- Nelson, W.G.; Bergen, B.J. The New Bedford Harbor Superfund site long–term monitoring program (1993–2009). Environ. Monit. Assess. 2012, 184, 7531–7550. [Google Scholar] [CrossRef] [PubMed]
- Kidd, K.A.; Blanchfield, P.J.; Mills, K.H.; Palace, V.P.; Evans, R.E.; Lazorchak, J.M.; Flick, R.W. Collapse of a fish population after exposure to a synthetic estrogen. Proc. Natl. Acad. Sci. USA 2007, 104, 8897–8901. [Google Scholar] [CrossRef] [PubMed]
- Oleksiak, M.F.; Karchner, S.I.; Jenny, M.J.; Franks, D.G.; Welch, D.B.M.; Hahn, M.E. Transcriptomic assessment of resistance to effects of an aryl hydrocarbon receptor (AHR) agonist in embryos of Atlantic killifish (Fundulus heteroclitus) from a marine Superfund site. BMC Genom. 2011, 12, 263. [Google Scholar] [CrossRef] [PubMed]
- Incardona, J.P.; Scholtz, N.L. The influence of heart developmental anatomy on cardiotoxicity–based adverse outcome pathways in fish. Aquat. Toxicol. 2016, 177, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Nacci, D.E.; Champlin, D.; Jayaraman, S. Adaptation of the estuarine fish Fundulus heteroclitus (Atlantic Killifish) to polychlorinated biphenyls (PCBs). Estuar. Coast. 2010, 33, 853–864. [Google Scholar] [CrossRef]
- Wirgin, I.; Roy, N.K.; Loftus, M.; Chambers, R.C.; Franks, D.G.; Hahn, M.E. Mechanistic basis of resistance to PCBs in Atlantic tomcod from the Hudson River. Science 2011, 331, 1322–1325. [Google Scholar] [CrossRef] [PubMed]
- Maguire_Group. Essential Fish Habitat (EFH) Assessment New Bedford/Fairhaven Harbor Massachusetts March 2002. Mass. Off. Coastl Zone Manag. 2002. [Google Scholar]
- Du, X.; Crawford, D.L.; Oleksiak, M.F. Effects of anthropogenic pollution on the oxidative phosphorylation pathway of hepatocytes from natural populations of Fundulus heteroclitus. Aquat. Toxicol. 2015, 165, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Crawford, D.L.; Nacci, D.; Oleksiak, M.F. Heritable adaptation of oxidative phosphorylation pathway in pollutant resistant Fundulus heteroclitus population. Aquat. Toxicol. 2016, 177, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Burnett, K.G.; Bain, L.J.; Baldwin, W.S.; Callard, G.V.; Di Giulio, R.T.; Evans, D.H.; Gomez-Chiarri, M.; Hahn, M.E.; Hoover, C.A.; Karchner, S.I.; et al. Fundulus as the premier teleost model in environmental biology: opportunities for new insights using genomics. Comp. Biochem. Physiol. Part D Genom. Proteom. 2007, 2, 257–286. [Google Scholar] [CrossRef] [PubMed]
- Oleksiak, M.F. Changes in gene expression due to chronic exposure to environmental pollutants. Aquat. Toxicol. 2008, 90, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, A.; Roach, J.L.; Zhang, S.; Galvez, F. Genomic mechanisms of evolved physiological plasticity in killifish distributed along an environmental salinity gradient. Proc. Natl. Acad. Sci. USA 2011, 108, 6193–6198. [Google Scholar] [CrossRef] [PubMed]
- Baris, T.Z.; Crawford, D.L.; Oleksiak, M.F. Acclimation and acute temperature effects on population differences in oxidative phosphorylation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 310, R185–R196. [Google Scholar] [CrossRef] [PubMed]
- Williams, L.M.; Oleksiak, M.F. Evolutionary and functional analyses of cytochrome P4501A promoter polymorphisms in natural populations. Mol. Ecol. 2011, 20, 5236–5247. [Google Scholar] [CrossRef] [PubMed]
- Reitzel, A.M.; Karchner, S.I.; Franks, D.G.; Evans, B.R.; Nacci, D.; Champlin, D.; Vieira, V.M.; Hahn, M.E. Genetic variation at aryl hydrocarbon receptor (AHR) loci in populations of Atlantic killifish (Fundulus heteroclitus) inhabiting polluted and reference habitats. BMC Evol. Biol. 2014, 14, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Roark, S.A.; Nacci, D.; Coiro, L.; Champlin, D.; Guttman, S.I. Population genetic structure of a nonmigratory estuarine fish (Fundulus heteroclitus) across a strong gradient of polychlorinated biphenyl contamination. Environ. Toxicol. Chem. 2005, 24, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Abraham, B.J. Species profiles, life histories and environmental requirements of coastal fishes and invertebrates (Mid–Atlantic). MUMMICHOG AND STRIPED KILLIFISH. DTIC Document (1985).
