Cystine/Glutamate Antiporter and Aripiprazole Compensate NMDA Antagonist-Induced Dysfunction of Thalamocortical L-Glutamatergic Transmission


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
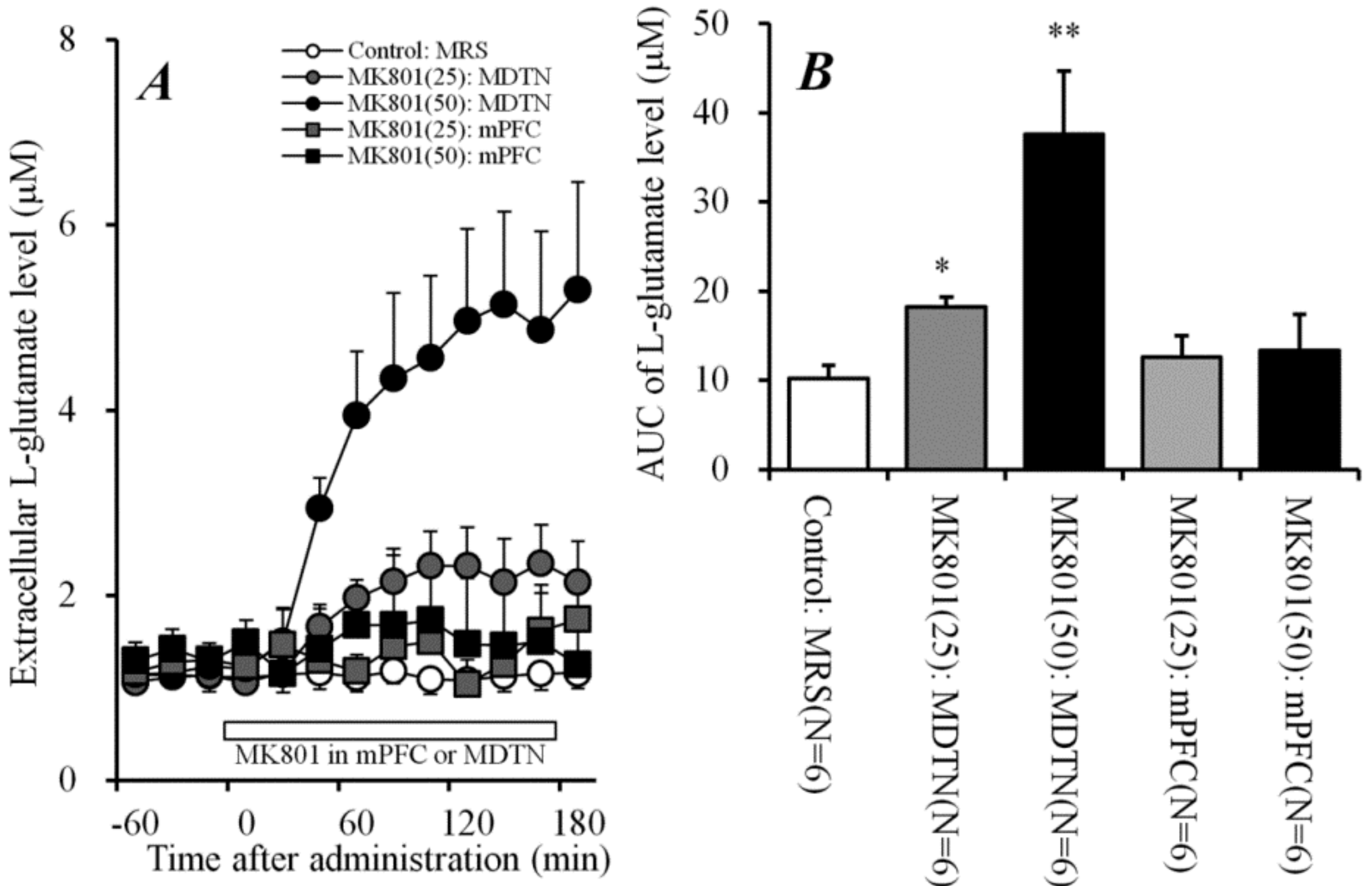
2.1. Effects of Systemic Administrations of MK801 and NAC on Extracellular L-Glutamate in mPFC (Study 1)
2.2. Concentration-Dependent Effects of Perfusion with MK801 into mPFC and MDTN on L-Glutamate Release in mPFC (Study 2)
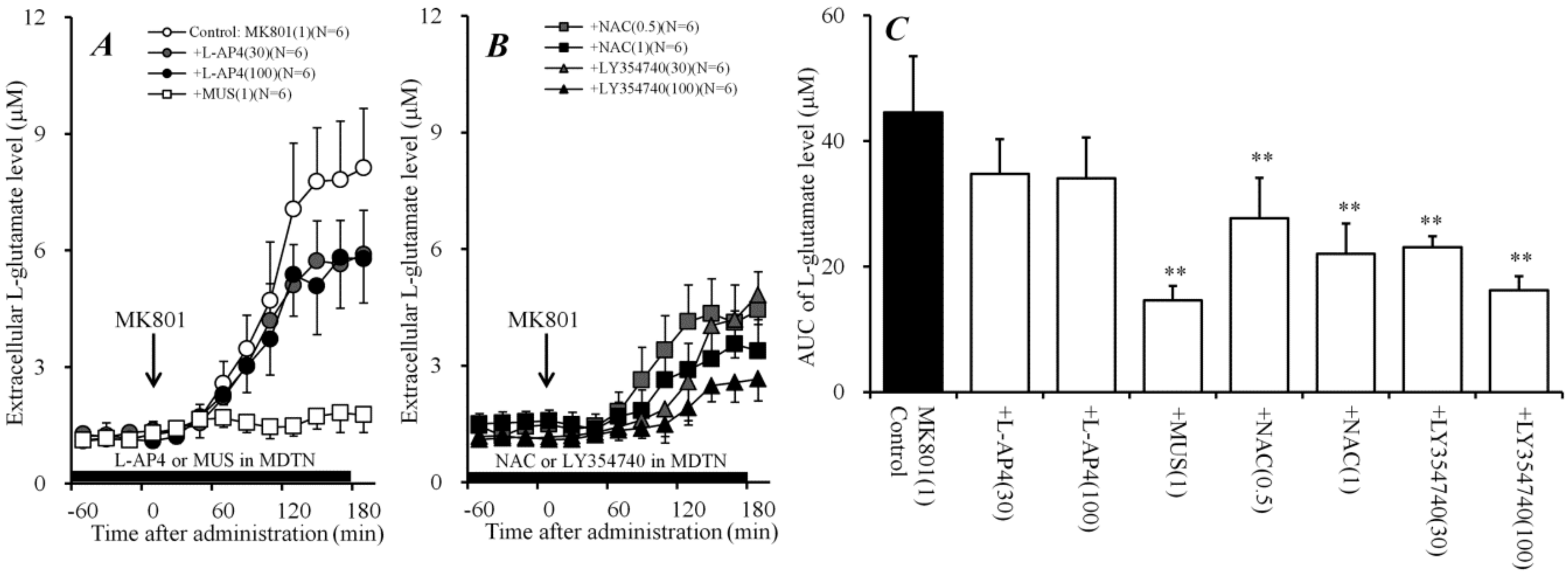
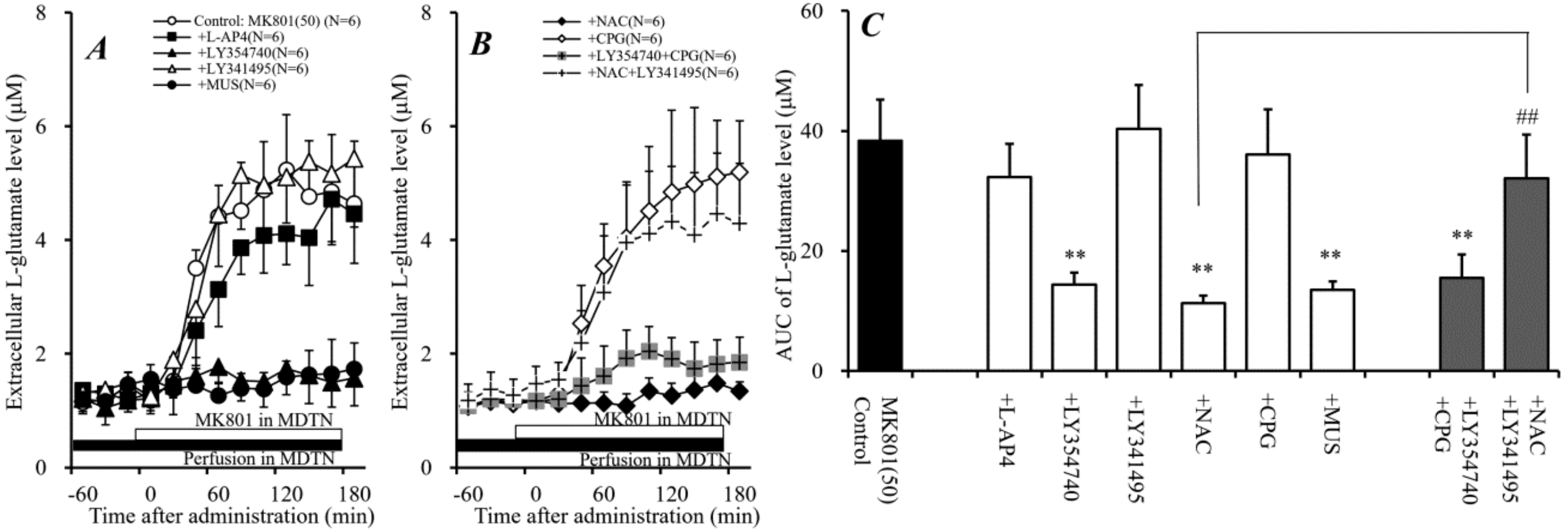
2.3. Effects of Perfusion of Activators of Sxc, mGluRs, and GABAA Receptor into MDTN on Systemic MK801-Evoked L-Glutamate Release (Study 3)
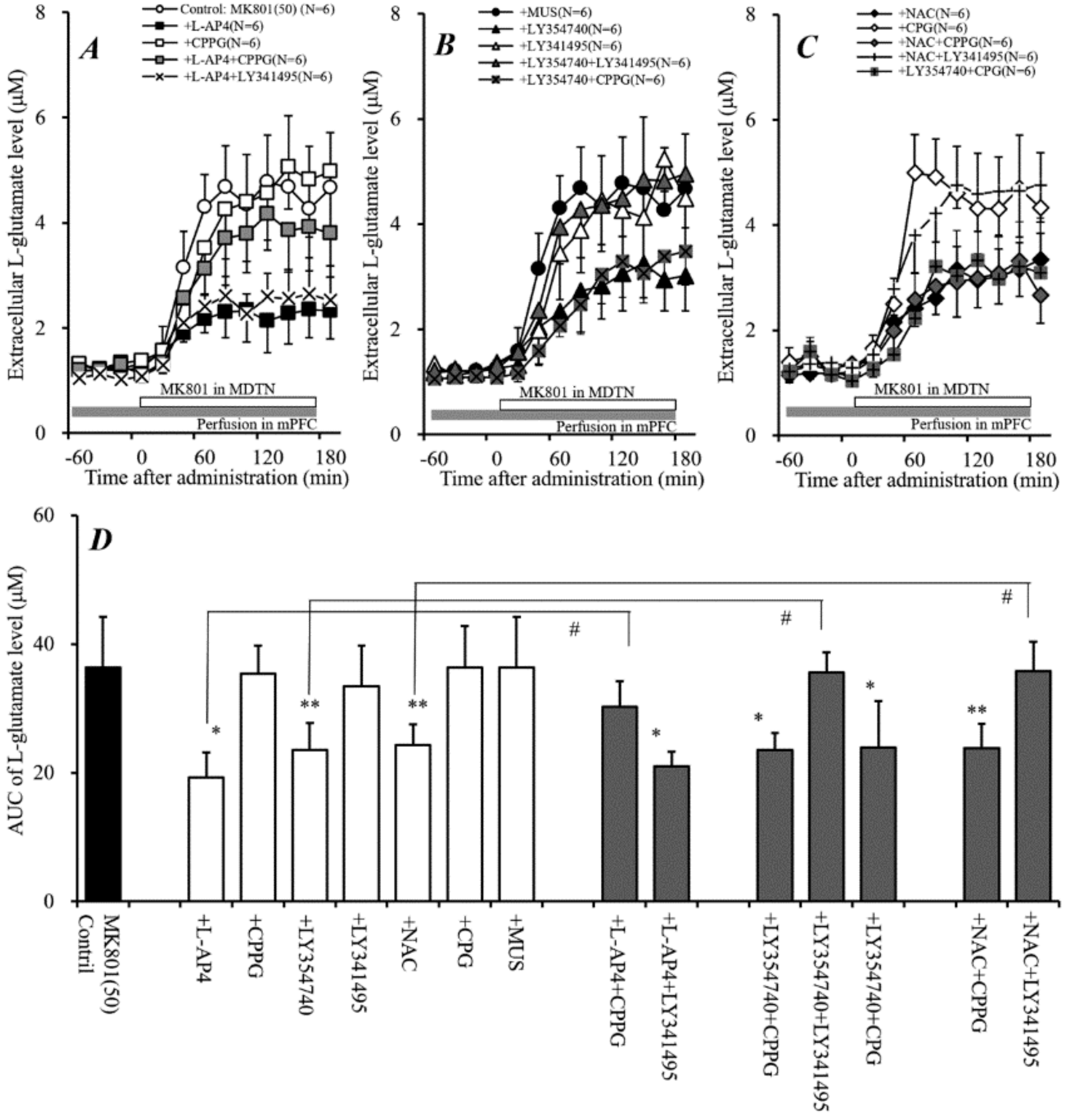
2.4. Effects of Perfusion of Activators of Sxc, mGluRs, and GABAA Receptor into mPFC on Systemic MK801-Evoked L-Glutamate Release (Study 4)
2.5. Effects of Local Administration of Modulators of Sxc, mGluRs, and GABAA Receptor into MDTN on Local MK801-Evoked L-Glutamate Release in mPFC (Study 5)
2.6. Effects of Local Administration of Modulators of Sxc, mGluRs, and GABAA Receptors into mPFC on Local MK801-Evoked L-Glutamate Release in mPFC (Study 6)
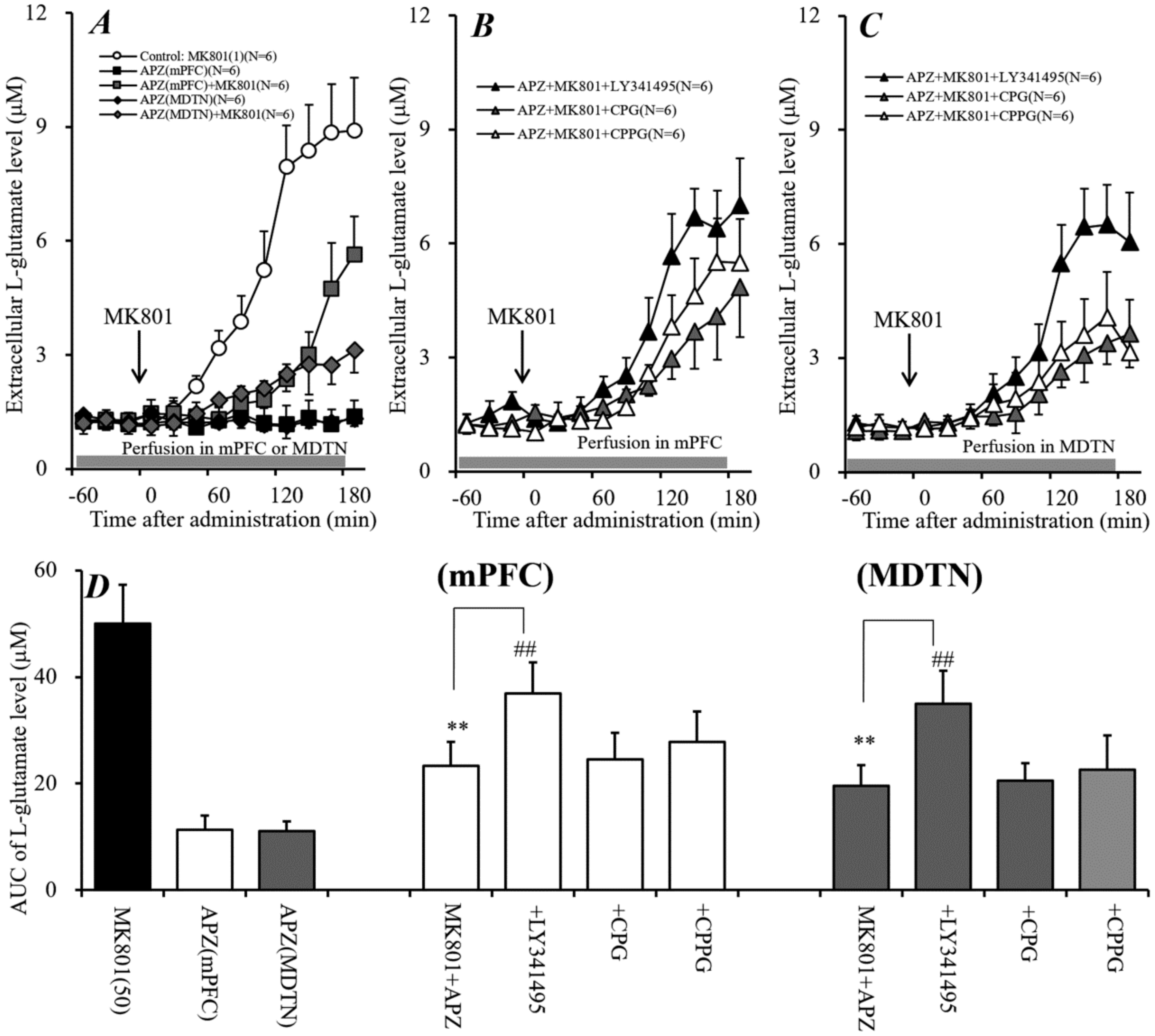
2.7. Interaction between Local Administration of APZ and mGluR Antagonists into mPFC and MDTN on Systemic MK801-Evoked L-Glutamate Release in mPFC (Study 7)
3. Discussion
3.1. Mechanism of Systemic MK801-Evoked L-Glutamate Release in mPFC
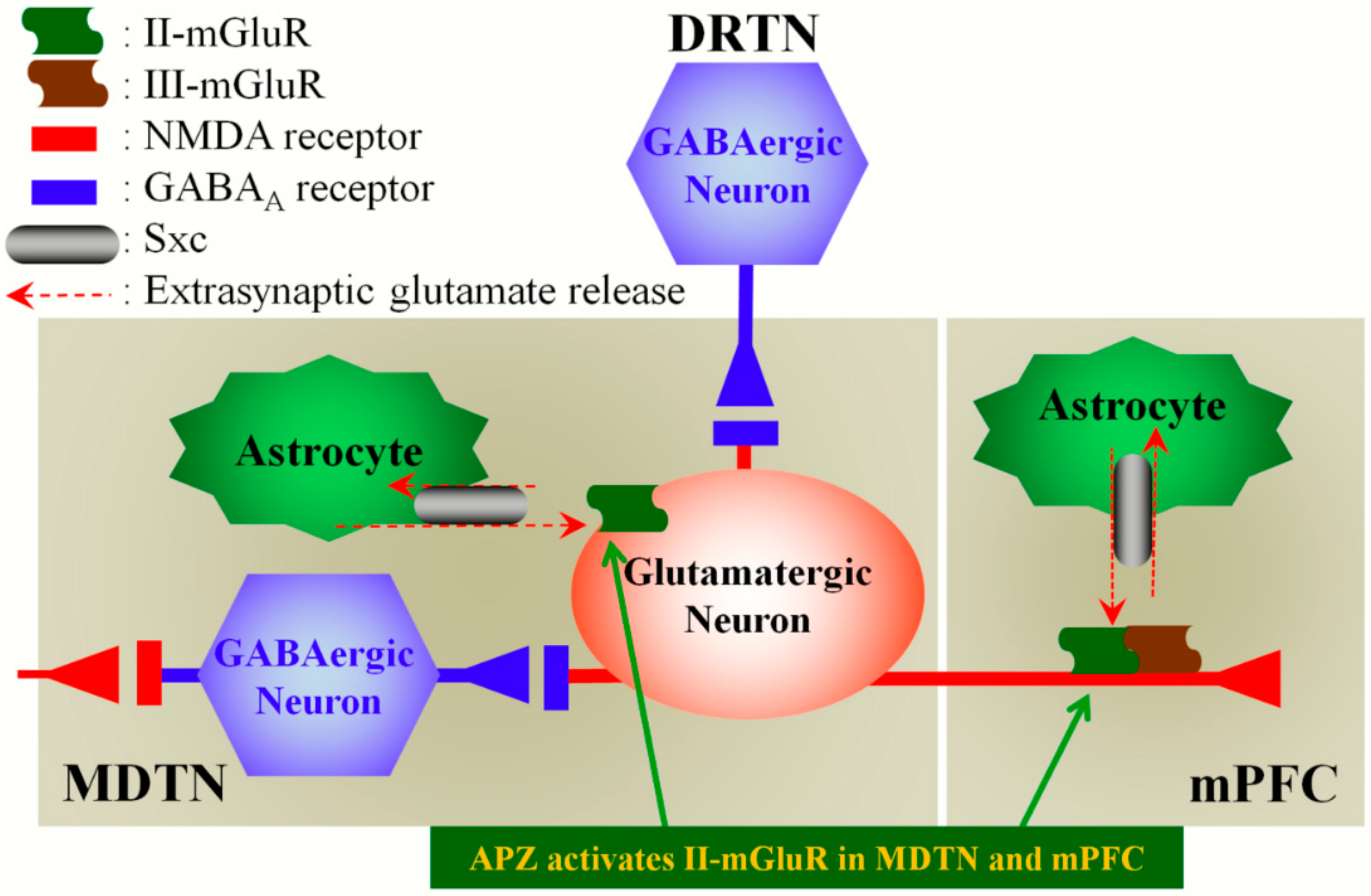
3.2. Regulation Mechanisms of MDTN–mPFC Glutamatergic Transmission
3.3. Mechanisms of Action of APZ
4. Materials and Methods
4.1. Chemical Agents
4.2. Preparation of Microdialysis System
4.3. Determination of Levels of L-Glutamate
4.4. Data Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| II-mGluR | group II metabotropic glutamate receptor |
| III-mGluR | group III metabotropic glutamate receptor |
| APZ | aripiprazole |
| CPG: | (S)-4-carboxyphenylglycine |
| CPP: | 3-(2-carboxypiperazin-4-yl)-propyl-1-phosphonic acid |
| CPPG: | (RS)-α-cyclopropyl-4-phosphonophenyl glycine |
| DRTN: | dorsal reticular thalamic nucleus |
| FRET: | fluorescence resonance energy transfer |
| L-AP4: | L-(+)-2-amino-4-phosphonobutyric acid |
| LMM: | linear mixed model |
| MAPK: | mitogen-activated protein kinase |
| MDTN: | mediodorsal thalamic nucleus |
