Protein Co-Aggregation Related to Amyloids: Methods of Investigation, Diversity, and Classification

 , , and
, , and
Abstract
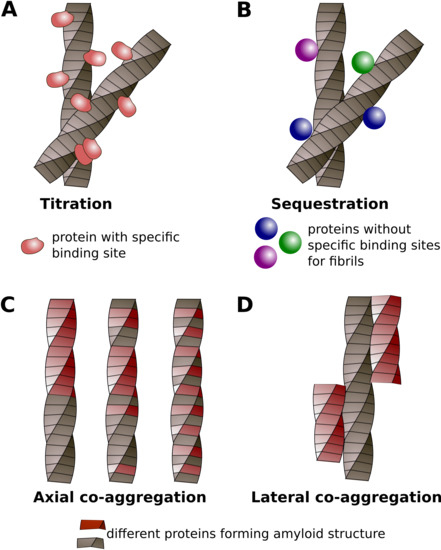
:
1. Introduction
2. Methods for Investigation of the Amyloid Interactome
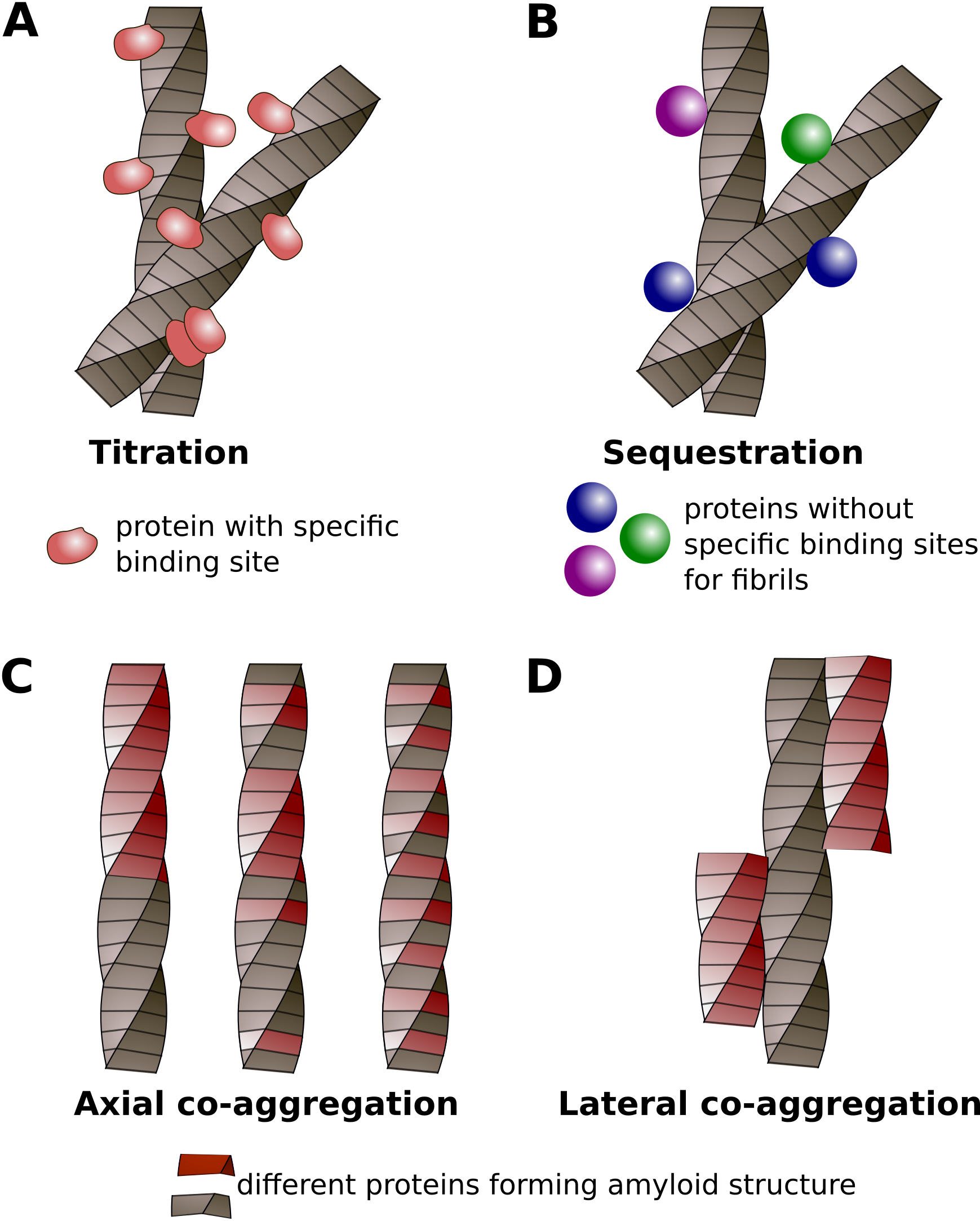
2.1. Co-Immunoprecipitation
2.2. Affinity Chromatography
2.3. Gel Filtration (Size Exclusion Chromatography) and Differential Centrifugation
2.4. Colocalization in Cells and Tissues
2.5. Electrophoresis
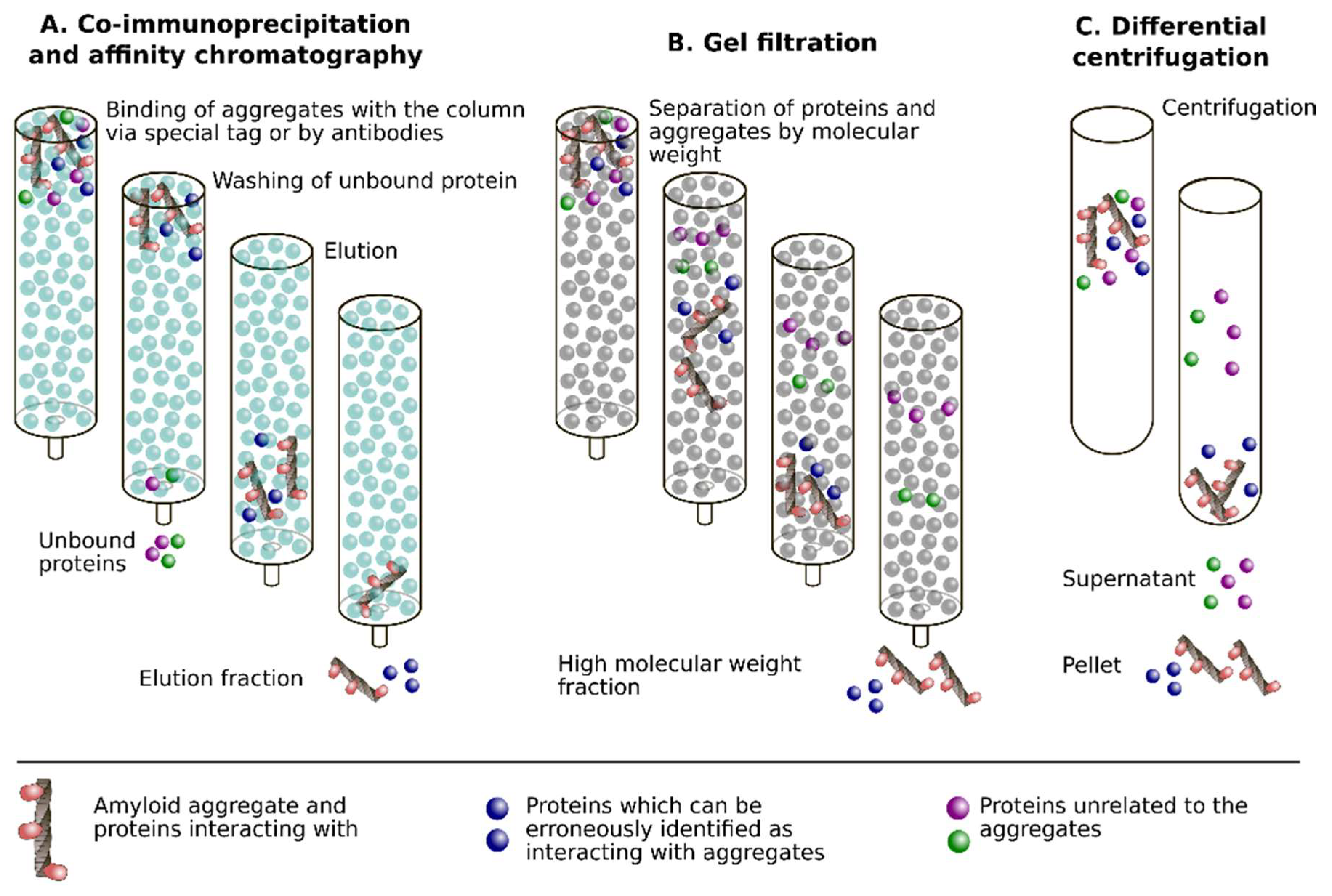
2.6. Cross-Seeding
2.7. Co-Incubation of Monomeric Proteins
2.8. Proteomic Analysis of the Amyloid Interactome
2.9. Transgenic Animals
2.10. The Yeast S. cerevisiae as a Model System
2.11. Computational Approaches
2.12. Biophysical Approaches
2.13. Common Limitations of the In Vitro Approaches
3. The Diversity of Amyloid Co-Aggregation Phenomenon
3.1. The Involvement of Protein Co-Aggregation in the Pathogenesis
3.1.1. Interactions between Pathological Amyloids and QN-Rich Proteins
3.1.2. Interactions between Pathological Amyloids and Non-QN-Rich Proteins
3.2. Yeast Prion Networks
3.3. Functional Amyloid Interactions
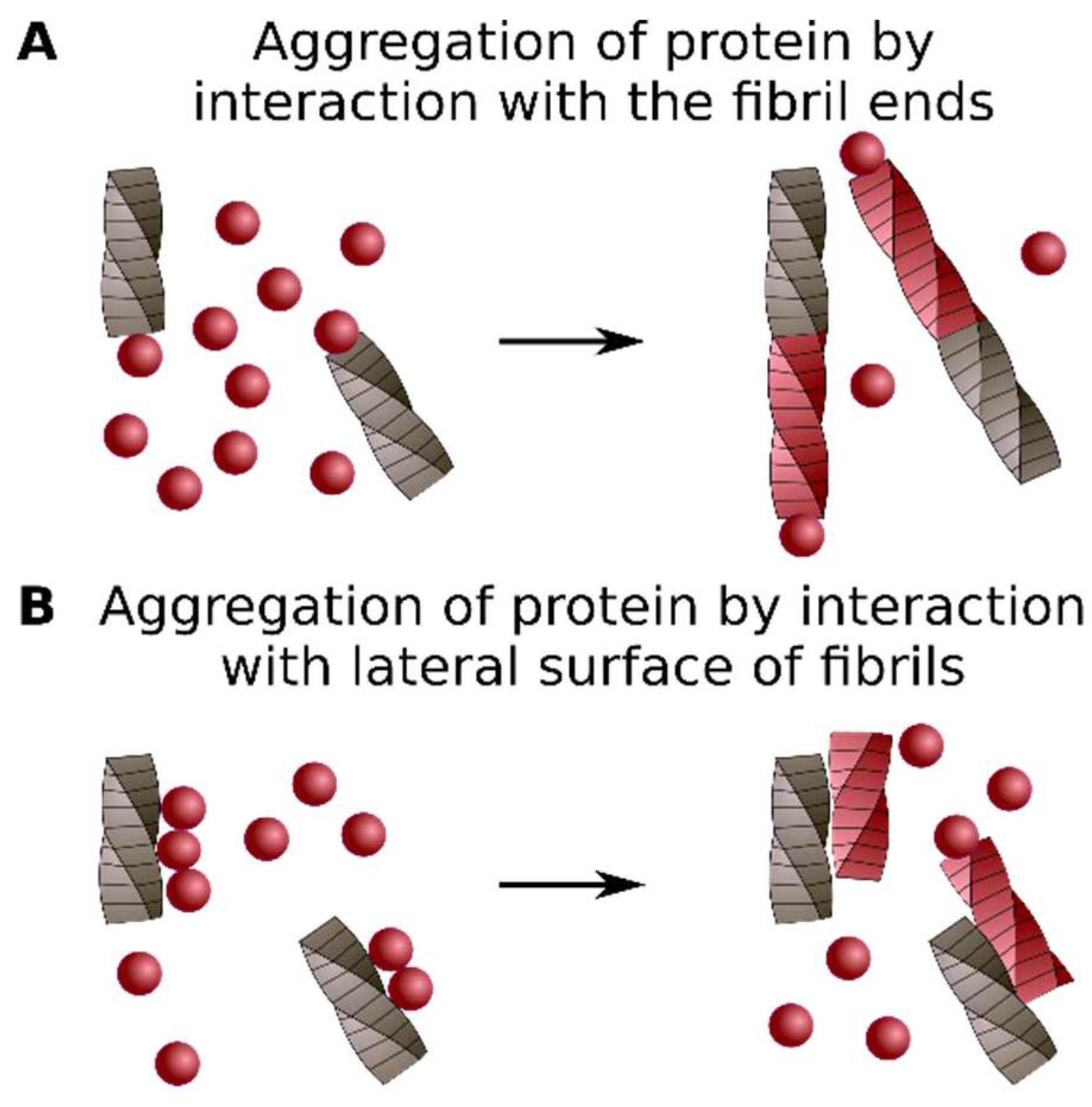
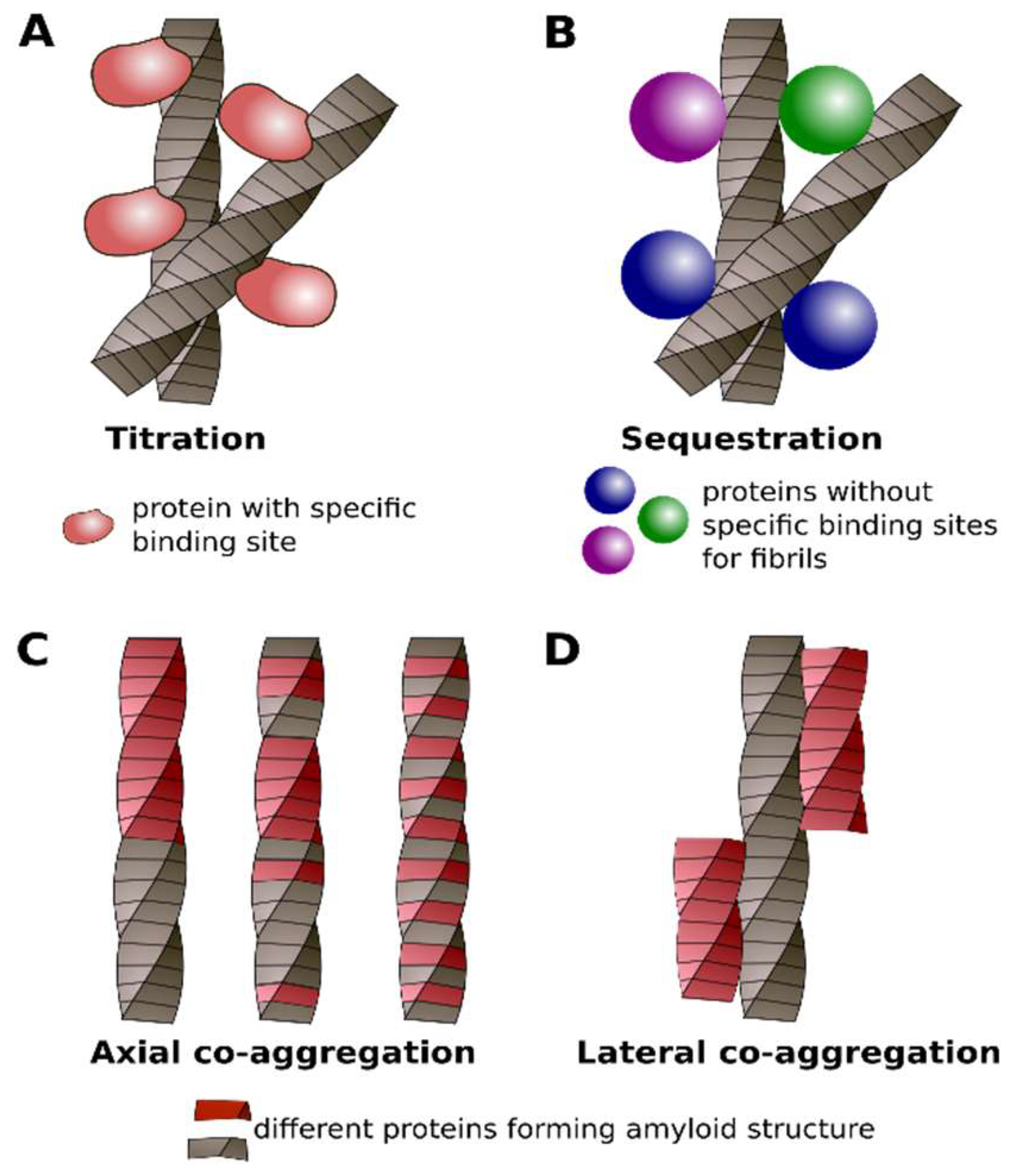
4. Classification of Protein Co-Aggregation
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 2D-GE | Two-dimensional gel electrophoresis |
| α-Syn | α-Synuclein |
| AD | Alzheimer’s disease |
| Aβ | Amyloid-β peptide |
| CBP | CREB-binding protein |
| CFP | Cyan fluorescent protein |
| CJD | Creutzfeldt Jakob disease |
| Co-IP | Co-immunoprecipitation |
| CR | Congo red |
| ESI-IMS-MS | Electrospray ionization-ion mobility spectrometry-mass spectrometry |
| FCCS | Fluorescence cross-correlation spectroscopy |
| FIDA | Fluorescent intensity distribution analysis technique |
| FRET | Förster resonance energy transfer |
| GFP | Green fluorescent protein |
| HD | Huntington’s disease |
| Htt | Huntingtin protein |
| HTT | Huntingtin gene |
| HPLC | High-performance liquid chromatography |
| IAPP | Islet amyloid polypeptide |
| ID | Intrinsically disordered (domains) |
| LBD | Lewy body disease |
| LCM | Laser capture microdissection |
| MLKL | Mixed-lineage kinase domain-like protein |
| MS | Mass-spectrometry |
| NMR | Nuclear magnetic resonance |
| PD | Parkinson’s disease |
| PrP | Prion protein: PrPC—normal isoform of the PrP: PrPSc—prion isoform of PrP |
| PSIA | Proteomic screening and identification of amyloids |
| RFP | Red fluorescent protein |
| Rnq1PrD | Prion domain of Rnq1 |
| Sarkosyl | Sodium lauroyl sarcosinate |
| SDS | Sodium dodecyl sulfate |
| SG | Stress granules |
| SPR | Surface plasmon resonance |
| Sup35NM | Amyloidogenic fragment of the Sup35, comprising N and M-domains of the protein |
| TAPI | Technique for amyloid purification and isolation |
| TBP | TATA-binding protein |
| TBP | TATA-binding protein |
| ThT | Thioflavin T |
| Tg | Transgenic animals |
| Y2H | Yeast two-hybrid system |
| YFP | Yellow fluorescent protein |
References
