Exploiting the Therapeutic Potential of Endogenous Immunomodulatory Systems in Multiple Sclerosis—Special Focus on the Peroxisome Proliferator-Activated Receptors (PPARs) and the Kynurenines


Abstract
:1. Introduction
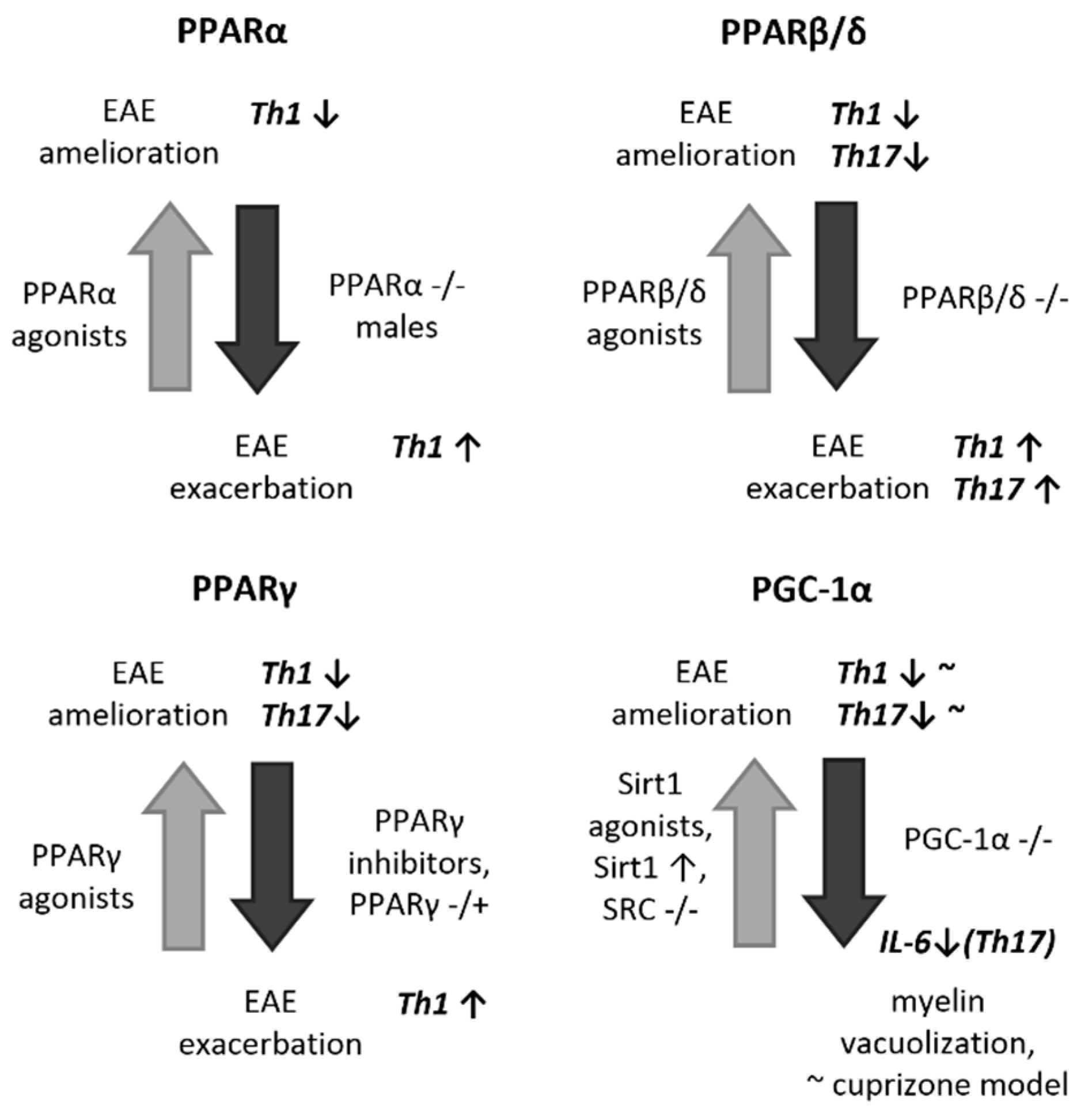
2. The PPAR System and PGC-1α
2.1. In Vitro Basis of the Role of PPARs and PGC-1α in Neuromodulation
2.2. In Vivo and Human Implications for a Protective Role of PPARs and PGC-1α in EAE/MS
2.3. Therapeutic and Diagnostic Perspectives of PPARs and PGC-1α in MS
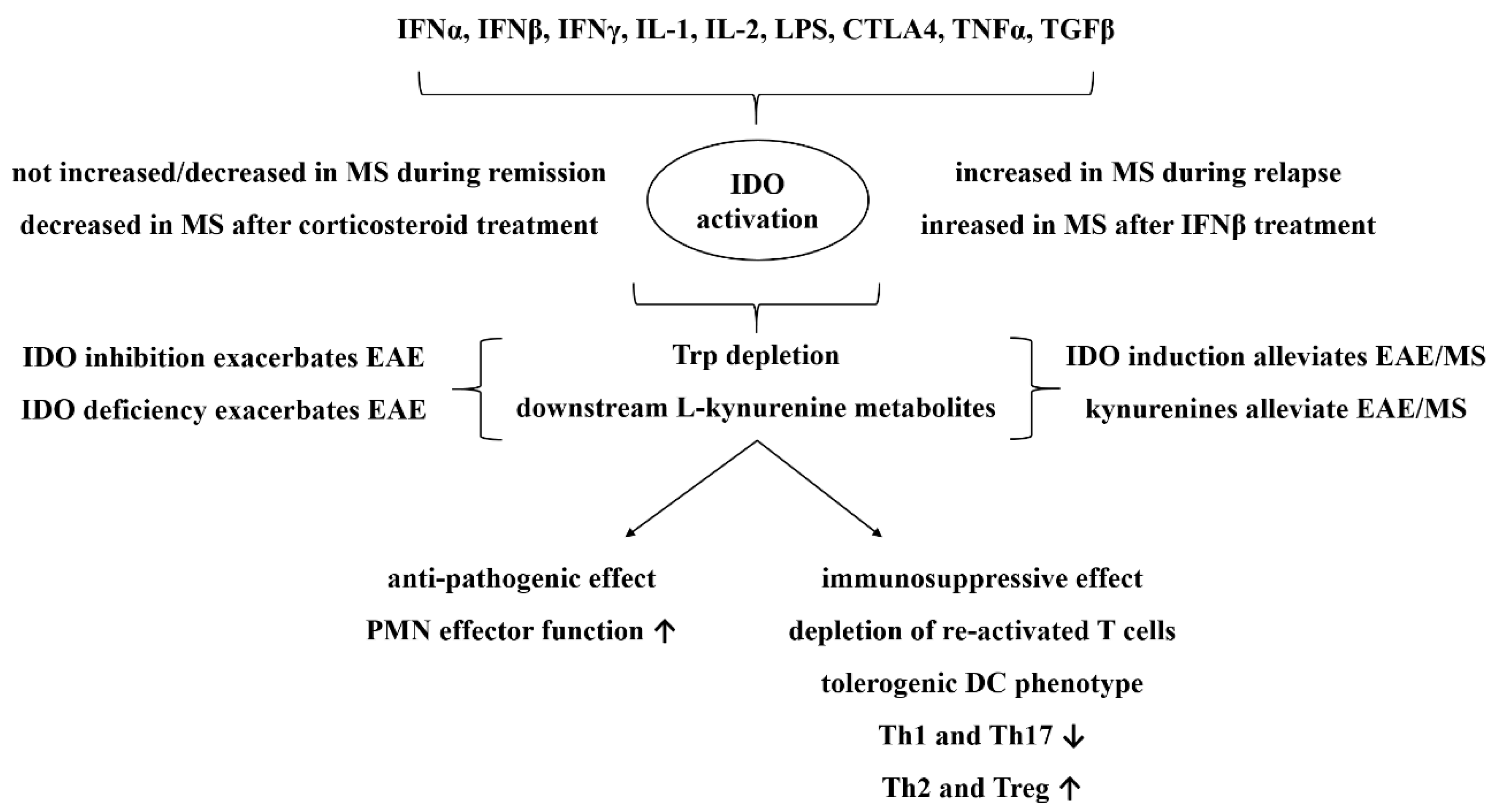
3. The Kynurenine Pathway
3.1. In Vitro Basis of the Role of the Kynurenine Pathway in Neuromodulation
3.2. In Vivo and Human Implications for a Protective role of the Kynurenine Pathway in EAE/MS
3.3. Therapeutic and Diagnostic Perspectives of the Kynurenine Pathway in MS
4. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Amedei, A.; Prisco, D.; D’Elios, M.M. Multiple sclerosis: The role of cytokines in pathogenesis and in therapies. Int. J. Mol. Sci. 2012, 13, 13438–13460. [Google Scholar] [CrossRef]
- Martin, R.; McFarland, H.F. Immunological aspects of experimental allergic encephalomyelitis and multiple sclerosis. Crit. Rev. Clin. Lab. Sci. 1995, 32, 121–182. [Google Scholar] [CrossRef] [PubMed]
- Gold, R.; Linington, C.; Lassmann, H. Understanding pathogenesis and therapy of multiple sclerosis via animal models: 70 years of merits and culprits in experimental autoimmune encephalomyelitis research. Brain 2006, 129 (Pt 8), 1953–1971. [Google Scholar] [CrossRef] [Green Version]
- Kimura, A.; Kishimoto, T. IL-6: Regulator of Treg/Th17 balance. Eur. J. Immunol. 2010, 40, 1830–1835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagelkerken, L. Role of Th1 and Th2 cells in autoimmune demyelinating disease. Braz. J. Med. Biol. Res. 1998, 31, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, T.; Horikawa, M.; Iwata, Y.; Tedder, T.F. Regulatory B cells (B10 cells) and regulatory T cells have independent roles in controlling experimental autoimmune encephalomyelitis initiation and late-phase immunopathogenesis. J. Immunol. 2010, 185, 2240–2252. [Google Scholar] [CrossRef] [PubMed]
- Mikita, J.; Dubourdieu-Cassagno, N.; Deloire, M.S.; Vekris, A.; Biran, M.; Raffard, G.; Brochet, B.; Canron, M.H.; Franconi, J.M.; Boiziau, C.; et al. Altered M1/M2 activation patterns of monocytes in severe relapsing experimental rat model of multiple sclerosis. Amelioration of clinical status by M2 activated monocyte administration. Mult. Scler. 2011, 17, 2–15. [Google Scholar] [CrossRef]
- Racke, M.K.; Gocke, A.R.; Muir, M.; Diab, A.; Drew, P.D.; Lovett-Racke, A.E. Nuclear receptors and autoimmune disease: The potential of PPAR agonists to treat multiple sclerosis. J. Nutr. 2006, 136, 700–703. [Google Scholar] [CrossRef]
- Miron, V.E.; Boyd, A.; Zhao, J.W.; Yuen, T.J.; Ruckh, J.M.; Shadrach, J.L.; van Wijngaarden, P.; Wagers, A.J.; Williams, A.; Franklin, R.J.M.; et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat. Neurosci. 2013, 16, 1211–1218. [Google Scholar] [CrossRef] [Green Version]
- Natarajan, C.; Bright, J.J. Peroxisome proliferator-activated receptor-gamma agonists inhibit experimental allergic encephalomyelitis by blocking IL-12 production, IL-12 signaling and Th1 differentiation. Genes Immun. 2002, 3, 59–70. [Google Scholar] [CrossRef] [Green Version]
- Kanakasabai, S.; Chearwae, W.; Walline, C.C.; Iams, W.; Adams, S.M.; Bright, J.J. Peroxisome proliferator-activated receptor delta agonists inhibit T helper type 1 (Th1) and Th17 responses in experimental allergic encephalomyelitis. Immunology 2010, 130, 572–588. [Google Scholar] [CrossRef] [PubMed]
- Okuda, Y.; Sakoda, S.; Fujimura, H.; Saeki, Y.; Kishimoto, T.; Yanagihara, T. IL-6 plays a crucial role in the induction phase of myelin oligodendrocyte glucoprotein 35-55 induced experimental autoimmune encephalomyelitis. J. Neuroimmunol. 1999, 101, 188–196. [Google Scholar] [CrossRef]
- Samoilova, E.B.; Horton, J.L.; Hilliard, B.; Liu, T.S.; Chen, Y. IL-6-deficient mice are resistant to experimental autoimmune encephalomyelitis: Roles of IL-6 in the activation and differentiation of autoreactive T cells. J. Immunol. 1998, 161, 6480–6486. [Google Scholar] [PubMed]
- Linker, R.A.; Luhder, F.; Kallen, K.J.; Lee, D.H.; Engelhardt, B.; Rose-John, S.; Gold, R. IL-6 transsignalling modulates the early effector phase of EAE and targets the blood-brain barrier. J. Neuroimmunol. 2008, 205, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Frohman, E.M.; Racke, M.K.; Raine, C.S. Multiple sclerosis—The plaque and its pathogenesis. N. Engl. J. Med. 2006, 354, 942–955. [Google Scholar] [CrossRef] [PubMed]
- Legroux, L.; Arbour, N. Multiple Sclerosis and T Lymphocytes: An Entangled Story. J. Neuroimmune Pharmacol. 2015, 10, 528–546. [Google Scholar] [CrossRef] [Green Version]
- Ohl, K.; Tenbrock, K.; Kipp, M. Oxidative stress in multiple sclerosis: Central and peripheral mode of action. Exp. Neurol. 2016, 277, 58–67. [Google Scholar] [CrossRef]
- LeVine, S.M. The role of reactive oxygen species in the pathogenesis of multiple sclerosis. Med. Hypotheses. 1992, 39, 271–274. [Google Scholar] [CrossRef]
- Miljkovic, D.; Timotijevic, G.; Stojkovic, M.M. Astrocytes in the tempest of multiple sclerosis. FEBS Lett. 2011, 585, 3781–3788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef]
- Werner, P.; Pitt, D.; Raine, C.S. Glutamate excitotoxicity—A mechanism for axonal damage and oligodendrocyte death in Multiple Sclerosis? J. Neural Transm. Suppl. 2000, 375–385. [Google Scholar]
- Ricote, M.; Li, A.C.; Willson, T.M.; Kelly, C.J.; Glass, C.K. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 1998, 391, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Straus, D.S.; Glass, C.K. Anti-inflammatory actions of PPAR ligands: New insights on cellular and molecular mechanisms. Trends Immunol. 2007, 28, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Dunn, S.E.; Bhat, R.; Straus, D.S.; Sobel, R.A.; Axtell, R.; Johnson, A.; Nguyen, K.; Mukundan, L.; Moshkova, M.; Dugas, J.C.; et al. Peroxisome proliferator-activated receptor delta limits the expansion of pathogenic Th cells during central nervous system autoimmunity. J. Exp. Med. 2010, 207, 1599–1608. [Google Scholar] [CrossRef] [PubMed]
- Diab, A.; Deng, C.; Smith, J.D.; Hussain, R.Z.; Phanavanh, B.; Lovett-Racke, A.E.; Drew, P.D.; Racke, M.K. Peroxisome proliferator-activated receptor-gamma agonist 15-deoxy-Delta(12,14)-prostaglandin J(2) ameliorates experimental autoimmune encephalomyelitis. J. Immunol. 2002, 168, 2508–2515. [Google Scholar] [CrossRef]
- Feinstein, D.L.; Galea, E.; Gavrilyuk, V.; Brosnan, C.F.; Whitacre, C.C.; Dumitrescu-Ozimek, L.; Landreth, G.E.; Pershadsingh, H.A.; Weinberg, G.; Heneka, M.T. Peroxisome proliferator-activated receptor-gamma agonists prevent experimental autoimmune encephalomyelitis. Ann. Neurol. 2002, 51, 694–702. [Google Scholar] [CrossRef]
- Bright, J.J.; Walline, C.C.; Kanakasabai, S.; Chakraborty, S. Targeting PPAR as a therapy to treat multiple sclerosis. Expert Opin. Ther. Targets 2008, 12, 1565–1575. [Google Scholar] [CrossRef]
- Xu, J.; Chavis, J.A.; Racke, M.K.; Drew, P.D. Peroxisome proliferator-activated receptor-alpha and retinoid X receptor agonists inhibit inflammatory responses of astrocytes. J. Neuroimmunol. 2006, 176, 95–105. [Google Scholar] [CrossRef]
- Xu, J.; Racke, M.K.; Drew, P.D. Peroxisome proliferator-activated receptor-alpha agonist fenofibrate regulates IL-12 family cytokine expression in the CNS: Relevance to multiple sclerosis. J. Neurochem. 2007, 103, 1801–1810. [Google Scholar] [CrossRef]