- Giska, I.; Babik, W.; van Gestel, C.A.; van Straalen, N.M.; Laskowski, R. Genome–wide genetic diversity of rove beetle populations along a metal pollution gradient. Ecotox. Environ. Safe 2015, 119, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Günther, T.; Coop, G. Robust identification of local adaptation from allele frequencies. Genetics 2013, 195, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Bickham, J.W. The four cornerstones of evolutionary toxicology. Ecotoxicology 2011, 20, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Narum, S.R.; Campbell, N.R.; Meyer, K.A.; Miller, M.R.; Hardy, R.W. Thermal adaptation and acclimation of ectotherms from differing aquatic climates. Mol. Ecol. 2013, 22, 3090–3097. [Google Scholar] [CrossRef] [PubMed]
- Ravinet, M.; Westram, A.; Johannesson, K.; Butlin, R.; Andre, C.; Panova, M. Shared and nonshared genomic divergence in parallel ecotypes of Littorina saxatilis at a local scale. Mol. Ecol. 2016, 25, 287–305. [Google Scholar] [CrossRef] [PubMed]
- van Straalen, N.; Timmermans, M. Genetic variation in toxicant–stressed populations: an evaluation of the “genetic erosion” hypothesis. Hum. Ecol. Risk Assess. 2002, 8, 983–1002. [Google Scholar] [CrossRef]
- Boorse, D.; Storlie, C. Diel migration of invertebrates and fishesin Dean Creek, Sapelo Island, GA (1993).
- Sexton, J.P.; Hangartner, S.B.; Hoffmann, A.A. Genetic isolation by environment or distance: which pattern of gene flow is most common? Evolution 2014, 68, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Sexton, J.P.; Strauss, S.Y.; Rice, K.J. Gene flow increases fitness at the warm edge of a species’ range. Proc. Natl. Acad. Sci. USA 2011, 108, 11704–11709. [Google Scholar] [CrossRef] [PubMed]
- Bijlsma, R.; Loeschcke, V. Genetic erosion impedes adaptive responses to stressful environments. Evol. Appl. 2012, 5, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Stinchcombe, J.R.; Hoekstra, H.E. Combining population genomics and quantitative genetics: finding the genes underlying ecologically important traits. Heredity 2008, 100, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Lotterhos, K.E.; Whitlock, M.C. Evaluation of demographic history and neutral parameterization on the performance of FST outlier tests. Mol. Ecol. 2014, 23, 2178–2192. [Google Scholar] [CrossRef] [PubMed]
- Whitlock, M.C. Modern approaches to local adaptation. Am. Nat. 2015, 186, S1–S4. [Google Scholar] [CrossRef] [PubMed]
- Whitlock, M.C.; Lotterhos, K.E. The relative power of genome scans to detect local adaptation depends on sampling design and statistical method. Mol. Ecol. 2015, 24, 1031–1046. [Google Scholar]
- Narum, S.R.; Hess, J.E. Comparison of F(ST) outlier tests for SNP loci under selection. Mol. Ecol. Res. 2011, 11, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Nosil, P.; Funk, D.J.; Ortiz–Barrientos, D. Divergent selection and heterogeneous genomic divergence. Mol. Ecol. 2009, 9, 375–402. [Google Scholar] [CrossRef] [PubMed]
