Catechol 1,2-Dioxygenase is an Analogue of Homogentisate 1,2-Dioxygenase in Pseudomonas chlororaphis Strain UFB2


Abstract
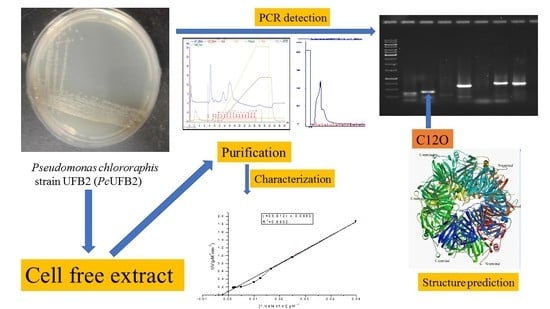
:
1. Introduction
2. Results
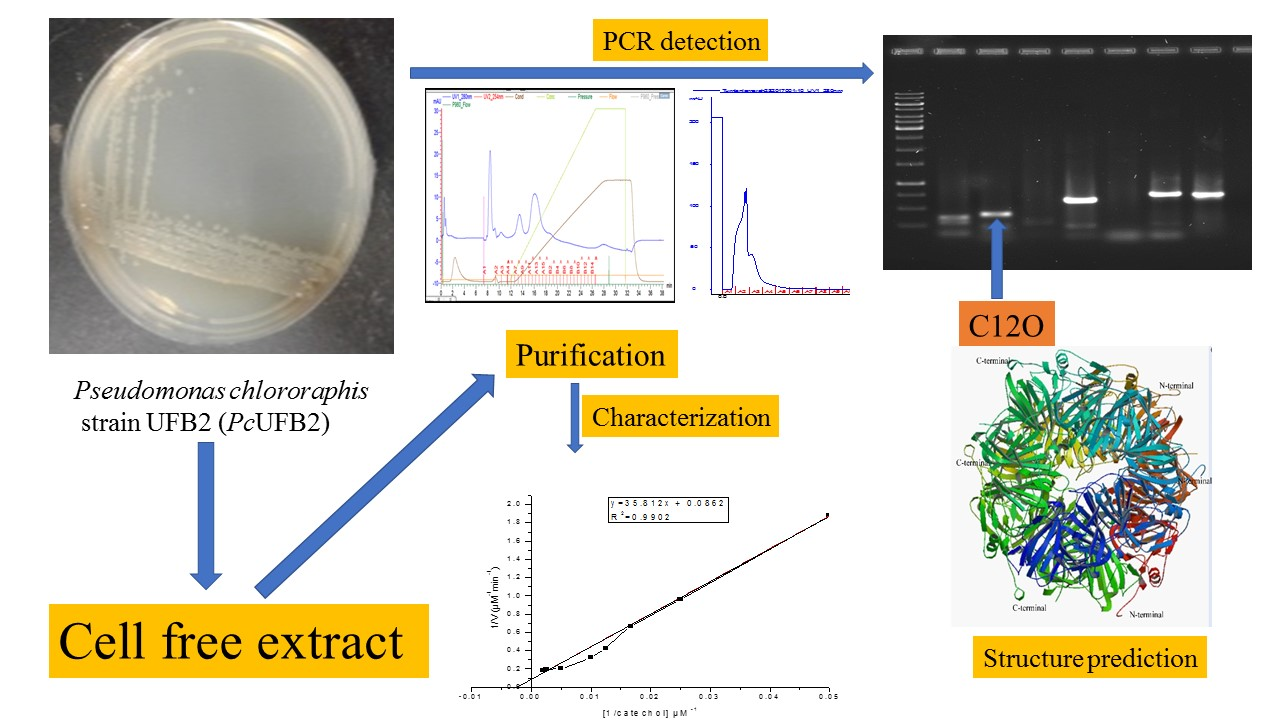
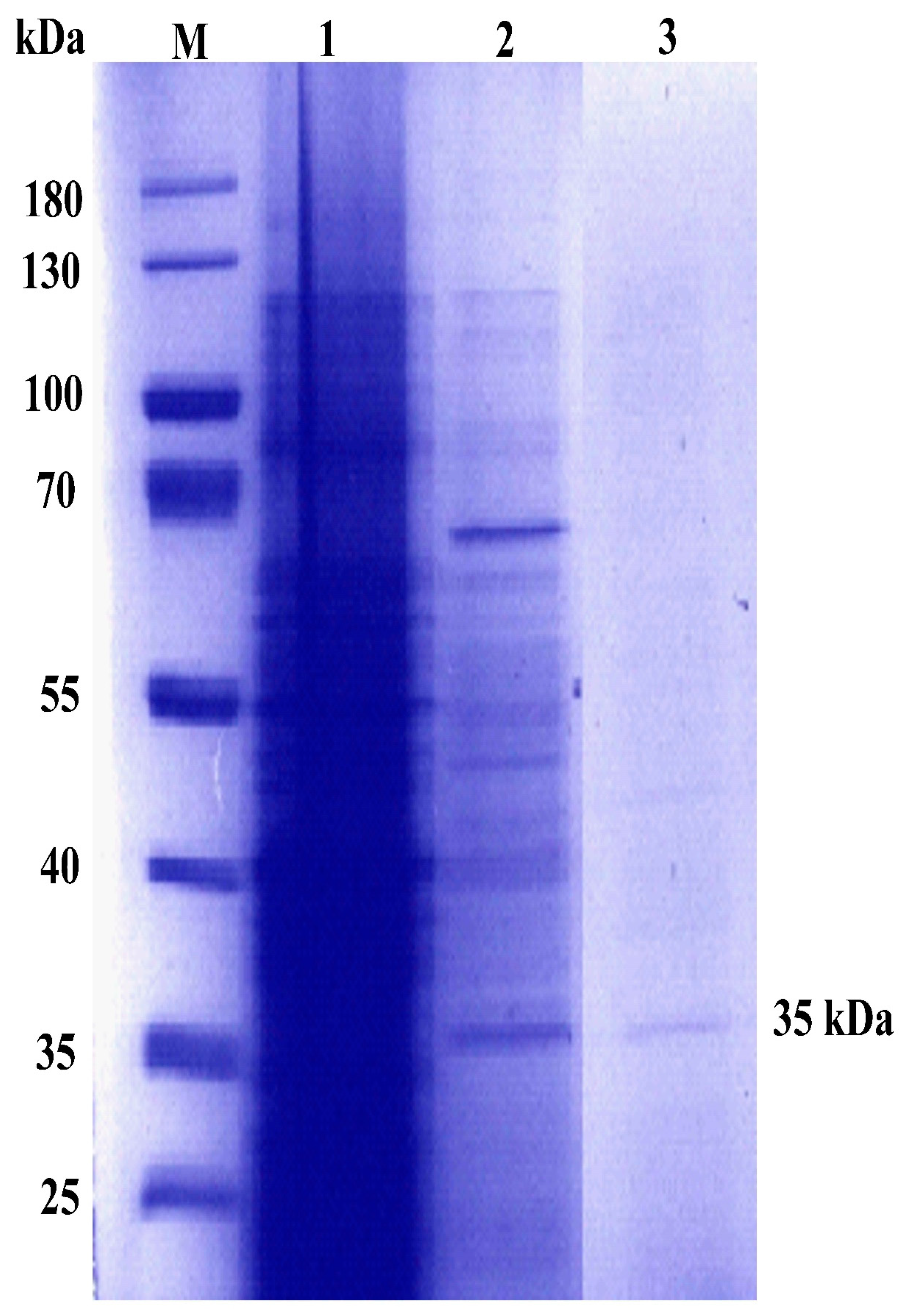
2.1. Production and Purification of C12O
2.2. Optimum pH and pH Stability of Purified C12O
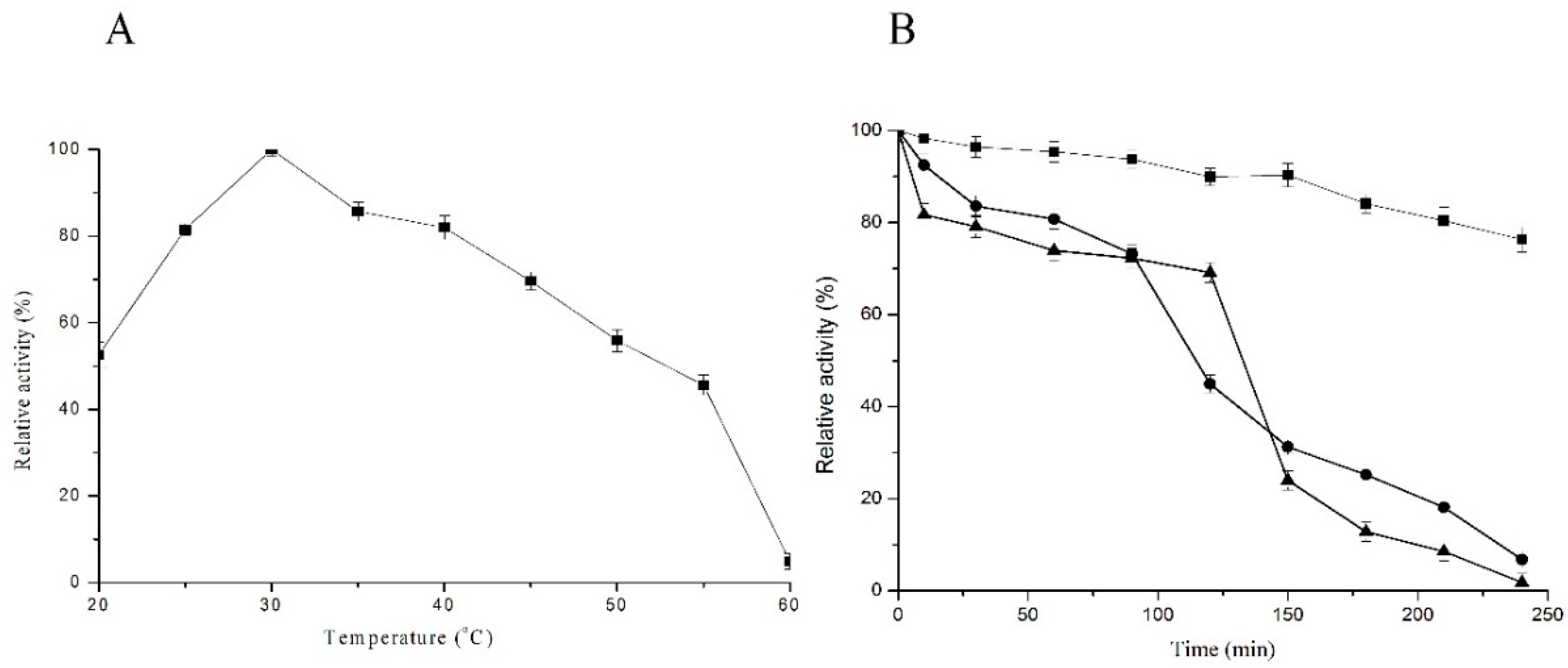
2.3. Optimum Temperature and Temperature Stability
2.4. The Kinetic Properties of C12O
2.5. Effects of Metals and Inhibitors on C12O Activity
2.6. Substrate Specificity of C12O
2.7. Amplification and Detection of C12O in PcUFB2
2.8. ES-MS and Amino Acid Sequence Determination
2.9. Template-Based Structure of Homogentisate 1,2-Dioxygenase
2.10. Predicted Biophysical Properties of C12O
3. Discussion
4. Materials and Methods
4.1. Sample Collection, Enrichment, and Isolation of Bacterial Isolates
4.2. Identification and Phylogenetic Analysis of the Bacterial Isolate
4.3. Preparation of Crude Extracts for Catechol 1,2-Dioxygenase (C12O) and Catechol 2,3-Dioxygenase (C23O) Activity
4.4. C12O and C23O Activity Assay
4.5. Purification of C12O
4.6. Determination of Optimum pH and Temperature
4.7. Temperature and pH Stability of C12O
4.8. Determination of the Enzyme Kinetic Parameters
4.9. Effects of Metals and Inhibitors on C12O Activity
4.10. Substrate Specificity of C12O
4.11. Determination of Amino Acid Sequences of the Purified C12O
4.12. Prediction of Biophysical Properties and Three-Dimensional Structure
4.13. Amplification and Detection of C12O in PcUFB2
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PsUFB2 | Pseudomonas chlororaphis strain UFB2 |
| C12O | Catechol 1,2-Dioxygenase |
| C23O | Catechol 2,3-Dioxygenase |
| H12D | Homogentisate 1,2-Dioxygenase |
References