| MK801 | 5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine |
| NMDAR | glutamate/NMDA receptor |
| mPFC: | medial pre-frontal cortex |
| MUS: | muscimol |
| NAC: | N-acetyl-L-cysteine |
| Sxc: | system xc- |
| UHPLC: | ultra-high-performance liquid-chromatography |
References
- Javitt, D.C. Glutamate and schizophrenia: Phencyclidine, n-methyl-d-aspartate receptors, and dopamine-glutamate interactions. Int. Rev. Neurobiol. 2007, 78, 69–108. [Google Scholar] [PubMed]
- Labrie, V.; Roder, J.C. The involvement of the nmda receptor d-serine/glycine site in the pathophysiology and treatment of schizophrenia. Neurosci. Biobehav. Rev. 2010, 34, 351–372. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, J.A.; Bymaster, F.P.; Meltzer, H.Y.; Deutch, A.Y.; Duncan, G.E.; Marx, C.E.; Aprille, J.R.; Dwyer, D.S.; Li, X.M.; Mahadik, S.P.; et al. Antipsychotic drugs: Comparison in animal models of efficacy, neurotransmitter regulation, and neuroprotection. Pharmacol. Rev. 2008, 60, 358–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meltzer, H.Y.; Huang, M. In vivo actions of atypical antipsychotic drug on serotonergic and dopaminergic systems. Prog. Brain Res. 2008, 172, 177–197. [Google Scholar] [PubMed]
- Yamamura, S.; Ohoyama, K.; Hamaguchi, T.; Nakagawa, M.; Suzuki, D.; Matsumoto, T.; Motomura, E.; Tanii, H.; Shiroyama, T.; Okada, M. Effects of zotepine on extracellular levels of monoamine, gaba and glutamate in rat prefrontal cortex. Br. J. Pharmacol. 2009, 157, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, S.; Ohoyama, K.; Hamaguchi, T.; Kashimoto, K.; Nakagawa, M.; Kanehara, S.; Suzuki, D.; Matsumoto, T.; Motomura, E.; Shiroyama, T.; et al. Effects of quetiapine on monoamine, gaba, and glutamate release in rat prefrontal cortex. Psychopharmacology (Berl) 2009, 206, 243–258. [Google Scholar] [CrossRef] [PubMed]
- Tanahashi, S.; Yamamura, S.; Nakagawa, M.; Motomura, E.; Okada, M. Dopamine d2 and serotonin 5-ht1a receptors mediate the actions of aripiprazole in mesocortical and mesoaccumbens transmission. Neuropharmacology 2012, 62, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Green, B. Focus on aripiprazole. Curr. Med. Res. Opin. 2004, 20, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, D.A.; Renock, S.; Arrington, E.; Chiodo, L.A.; Liu, L.X.; Sibley, D.R.; Roth, B.L.; Mailman, R. Aripiprazole, a novel atypical antipsychotic drug with a unique and robust pharmacology. Neuropsychopharmacology 2003, 28, 1400–1411. [Google Scholar] [CrossRef] [PubMed]