- Xu, J.; Storer, P.D.; Chavis, J.A.; Racke, M.K.; Drew, P.D. Agonists for the peroxisome proliferator-activated receptor-alpha and the retinoid X receptor inhibit inflammatory responses of microglia. J. Neurosci. Res. 2005, 81, 403–411. [Google Scholar] [CrossRef]
- Combs, C.K.; Bates, P.; Karlo, J.C.; Landreth, G.E. Regulation of beta-amyloid stimulated proinflammatory responses by peroxisome proliferator-activated receptor alpha. Neurochem. Int. 2001, 39, 449–457. [Google Scholar] [CrossRef]
- Dasgupta, S.; Roy, A.; Jana, M.; Hartley, D.M.; Pahan, K. Gemfibrozil ameliorates relapsing-remitting experimental autoimmune encephalomyelitis independent of peroxisome proliferator-activated receptor-alpha. Mol. Pharmacol. 2007, 72, 934–946. [Google Scholar] [CrossRef] [PubMed]
- Lovett-Racke, A.E.; Hussain, R.Z.; Northrop, S.; Choy, J.; Rocchini, A.; Matthes, L.; Chavis, J.A.; Diab, A.; Drew, P.D.; Racke, M.K. Peroxisome proliferator-activated receptor alpha agonists as therapy for autoimmune disease. J. Immunol. 2004, 172, 5790–5798. [Google Scholar] [CrossRef] [PubMed]
- Menendez-Gutierrez, M.P.; Roszer, T.; Ricote, M. Biology and therapeutic applications of peroxisome proliferator- activated receptors. Curr. Top. Med. Chem. 2012, 12, 548–584. [Google Scholar] [CrossRef] [PubMed]
- Storer, P.D.; Xu, J.; Chavis, J.; Drew, P.D. Peroxisome proliferator-activated receptor-gamma agonists inhibit the activation of microglia and astrocytes: Implications for multiple sclerosis. J. Neuroimmunol. 2005, 161, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Chearwae, W.; Bright, J.J. 15-deoxy-Delta(12,14)-prostaglandin J(2) and curcumin modulate the expression of toll-like receptors 4 and 9 in autoimmune T lymphocyte. J. Clin. Immunol. 2008, 28, 558–570. [Google Scholar] [CrossRef] [PubMed]
- Peiris, M.; Monteith, G.R.; Roberts-Thomson, S.J.; Cabot, P.J. A model of experimental autoimmune encephalomyelitis (EAE) in C57BL/6 mice for the characterisation of intervention therapies. J. Neurosci. Methods 2007, 163, 245–254. [Google Scholar] [CrossRef]
- Schintu, N.; Frau, L.; Ibba, M.; Caboni, P.; Garau, A.; Carboni, E.; Carta, A.R. PPAR-gamma-mediated neuroprotection in a chronic mouse model of Parkinson’s disease. Eur. J. Neurosci. 2009, 29, 954–963. [Google Scholar] [CrossRef]
- Niino, M.; Iwabuchi, K.; Kikuchi, S.; Ato, M.; Morohashi, T.; Ogata, A.; Tashiro, K.; Onoe, K. Amelioration of experimental autoimmune encephalomyelitis in C57BL/6 mice by an agonist of peroxisome proliferator-activated receptor-gamma. J. Neuroimmunol. 2001, 116, 40–48. [Google Scholar] [CrossRef]
- Nishijima, C.; Kimoto, K.; Arakawa, Y. Survival activity of troglitazone in rat motoneurones. J. Neurochem. 2001, 76, 383–390. [Google Scholar] [CrossRef] [Green Version]
- Kielian, T.; Syed, M.M.; Liu, S.; Phulwani, N.K.; Phillips, N.; Wagoner, G.; Drew, P.D.; Esen, N. The synthetic peroxisome proliferator-activated receptor-gamma agonist ciglitazone attenuates neuroinflammation and accelerates encapsulation in bacterial brain abscesses. J. Immunol. 2008, 180, 5004–5016. [Google Scholar] [CrossRef] [PubMed]
- Sznaidman, M.L.; Haffner, C.D.; Maloney, P.R.; Fivush, A.; Chao, E.; Goreham, D.; Sierra, M.L.; LeGrumelec, C.; Xu, H.E.; Montana, V.G.; et al. Novel selective small molecule agonists for peroxisome proliferator-activated receptor delta (PPARdelta)—Synthesis and biological activity. Bioorg. Med. Chem. Lett. 2003, 13, 1517–1521. [Google Scholar] [CrossRef]
- Polak, P.E.; Kalinin, S.; Dello Russo, C.; Gavrilyuk, V.; Sharp, A.; Peters, J.M.; Richardson, J.; Willson, T.M.; Weinberg, G.; Feinstein, D.L. Protective effects of a peroxisome proliferator-activated receptor-beta/delta agonist in experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2005, 168, 65–75. [Google Scholar] [CrossRef]
- Oliver, W.R., Jr.; Shenk, J.L.; Snaith, M.R.; Russell, C.S.; Plunket, K.D.; Bodkin, N.L.; Lewis, M.C.; Winegar, D.A.; Sznaidman, M.L.; Lambert, M.H.; et al. A selective peroxisome proliferator-activated receptor delta agonist promotes reverse cholesterol transport. Proc. Natl. Acad. Sci. USA 2001, 98, 5306–5311. [Google Scholar] [CrossRef] [PubMed]
- Inestrosa, N.C.; Godoy, J.A.; Quintanilla, R.A.; Koenig, C.S.; Bronfman, M. Peroxisome proliferator-activated receptor gamma is expressed in hippocampal neurons and its activation prevents beta-amyloid neurodegeneration: Role of Wnt signaling. Exp. Cell Res. 2005, 304, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.C.; Ding, X.; Daynes, R.A. Nuclear receptor peroxisome proliferator-activated receptor alpha (PPARalpha) is expressed in resting murine lymphocytes. The PPARalpha in T and B lymphocytes is both transactivation and transrepression competent. J. Biol. Chem. 2002, 277, 6838–6845. [Google Scholar] [CrossRef] [PubMed]
- Jana, M.; Pahan, K. Gemfibrozil, a lipid lowering drug, inhibits the activation of primary human microglia via peroxisome proliferator-activated receptor beta. Neurochem. Res. 2012, 37, 1718–1729. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, A.; Minghetti, L. Regulation of Glial Cell Functions by PPAR-gamma Natural and Synthetic Agonists. PPAR Res. 2008, 2008, 864140. [Google Scholar] [CrossRef]
- Harris, S.G.; Phipps, R.P. The nuclear receptor PPAR gamma is expressed by mouse T lymphocytes and PPAR gamma agonists induce apoptosis. Eur. J. Immunol. 2001, 31, 1098–1105. [Google Scholar] [CrossRef] [Green Version]
- Padilla, J.; Leung, E.; Phipps, R.P. Human B lymphocytes and B lymphomas express PPAR-gamma and are killed by PPAR-gamma agonists. Clin. Immunol. 2002, 103, 22–33. [Google Scholar] [CrossRef]
- Yang, Y.; Lovett-Racke, A.E.; Racke, M.K. Regulation of Immune Responses and Autoimmune Encephalomyelitis by PPARs. PPAR Res. 2010, 2010, 104705. [Google Scholar] [CrossRef] [PubMed]
- Klotz, L.; Diehl, L.; Dani, I.; Neumann, H.; von Oppen, N.; Dolf, A.; Endl, E.; Klockgether, T.; Engelhardt, B.; Knolle, P. Brain endothelial PPARgamma controls inflammation-induced CD4+ T cell adhesion and transmigration in vitro. J. Neuroimmunol. 2007, 190, 34–43. [Google Scholar] [CrossRef]
- Daynes, R.A.; Jones, D.C. Emerging roles of PPARs in inflammation and immunity. Nat. Rev. Immunol. 2002, 2, 748–759. [Google Scholar] [CrossRef]
- Xu, J.; Drew, P.D. Peroxisome proliferator-activated receptor-gamma agonists suppress the production of IL-12 family cytokines by activated glia. J. Immunol. 2007, 178, 1904–1913. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Tsai, C.P.; Lee, C.L.; Chen, S.Y.; Lin, G.J.; Yen, M.H.; Sytwu, H.K.; Chen, S.J. alpha-Lipoic acid enhances endogenous peroxisome-proliferator-activated receptor-gamma to ameliorate experimental autoimmune encephalomyelitis in mice. Clin. Sci. (Lond) 2013, 125, 329–340. [Google Scholar] [CrossRef]
- Klotz, L.; Burgdorf, S.; Dani, I.; Saijo, K.; Flossdorf, J.; Hucke, S.; Alferink, J.; Nowak, N.; Beyer, M.; Mayer, G.; et al. The nuclear receptor PPAR gamma selectively inhibits Th17 differentiation in a T cell-intrinsic fashion and suppresses CNS autoimmunity. J. Exp. Med. 2009, 206, 2079–2089. [Google Scholar] [CrossRef]
- Chen, T.; Tibbitt, C.A.; Feng, X.; Stark, J.M.; Rohrbeck, L.; Rausch, L.; Sedimbi, S.K.; Karlsson, M.C.I.; Lambrecht, B.N.; Karlsson Hedestam, G.B.; et al. PPAR-gamma promotes type 2 immune responses in allergy and nematode infection. Sci. Immunol. 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Saubermann, L.J.; Nakajima, A.; Wada, K.; Zhao, S.; Terauchi, Y.; Kadowaki, T.; Aburatani, H.; Matsuhashi, N.; Nagai, R.; Blumberg, R.S. Peroxisome proliferator-activated receptor gamma agonist ligands stimulate a Th2 cytokine response and prevent acute colitis. Inflamm. Bowel Dis. 2002, 8, 330–339. [Google Scholar] [CrossRef]