- Igbinosa, E.O.; Odjadjare, E.E.; Chigor, V.N.; Igbinosa, I.H.; Emoghene, A.O.; Ekhaise, F.O.; Igiehon, N.O.; Idemudia, O.G. Toxicological profile of chlorophenols and their derivatives in the environment: The public health perspective. Sci. World J. 2013, 2013, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Das, N.; Chandran, P. Microbial degradation of petroleum hydrocarbon contaminants: An overview. Biotechnol. Res. Int. 2011, 2011, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Karigar, C.S.; Rao, S.S. Role of microbial enzymes in the bioremediation of pollutants: A review. Enzyme Res. 2011, 2011, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mahiudddin, M.; Fakhruddin, A.N.M.; Al-Mahin, A. Degradation of phenol via meta- cleavage pathway by Pseudomonas fluorescens PU1. ISRN Microbiol. 2012, 2012, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Arora, P.K.; Bae, H. Bacterial degradation of chlorophenols and their derivatives. Microb. Cell Fact. 2014, 13, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, Y.; Yang, S.; Xie, Z.; Cheng, L. Identification and characterization of phenol hydroxylase from phenol-degrading Candida tropicalis strain JH8. Can. J. Microbiol. 2014, 60, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Powlowski, J.; Shingler, V. Genetics and biochemistry of phenol degradation by Pseudomonas sp. CF600. Biodegradation 1994, 5, 219–236. [Google Scholar] [CrossRef]
- Harayama, S.; Kok, M.; Neidle, E.L. Functional and evolutionary relationships among diverse oxygenases. Annu. Rev. Microbiol. 1992, 46, 565–601. [Google Scholar] [CrossRef]
- Cerniglia, C.E. Microbial metabolism of polycyclic aromatic hydrocarbons. Adv. Appl. Microbiol. 1984, 30, 31–71. [Google Scholar]
- Shumkova, E.S.; Solyanikova, I.P.; Plotnikova, E.G.; Golovleva, L.A. Phenol degradation by Rhodococcus opacus strain 1G. Appl. Biochem. Microbiol. 2009, 45, 43–49. [Google Scholar] [CrossRef]
- Silva, A.S.; Camargo, F.A.; Andreazza, R.; Jacques, R.J.; Baldoni, D.B.; Bento, F.M. Enzymatic activity of catechol 1,2-dioxygenase and catechol 2,3-dioxygenase produced by Gordonia polyisoprenivorans. Quim. Nova 2012, 35, 1587–1592. [Google Scholar] [CrossRef]