- Bruins Slot, L.A.; Kleven, M.S.; Newman-Tancredi, A. Effects of novel antipsychotics with mixed d(2) antagonist/5-ht(1a) agonist properties on pcp-induced social interaction deficits in the rat. Neuropharmacology 2005, 49, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Amano, T.; Matsubayashi, H.; Momiyama, T.; Ishihara, K.; Todo, N.; Sasa, M. Antagonizing effects of a novel antipsychotic quinolinone derivative (opc-14597) on dopaminergic inhibition of neuronal activities in the nucleus accumbens. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 1995, 19, 105–116. [Google Scholar] [CrossRef]
- Malhotra, A.K.; Pinals, D.A.; Weingartner, H.; Sirocco, K.; Missar, C.D.; Pickar, D.; Breier, A. Nmda receptor function and human cognition: The effects of ketamine in healthy volunteers. Neuropsychopharmacology 1996, 14, 301–307. [Google Scholar] [CrossRef]
- Krystal, J.H.; Karper, L.P.; Seibyl, J.P.; Freeman, G.K.; Delaney, R.; Bremner, J.D.; Heninger, G.R.; Bowers, M.B., Jr.; Charney, D.S. Subanesthetic effects of the noncompetitive nmda antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch. Gen. Psychiatry 1994, 51, 199–214. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, A.K.; Pinals, D.A.; Adler, C.M.; Elman, I.; Clifton, A.; Pickar, D.; Breier, A. Ketamine-induced exacerbation of psychotic symptoms and cognitive impairment in neuroleptic-free schizophrenics. Neuropsychopharmacology 1997, 17, 141–150. [Google Scholar] [CrossRef]
- Krystal, J.H.; D’Souza, D.C.; Mathalon, D.; Perry, E.; Belger, A.; Hoffman, R. Nmda receptor antagonist effects, cortical glutamatergic function, and schizophrenia: Toward a paradigm shift in medication development. Psychopharmacology (Berl) 2003, 169, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Lorrain, D.S.; Schaffhauser, H.; Campbell, U.C.; Baccei, C.S.; Correa, L.D.; Rowe, B.; Rodriguez, D.E.; Anderson, J.J.; Varney, M.A.; Pinkerton, A.B.; et al. Group ii mglu receptor activation suppresses norepinephrine release in the ventral hippocampus and locomotor responses to acute ketamine challenge. Neuropsychopharmacology 2003, 28, 1622–1632. [Google Scholar] [CrossRef] [PubMed]