- Sipe, J.D.; Cohen, A.S. Review: History of the amyloid fibril. J. Struct. Biol. 2000, 130, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Fändrich, M. On the structural definition of amyloid fibrils and other polypeptide aggregates. Cell. Mol. Life Sci. 2007, 64, 2066–2078. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, D.; Jucker, M. The amyloid state of proteins in human diseases. Cell 2012, 148, 1188–1203. [Google Scholar] [CrossRef] [PubMed]
- Sipe, J.D.; Benson, M.D.; Buxbaum, J.N.; Ikeda, S.-I.; Merlini, G.; Saraiva, M.J.M.; Westermark, P. Amyloid fibril proteins and amyloidosis: Chemical identification and clinical classification International Society of Amyloidosis 2016 Nomenclature Guidelines. Amyloid 2016, 23, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Saunders, S.E.; Bartelt-Hunt, S.L.; Bartz, J.C. Prions in the environment: Occurrence, fate and mitigation. Prion 2008, 2, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Toyama, B.H.; Weissman, J.S. Amyloid Structure: Conformational diversity and consequences. Annu. Rev. Biochem. 2011, 80, 557–585. [Google Scholar] [CrossRef] [PubMed]
- Van Gerven, N.; Klein, R.D.; Hultgren, S.J.; Remaut, H. Bacterial amyloid formation: Structural insights into curli biogensis. Trends Microbiol. 2015, 23, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Taglialegna, A.; Lasa, I.; Valle, J. Amyloid structures as biofilm matrix scaffolds. J. Bacteriol. 2016, 198, 2579–2588. [Google Scholar] [CrossRef] [PubMed]
- Dueholm, M.S.; Larsen, P.; Finster, K.; Stenvang, M.R.; Christiansen, G.; Vad, B.S.; Bøggild, A.; Otzen, D.E.; Nielsen, P.H. The tubular sheaths encasing Methanosaeta thermophila filaments are functional amyloids. J. Biol. Chem. 2015, 290, 20590–20600. [Google Scholar] [CrossRef] [PubMed]
- Dragoš, A.; Kovács, Á.T.; Claessen, D. The role of functional amyloids in multicellular growth and development of gram-positive bacteria. Biomolecules 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Bieler, S.; Estrada, L.; Lagos, R.; Baeza, M.; Castilla, J.; Soto, C. Amyloid formation modulates the biological activity of a bacterial protein. J. Biol. Chem. 2005, 280, 26880–26885. [Google Scholar] [CrossRef] [PubMed]
- Bavdek, A.; Kostanjšek, R.; Antonini, V.; Lakey, J.H.; Dalla Serra, M.; Gilbert, R.J.C.; Anderluh, G. PH dependence of listeriolysin O aggregation and pore-forming ability. FEBS J. 2012, 279, 126–141. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.P.; Hewitt, E.W. Why are functional amyloids non-toxic in humans? Biomolecules 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Roberts, B.T.; Wickner, R.B. Heritable activity: A prion that propagates by covalent autoactivation. Genes Dev. 2003, 17, 2083–2087. [Google Scholar] [CrossRef] [PubMed]
- Yuan, A.H.; Hochschild, A. A bacterial global regulator forms a prion. Science 2017, 355, 198–201. [Google Scholar] [CrossRef] [PubMed]
- Liebman, S.W.; Chernoff, Y.O. Prions in yeast. Genetics 2012, 191, 1041–1072. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Biology and genetics of prions causing neurodegeneration. Annu. Rev. Genet. 2013, 47, 601–623. [Google Scholar] [CrossRef] [PubMed]
- Soto, C. Transmissible proteins: Expanding the prion heresy. Cell 2012, 149, 968–977. [Google Scholar] [CrossRef] [PubMed]