- Sridevi, V.; Lakshmi, M.; Manasa, M.; Sravani, M. Metabolic pathways for the biodegradation of phenol. Int. J. Eng. Sci. Adv. Technol. 2012, 2, 695–705. [Google Scholar]
- Krastanov, A.; Alexieva, Z.; Yemendzhiev, H. Microbial degradation of phenol and phenolic derivatives. Eng. Life Sci. 2013, 13, 76–87. [Google Scholar] [CrossRef]
- Tsai, S.-C.; Li, Y.-K. Purification and characterization of a catechol 1,2-dioxygenase from a phenol degrading Candida albicans TL3. Arch. Microbiol. 2007, 187, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Al-Hakim, M.H.; Hasan, R.; Ali, M.H.; Rabbee, M.F.; Marufatuzzahan, H.M.; Joy, Z.F. In-silico characterization and homology modeling of catechol 1,2 dioxygenase involved in processing of catechol- an intermediate of aromatic compound degradation pathway. Glob. J. Sci. Front. Res. G Bio-Tech Genet. 2015, 15, 1–13. [Google Scholar]
- Bhat, M.A.; Ishida, T.; Horiike, K.; Vaidyanathan, C.S.; Nozaki, M. Purification of 3,5-dichlorocatechol 1,2-dioxygenase, a nonheme iron dioxygenase and a key enzyme in the biodegradation of a herbicide, 2,4-dichlorophenoxyacetic acid (2,4-D), from Pseudomonas cepacia CSV90. Arch. Biochem. Biophys. 1993, 300, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Lofrano, G.; Rizzo, L.; Grassi, M.; Belgiorno, V. Advanced oxidation of catechol: A comparison among photocatalysis, Fenton and photo-Fenton processes. Desalination 2009, 249, 878–883. [Google Scholar] [CrossRef]
- Subramanyam, R.; Mishra, I.M. Biodegradation of catechol (2-hydroxy phenol) bearing wastewater in an UASB reactor. Chemosphere 2007, 69, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Titus, G.P.; Mueller, H.A.; Burgner, J.; Rodríguez de Córdoba, S.; Peñalva, M.A.; Timm, D.E. Crystal structure of human homogentisate dioxygenase. Nat. Struct. Biol. 2000, 7, 542. [Google Scholar] [PubMed]
- Cha, C.-J. Catechol 1,2-dioxygenase from Rhodococcus rhodochrous N75 capable of metabolizing alkyl-substituted catechols. J. Microbiol. Biotechnol. 2006, 16, 778–785. [Google Scholar]