- Moghaddam, B.; Adams, B.W. Reversal of phencyclidine effects by a group ii metabotropic glutamate receptor agonist in rats. Science 1998, 281, 1349–1352. [Google Scholar] [CrossRef] [PubMed]
- Homayoun, H.; Jackson, M.E.; Moghaddam, B. Activation of metabotropic glutamate 2/3 receptors reverses the effects of nmda receptor hypofunction on prefrontal cortex unit activity in awake rats. J. Neurophysiol. 2005, 93, 1989–2001. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.A.; Madayag, A.; Kristiansen, L.V.; Meador-Woodruff, J.H.; Haroutunian, V.; Raju, I. Contribution of cystine-glutamate antiporters to the psychotomimetic effects of phencyclidine. Neuropsychopharmacology 2008, 33, 1760–1772. [Google Scholar] [CrossRef] [PubMed]
- Tanahashi, S.; Yamamura, S.; Nakagawa, M.; Motomura, E.; Okada, M. Clozapine, but not haloperidol, enhances glial d-serine and l-glutamate release in rat frontal cortex and primary cultured astrocytes. Br. J. Pharmacol. 2012, 165, 1543–1555. [Google Scholar] [CrossRef] [PubMed]
- Lorrain, D.S.; Baccei, C.S.; Bristow, L.J.; Anderson, J.J.; Varney, M.A. Effects of ketamine and n-methyl-d-aspartate on glutamate and dopamine release in the rat prefrontal cortex: Modulation by a group ii selective metabotropic glutamate receptor agonist ly379268. Neuroscience 2003, 117, 697–706. [Google Scholar] [CrossRef]
- Lopez-Gil, X.; Babot, Z.; Amargos-Bosch, M.; Sunol, C.; Artigas, F.; Adell, A. Clozapine and haloperidol differently suppress the mk-801-increased glutamatergic and serotonergic transmission in the medial prefrontal cortex of the rat. Neuropsychopharmacology 2007, 32, 2087–2097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotman, C.W.; Monaghan, D.T. Anatomical organization of excitatory amino acid receptors and their properties. Adv. Exp. Med. Biol. 1986, 203, 237–252. [Google Scholar] [PubMed]
- Jay, T.M.; Thierry, A.M.; Wiklund, L.; Glowinski, J. Excitatory amino acid pathway from the hippocampus to the prefrontal cortex. Contribution of ampa receptors in hippocampo-prefrontal cortex transmission. Eur. J. Neurosci. 1992, 4, 1285–1295. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, M.; Yokofujita, J.; Murakami, K. An ultrastructural study of the neural circuit between the prefrontal cortex and the mediodorsal nucleus of the thalamus. Prog. Neurobiol. 1998, 54, 417–458. [Google Scholar] [CrossRef]