- Cunard, R.; Ricote, M.; DiCampli, D.; Archer, D.C.; Kahn, D.A.; Glass, C.K.; Kelly, C.J. Regulation of cytokine expression by ligands of peroxisome proliferator activated receptors. J. Immunol. 2002, 168, 2795–2802. [Google Scholar] [CrossRef]
- Clark, R.B.; Bishop-Bailey, D.; Estrada-Hernandez, T.; Hla, T.; Puddington, L.; Padula, S.J. The nuclear receptor PPAR gamma and immunoregulation: PPAR gamma mediates inhibition of helper T cell responses. J. Immunol. 2000, 164, 1364–1371. [Google Scholar] [CrossRef]
- Chinetti, G.; Griglio, S.; Antonucci, M.; Torra, I.P.; Delerive, P.; Majd, Z.; Fruchart, J.C.; Chapman, J.; Najib, J.; Staels, B. Activation of proliferator-activated receptors alpha and gamma induces apoptosis of human monocyte-derived macrophages. J. Biol. Chem. 1998, 273, 25573–25580. [Google Scholar] [CrossRef] [PubMed]
- Klotz, L.; Dani, I.; Edenhofer, F.; Nolden, L.; Evert, B.; Paul, B.; Kolanus, W.; Klockgether, T.; Knolle, P.; Diehl, L. Peroxisome proliferator-activated receptor gamma control of dendritic cell function contributes to development of CD4+ T cell anergy. J. Immunol. 2007, 178, 2122–2131. [Google Scholar] [CrossRef] [PubMed]
- Bouhlel, M.A.; Brozek, J.; Derudas, B.; Zawadzki, C.; Jude, B.; Staels, B.; Chinetti-Gbaguidi, G. Unlike PPARgamma, PPARalpha or PPARbeta/delta activation does not promote human monocyte differentiation toward alternative macrophages. Biochem. Biophys. Res. Commun. 2009, 386, 459–462. [Google Scholar] [CrossRef] [PubMed]
- Szanto, A.; Balint, B.L.; Nagy, Z.S.; Barta, E.; Dezso, B.; Pap, A.; Szeles, L.; Poliska, S.; Oros, M.; Evans, R.M.; et al. STAT6 transcription factor is a facilitator of the nuclear receptor PPARgamma-regulated gene expression in macrophages and dendritic cells. Immunity 2010, 33, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.; Taniguchi, T.; Kimura, H.; Nomura, Y.; Gebicke-Haerter, P.J. Interleukin-4-inhibited mRNA expression in mixed rat glial and in isolated microglial cultures. J. Neuroimmunol. 2000, 106, 95–104. [Google Scholar] [CrossRef]
- Ricote, M.; Welch, J.S.; Glass, C.K. Regulation of macrophage gene expression by the peroxisome proliferator-activated receptor-gamma. Horm. Res. 2000, 54, 275–280. [Google Scholar] [PubMed]
- Huang, J.T.; Welch, J.S.; Ricote, M.; Binder, C.J.; Willson, T.M.; Kelly, C.; Witztum, J.L.; Funk, C.D.; Conrad, D.; Glass, C.K. Interleukin-4-dependent production of PPAR-gamma ligands in macrophages by 12/15-lipoxygenase. Nature 1999, 400, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Lovren, F.; Pan, Y.; Quan, A.; Szmitko, P.E.; Singh, K.K.; Shukla, P.C.; Gupta, M.; Chan, L.; Al-Omran, M.; Teoh, H.; et al. Adiponectin primes human monocytes into alternative anti-inflammatory M2 macrophages. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H656–H663. [Google Scholar] [CrossRef] [Green Version]
- Duvanel, C.B.; Honegger, P.; Pershadsingh, H.; Feinstein, D.; Matthieu, J.M. Inhibition of glial cell proinflammatory activities by peroxisome proliferator-activated receptor gamma agonist confers partial protection during antimyelin oligodendrocyte glycoprotein demyelination in vitro. J. Neurosci. Res. 2003, 71, 246–255. [Google Scholar] [CrossRef]
- Kanakasabai, S.; Pestereva, E.; Chearwae, W.; Gupta, S.K.; Ansari, S.; Bright, J.J. PPARgamma agonists promote oligodendrocyte differentiation of neural stem cells by modulating stemness and differentiation genes. PLoS ONE 2012, 7, e50500. [Google Scholar] [CrossRef]
- Bernardo, A.; Bianchi, D.; Magnaghi, V.; Minghetti, L. Peroxisome proliferator-activated receptor-gamma agonists promote differentiation and antioxidant defenses of oligodendrocyte progenitor cells. J. Neuropathol. Exp. Neurol. 2009, 68, 797–808. [Google Scholar] [CrossRef] [PubMed]
- De Nuccio, C.; Bernardo, A.; De Simone, R.; Mancuso, E.; Magnaghi, V.; Visentin, S.; Minghetti, L. Peroxisome proliferator-activated receptor gamma agonists accelerate oligodendrocyte maturation and influence mitochondrial functions and oscillatory Ca(2+) waves. J. Neuropathol. Exp. Neurol. 2011, 70, 900–912. [Google Scholar] [CrossRef] [PubMed]
- Roth, A.D.; Leisewitz, A.V.; Jung, J.E.; Cassina, P.; Barbeito, L.; Inestrosa, N.C.; Bronfman, M. PPAR gamma activators induce growth arrest and process extension in B12 oligodendrocyte-like cells and terminal differentiation of cultured oligodendrocytes. J. Neurosci. Res. 2003, 72, 425–435. [Google Scholar] [CrossRef]
- Vallee, A.; Vallee, J.N.; Guillevin, R.; Lecarpentier, Y. Interactions Between the Canonical WNT/Beta-Catenin Pathway and PPAR Gamma on Neuroinflammation, Demyelination, and Remyelination in Multiple Sclerosis. Cell. Mol. Neurobiol. 2018, 38, 783–795. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Kwon, K.J.; Park, J.Y.; Lee, S.H.; Moon, C.H.; Baik, E.J. Effects of peroxisome proliferator-activated receptor agonists on LPS-induced neuronal death in mixed cortical neurons: Associated with iNOS and COX-2. Brain Res. 2002, 941, 1–10. [Google Scholar] [CrossRef]
- Luna-Medina, R.; Cortes-Canteli, M.; Alonso, M.; Santos, A.; Martinez, A.; Perez-Castillo, A. Regulation of inflammatory response in neural cells in vitro by thiadiazolidinones derivatives through peroxisome proliferator-activated receptor gamma activation. J. Biol. Chem. 2005, 280, 21453–21462. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Ou, Z.; Grotta, J.C.; Waxham, N.; Aronowski, J. Peroxisome-proliferator-activated receptor-gamma (PPARgamma) activation protects neurons from NMDA excitotoxicity. Brain Res. 2006, 1073–1074, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Brodbeck, J.; Balestra, M.E.; Saunders, A.M.; Roses, A.D.; Mahley, R.W.; Huang, Y. Rosiglitazone increases dendritic spine density and rescues spine loss caused by apolipoprotein E4 in primary cortical neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 1343–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, S.A.; Monteith, G.R.; Holman, N.A.; Robinson, J.A.; May, F.J.; Roberts-Thomson, S.J. Effects of peroxisome proliferator-activated receptor gamma ligands ciglitazone and 15-deoxy-delta 12,14-prostaglandin J2 on rat cultured cerebellar granule neuronal viability. J. Neurosci. Res. 2003, 72, 747–755. [Google Scholar] [CrossRef]
- Dreyer, C.; Krey, G.; Keller, H.; Givel, F.; Helftenbein, G.; Wahli, W. Control of the peroxisomal beta-oxidation pathway by a novel family of nuclear hormone receptors. Cell 1992, 68, 879–887. [Google Scholar] [CrossRef]
- Rusyn, I.; Rose, M.L.; Bojes, H.K.; Thurman, R.G. Novel role of oxidants in the molecular mechanism of action of peroxisome proliferators. Antioxid. Redox Signal. 2000, 2, 607–621. [Google Scholar] [CrossRef] [PubMed]
- Granneman, J.; Skoff, R.; Yang, X. Member of the peroxisome proliferator-activated receptor family of transcription factors is differentially expressed by oligodendrocytes. J. Neurosci. Res. 1998, 51, 563–573. [Google Scholar] [CrossRef]