- Kemper, K.E.; Saxton, S.J.; Bolormaa, S.; Hayes, B.J.; Goddard, M.E. Selection for complex traits leaves little or no classic signatures of selection. BMC Genomics 2014, 15, 246. [Google Scholar] [CrossRef] [PubMed]
- Le Corre, V.; Kremer, A. The genetic differentiation at quantitative trait loci under local adaptation. Mol. Ecol. 2012, 20, 1548–1566. [Google Scholar] [CrossRef] [PubMed]
- De Mita, S.; Thuillet, A.C.; Gay, L.; Ahmadi, N.; Manel, S.; Ronfort, J.; Vigouroux, Y. Detecting selection along environmental gradients: analysis of eight methods and their effectiveness for outbreeding and selfing populations. Mol. Ecol. 2013, 22, 1383–1399. [Google Scholar] [CrossRef] [PubMed]
- Stankowski, S.; Sobel, J.M.; Streisfeld, M.A. Geographic cline analysis as a tool for studying genome–wide variation: a case study of pollinator–mediated divergence in a monkeyflower. Mol. Ecol. 2017, 26, 107–122. [Google Scholar] [CrossRef] [PubMed]
- Babin, C.; Gagnaire, P.A.; Pavey, S.A.; Bernatchez, L. RAD–Seq reveals patterns of additive polygenic variation caused by spatially–varying selection in the American Eel (Anguilla rostrata). Genome Biol. Evol. 2017, 9, 2974–2986. [Google Scholar] [CrossRef] [PubMed]
- Gascon, M.; Morales, E.; Sunyer, J.; Vrijheid, M. Effects of persistent organic pollutants on the developing respiratory and immune systems: a systematic review. Environ. Int. 2013, 52, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.W.; Novak, R.F.; Anderson, H.A.; Birnbaum, L.S.; Blystone, C.; DeVito, M.; Jacobs, D.; Lee, D.H. Evaluation of the association between persistent organic pollutants (POPs) and diabetes in epidemiological studies: a national toxicology program workshop review. Environ. Health Persp. 2013, 121, 774–783. [Google Scholar] [CrossRef] [PubMed]
- Perkins, J.T.; Petriello, M.C.; Newsome, B.J.; Hennig, B. Polychlorinated biphenyls and links to cardiovascular disease. Environ. Sci. Pollut. Res. Int. 2016, 23, 2160–2172. [Google Scholar] [CrossRef] [PubMed]
- Proestou, D.A.; Flight, P.; Champlin, D.; Nacci, D. Targeted approach to identify genetic loci associated with evolved dioxin tolerance in the Atlantic Killifish (Fundulus heteroclitus). BMC Evol. Biol. 2014, 14, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Hutt, D.M.; Baltz, J.M.; Ngsee, J.K. Synaptotagmin VI and VIII and syntaxin 2 are essential for the mouse sperm acrosome reaction. J. Biol. Chem. 2005, 208, 20197–20203. [Google Scholar] [CrossRef] [PubMed]
- Souza, M.S.; Hallgren, P.; Balseiro, E.; Hansson, L.A. Low concentrations, potential ecological consequences: synthetic estrogens alter life–history and demographic structures of aquatic invertebrates. Environ. Pollut. 2013, 178, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Jarque, S.; Quiros, L.; Grimalt, J.O.; Gallego, E.; Catalan, J.; Lackner, R.; Piña, B. Background fish feminization effects in European remote sites. Sci. Rep. 2015, 5, 11292. [Google Scholar] [CrossRef] [PubMed]