- Espargaró, A.; Busquets, M.A.; Estelrich, J.; Sabate, R. Key points concerning amyloid infectivity and prion-like neuronal invasion. Front. Mol. Neurosci. 2016, 9, 29. [Google Scholar] [CrossRef] [PubMed]
- Victoria, G.S.; Zurzolo, C. The spread of prion-like proteins by lysosomes and tunneling nanotubes: Implications for neurodegenerative diseases. J. Cell Biol. 2017, 216, 2633–2644. [Google Scholar] [CrossRef] [PubMed]
- Wickner, R.B.; Kelly, A.C. Prions are affected by evolution at two levels. Cell. Mol. Life Sci. 2016, 73, 1131–1144. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Looking at the recent advances in understanding α-synuclein and its aggregation through the proteoform prism. F1000Research 2017, 6, 525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales, R.; Estrada, L.D.; Diaz-Espinoza, R.; Morales-Scheihing, D.; Jara, M.C.; Castilla, J.; Soto, C. Molecular cross talk between misfolded proteins in animal models of Alzheimer’s and prion diseases. J. Neurosci. 2010, 30, 4528–4535. [Google Scholar] [CrossRef] [PubMed]
- Duka, T.; Rusnak, M.; Drolet, R.E.; Duka, V.; Wersinger, C.; Goudreau, J.L.; Sidhu, A. Alpha-synuclein induces hyperphosphorylation of Au in the MPTP model of Parkinsonism. FASEB J. 2006, 20, 2302–2312. [Google Scholar] [CrossRef] [PubMed]
- Tsigelny, I.F.; Crews, L.; Desplats, P.; Shaked, G.M.; Sharikov, Y.; Mizuno, H.; Spencer, B.; Rockenstein, E.; Trejo, M.; Platoshyn, O.; et al. Mechanisms of hybrid oligomer formation in the pathogenesis of combined Alzheimer’s and Parkinson’s diseases. PLoS ONE 2008, 3, e3135. [Google Scholar] [CrossRef] [PubMed]
- Calero, M.; Rostagno, A.; Ghiso, J. Amyloid Proteins. In Methods in Molecular Biology; Sigurdsson, E.M., Calero, M., Gasset, M., Eds.; Humana Press: Totowa, NJ, USA, 2012; Volume 849, ISBN 978-1-61779-550-3. [Google Scholar]
- Jensen, P.H.; Hager, H.; Nielsen, M.S.; Højrup, P.; Gliemann, J.; Jakes, R. α-Synuclein binds to Tau and stimulates the protein kinase A-catalyzed Tau phosphorylation of serine residues 262 and 356. J. Biol. Chem. 1999, 274, 25481–25489. [Google Scholar] [CrossRef] [PubMed]
- Cox, B.S. Ψ, A cytoplasmic suppressor of super-suppressor in yeast. Heredity 1965, 20, 505–521. [Google Scholar] [CrossRef] [Green Version]
- Wickner, R. [URE3] as an altered URE2 protein: Evidence for a prion analog in Saccharomyces cerevisiae. Science 1994, 264, 566–569. [Google Scholar] [CrossRef] [PubMed]
- Salnikova, A.B.; Kryndushkin, D.S.; Smirnov, V.N.; Kushnirov, V.V.; Ter-Avanesyan, M.D. Nonsense suppression in yeast cells overproducing Sup35 (eRF3) Is caused by its non-heritable amyloids. J. Biol. Chem. 2005, 280, 8808–8812. [Google Scholar] [CrossRef] [PubMed]
- Keefer, K.M.; Stein, K.C.; True, H.L. Heterologous prion-forming proteins interact to cross-seed aggregation in Saccharomyces cerevisiae. Sci. Rep. 2017, 7, 5853. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.Q.; Xiao, X.; Yuan, J.; Puoti, G.; Fujioka, H.; Wang, X.; Richardson, S.; Zhou, X.; Zou, R.; Li, S.; et al. Amyloid-β42 interacts mainly with insoluble prion protein in the Alzheimer brain. J. Biol. Chem. 2011, 286, 15095–15105. [Google Scholar] [CrossRef] [PubMed]