- Cimini, A.; Bernardo, A.; Cifone, M.G.; Di Marzio, L.; Di Loreto, S. TNFalpha downregulates PPARdelta expression in oligodendrocyte progenitor cells: Implications for demyelinating diseases. Glia 2003, 41, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Moreno, S.; Farioli-Vecchioli, S.; Ceru, M.P. Immunolocalization of peroxisome proliferator-activated receptors and retinoid X receptors in the adult rat CNS. Neuroscience 2004, 123, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Warden, A.; Truitt, J.; Merriman, M.; Ponomareva, O.; Jameson, K.; Ferguson, L.B.; Mayfield, R.D.; Harris, R.A. Localization of PPAR isotypes in the adult mouse and human brain. Sci. Rep. 2016, 6, 27618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basu-Modak, S.; Braissant, O.; Escher, P.; Desvergne, B.; Honegger, P.; Wahli, W. Peroxisome proliferator-activated receptor beta regulates acyl-CoA synthetase 2 in reaggregated rat brain cell cultures. J. Biol. Chem. 1999, 274, 35881–35888. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, M.A.; Petersen, R.K.; Kristiansen, K.; Lange, M.; Lillevang, S.T. Peroxisome proliferator-activated receptor alpha, delta, gamma1 and gamma2 expressions are present in human monocyte-derived dendritic cells and modulate dendritic cell maturation by addition of subtype-specific ligands. Scand. J. Immunol. 2006, 63, 330–337. [Google Scholar] [CrossRef]
- Dunn, S.E.; Ousman, S.S.; Sobel, R.A.; Zuniga, L.; Baranzini, S.E.; Youssef, S.; Crowell, A.; Loh, J.; Oksenberg, J.; Steinman, L. Peroxisome proliferator-activated receptor (PPAR)alpha expression in T cells mediates gender differences in development of T cell-mediated autoimmunity. J. Exp. Med. 2007, 204, 321–330. [Google Scholar] [CrossRef]
- Zhang, M.A.; Rego, D.; Moshkova, M.; Kebir, H.; Chruscinski, A.; Nguyen, H.; Akkermann, R.; Stanczyk, F.Z.; Prat, A.; Steinman, L.; et al. Peroxisome proliferator-activated receptor (PPAR)alpha and -gamma regulate IFNgamma and IL-17A production by human T cells in a sex-specific way. Proc. Natl. Acad. Sci. USA 2012, 109, 9505–9510. [Google Scholar] [CrossRef]
- Nagy, Z.S.; Czimmerer, Z.; Szanto, A.; Nagy, L. Pro-inflammatory cytokines negatively regulate PPARgamma mediated gene expression in both human and murine macrophages via multiple mechanisms. Immunobiology 2013, 218, 1336–1344. [Google Scholar] [CrossRef]
- Woods, J.W.; Tanen, M.; Figueroa, D.J.; Biswas, C.; Zycband, E.; Moller, D.E.; Austin, C.P.; Berger, J.P. Localization of PPARdelta in murine central nervous system: Expression in oligodendrocytes and neurons. Brain Res. 2003, 975, 10–21. [Google Scholar] [CrossRef]
- Vittoria Simonini, M.; Polak, P.E.; Boullerne, A.I.; Peters, J.M.; Richardson, J.C.; Feinstein, D.L. Regulation of oligodendrocyte progenitor cell maturation by PPARdelta: Effects on bone morphogenetic proteins. ASN Neuro 2010, 2, e00025. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.; Reilly, S.M.; Karabacak, V.; Gangl, M.R.; Fitzgerald, K.; Hatano, B.; Lee, C.H. Adipocyte-derived Th2 cytokines and myeloid PPARdelta regulate macrophage polarization and insulin sensitivity. Cell Metab. 2008, 7, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Red Eagle, A.; Vats, D.; Morel, C.R.; Goforth, M.H.; Subramanian, V.; Mukundan, L.; Ferrante, A.W.; Chawla, A. Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity-induced insulin resistance. Cell Metab. 2008, 7, 496–507. [Google Scholar] [CrossRef]
- Defaux, A.; Zurich, M.G.; Braissant, O.; Honegger, P.; Monnet-Tschudi, F. Effects of the PPAR-beta agonist GW501516 in an in vitro model of brain inflammation and antibody-induced demyelination. J. Neuroinflamm. 2009, 6, 15. [Google Scholar] [CrossRef] [PubMed]
- Hondares, E.; Rosell, M.; Diaz-Delfin, J.; Olmos, Y.; Monsalve, M.; Iglesias, R.; Villarroya, F.; Giralt, M. Peroxisome proliferator-activated receptor alpha (PPARalpha) induces PPARgamma coactivator 1alpha (PGC-1alpha) gene expression and contributes to thermogenic activation of brown fat: Involvement of PRDM16. J. Biol. Chem. 2011, 286, 43112–43122. [Google Scholar] [CrossRef] [PubMed]
- Karimfar, M.H.; Haghani, K.; Babakhani, A.; Bakhtiyari, S. Rosiglitazone, but not epigallocatechin-3-gallate, attenuates the decrease in PGC-1alpha protein levels in palmitate-induced insulin-resistant C2C12 cells. Lipids 2015, 50, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Hondares, E.; Pineda-Torra, I.; Iglesias, R.; Staels, B.; Villarroya, F.; Giralt, M. PPARdelta, but not PPARalpha, activates PGC-1alpha gene transcription in muscle. Biochem. Biophys. Res. Commun. 2007, 354, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
- Szalardy, L.; Zadori, D.; Klivenyi, P.; Toldi, J.; Vecsei, L. Electron Transport Disturbances and Neurodegeneration: From Albert Szent-Gyorgyi’s Concept (Szeged) till Novel Approaches to Boost Mitochondrial Bioenergetics. Oxid. Med. Cell. Longev. 2015, 2015, 498401. [Google Scholar] [CrossRef]
- Nijland, P.G.; Michailidou, I.; Witte, M.E.; Mizee, M.R.; van der Pol, S.M.; van Het Hof, B.; Reijerkerk, A.; Pellerin, L.; van der Valk, P.; de Vries, H.E.; et al. Cellular distribution of glucose and monocarboxylate transporters in human brain white matter and multiple sclerosis lesions. Glia 2014, 62, 1125–1141. [Google Scholar] [CrossRef]
- van Horssen, J.; Schreibelt, G.; Drexhage, J.; Hazes, T.; Dijkstra, C.D.; van der Valk, P.; de Vries, H.E. Severe oxidative damage in multiple sclerosis lesions coincides with enhanced antioxidant enzyme expression. Free Radic. Biol. Med. 2008, 45, 1729–1737. [Google Scholar] [CrossRef] [PubMed]
- Szalardy, L.; Zadori, D.; Plangar, I.; Vecsei, L.; Weydt, P.; Ludolph, A.C.; Klivenyi, P.; Kovacs, G.G. Neuropathology of partial PGC-1alpha deficiency recapitulates features of mitochondrial encephalopathies but not of neurodegenerative diseases. Neurodegener. Dis. 2013, 12, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Szalardy, L.; Molnar, M.; Torok, R.; Zadori, D.; Vecsei, L.; Klivenyi, P.; Liberski, P.; Kovacs, G.G. Histopathological comparison of Kearns-Sayre syndrome and PGC-1alpha-deficient mice suggests a novel concept for vacuole formation in mitochondrial encephalopathy. Folia Neuropathol. 2016, 54, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Szalardy, L.; Molnar, M.; Torok, R.; Zadori, D.; Kovacs, G.G.; Vecsei, L.; Klivenyi, P. Lack of age-related clinical progression in PGC-1alpha-deficient mice - implications for mitochondrial encephalopathies. Behav. Brain Res. 2016, 313, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Nijland, P.G.; Witte, M.E.; van het Hof, B.; van der Pol, S.; Bauer, J.; Lassmann, H.; van der Valk, P.; de Vries, H.E.; van Horssen, J. Astroglial PGC-1alpha increases mitochondrial antioxidant capacity and suppresses inflammation: Implications for multiple sclerosis. Acta Neuropathol. Commun. 2014, 2, 170. [Google Scholar] [CrossRef] [PubMed]
- Handschin, C.; Choi, C.S.; Chin, S.; Kim, S.; Kawamori, D.; Kurpad, A.J.; Neubauer, N.; Hu, J.; Mootha, V.K.; Kim, Y.B.; et al. Abnormal glucose homeostasis in skeletal muscle-specific PGC-1alpha knockout mice reveals skeletal muscle-pancreatic beta cell crosstalk. J. Clin. Invest. 2007, 117, 3463–3474. [Google Scholar] [CrossRef] [PubMed]
- Eisele, P.S.; Furrer, R.; Beer, M.; Handschin, C. The PGC-1 coactivators promote an anti-inflammatory environment in skeletal muscle in vivo. Biochem. Biophys. Res. Commun. 2015, 464, 692–697. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, C.; Lieggi, N.T.; Barry, D.; Mooney, D.; de Gaetano, M.; James, W.G.; McClelland, S.; Barry, M.C.; Escoubet-Lozach, L.; Li, A.C.; et al. Macrophage PPAR gamma Co-activator-1 alpha participates in repressing foam cell formation and atherosclerosis in response to conjugated linoleic acid. EMBO Mol. Med. 2013, 5, 1443–1457. [Google Scholar] [CrossRef]
- Ferrer, M.D.; Tauler, P.; Sureda, A.; Tur, J.A.; Pons, A. Antioxidant regulatory mechanisms in neutrophils and lymphocytes after intense exercise. J. Sports Sci. 2009, 27, 49–58. [Google Scholar] [CrossRef]
- Fabregat-Andres, O.; Tierrez, A.; Mata, M.; Estornell-Erill, J.; Ridocci-Soriano, F.; Monsalve, M. Induction of PGC-1alpha expression can be detected in blood samples of patients with ST-segment elevation acute myocardial infarction. PLoS ONE 2011, 6, e26913. [Google Scholar] [CrossRef]