- Young, K.A.; Holcomb, L.A.; Yazdani, U.; Hicks, P.B.; German, D.C. Elevated neuron number in the limbic thalamus in major depression. Am. J. Psychiatry 2004, 161, 1270–1277. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.J.; Weinberger, D.R. Schizophrenia genes, gene expression, and neuropathology: On the matter of their convergence. Mol. Psychiatry 2005, 10, 40–68. [Google Scholar] [CrossRef] [PubMed]
- Bertram, E.H.; Onat, F.Y.; Ozkara, C.; Moshe, S.L.; Avanzini, G. Workshop on idiopathic generalized epilepsies: Bridging basic science and clinical research (October 3-6, 2007; Antalya, Turkey). Epilepsia 2008, 49, 1969–1972. [Google Scholar] [CrossRef] [PubMed]
- Skvarc, D.R.; Dean, O.M.; Byrne, L.K.; Gray, L.; Lane, S.; Lewis, M.; Fernandes, B.S.; Berk, M.; Marriott, A. The effect of n-acetylcysteine (nac) on human cognition—A systematic review. Neurosci. Biobehav. Rev. 2017, 78, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Massie, A.; Boillee, S.; Hewett, S.; Knackstedt, L.; Lewerenz, J. Main path and byways: Non-vesicular glutamate release by system xc(-) as an important modifier of glutamatergic neurotransmission. J. Neurochem. 2015, 135, 1062–1079. [Google Scholar] [CrossRef] [PubMed]
- Moran, M.M.; McFarland, K.; Melendez, R.I.; Kalivas, P.W.; Seamans, J.K. Cystine/glutamate exchange regulates metabotropic glutamate receptor presynaptic inhibition of excitatory transmission and vulnerability to cocaine seeking. J. Neurosci. 2005, 25, 6389–6393. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.A.; Xi, Z.X.; Shen, H.; Swanson, C.J.; Kalivas, P.W. The origin and neuronal function of in vivo nonsynaptic glutamate. J. Neurosci. 2002, 22, 9134–9141. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Hinoi, E.; Takemori, A.; Nakamichi, N.; Yoneda, Y. Glutamate inhibits chondral mineralization through apoptotic cell death mediated by retrograde operation of the cystine/glutamate antiporter. J. Boil. Chem. 2006, 281, 24553–24565. [Google Scholar] [CrossRef] [PubMed]
- Ishii, D.; Matsuzawa, D.; Kanahara, N.; Matsuda, S.; Sutoh, C.; Ohtsuka, H.; Nakazawa, K.; Kohno, M.; Hashimoto, K.; Iyo, M.; et al. Effects of aripiprazole on mk-801-induced prepulse inhibition deficits and mitogen-activated protein kinase signal transduction pathway. Neurosci. Lett. 2010, 471, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Carli, M.; Calcagno, E.; Mainolfi, P.; Mainini, E.; Invernizzi, R.W. Effects of aripiprazole, olanzapine, and haloperidol in a model of cognitive deficit of schizophrenia in rats: Relationship with glutamate release in the medial prefrontal cortex. Psychopharmacology (Berl) 2011, 214, 639–652. [Google Scholar] [CrossRef] [PubMed]