- Shaw, B.F.; Lelie, H.L.; Durazo, A.; Nersissian, A.M.; Xu, G.; Chan, P.K.; Gralla, E.B.; Tiwari, A.; Hayward, L.J.; Borchelt, D.R.; et al. Detergent-insoluble aggregates associated with amyotrophic lateral sclerosis in transgenic mice contain primarily full-length, unmodified superoxide dismutase-1. J. Biol. Chem. 2008, 283, 8340–8350. [Google Scholar] [CrossRef] [PubMed]
- Bagriantsev, S.N.; Gracheva, E.O.; Richmond, J.E.; Liebman, S.W. Variant-specific [PSI+] infection Is transmitted by Sup35 polymers within [PSI+] aggregates with heterogeneous protein composition. Mol. Biol. Cell 2008, 19, 2433–2443. [Google Scholar] [CrossRef] [PubMed]
- Nevzglyadova, O.V.; Artemov, A.V.; Mittenberg, A.G.; Solovyov, K.V.; Kostyleva, E.I.; Mikhailova, E.V.; Kuznetsova, I.M.; Turoverov, K.K.; Soidla, T.R. Prion-associated proteins in yeast: Comparative analysis of isogenic [PSI+] and [psi−] strains. Yeast 2009, 26, 611–631. [Google Scholar] [CrossRef] [PubMed]
- Derkatch, I.L.; Uptain, S.M.; Outeiro, T.F.; Krishnan, R.; Lindquist, S.L.; Liebman, S.W. Effects of Q/N-rich, polyQ, and non-polyQ amyloids on the de novo formation of the [PSI+] prion in yeast and aggregation of Sup35 in vitro. Proc. Natl. Acad. Sci. USA 2004, 101, 12934–12939. [Google Scholar] [CrossRef] [PubMed]
- Arslan, F.; Hong, J.Y.; Kanneganti, V.; Park, S.-K.; Liebman, S.W. Heterologous aggregates promote de novo prion appearance via more than one mechanism. PLoS Genet. 2015, 11, e1004814. [Google Scholar] [CrossRef] [PubMed]
- Saibil, H.R.; Seybert, A.; Habermann, A.; Winkler, J.; Eltsov, M.; Perkovic, M.; Castano-Diez, D.; Scheffer, M.P.; Haselmann, U.; Chlanda, P.; et al. Heritable yeast prions have a highly organized three-dimensional architecture with interfiber structures. Proc. Natl. Acad. Sci. USA 2012, 109, 14906–14911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazono, M.; Kitamoto, T.; Iwaki, T.; Tateishi, J. Colocalization of prion protein and β protein in the same amyloid plaques in patients with Gerstmann-Sträussler syndrome. Acta Neuropathol. 1992, 83, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Giasson, B.I.; Forman, M.S.; Higuchi, M.; Golbe, L.I.; Graves, C.L.; Kotzbauer, P.T.; Trojanowski, J.Q.; Lee, V.M.-Y. Initiation and synergistic fibrillization of Tau and alpha-synuclein. Science 2003, 300, 636–640. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, Y.; Kaneko, K.; Matsumoto, G.; Kurosawa, M.; Nukina, N. Cross-seeding fibrillation of Q/N-rich proteins offers new pathomechanism of polyglutamine diseases. J. Neurosci. 2009, 29, 5153–5162. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Fu, X.; Ge, F.; Zhang, B.; Yao, J.; Zhang, H.; Qian, J.; Tomozawa, H.; Naiki, H.; Sawashita, J.; et al. Cross-seeding and cross-competition in mouse apolipoprotein A-II amyloid fibrils and protein A amyloid fibrils. Am. J. Pathol. 2007, 171, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, B.; Stancu, I.C.; Buist, A.; Bird, M.; Wang, P.; Vanoosthuyse, A.; Van Kolen, K.; Verheyen, A.; Kienlen-Campard, P.; Octave, J.N.; et al. Heterotypic seeding of Tau fibrillization by pre-aggregated Abeta provides potent seeds for prion-like seeding and propagation of Tau-pathology in vivo. Acta Neuropathol. 2016, 131, 549–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutz, M.I.; Schwaiger, C.; Hochreiter, B.; Kovacs, G.G.; Schmid, J.A. Novel approach for accurate tissue-based protein colocalization and proximity microscopy. Sci. Rep. 2017, 7, 2668. [Google Scholar] [CrossRef] [PubMed]