- Klotz, L.; Schmidt, S.; Heun, R.; Klockgether, T.; Kolsch, H. Association of the PPARgamma gene polymorphism Pro12Ala with delayed onset of multiple sclerosis. Neurosci. Lett. 2009, 449, 81–83. [Google Scholar] [CrossRef] [PubMed]
- Paintlia, A.S.; Paintlia, M.K.; Singh, A.K.; Orak, J.K.; Singh, I. Activation of PPAR-gamma and PTEN cascade participates in lovastatin-mediated accelerated differentiation of oligodendrocyte progenitor cells. Glia 2010, 58, 1669–1685. [Google Scholar] [CrossRef] [PubMed]
- Galuppo, M.; Giacoppo, S.; De Nicola, G.R.; Iori, R.; Navarra, M.; Lombardo, G.E.; Bramanti, P.; Mazzon, E. Antiinflammatory activity of glucomoringin isothiocyanate in a mouse model of experimental autoimmune encephalomyelitis. Fitoterapia 2014, 95, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Giacoppo, S.; Soundara Rajan, T.; De Nicola, G.R.; Iori, R.; Bramanti, P.; Mazzon, E. Moringin activates Wnt canonical pathway by inhibiting GSK3beta in a mouse model of experimental autoimmune encephalomyelitis. Drug Des. Dev. Ther. 2016, 10, 3291–3304. [Google Scholar] [CrossRef] [PubMed]
- Swafford, D.; Manicassamy, S. Wnt signaling in dendritic cells: Its role in regulation of immunity and tolerance. Discov. Med. 2015, 19, 303–310. [Google Scholar] [PubMed]
- Natarajan, C.; Muthian, G.; Barak, Y.; Evans, R.M.; Bright, J.J. Peroxisome proliferator-activated receptor-gamma-deficient heterozygous mice develop an exacerbated neural antigen-induced Th1 response and experimental allergic encephalomyelitis. J. Immunol. 2003, 171, 5743–5750. [Google Scholar] [PubMed]
- Raikwar, H.P.; Muthian, G.; Rajasingh, J.; Johnson, C.; Bright, J.J. PPARgamma antagonists exacerbate neural antigen-specific Th1 response and experimental allergic encephalomyelitis. J. Neuroimmunol. 2005, 167, 99–107. [Google Scholar] [CrossRef]
- Raikwar, H.P.; Muthian, G.; Rajasingh, J.; Johnson, C.N.; Bright, J.J. PPARgamma antagonists reverse the inhibition of neural antigen-specific Th1 response and experimental allergic encephalomyelitis by Ciglitazone and 15-deoxy-Delta12,14-prostaglandin J2. J. Neuroimmunol. 2006, 178, 76–86. [Google Scholar] [CrossRef]
- Diab, A.; Hussain, R.Z.; Lovett-Racke, A.E.; Chavis, J.A.; Drew, P.D.; Racke, M.K. Ligands for the peroxisome proliferator-activated receptor-gamma and the retinoid X receptor exert additive anti-inflammatory effects on experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2004, 148, 116–126. [Google Scholar] [CrossRef]
- Szalardy, L.; Zadori, D.; Tanczos, E.; Simu, M.; Bencsik, K.; Vecsei, L.; Klivenyi, P. Elevated levels of PPAR-gamma in the cerebrospinal fluid of patients with multiple sclerosis. Neurosci. Lett. 2013, 554, 131–134. [Google Scholar] [CrossRef] [Green Version]
- Gocke, A.R.; Hussain, R.Z.; Yang, Y.; Peng, H.; Weiner, J.; Ben, L.H.; Drew, P.D.; Stuve, O.; Lovett-Racke, A.E.; Racke, M.K. Transcriptional modulation of the immune response by peroxisome proliferator-activated receptor-{alpha} agonists in autoimmune disease. J. Immunol. 2009, 182, 4479–4487. [Google Scholar] [CrossRef] [PubMed]
- Szalardy, L.; Zadori, D.; Bencsik, K.; Vecsei, L.; Klivenyi, P. Unlike PPARgamma, neither other PPARs nor PGC-1alpha is elevated in the cerebrospinal fluid of patients with multiple sclerosis. Neurosci. Lett. 2017, 651, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Kanakasabai, S.; Walline, C.C.; Chakraborty, S.; Bright, J.J. PPARdelta deficient mice develop elevated Th1/Th17 responses and prolonged experimental autoimmune encephalomyelitis. Brain Res. 2011, 1376, 101–112. [Google Scholar] [CrossRef]
- Xiao, Y.; Xu, J.; Wang, S.; Mao, C.; Jin, M.; Ning, G.; Zhang, Y. Genetic ablation of steroid receptor coactivator-3 promotes PPAR-beta-mediated alternative activation of microglia in experimental autoimmune encephalomyelitis. Glia 2010, 58, 932–942. [Google Scholar]
- Bogie, J.F.; Jorissen, W.; Mailleux, J.; Nijland, P.G.; Zelcer, N.; Vanmierlo, T.; Van Horssen, J.; Stinissen, P.; Hellings, N.; Hendriks, J.J. Myelin alters the inflammatory phenotype of macrophages by activating PPARs. Acta Neuropathol. Commun. 2013, 1, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unoda, K.; Doi, Y.; Nakajima, H.; Yamane, K.; Hosokawa, T.; Ishida, S.; Kimura, F.; Hanafusa, T. Eicosapentaenoic acid (EPA) induces peroxisome proliferator-activated receptors and ameliorates experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2013, 256, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Bartholomaus, I.; Kawakami, N.; Odoardi, F.; Schlager, C.; Miljkovic, D.; Ellwart, J.W.; Klinkert, W.E.; Flugel-Koch, C.; Issekutz, T.B.; Wekerle, H.; et al. Effector T cell interactions with meningeal vascular structures in nascent autoimmune CNS lesions. Nature 2009, 462, 94–98. [Google Scholar] [CrossRef]
- Shinto, L.; Marracci, G.; Bumgarner, L.; Yadav, V. The effects of omega-3 Fatty acids on matrix metalloproteinase-9 production and cell migration in human immune cells: Implications for multiple sclerosis. Autoimmune Dis. 2011, 2011, 134592. [Google Scholar] [CrossRef]
- Ye, P.; Li, J.; Wang, S.; Xie, A.; Sun, W.; Xia, J. Eicosapentaenoic acid disrupts the balance between Tregs and IL-17+ T cells through PPARgamma nuclear receptor activation and protects cardiac allografts. J. Surg. Res. 2012, 173, 161–170. [Google Scholar] [CrossRef]
- Shindler, K.S.; Ventura, E.; Dutt, M.; Elliott, P.; Fitzgerald, D.C.; Rostami, A. Oral resveratrol reduces neuronal damage in a model of multiple sclerosis. J. Neuroophthalmol. 2010, 30, 328–339. [Google Scholar] [CrossRef]
- Fonseca-Kelly, Z.; Nassrallah, M.; Uribe, J.; Khan, R.S.; Dine, K.; Dutt, M.; Shindler, K.S. Resveratrol neuroprotection in a chronic mouse model of multiple sclerosis. Front. Neurol. 2012, 3, 84. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Li, S.P.; Fu, J.S.; Zhang, S.; Bai, L.; Guo, L. Resveratrol defends blood-brain barrier integrity in experimental autoimmune encephalomyelitis mice. J. Neurophysiol. 2016, 116, 2173–2179. [Google Scholar] [CrossRef] [PubMed]
- Imler, T.J., Jr.; Petro, T.M. Decreased severity of experimental autoimmune encephalomyelitis during resveratrol administration is associated with increased IL-17+IL-10+ T cells, CD4(-) IFN-gamma+ cells, and decreased macrophage IL-6 expression. Int. Immunopharmacol. 2009, 9, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Sato, F.; Martinez, N.E.; Shahid, M.; Rose, J.W.; Carlson, N.G.; Tsunoda, I. Resveratrol exacerbates both autoimmune and viral models of multiple sclerosis. Am. J. Pathol. 2013, 183, 1390–1396. [Google Scholar] [CrossRef] [PubMed]
- Khan, R.S.; Dine, K.; Das Sarma, J.; Shindler, K.S. SIRT1 activating compounds reduce oxidative stress mediated neuronal loss in viral induced CNS demyelinating disease. Acta Neuropathol. Commun. 2014, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Shindler, K.S.; Ventura, E.; Rex, T.S.; Elliott, P.; Rostami, A. SIRT1 activation confers neuroprotection in experimental optic neuritis. Invest. Ophthalmol. Vis. Sci. 2007, 48, 3602–3609. [Google Scholar] [CrossRef] [PubMed]
- Nimmagadda, V.K.; Bever, C.T.; Vattikunta, N.R.; Talat, S.; Ahmad, V.; Nagalla, N.K.; Trisler, D.; Judge, S.I.; Royal, W., 3rd; Chandrasekaran, K.; et al. Overexpression of SIRT1 protein in neurons protects against experimental autoimmune encephalomyelitis through activation of multiple SIRT1 targets. J. Immunol. 2013, 190, 4595–4607. [Google Scholar] [CrossRef]