- Dawson, N.; Morris, B.J.; Pratt, J.A. Subanaesthetic ketamine treatment alters prefrontal cortex connectivity with thalamus and ascending subcortical systems. Schizophr. Bull. 2013, 39, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Vertes, R.P.; Linley, S.B.; Hoover, W.B. Limbic circuitry of the midline thalamus. Neurosci. Biobehav. Rev. 2015, 54, 89–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoepp, D.D.; Jane, D.E.; Monn, J.A. Pharmacological agents acting at subtypes of metabotropic glutamate receptors. Neuropharmacology 1999, 38, 1431–1476. [Google Scholar] [CrossRef]
- Niswender, C.M.; Conn, P.J. Metabotropic glutamate receptors: Physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 295–322. [Google Scholar] [CrossRef] [PubMed]
- Copeland, C.S.; Neale, S.A.; Salt, T.E. Neuronal activity patterns in the mediodorsal thalamus and related cognitive circuits are modulated by metabotropic glutamate receptors. Neuropharmacology 2015, 92, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Copeland, C.S.; Wall, T.M.; Sims, R.E.; Neale, S.A.; Nisenbaum, E.; Parri, H.R.; Salt, T.E. Astrocytes modulate thalamic sensory processing via mglu2 receptor activation. Neuropharmacology 2017, 121, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Herman, E.J.; Bubser, M.; Conn, P.J.; Jones, C.K. Metabotropic glutamate receptors for new treatments in schizophrenia. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2012; pp. 297–365. [Google Scholar]
- Kalivas, P.W. The glutamate homeostasis hypothesis of addiction. Nat. Rev. Neurosci. 2009, 10, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Tadori, Y.; Miwa, T.; Tottori, K.; Burris, K.D.; Stark, A.; Mori, T.; Kikuchi, T. Aripiprazole’s low intrinsic activities at human dopamine d2l and d2s receptors render it a unique antipsychotic. Eur. J. Pharmacol. 