- Salama, J.; Chakraborty, T.R.; Ng, L.; Gore, A.C. Effects of Polychlorinated Biphenyls on Estrogen Receptor–ß Expression in the Anteroventral Periventricular Nucleus. Environ. Health Persp. 2003, 111, 1278–1282. [Google Scholar]
- Lin, C.H.; Huang, C.L.; Chuang, M.C.; Wang, Y.J.; Chen, D.R.; Chen, S.T.; Lin, P.H. Protective role of estrogen receptor–alpha on lower chlorinated PCB congener–induced DNA damage and repair in human tumoral breast cells. Toxicol. Lett. 2009, 188, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Greytak, S.R.; Callard, G.V. Cloning of three estrogen receptors (ER) from killifish (Fundulus heteroclitus): differences in populations from polluted and reference environments. Gen. Comp. Endocr. 2007, 150, 174–188. [Google Scholar] [CrossRef] [PubMed]
- Greytak, S.R.; Tarrant, A.M.; Nacci, D.; Hahn, M.E.; Callard, G.V. Estrogen responses in killifish (Fundulus heteroclitus) from polluted and unpolluted environments are site– and gene–specific. Aquat. Toxicol. 2010, 99, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Uno, T.; Ishizuka, M.; Itakura, T. Cytochrome P450 (CYP) in fish. Environ. Toxicol. Pharmacol. 2012, 34, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C. Polyunsaturated fatty acids and inflammatory processes: New twists in an old tale. Biochimie 2009, 91, 791–795. [Google Scholar] [CrossRef] [PubMed]
- Powell, W.H.; Bright, R.; Bello, S.M.; Hahn, M.E. Developmental and Tissue–Specific Expression of AHR1, AHR2, and ARNT2 in Dioxin–Sensitive and –Resistant Populations of the Marine Fish Fundulus heteroclitus. Toxicoll. Sci. 2000, 57, 229–239. [Google Scholar] [CrossRef]
- Nacci, D.; Huber, M.; Champlin, D.; Jayaraman, S.; Cohen, S.; Gauger, E.; Fong, A.; Gomez–Chiari, M. Evolution of tolerance to PCBs and susceptibility to a bacterial pathogen (Vibrio harveyi) in Atlantic killifish (Fundulus heteroclitus) from New Bedford (MA, USA) harbour. Environ. Poll. 2009, 157, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, N.V.; Dewaard, J.R.; Hebert, P.D.N. An inexpensive, automation–friendly protocol for recovering high–quality DNA. Mol. Ecol. Notes 2006, 6, 998–1002. [Google Scholar] [CrossRef]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevenssss, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [PubMed]
- Reid, N.M.; Jackson, C.E.; Gilbert, D.; Minx, P.; Montague, M.J.; Hampton, T.H.; Helfrich, L.W.; King, B.L.; Nacci, D.E.; Aluru, N.; et al. The Landscape of Extreme Genomic Variation in the Highly Adaptable Atlantic Killifish. Genome Biol. Evol. 2017, 9, 659–676. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Lischer, H.E. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Res. 2010, 10, 564–567. [Google Scholar]
- Nunez, J.C.; Seale, T.P.; Fraser, M.A.; Burton, T.L.; Fortson, T.L.; Hoover, D.; Travis, J.; Oleksiak, M.F.; Crawford, D.L. Population Genomics of the Euryhaline Teleost Poecilia latipinna. PLoS ONE 2015, 10, e0137077. [Google Scholar] [CrossRef] [PubMed]