- Nadaf, N.; Ghosh, J. Purification and characterization of catechol 1,2-dioxygenase from Rhodococcus sp. NCIM 2891. Res. J. Environ. Earth Sci. 2011, 3, 608–613. [Google Scholar]
- Lin, J.; Milase, R.N. Purification and characterization of catechol 1,2-dioxygenase from Acinetobacter sp. Y64 strain and Escherichia coli transformants. Protein J. 2015, 34, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Guzik, U.; Greń, I.; Hupert-Kocurek, K.; Wojcieszyńska, D. Catechol 1,2-dioxygenase from the new aromatic compounds—Degrading Pseudomonas putida strain N6. Int. Biodeterior. Biodegrad. 2011, 65, 504–512. [Google Scholar] [CrossRef]
- Wang, C.-L.; You, S.-L.; Wang, S.-L. Purification and characterization of a novel catechol 1,2-dioxygenase from Pseudomonas aeruginosa with benzoic acid as a carbon source. Process Biochem. 2006, 41, 1594–1601. [Google Scholar] [CrossRef]
- Guzik, U.; Hupert-Kocurek, K.; Sitnik, M.; Wojcieszyńska, D. High activity catechol 1,2-dioxygenase from Stenotrophomonas maltophilia strain KB2 as a useful tool in cis, cis-muconic acid production. Antonie Van Leeuwenhoek 2013, 103, 1297–1307. [Google Scholar] [CrossRef] [PubMed]
- Solyanikova, I.P.; Konovalova, E.I.; Golovleva, L.A. Methylcatechol 1,2-dioxygenase of Rhodococcus opacus 6a is a new type of the catechol-cleaving enzyme. Biochemistry 2009, 74, 994–1001. [Google Scholar] [CrossRef] [PubMed]
- Pandeeti, E.V.P.; Siddavattam, D. Purification and characterization of catechol 1,2-dioxygenase from Acinetobacter sp. DS002 and cloning, sequencing of partial catA gene. Indian J. Microbiol. 2011, 51, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Stoilova, I.; Krastanov, A.; Stanchev, V.; Daniel, D.; Gerginova, M.; Alexieva, Z. Biodegradation of high amounts of phenol, catechol, 2,4-dichlorophenol and 2,6-dimethoxyphenol by Aspergillus awamori cells. Enzyme Microb. Technol. 2006, 39, 1036–1041. [Google Scholar] [CrossRef]