- Makar, T.K.; Nimmagadda, V.K.; Patibandla, G.K.; Le, T.; Judge, S.I.; Trisler, D.; Bever, C.T. Use of engineered bone marrow stem cells to deliver brain derived neurotrophic factor under the control of a tetracycline sensitive response element in experimental allergic encephalomyelitis. J. Neuroimmunol. 2012, 252, 1–15. [Google Scholar] [CrossRef]
- Lim, H.W.; Kang, S.G.; Ryu, J.K.; Schilling, B.; Fei, M.; Lee, I.S.; Kehasse, A.; Shirakawa, K.; Yokoyama, M.; Schnolzer, M.; et al. SIRT1 deacetylates RORgammat and enhances Th17 cell generation. J. Exp. Med. 2015, 212, 607–617. [Google Scholar] [CrossRef]
- Xiang, Z.; Valenza, M.; Cui, L.; Leoni, V.; Jeong, H.K.; Brilli, E.; Zhang, J.; Peng, Q.; Duan, W.; Reeves, S.A.; et al. Peroxisome-proliferator-activated receptor gamma coactivator 1 alpha contributes to dysmyelination in experimental models of Huntington’s disease. J. Neurosci. 2011, 31, 9544–9553. [Google Scholar] [CrossRef]
- Suzuki, K.; Kikkawa, Y. Status spongiosus of CNS and hepatic changes induced by cuprizone (biscyclohexanone oxalyldihydrazone). Am. J. Pathol. 1969, 54, 307–325. [Google Scholar] [PubMed]
- Tegla, C.A.; Azimzadeh, P.; Andrian-Albescu, M.; Martin, A.; Cudrici, C.D.; Trippe, R., 3rd; Sugarman, A.; Chen, H.; Boodhoo, D.; Vlaicu, S.I.; et al. SIRT1 is decreased during relapses in patients with multiple sclerosis. Exp. Mol. Pathol. 2014, 96, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Witte, M.E.; Nijland, P.G.; Drexhage, J.A.; Gerritsen, W.; Geerts, D.; van Het Hof, B.; Reijerkerk, A.; de Vries, H.E.; van der Valk, P.; van Horssen, J. Reduced expression of PGC-1alpha partly underlies mitochondrial changes and correlates with neuronal loss in multiple sclerosis cortex. Acta Neuropathol. 2013, 125, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Pershadsingh, H.A.; Heneka, M.T.; Saini, R.; Amin, N.M.; Broeske, D.J.; Feinstein, D.L. Effect of pioglitazone treatment in a patient with secondary multiple sclerosis. J. Neuroinflamm. 2004, 1, 3. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, C.C.; Shukla, D.K.; Stebbins, G.T.; Skias, D.D.; Jeffery, D.R.; Stefoski, D.; Katsamakis, G.; Feinstein, D.L. A pilot test of pioglitazone as an add-on in patients with relapsing remitting multiple sclerosis. J. Neuroimmunol. 2009, 211, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Shukla, D.K.; Kaiser, C.C.; Stebbins, G.T.; Feinstein, D.L. Effects of pioglitazone on diffusion tensor imaging indices in multiple sclerosis patients. Neurosci. Lett. 2010, 472, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Negrotto, L.; Farez, M.F.; Correale, J. Immunologic Effects of Metformin and Pioglitazone Treatment on Metabolic Syndrome and Multiple Sclerosis. JAMA Neurol. 2016, 73, 520–528. [Google Scholar] [CrossRef]
- Torkildsen, O.; Wergeland, S.; Bakke, S.; Beiske, A.G.; Bjerve, K.S.; Hovdal, H.; Midgard, R.; Lilleas, F.; Pedersen, T.; Bjornara, B.; et al. omega-3 fatty acid treatment in multiple sclerosis (OFAMS Study): A randomized, double-blind, placebo-controlled trial. Arch. Neurol. 2012, 69, 1044–1051. [Google Scholar] [CrossRef]
- Wolf, H. The effect of hormones and vitamin B6 on urinary excretion of metabolites of the kynurenine pathway. Scand. J. Clin. Lab. Invest. Suppl. 1974, 136, 1–186. [Google Scholar]
- Herve, P.; Launay, J.M.; Scrobohaci, M.L.; Brenot, F.; Simonneau, G.; Petitpretz, P.; Poubeau, P.; Cerrina, J.; Duroux, P.; Drouet, L. Increased plasma serotonin in primary pulmonary hypertension. Am. J. Med. 1995, 99, 249–254. [Google Scholar] [CrossRef]
- Vecsei, L.; Szalardy, L.; Fulop, F.; Toldi, J. Kynurenines in the CNS: Recent advances and new questions. Nat. Rev. Drug Discov. 2013, 12, 64–82. [Google Scholar] [CrossRef] [PubMed]
- Perkins, M.N.; Stone, T.W. An iontophoretic investigation of the actions of convulsant kynurenines and their interaction with the endogenous excitant quinolinic acid. Brain Res. 1982, 247, 184–187. [Google Scholar] [CrossRef]
- Lim, C.K.; Brew, B.J.; Sundaram, G.; Guillemin, G.J. Understanding the roles of the kynurenine pathway in multiple sclerosis progression. Int. J. Tryptophan Res. 2010, 3, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Foster, A.C.; Vezzani, A.; French, E.D.; Schwarcz, R. Kynurenic acid blocks neurotoxicity and seizures induced in rats by the related brain metabolite quinolinic acid. Neurosci. Lett. 1984, 48, 273–278. [Google Scholar] [CrossRef]
- Lugo-Huitron, R.; Blanco-Ayala, T.; Ugalde-Muniz, P.; Carrillo-Mora, P.; Pedraza-Chaverri, J.; Silva-Adaya, D.; Maldonado, P.D.; Torres, I.; Pinzon, E.; Ortiz-Islas, E.; et al. On the antioxidant properties of kynurenic acid: Free radical scavenging activity and inhibition of oxidative stress. Neurotoxicol. Teratol. 2011, 33, 538–547. [Google Scholar] [CrossRef] [PubMed]
- Hartai, Z.; Klivenyi, P.; Janaky, T.; Penke, B.; Dux, L.; Vecsei, L. Kynurenine metabolism in multiple sclerosis. Acta Neurol. Scand. 2005, 112, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Braidy, N.; Grant, R.; Adams, S.; Brew, B.J.; Guillemin, G.J. Mechanism for quinolinic acid cytotoxicity in human astrocytes and neurons. Neurotox. Res. 2009, 16, 77–86. [Google Scholar] [CrossRef]
- Szalardy, L.; Klivenyi, P.; Zadori, D.; Fulop, F.; Toldi, J.; Vecsei, L. Mitochondrial disturbances, tryptophan metabolites and neurodegeneration: Medicinal chemistry aspects. Curr. Med. Chem. 2012, 19, 1899–1920. [Google Scholar] [CrossRef]
- Guidetti, P.; Wu, H.Q.; Schwarcz, R. In situ produced 7-chlorokynurenate provides protection against quinolinate- and malonate-induced neurotoxicity in the rat striatum. Exp. Neurol. 2000, 163, 123–130. [Google Scholar] [CrossRef]
- Okuda, S.; Nishiyama, N.; Saito, H.; Katsuki, H. Hydrogen peroxide-mediated neuronal cell death induced by an endogenous neurotoxin, 3-hydroxykynurenine. Proc. Natl. Acad. Sci. USA 1996, 93, 12553–12558. [Google Scholar] [CrossRef]
- Okuda, S.; Nishiyama, N.; Saito, H.; Katsuki, H. 3-Hydroxykynurenine, an endogenous oxidative stress generator, causes neuronal cell death with apoptotic features and region selectivity. J. Neurochem. 1998, 70, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Dykens, J.A.; Sullivan, S.G.; Stern, A. Oxidative reactivity of the tryptophan metabolites 3-hydroxyanthranilate, cinnabarinate, quinolinate and picolinate. Biochem. Pharmacol. 1987, 36, 211–217. [Google Scholar] [CrossRef]
- Mandi, Y.; Vecsei, L. The kynurenine system and immunoregulation. J. Neural Transm. 2012, 119, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Kwidzinski, E.; Bechmann, I. IDO expression in the brain: A double-edged sword. J. Mol. Med. 2007, 85, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Pfefferkorn, E.R. Interferon gamma blocks the growth of Toxoplasma gondii in human fibroblasts by inducing the host cells to degrade tryptophan. Proc. Natl. Acad. Sci. USA 1984, 81, 908–912. [Google Scholar] [CrossRef] [PubMed]
- Forouzandeh, F.; Jalili, R.B.; Germain, M.; Duronio, V.; Ghahary, A. Differential immunosuppressive effect of indoleamine 2,3-dioxygenase (IDO) on primary human CD4+ and CD8+ T cells. Mol. Cell. Biochem. 2008, 309, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 2010, 185, 3190–3198. [Google Scholar] [CrossRef]
- Grohmann, U.; Bianchi, R.; Belladonna, M.L.; Silla, S.; Fallarino, F.; Fioretti, M.C.; Puccetti, P. IFN-gamma inhibits presentation of a tumor/self peptide by CD8 alpha- dendritic cells via potentiation of the CD8 alpha+ subset. J. Immunol. 2000, 165, 1357–1363. [Google Scholar] [CrossRef]
- Dai, X.; Zhu, B.T. Suppression of T-cell response and prolongation of allograft survival in a rat model by tryptophan catabolites. Eur. J. Pharmacol. 2009, 606, 225–232. [Google Scholar] [CrossRef]