- Esposito, A.; Dohm, C.P.; Kermer, P.; Bähr, M.; Wouters, F.S. α-Synuclein and its disease-related mutants interact differentially with the microtubule protein tau and associate with the actin cytoskeleton. Neurobiol. Dis. 2007, 26, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Badiola, N.; de Oliveira, R.M.; Herrera, F.; Guardia-Laguarta, C.; Gonçalves, S.A.; Pera, M.; Suárez-Calvet, M.; Clarimon, J.; Outeiro, T.F.; Lleó, A. Tau enhances α-synuclein aggregation and toxicity in cellular models of synucleinopathy. PLoS ONE 2011, 6, e26609. [Google Scholar] [CrossRef] [PubMed]
- Rubel, A.A.; Ryzhova, T.A.; Antonets, K.S.; Chernoff, Y.O.; Galkin, A. Identification of PrP sequences essential for the interaction between the PrP polymers and Aβ peptide in a yeast-based assay. Prion 2013, 7, 1–8. [Google Scholar] [CrossRef]
- Pack, C.G.; Inoue, Y.; Higurashi, T.; Kawai-Noma, S.; Hayashi, D.; Craig, E.; Taguchi, H. Heterogeneous interaction network of yeast prions and remodeling factors detected in live cells. BMB Rep. 2017, 50, 478. [Google Scholar] [CrossRef] [PubMed]
- Nübling, G.; Bader, B.; Levin, J.; Hildebrandt, J.; Kretzschmar, H.; Giese, A. Synergistic influence of phosphorylation and metal ions on tau oligomer formation and coaggregation with α-synuclein at the single molecule level. Mol. Neurodegener. 2012, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kryndushkin, D.S.; Alexandrov, I.M.; Ter-Avanesyan, M.D.; Kushnirov, V.V. Yeast [PSI+] prion aggregates are formed by small Sup35 polymers fragmented by Hsp104. J. Biol. Chem. 2003, 278, 49636–49643. [Google Scholar] [CrossRef] [PubMed]
- Halfmann, R.; Lindquist, S. Screening for Amyloid Aggregation by Semi-Denaturing Detergent-Agarose Gel Electrophoresis. NIH Public Access 2009, 17, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Urakov, V.N.; Mitkevich, O.V.; Safenkova, I.V.; Ter-Avanesyan, M.D. Ribosome-bound Pub1 modulates stop codon decoding during translation termination in yeast. FEBS J. 2017, 284, 1914–1930. [Google Scholar] [CrossRef] [PubMed]
- Matveenko, A.G.; Drozdova, P.B.; Belousov, M.V.; Moskalenko, S.E.; Bondarev, S.A.; Barbitoff, Y.A.; Nizhnikov, A.A.; Zhouravleva, G.A. SFP1-mediated prion-dependent lethality is caused by increased Sup35 aggregation and alleviated by Sis1. Genes Cells 2016, 21, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Morales, R.; Moreno-Gonzalez, I.; Soto, C. Cross-seeding of misfolded proteins: Implications for etiology and pathogenesis of protein misfolding diseases. PLoS Pathog. 2013, 9, e1003537. [Google Scholar] [CrossRef] [PubMed]
- Villar-Piqué, A.; Schmitz, M.; Candelise, N.; Ventura, S.; Llorens, F.; Zerr, I. Molecular and Clinical Aspects of Protein Aggregation Assays in Neurodegenerative Diseases. Mol. Neurobiol. 2018, 55, 7588–7605. [Google Scholar] [CrossRef] [PubMed]