- Pakala, S.B.; Gorla, P.; Pinjari, A.B.; Krovidi, R.K.; Baru, R.; Yanamandra, M.; Merrick, M.; Siddavattam, D. Biodegradation of methyl parathion and p-nitrophenol: Evidence for the presence of a p-nitrophenol 2-hydroxylase in a Gram-negative Serratia sp. strain DS001. Appl. Microbiol. Biotechnol. 2007, 73, 1452–1462. [Google Scholar] [CrossRef]
- Jeoung, J.-H.; Bommer, M.; Lin, T.-Y.; Dobbek, H. Visualizing the substrate-, superoxo-, alkylperoxo-, and product-bound states at the nonheme Fe (II) site of homogentisate dioxygenase. Proc. Natl. Acad. Sci. USA 2013, 110, 12625–12630. [Google Scholar] [CrossRef]
- Mendez, V.; Agullo, L.; Gonzalez, M.; Seeger, M. The homogentisate and homoprotocatechuate central pathways are involved in 3- and 4-hydroxyphenylacetate degradation by Burkholderia xenovorans LB400. PLoS ONE 2011, 6, e17583. [Google Scholar] [CrossRef] [PubMed]
- Olaniran, A.O.; Singh, L.; Kumar, A.; Mokoena, P.; Pillay, B. Aerobic degradation of 2,4-dichlorophenoxyacetic acid and other chlorophenols by Pseudomonas strains indigenous to contaminated soil in South Africa: Growth kinetics and degradation pathway. Appl. Biochem. Microbiol. 2017, 53, 209–216. [Google Scholar] [CrossRef]
- Marchesi, J.R.; Sato, T.; Weightman, A.J.; Martin, T.A.; Fry, J.C.; Hiom, S.J.; Wade, W.G. Design and evaluation of useful bacterium-specific PCR primers that amplify genes coding for bacterial 16S rRNA. Appl. Environ. Microbiol. 1998, 64, 795–799. [Google Scholar] [PubMed]
- Kumar, A.; Khan, F.I.; Olaniran, A.O. Chloroacetaldehyde dehydrogenase from Ancylobacter aquaticus UV5: Cloning, expression, characterization and molecular modeling. Int. J. Biol. Macromol. 2018, 114, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Cañón, J.M.; Peñalva, M.A. Spectrophotometric determination of homogentisate using Aspergillus nidulans homogentisate dioxygenase. Anal. Biochem. 1997, 245, 218–221. [Google Scholar] [CrossRef] [PubMed]