- Terness, P.; Bauer, T.M.; Rose, L.; Dufter, C.; Watzlik, A.; Simon, H.; Opelz, G. Inhibition of allogeneic T cell proliferation by indoleamine 2,3-dioxygenase-expressing dendritic cells: Mediation of suppression by tryptophan metabolites. J. Exp. Med. 2002, 196, 447–457. [Google Scholar] [CrossRef]
- Fallarino, F.; Grohmann, U.; Vacca, C.; Bianchi, R.; Orabona, C.; Spreca, A.; Fioretti, M.C.; Puccetti, P. T cell apoptosis by tryptophan catabolism. Cell Death Differ. 2002, 9, 1069–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallarino, F.; Grohmann, U.; Vacca, C.; Orabona, C.; Spreca, A.; Fioretti, M.C.; Puccetti, P. T cell apoptosis by kynurenines. Adv. Exp. Med. Biol. 2003, 527, 183–190. [Google Scholar] [PubMed]
- Bauer, T.M.; Jiga, L.P.; Chuang, J.J.; Randazzo, M.; Opelz, G.; Terness, P. Studying the immunosuppressive role of indoleamine 2,3-dioxygenase: Tryptophan metabolites suppress rat allogeneic T-cell responses in vitro and in vivo. Transpl. Int. 2005, 18, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Belladonna, M.L.; Grohmann, U.; Guidetti, P.; Volpi, C.; Bianchi, R.; Fioretti, M.C.; Schwarcz, R.; Fallarino, F.; Puccetti, P. Kynurenine pathway enzymes in dendritic cells initiate tolerogenesis in the absence of functional IDO. J. Immunol. 2006, 177, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Quintana, F.J.; Murugaiyan, G.; Farez, M.F.; Mitsdoerffer, M.; Tukpah, A.M.; Burns, E.J.; Weiner, H.L. An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2010, 107, 20768–20773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, P.; Hu, G.H.; Kang, H.Y.; Yao, H.B.; Kou, W.; Liu, H.; Zhang, C.; Hong, S.L. An aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress the Th17 response in allergic rhinitis patients. Lab. Invest. 2014, 94, 528–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, C.F.; Goth, S.R.; Dong, B.; Pessah, I.N.; Matsumura, F. Aryl hydrocarbon receptor signaling mediates expression of indoleamine 2,3-dioxygenase. Biochem. Biophys. Res. Commun. 2008, 375, 331–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintana, F.J.; Basso, A.S.; Iglesias, A.H.; Korn, T.; Farez, M.F.; Bettelli, E.; Caccamo, M.; Oukka, M.; Weiner, H.L. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 2008, 453, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Veldhoen, M.; Hirota, K.; Westendorf, A.M.; Buer, J.; Dumoutier, L.; Renauld, J.C.; Stockinger, B. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 2008, 453, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Maaetoft-Udsen, K.; Shimoda, L.M.; Frokiaer, H.; Turner, H. Aryl hydrocarbon receptor ligand effects in RBL2H3 cells. J. Immunotoxicol. 2012, 9, 327–337. [Google Scholar] [CrossRef]
- Steiner, L.; Gold, M.; Mengel, D.; Dodel, R.; Bach, J.P. The endogenous alpha7 nicotinic acetylcholine receptor antagonist kynurenic acid modulates amyloid-beta-induced inflammation in BV-2 microglial cells. J. Neurol. Sci. 2014, 344, 94–99. [Google Scholar] [CrossRef] [PubMed]
- DiNatale, B.C.; Murray, I.A.; Schroeder, J.C.; Flaveny, C.A.; Lahoti, T.S.; Laurenzana, E.M.; Omiecinski, C.J.; Perdew, G.H. Kynurenic acid is a potent endogenous aryl hydrocarbon receptor ligand that synergistically induces interleukin-6 in the presence of inflammatory signaling. Toxicol. Sci. 2010, 115, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Salimi Elizei, S.; Poormasjedi-Meibod, M.S.; Wang, X.; Kheirandish, M.; Ghahary, A. Kynurenic acid downregulates IL-17/1L-23 axis in vitro. Mol. Cell. Biochem. 2017, 431, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Agudelo, L.Z.; Femenia, T.; Orhan, F.; Porsmyr-Palmertz, M.; Goiny, M.; Martinez-Redondo, V.; Correia, J.C.; Izadi, M.; Bhat, M.; Schuppe-Koistinen, I.; et al. Skeletal muscle PGC-1alpha1 modulates kynurenine metabolism and mediates resilience to stress-induced depression. Cell 2014, 159, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Szalardy, L.; Molnar, M.F.; Zadori, D.; Cseh, E.K.; Veres, G.; Kovacs, G.G.; Vecsei, L.; Klivenyi, P. Non-motor Behavioral Alterations of PGC-1alpha-Deficient Mice—A Peculiar Phenotype With Slight Male Preponderance and No Apparent Progression. Front. Behav. Neurosci. 2018, 12, 180. [Google Scholar] [CrossRef] [PubMed]
- Agudelo, L.Z.; Ferreira, D.M.S.; Cervenka, I.; Bryzgalova, G.; Dadvar, S.; Jannig, P.R.; Pettersson-Klein, A.T.; Lakshmikanth, T.; Sustarsic, E.G.; Porsmyr-Palmertz, M.; et al. Kynurenic Acid and Gpr35 Regulate Adipose Tissue Energy Homeostasis and Inflammation. Cell Metab. 2018, 27, 378–392.e5. [Google Scholar] [CrossRef]
- Kwidzinski, E.; Bunse, J.; Kovac, A.D.; Ullrich, O.; Zipp, F.; Nitsch, R.; Bechmann, I. IDO (indolamine 2,3-dioxygenase) expression and function in the CNS. Adv. Exp. Med. Biol. 2003, 527, 113–118. [Google Scholar]
- Kwidzinski, E.; Bunse, J.; Aktas, O.; Richter, D.; Mutlu, L.; Zipp, F.; Nitsch, R.; Bechmann, I. Indolamine 2,3-dioxygenase is expressed in the CNS and down-regulates autoimmune inflammation. FASEB J. 2005, 19, 1347–1349. [Google Scholar] [CrossRef]
- Flanagan, E.M.; Erickson, J.B.; Viveros, O.H.; Chang, S.Y.; Reinhard, J.F., Jr. Neurotoxin quinolinic acid is selectively elevated in spinal cords of rats with experimental allergic encephalomyelitis. J. Neurochem. 1995, 64, 1192–1196. [Google Scholar] [CrossRef]
- Chiarugi, A.; Cozzi, A.; Ballerini, C.; Massacesi, L.; Moroni, F. Kynurenine 3-mono-oxygenase activity and neurotoxic kynurenine metabolites increase in the spinal cord of rats with experimental allergic encephalomyelitis. Neuroscience 2001, 102, 687–695. [Google Scholar] [CrossRef]
- Yan, Y.; Zhang, G.X.; Gran, B.; Fallarino, F.; Yu, S.; Li, H.; Cullimore, M.L.; Rostami, A.; Xu, H. IDO upregulates regulatory T cells via tryptophan catabolite and suppresses encephalitogenic T cell responses in experimental autoimmune encephalomyelitis. J. Immunol. 2010, 185, 5953–5961. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, K.; Zou, J.P.; Tschetter, J.R.; Ward, J.M.; Shearer, G.M. Effect of indoleamine 2,3-dioxygenase on induction of experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2002, 129, 186–196. [Google Scholar] [CrossRef]
- Xiao, B.G.; Wu, X.C.; Yang, J.S.; Xu, L.Y.; Liu, X.; Huang, Y.M.; Bjelke, B.; Link, H. Therapeutic potential of IFN-gamma-modified dendritic cells in acute and chronic experimental allergic encephalomyelitis. Int. Immunol. 2004, 16, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.G.; Liu, X.; Link, H. Antigen-specific T cell functions are suppressed over the estrogen-dendritic cell-indoleamine 2,3-dioxygenase axis. Steroids 2004, 69, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Fazio, F.; Zappulla, C.; Notartomaso, S.; Busceti, C.; Bessede, A.; Scarselli, P.; Vacca, C.; Gargaro, M.; Volpi, C.; Allegrucci, M.; et al. Cinnabarinic acid, an endogenous agonist of type-4 metabotropic glutamate receptor, suppresses experimental autoimmune encephalomyelitis in mice. Neuropharmacology 2014, 81, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; Ho, P.P.; Youssef, S.; Fontoura, P.; Garren, H.; Hur, E.M.; Gupta, R.; Lee, L.Y.; Kidd, B.A.; Robinson, W.H.; et al. Treatment of autoimmune neuroinflammation with a synthetic tryptophan metabolite. Science 2005, 310, 850–855. [Google Scholar] [CrossRef]
- Zadori, D.; Nyiri, G.; Szonyi, A.; Szatmari, I.; Fulop, F.; Toldi, J.; Freund, T.F.; Vecsei, L.; Klivenyi, P. Neuroprotective effects of a novel kynurenic acid analogue in a transgenic mouse model of Huntington’s disease. J. Neural Transm. 2011, 118, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Monaco, F.; Fumero, S.; Mondino, A.; Mutani, R. Plasma and cerebrospinal fluid tryptophan in multiple sclerosis and degenerative diseases. J. Neurol. Neurosurg. Psychiatry 1979, 42, 640–641. [Google Scholar] [CrossRef] [PubMed]