2005, 515, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Snigdha, S.; Neill, J.C. Improvement of phencyclidine-induced social behaviour deficits in rats: Involvement of 5-ht1a receptors. Behav. Brain Res. 2008, 191, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Wieronska, J.M.; Slawinska, A.; Stachowicz, K.; Lason-Tyburkiewicz, M.; Gruca, P.; Papp, M.; Pilc, A. The reversal of cognitive, but not negative or positive symptoms of schizophrenia, by the mglu(2)/(3) receptor agonist, ly379268, is 5-ht(1)a dependent. Behav. Brain Res. 2013, 256, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Abekawa, T.; Ito, K.; Koyama, T. Role of the simultaneous enhancement of nmda and dopamine d1 receptor-mediated neurotransmission in the effects of clozapine on phencyclidine-induced acute increases in glutamate levels in the rat medial prefrontal cortex. N-S Arch. Pharmacol. 2006, 374, 177–193. [Google Scholar] [CrossRef] [PubMed]
- Calcagno, E.; Carli, M.; Invernizzi, R.W. The 5-ht(1a) receptor agonist 8-oh-dpat prevents prefrontocortical glutamate and serotonin release in response to blockade of cortical nmda receptors. J. Neurochem. 2006, 96, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Sadowska-Bartosz, I.; Galiniak, S.; Bartosz, G.; Zuberek, M.; Grzelak, A.; Dietrich-Muszalska, A. Antioxidant properties of atypical antipsychotic drugs used in the treatment of schizophrenia. Schizophr. Res. 2016, 176, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Kahn, L.; Alonso, G.; Robbe, D.; Bockaert, J.; Manzoni, O.J. Group 2 metabotropic glutamate receptors induced long term depression in mouse striatal slices. Neurosci. Lett. 2001, 316, 178–182. [Google Scholar] [CrossRef]
- Ferraguti, F.; Baldani-Guerra, B.; Corsi, M.; Nakanishi, S.; Corti, C. Activation of the extracellular signal-regulated kinase 2 by metabotropic glutamate receptors. Eur. J. Neurosci. 1999, 11, 2073–2082. [Google Scholar] [CrossRef] [PubMed]
- Urban, J.D.; Vargas, G.A.; Von Zastrow, M.; Mailman, R.B. Aripiprazole has functionally selective actions at dopamine d2 receptor-mediated signaling pathways. Neuropsychopharmacology 2007, 32, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Alexander, S.P.; Peters, J.A.; Kelly, E.; Marrion, N.V.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; Sharman, J.L.; Southan, C.; Davies, J.A.; et al. The concise guide to pharmacology 2017/18: Ligand-gated ion channels. Br. J. Pharmacol. 2017, 174 (Suppl. 1), S130–S159. [Google Scholar] [CrossRef] [PubMed]