- Borowski, T.; Georgiev, V.; Siegbahn, P.E.M. Catalytic reaction mechanism of homogentisate dioxygenase: A hybrid DFT study. J. Am. Chem. Soc. 2005, 127, 17303–17314. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Step | Total Activity (U/mL) | Total Protein (mg/mL) | Specific Activity (U/mg) | Yield (%) | Purification Fold |
|---|---|---|---|---|---|
| Crude | 10.34 | 4.64 | 2.23 | 100 | 1 |
| Anion Exchange Chromatography | 5.16 | 1.75 | 2.91 | 57.7 | 1.3 |
| Gel Filtration Chromatography | 2.02 | 0.59 | 3.42 | 13.02 | 1.5 |
| Metal/Inhibitor/Detergent | Residual Activity (%) * |
|---|---|
| None (Control) | 100.00 ± 0.04 |
| β-Mercaptoethanol | 59.00 ± 0.03 |
| EDTA | 58.00 ± 0.02 |
| CuSO4 | 33.00 ± 0.01 |
| HgCl2 | 15.00 ± 0.02 |
| Tween 20 | 80.00 ± 0.02 |
| Tween 80 | 96.00 ± 0.02 |
| SDS | 10.00 ± 0.01 |
| Substrates | Residual Activity (%) * |
|---|---|
| Catechol | 100.00 ± 0.01 |
| 4-Nitrocatechol | 25.00 ± 0.00 |
| 4-Methylcatechol | 0.00 ± 0.00 |
| 3-Methylcatechol | 0.00 ± 0.00 |
| 1,2,4-Benzenetriol | 21.00 ± 0.02 |
| Phenol | 72.00 ± 0.02 |
| 2,4-Dichlorophenol | 51.00 ± 0.00 |
| Homogentisate | 60 nmol−1·min−1·mg−1 # |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Setlhare, B.; Kumar, A.; Mokoena, M.P.; Olaniran, A.O. Catechol 1,2-Dioxygenase is an Analogue of Homogentisate 1,2-Dioxygenase in Pseudomonas chlororaphis Strain UFB2. Int. J. Mol. Sci. 2019, 20, 61. https://doi.org/10.3390/ijms20010061
Setlhare B, Kumar A, Mokoena MP, Olaniran AO. Catechol 1,2-Dioxygenase is an Analogue of Homogentisate 1,2-Dioxygenase in Pseudomonas chlororaphis Strain UFB2. International Journal of Molecular Sciences. 2019; 20(1):61. https://doi.org/10.3390/ijms20010061
Chicago/Turabian StyleSetlhare, Boitumelo, Ajit Kumar, Mduduzi P. Mokoena, and Ademola O. Olaniran. 2019. "Catechol 1,2-Dioxygenase is an Analogue of Homogentisate 1,2-Dioxygenase in Pseudomonas chlororaphis Strain UFB2" International Journal of Molecular Sciences 20, no. 1: 61. https://doi.org/10.3390/ijms20010061