- Sadowska-Bartosz, I.; Adamczyk-Sowa, M.; Gajewska, A.; Bartosz, G. Oxidative modification of blood serum proteins in multiple sclerosis after interferon or mitoxantrone treatment. J. Neuroimmunol. 2014, 266, 67–74. [Google Scholar] [CrossRef]
- Ott, M.; Demisch, L.; Engelhardt, W.; Fischer, P.A. Interleukin-2, soluble interleukin-2-receptor, neopterin, L-tryptophan and beta 2-microglobulin levels in CSF and serum of patients with relapsing-remitting or chronic-progressive multiple sclerosis. J. Neurol. 1993, 241, 108–114. [Google Scholar] [CrossRef]
- Rudzite, V.; Berzinsh, J.; Grivane, I.; Fuchs, D.; Baier-Bitterlich, G.; Wachter, H. Serum tryptophan, kynurenine, and neopterin in patients with Guillain-Barre-syndrome (GBS) and multiple sclerosis (MS). Adv. Exp. Med. Biol. 1996, 398, 183–187. [Google Scholar] [PubMed]
- Amirkhani, A.; Rajda, C.; Arvidsson, B.; Bencsik, K.; Boda, K.; Seres, E.; Markides, K.E.; Vecsei, L.; Bergquist, J. Interferon-beta affects the tryptophan metabolism in multiple sclerosis patients. Eur. J. Neurol. 2005, 12, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, R.; Hernis, A.; Agostini, S.; Rovaris, M.; Caputo, D.; Fuchs, D.; Clerici, M. Indoleamine 2,3 Dioxygenase (IDO) Expression and Activity in Relapsing-Remitting Multiple Sclerosis. PLoS ONE 2015, 10, e0130715. [Google Scholar] [CrossRef] [PubMed]
- Rothhammer, V.; Borucki, D.M.; Garcia Sanchez, M.I.; Mazzola, M.A.; Hemond, C.C.; Regev, K.; Paul, A.; Kivisakk, P.; Bakshi, R.; Izquierdo, G.; et al. Dynamic regulation of serum aryl hydrocarbon receptor agonists in MS. Neurol. Neuroimmunol. Neuroinflamm. 2017, 4, e359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, C.K.; Bilgin, A.; Lovejoy, D.B.; Tan, V.; Bustamante, S.; Taylor, B.V.; Bessede, A.; Brew, B.J.; Guillemin, G.J. Kynurenine pathway metabolomics predicts and provides mechanistic insight into multiple sclerosis progression. Sci. Rep. 2017, 7, 41473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aeinehband, S.; Brenner, P.; Stahl, S.; Bhat, M.; Fidock, M.D.; Khademi, M.; Olsson, T.; Engberg, G.; Jokinen, J.; Erhardt, S.; et al. Cerebrospinal fluid kynurenines in multiple sclerosis; relation to disease course and neurocognitive symptoms. Brain Behav. Immun. 2016, 51, 47–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rejdak, K.; Petzold, A.; Kocki, T.; Kurzepa, J.; Grieb, P.; Turski, W.A.; Stelmasiak, Z. Astrocytic activation in relation to inflammatory markers during clinical exacerbation of relapsing-remitting multiple sclerosis. J. Neural Transm. 2007, 114, 1011–1015. [Google Scholar] [CrossRef]
- Rejdak, K.; Bartosik-Psujek, H.; Dobosz, B.; Kocki, T.; Grieb, P.; Giovannoni, G.; Turski, W.A.; Stelmasiak, Z. Decreased level of kynurenic acid in cerebrospinal fluid of relapsing-onset multiple sclerosis patients. Neurosci. Lett. 2002, 331, 63–65. [Google Scholar] [CrossRef]
- Kaye, J.; Piryatinsky, V.; Birnberg, T.; Hingaly, T.; Raymond, E.; Kashi, R.; Amit-Romach, E.; Caballero, I.S.; Towfic, F.; Ator, M.A.; et al. Laquinimod arrests experimental autoimmune encephalomyelitis by activating the aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. USA 2016, 113, E6145–E6152. [Google Scholar] [CrossRef]
- Berg, J.; Mahmoudjanlou, Y.; Duscha, A.; Massa, M.G.; Thone, J.; Esser, C.; Gold, R.; Haghikia, A. The immunomodulatory effect of laquinimod in CNS autoimmunity is mediated by the aryl hydrocarbon receptor. J. Neuroimmunol. 2016, 298, 9–15. [Google Scholar] [CrossRef]
- Aly, L.; Hemmer, B.; Korn, T. From Leflunomide to Teriflunomide: Drug Development and Immunosuppressive Oral Drugs in the Treatment of Multiple Sclerosis. Curr. Neuropharmacol. 2017, 15, 874–891. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.P.; Harp, C.T.; Noronha, A.; Miller, S.D. The experimental autoimmune encephalomyelitis (EAE) model of MS: Utility for understanding disease pathophysiology and treatment. Handb. Clin. Neurol. 2014, 122, 173–189. [Google Scholar] [PubMed]
- Tan, I.L.; Lycklama a Nijeholt, G.J.; Polman, C.H.; Ader, H.J.; Barkhof, F. Linomide in the treatment of multiple sclerosis: MRI results from prematurely terminated phase-III trials. Mult. Scler. 2000, 6, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Kieseier, B.C. Defining a role for laquinimod in multiple sclerosis. Ther. Adv. Neurol. Disord 2014, 7, 195–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teva and Active Biotech Announce CONCERTO Trial of Laquinimod in RRMS Did Not Meet Primary Endpoint. Available online: https://www.tevapharm.com/news/teva_and_active_biotech_announce_concerto_trial_of_laquinimod_in_rrms_did_not_meet_primary_endpoint_05_17.aspx (accessed on 12 January 2019).
- O’Donnell, E.F.; Saili, K.S.; Koch, D.C.; Kopparapu, P.R.; Farrer, D.; Bisson, W.H.; Mathew, L.K.; Sengupta, S.; Kerkvliet, N.I.; Tanguay, R.L.; et al. The anti-inflammatory drug leflunomide is an agonist of the aryl hydrocarbon receptor. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Antochi, F. Teriflunomide—A new oral agent for multiple sclerosis treatment. Maedica 2013, 8, 404–405. [Google Scholar] [PubMed]



{kind=link}
{kind=link}
| Isoform | Type | Molecule | Role | References |
|---|---|---|---|---|
| PPARα | synthetic | fenofibrate | Suppresses T cell proliferation and IL-1β, TNFα, and IL-6 production, and increases IL-4 production. | [27,28,29,30,31] |
| gemfibrozil | Inhibits mononuclear cell infiltration and Th1 differentiation. | [27,28,30,31,32] | ||
| WY14463 | Inhibits IFNγ, IL-6, and TNFα production in T cells. | [27,28,30,31,33] | ||
| PPARγ | natural | 15dPGJ2 | Inhibits T cell proliferation, IL-1β, IL-4, IL-6, IL-10, IL-12, IFNγ, MCP1, NO, TNFα, and TLR4/TLR9 production, and Th1 differentiation. | [10,25,27,34,35,36] |
| synthetic | GW7845 | Reduces cytokine and chemokine secretion, and leukocyte infiltration. | [27,37] | |
| rosiglitazone | Reduces T cell infiltration into the brain. | [26,27,38] | ||
| troglitazone | Suppresses IL1-β and TNFα. | [27,39] | ||
| pioglitazone | Reduces INFγ and T cell response. | [27,35,40] | ||
| ciglitazone | Inhibits IL-12 production of macrophage/microglial cells. | [27,29,41] | ||
| PPARβ/δ | synthetic | GW501516 | Inhibits EAE by modulating the development of Th1 and Th17 responses and decreases the production of IFNγ and IL-17 in the CNS. | [11] |
| GW610742 | Reduces inflammation in the CNS. | [27,42,43,44] | ||
| L-165041 | Inhibits EAE by modulating the development of Th1 and Th17 responses and decreases the production of IFNγ and IL-17 in the CNS. | [11] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fakan, B.; Szalardy, L.; Vecsei, L. Exploiting the Therapeutic Potential of Endogenous Immunomodulatory Systems in Multiple Sclerosis—Special Focus on the Peroxisome Proliferator-Activated Receptors (PPARs) and the Kynurenines. Int. J. Mol. Sci. 2019, 20, 426. https://doi.org/10.3390/ijms20020426
Fakan B, Szalardy L, Vecsei L. Exploiting the Therapeutic Potential of Endogenous Immunomodulatory Systems in Multiple Sclerosis—Special Focus on the Peroxisome Proliferator-Activated Receptors (PPARs) and the Kynurenines. International Journal of Molecular Sciences. 2019; 20(2):426. https://doi.org/10.3390/ijms20020426
Chicago/Turabian StyleFakan, Bernadett, Levente Szalardy, and Laszlo Vecsei. 2019. "Exploiting the Therapeutic Potential of Endogenous Immunomodulatory Systems in Multiple Sclerosis—Special Focus on the Peroxisome Proliferator-Activated Receptors (PPARs) and the Kynurenines" International Journal of Molecular Sciences 20, no. 2: 426. https://doi.org/10.3390/ijms20020426