- Krebs, M.R.H.; Morozova-Roche, L.A.; Daniel, K.; Robinson, C.V.; Dobson, C.M. Observation of sequence specificity in the seeding of protein amyloid fibrils. Protein Sci. 2004, 13, 1933–1938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, R.; Luo, Y.; Wei, G.; Nussinov, R.; Ma, B. Aβ “stretching-and-packing” cross-seeding mechanism can trigger Tau protein aggregation. J. Phys. Chem. Lett. 2015, 6, 3276–3282. [Google Scholar] [CrossRef]
- LeVine, H. Quantification of β-sheet amyloid fibril structures with thioflavin T. Methods Enzymol. 1999, 309, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Horvath, I.; Rocha, S.; Wittung-Stafshede, P. In vitro Analysis of α-Synuclein Amyloid Formation and Cross-Reactivity. In Amyloid Proteins; Humana Press: New York, NY, USA, 2018; Volume 1779, pp. 73–83. ISBN 9781493978168. [Google Scholar]
- Krebs, M.R.H.; Bromley, E.H.C.; Donald, A.M. The binding of thioflavin-T to amyloid fibrils: Localisation and implications. J. Struct. Biol. 2005, 149, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsova, I.M.; Sulatskaya, A.I.; Uversky, V.N.; Turoverov, K.K. A new trend in the experimental methodology for the analysis of the thioflavin T binding to amyloid fibrils. Mol. Neurobiol. 2012, 45, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Streets, A.M.; Sourigues, Y.; Kopito, R.R.; Melki, R.; Quake, S.R. Simultaneous measurement of amyloid fibril formation by dynamic light scattering and fluorescence reveals complex aggregation kinetics. PLoS ONE 2013, 8, e54541. [Google Scholar] [CrossRef] [PubMed]
- O’Nuallain, B.; Williams, A.D.; Westermark, P.; Wetzel, R. Seeding specificity in amyloid growth induced by heterologous fibrils. J. Biol. Chem. 2004, 279, 17490–17499. [Google Scholar] [CrossRef] [PubMed]
- O’Nuallain, B.; Wetzel, R. Conformational Abs recognizing a generic amyloid fibril epitope. Proc. Natl. Acad. Sci. USA 2002, 99, 1485–1490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, K.; Takahashi, R.; Ikeda, T.; Yamada, M. Cross-seeding effects of amyloid β-protein and α-synuclein. J. Neurochem. 2012, 122, 883–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitrenko, Y.A.; Gracheva, E.O.; Richmond, J.E.; Liebman, S.W. Visualization of aggregation of the Rnq1 prion domain and cross-seeding interactions with Sup35NM. J. Biol. Chem. 2007, 282, 1779–1787. [Google Scholar] [CrossRef] [PubMed]
- Sharma, J.; Liebman, S.W. Exploring the basis of [PIN+] variant differences in [PSI+] induction. J. Mol. Biol. 2013, 425, 3046–3059. [Google Scholar] [CrossRef] [PubMed]
- Waxman, E.A.; Giasson, B.I. Induction of intracellular Tau aggregation is promoted by α-synuclein seeds and provides novel insights into the hyperphosphorylation of Tau. J. Neurosci. 2011, 31, 7604–7618. [Google Scholar] [CrossRef] [PubMed]
- Sarell, C.J.; Stockley, P.G.; Radford, S.E. Assessing the causes and consequences of co-polymerization in amyloid formation. Prion 2013, 7, 359–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younan, N.D.; Sarell, C.J.; Davies, P.; Brown, D.R.; Viles, J.H. The cellular prion protein traps Alzheimer’s Aβ in an oligomeric form and disassembles amyloid fibers. FASEB J. 2013, 27, 1847–1858. [Google Scholar] [CrossRef] [PubMed]