- Crawford, D.L.; Oleksiak, M.F. Ecological population genomics in the marine environment. Brief Funct. Genomics 2016, elw008. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Antao, T.; Lopes, A.; Lopes, R.J.; Beja–Pereira, A.; Luikart, G. LOSITAN: a workbench to detect molecular adaptation based on a Fst–outlier method. BMC Bioinformatics 2008, 9, 323. [Google Scholar] [CrossRef] [PubMed]
- Jombart, T.; Devillard, S.; Balloux, F. Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genetics 2010, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnely, P. Inference of Population Structure Using Multilocus Genotype Data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Piry, S.; Alapetite, A.; Cornuet, J.M.; Paetkau, D.; Baudouin, L.; Estoup, A. GENECLASS2: a software for genetic assignment and first–generation migrant detection. J. Hered. 2004, 95, 536–539. [Google Scholar] [CrossRef] [PubMed]
- Beerli, P. Comparison of Bayesian and maximum–likelihood inference of population genetic parameters. Bioinformatics 2006, 22, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Weir, B.; Cockerham, C.C. Estimating F–statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [PubMed]
- Jombart, T.; Ahmed, I. Adegenet 1.3–1: new tools for the analysis of genome–wide SNP data. Bioinformatics 2011, 27, 3070–3071. [Google Scholar] [CrossRef] [PubMed]
- Earl, D.A.; vonHoldt, B.M. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2011, 4, 359–361. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 2011, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Res. 2015, 15, 1179–1191. [Google Scholar] [CrossRef] [PubMed]
- Rannala, B.; Mountain, J.L. Detecting immigration by using multilocus genotypes. Proc. Natl. Acad. Sci. USA 1997, 94, 9197–9201. [Google Scholar] [CrossRef]
- Excoffier, L.; Hofer, T.; Foll, M. Detecting loci under selection in a hierarchically structured population. Heredity 2009, 103, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. "BLAST+: architecture and applications. BMC Bioinformatics 2008, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PIL | FAH | HST | MAT | |
|---|---|---|---|---|
| PIL | 0 | 0.0303 ** | 0.0034 | 0.0111 * |
| FAH | −0.0039 | 0 | 0.0250 *** | 0.0221 *** |
| HST | −0.0048 | −0.0053 | 0 | 0.0158 ** |
| MAT | −0.004 | −0.0052 | −0.0057 | 0 |
| KEGG Pathway Terms | N | % | p-Value | Category |
|---|---|---|---|---|
| Nitrogen metabolism | 11 | 2.5 | 3.40 × 10−12 | a |
| PPAR signaling pathway | 11 | 2.5 | 1.30 × 10−5 | a |
| Mineral absorption | 9 | 2.1 | 2.70 × 10−5 | a |
| Neurotrophin signaling pathway | 14 | 3.2 | 2.40 × 10−5 | b |
| Regulation of actin cytoskeleton | 18 | 4.2 | 6.30 × 10−5 | c |
| ErbB signaling pathway | 11 | 2.5 | 1.30 × 10−4 | b, d |
| Adherens junction | 8 | 1.9 | 3.30 × 10−3 | c |
| Ras signaling pathway | 15 | 3.5 | 3.80 × 10−3 | b, c |
| MAPK signaling pathway | 16 | 3.7 | 4.20 × 10−3 | b, h |
| Fc gamma R-mediated phagocytosis | 8 | 1.9 | 8.30 × 10−3 | b, c, f |
| Proteoglycans in cancer | 13 | 3 | 9.20 × 10−3 | b, c, d |
| Tight junction | 8 | 1.9 | 1.00 × 10−2 | b, c |
| Epithelial cell signaling in Helicobacter pylori infection | 7 | 1.6 | 1.00 × 10−2 | b, f, h |