- Alexander, S.P.; Kelly, E.; Marrion, N.V.; Peters, J.A.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; Sharman, J.L.; Southan, C.; Davies, J.A.; et al. The concise guide to pharmacology 2017/18: Transporters. Br. J. Pharmacol. 2017, 174 (Suppl. 1), S360–S446. [Google Scholar] [CrossRef] [PubMed]
- Alexander, S.P.; Christopoulos, A.; Davenport, A.P.; Kelly, E.; Marrion, N.V.; Peters, J.A.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; Sharman, J.L.; et al. The concise guide to pharmacology 2017/18: G protein-coupled receptors. Br. J. Pharmacol. 2017, 174 (Suppl. 1), S17–S129. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.A.; Warren, B.A.; Rhoderick, J.F.; Bridges, R.J. Differentiation of substrate and non-substrate inhibitors of transport system xc(-): An obligate exchanger of l-glutamate and l-cystine. Neuropharmacology 2004, 46, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, S.; Hoshikawa, M.; Kato, D.; Saito, H.; Suzuki, N.; Niwa, O.; Okada, M. Ono-2506 inhibits spike-wave discharges in a genetic animal model without affecting traditional convulsive tests via gliotransmission regulation. Br. J. Pharmacol. 2013, 168, 1088–1100. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.C.; Drummond, G.B.; McLachlan, E.M.; Kilkenny, C.; Wainwright, C.L. Guidelines for reporting experiments involving animals: The arrive guidelines. Br. J. Pharmacol. 2010, 160, 1573–1576. [Google Scholar] [CrossRef] [PubMed]
- Paxinos, G.; Watson, C. The Rat Brain: In Stereotoxic Coordinates, 6th ed.; Academic Press: San Diego, CA, USA, 2007. [Google Scholar]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fukuyama, K.; Hasegawa, T.; Okada, M. Cystine/Glutamate Antiporter and Aripiprazole Compensate NMDA Antagonist-Induced Dysfunction of Thalamocortical L-Glutamatergic Transmission. Int. J. Mol. Sci. 2018, 19, 3645. https://doi.org/10.3390/ijms19113645
Fukuyama K, Hasegawa T, Okada M. Cystine/Glutamate Antiporter and Aripiprazole Compensate NMDA Antagonist-Induced Dysfunction of Thalamocortical L-Glutamatergic Transmission. International Journal of Molecular Sciences. 2018; 19(11):3645. https://doi.org/10.3390/ijms19113645
Chicago/Turabian StyleFukuyama, Kouji, Toshiki Hasegawa, and Motohiro Okada. 2018. "Cystine/Glutamate Antiporter and Aripiprazole Compensate NMDA Antagonist-Induced Dysfunction of Thalamocortical L-Glutamatergic Transmission" International Journal of Molecular Sciences 19, no. 11: 3645. https://doi.org/10.3390/ijms19113645