- Young, L.M.; Tu, L.-H.; Raleigh, D.P.; Ashcroft, A.E.; Radford, S.E. Understanding co-polymerization in amyloid formation by direct observation of mixed oligomers. Chem. Sci. 2017, 8, 5030–5040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pienaar, I.S.; Daniels, W.M.U.; Götz, J. Neuroproteomics as a promising tool in Parkinson’s disease research. J. Neural Transm. 2008, 115, 1413–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craft, G.E.; Chen, A.; Nairn, A.C. Recent advances in quantitative neuroproteomics. Methods 2013, 61, 186–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shevchenko, G.; Konzer, A.; Musunuri, S.; Bergquist, J. Neuroproteomics tools in clinical practice. Biochim. Biophys. Acta Proteins Proteom. 2015, 1854, 705–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schonberger, S.J.; Edgar, P.F.; Kydd, R.; Faull, R.L.M.; Cooper, G.J.S. Proteomic analysis of the brain in Alzheimer’s disease: Molecular phenotype of a complex disease process. Proteomics 2001, 1, 1519–1528. [Google Scholar] [CrossRef]
- Tsuji, T.; Shiozaki, A.; Kohno, R.; Yoshizato, K.; Shimohama, S. Proteomic profiling and neurodegeneration in Alzheimer’s disease. Neurochem. Res. 2002, 27, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.; Cheng, D.; Wang, J.; Duong, D.M.; Losik, T.G.; Gearing, M.; Rees, H.D.; Lah, J.J.; Levey, A.I.; Peng, J. Proteomic characterization of postmortem amyloid plaques isolated by laser capture microdissection. J. Biol. Chem. 2004, 279, 37061–37068. [Google Scholar] [CrossRef] [PubMed]
- Minjarez, B.; Rustarazo, M.L.V.; Sanchez Del Pino, M.M.; González-Robles, A.; Sosa-Melgarejo, J.A.; Luna-Muñoz, J.; Mena, R.; Luna-Arias, J.P. Identification of polypeptides in neurofibrillary tangles and total homogenates of brains with Alzheimer’s disease by tandem mass spectrometry. J. Alzheimers Dis. 2013, 34, 239–262. [Google Scholar] [CrossRef] [PubMed]
- Vrana, J.A.; Gamez, J.D.; Madden, B.J.; Theis, J.D.; Bergen, H.R.; Dogan, A. Classification of amyloidosis by laser microdissection and mass spectrometry-based proteomic analysis in clinical biopsy specimens. Blood 2009, 114, 4957–4959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sethi, S.; Vrana, J.A.; Theis, J.D.; Leung, N.; Sethi, A.; Nasr, S.H.; Fervenza, F.C.; Cornell, L.D.; Fidler, M.E.; Dogan, A. Laser microdissection and mass spectrometry-based proteomics aids the diagnosis and typing of renal amyloidosis. Kidney Int. 2012, 82, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Gu, G.; Goodlett, D.R.; Zhang, T.; Pan, C.; Montine, T.J.; Montine, K.S.; Aebersold, R.H.; Zhang, J. Analysis of α-synuclein-associated proteins by quantitative proteomics. J. Biol. Chem. 2004, 279, 39155–39164. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Li, G.J.; Davis, J.; Zhu, D.; Wang, Y.; Pan, C.; Zhang, J. Identification of novel proteins associated with both α-synuclein and DJ-1. Mol. Cell. Proteom. 2007, 6, 845–859. [Google Scholar] [CrossRef] [PubMed]