| Pathogenic Escherichia coli infection | 6 | 1.4 | 1.30 × 10−2 | c, f, h |
| Rap1 signaling pathway | 13 | 3 | 1.30 × 10−2 | b, c |
| Leukocyte transendothelial migration | 9 | 2.1 | 1.40 × 10−2 | c, f, h |
| Endocytosis | 14 | 3.2 | 1.60 × 10−2 | a, c |
| cAMP signaling pathway | 12 | 2.8 | 2.10 × 10−2 | b, c, e, f, g |
| Salmonella infection | 7 | 1.5 | 4.60 × 10−2 | b, f, h |
| Renal cell carcinoma | 6 | 1.4 | 3.50 × 10−2 | b, d |
| Arhythmogenic right ventricular cardiomyopathy (ARVC) | 6 | 1.4 | 4.20 × 10−2 | c, f, h |
| Cell adhesion molecules (CAMs) | 9 | 2.1 | 4.20 × 10−2 | c, f |
| Inflammatory mediator regulation of TRP channels | 7 | 1.6 | 4.30 × 10−2 | h |
| Estrogen signaling pathway | 7 | 1.6 | 4.60 × 10−3 | b, g |
| T cell receptor signaling pathway | 7 | 1.6 | 5.80 × 10−2 | b, f, h |
| Choline metabolism in cancer | 7 | 1.6 | 6.00 × 10−2 | b, c, h |
| Dopaminergic synapse | 8 | 1.9 | 6.40 × 10−2 | i |
| Bacterial invasion of epithelial cells | 6 | 1.4 | 6.40 × 10−2 | c, f, h |
| Non-small cell lung cancer | 5 | 1.8 | 6.80 × 10−2 | b, d |
| Regulation of lipolysis in adipocytes | 5 | 1.8 | 5.80 × 10−2 | a |
| Hepatitis C | 8 | 1.9 | 7.50 × 10−2 | d, h |
| Apoptosis | 5 | 1.2 | 9.10 × 10−2 | b, d |
| Shigellosis | 5 | 1.2 | 1.00 × 10−1 | d, h |
| Central carbon metabolism in cancer | 5 | 1.2 | 1.00 × 10−1 | d, h |
| Disease Category | N | % | P-Value | FDR |
|---|---|---|---|---|
| Metabolic | 167 | 34.9 | 1.80 × 10−3 | 4.60 × 10−3 |
| Cardiovascular | 137 | 28.7 | 4.50 × 10−3 | 1.00 × 10−2 |
| Chemo-dependency | 115 | 24.1 | 1.90 × 10−2 | 2.80 × 10−2 |
| Pharmacogenomic | 109 | 22.8 | 2.40 × 10−6 | 4.30 × 10−5 |
| Neurological | 106 | 22.2 | 1.80 × 10−4 | 8.00 × 10−4 |
| Cancer | 103 | 21.5 | 1.70 × 10−2 | 2.80 × 10−2 |
| Psych | 81 | 16.9 | 4.10 × 10−5 | 2.50 × 10−4 |
| Unknown | 66 | 13.8 | 3.10 × 10−4 | 1.10 × 10−3 |
| Developmental | 60 | 12.6 | 1.10 × 10−3 | 3.40 × 10−3 |
| Other | 58 | 12.1 | 1.10 × 10−2 | 1.90 × 10−2 |
| Renal | 54 | 11.3 | 5.70 × 10−3 | 1.10 × 10−2 |
| Reproduction | 37 | 7.7 | 2.10 × 10−2 | 2.90 × 10−2 |
| Normal-variation | 33 | 6.9 | 7.00 × 10−6 | 6.30 × 10−5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruggeri, P.; Du, X.; Crawford, D.L.; Oleksiak, M.F. Evolutionary Toxicogenomics of the Striped Killifish (Fundulus majalis) in the New Bedford Harbor (Massachusetts, USA). Int. J. Mol. Sci. 2019, 20, 1129. https://doi.org/10.3390/ijms20051129
Ruggeri P, Du X, Crawford DL, Oleksiak MF. Evolutionary Toxicogenomics of the Striped Killifish (Fundulus majalis) in the New Bedford Harbor (Massachusetts, USA). International Journal of Molecular Sciences. 2019; 20(5):1129. https://doi.org/10.3390/ijms20051129
Chicago/Turabian StyleRuggeri, Paolo, Xiao Du, Douglas L. Crawford, and Marjorie F. Oleksiak. 2019. "Evolutionary Toxicogenomics of the Striped Killifish (Fundulus majalis) in the New Bedford Harbor (Massachusetts, USA)" International Journal of Molecular Sciences 20, no. 5: 1129. https://doi.org/10.3390/ijms20051129