- McFarland, M.A.; Ellis, C.E.; Markey, S.P.; Nussbaum, R.L. Proteomics analysis identifies phosphorylation-dependent α-synuclein protein interactions. Mol. Cell. Proteom. 2008, 7, 2123–2137. [Google Scholar] [CrossRef] [PubMed]
- Ayyadevara, S.; Balasubramaniam, M.; Parcon, P.A.; Barger, S.W.; Griffin, W.S.T.; Alla, R.; Tackett, A.J.; Mackintosh, S.G.; Petricoin, E.; Zhou, W.; et al. Proteins that mediate protein aggregation and cytotoxicity distinguish Alzheimer’s hippocampus from normal controls. Aging Cell 2016, 15, 924–939. [Google Scholar] [CrossRef] [PubMed]
- Olzscha, H.; Schermann, S.M.; Woerner, A.C.; Pinkert, S.; Hecht, M.H.; Tartaglia, G.G.; Vendruscolo, M.; Hayer-Hartl, M.; Hartl, F.U.; Vabulas, R.M. Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell 2011, 144, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Ratovitski, T.; Chighladze, E.; Arbez, N.; Boronina, T.; Herbrich, S.; Cole, R.N.; Ross, C.A. Huntingtin protein interactions altered by polyglutamine expansion as determined by quantitative proteomic analysis. Cell Cycle 2012, 11, 2006–2021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betzer, C.; Movius, A.J.; Shi, M.; Gai, W.-P.; Zhang, J.; Jensen, P.H. Identification of synaptosomal proteins binding to monomeric and oligomeric α-synuclein. PLoS ONE 2015, 10, e0116473. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Stevens, S.M.; Moore, B.D.; McClung, S.; Borchelt, D.R. Cytosolic proteins lose solubility as amyloid deposits in a transgenic mouse model of alzheimer-type amyloidosis. Hum. Mol. Genet. 2013, 22, 2765–2774. [Google Scholar] [CrossRef] [PubMed]
- Hosp, F.; Gutiérrez-Ángel, S.; Schaefer, M.H.; Cox, J.; Meissner, F.; Hipp, M.S.; Hartl, F.-U.; Klein, R.; Dudanova, I.; Mann, M. Spatiotemporal proteomic profiling of Huntington’s disease inclusions reveals widespread loss of protein function. Cell Rep. 2017, 21, 2291–2303. [Google Scholar] [CrossRef] [PubMed]
- Ayyadevara, S.; Balasubramaniam, M.; Gao, Y.; Yu, L.R.; Alla, R.; Shmookler Reis, R. Proteins in aggregates functionally impact multiple neurodegenerative disease models by forming proteasome-blocking complexes. Aging Cell 2015, 14, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Kryndushkin, D.; Pripuzova, N.; Burnett, B.G.; Shewmaker, F. Non-targeted identification of prions and amyloid-forming proteins from yeast and mammalian cells. J. Biol. Chem. 2013, 288, 27100–27111. [Google Scholar] [CrossRef] [PubMed]
- Kryndushkin, D.; Wear, M.P.; Shewmaker, F. Amyloid cannot resist identification. Prion 2013, 7, 464–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wear, M.P.; Kryndushkin, D.; O’Meally, R.; Sonnenberg, J.L.; Cole, R.N.; Shewmaker, F.P.; Nagai, Y. Proteins with intrinsically disordered domains are preferentially recruited to polyglutamine aggregates. PLoS ONE 2015, 10, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Nizhnikov, A.A.; Alexandrov, A.I.; Ryzhova, T.A.; Mitkevich, O.V.; Dergalev, A.A.; Ter-Avanesyan, M.D.; Galkin, A.P. Proteomic screening for amyloid proteins. PLoS ONE 2014, 9, e116003. [Google Scholar] [CrossRef] [PubMed]
- Antonets, K.S.; Volkov, K.V.; Maltseva, A.L.; Arshakian, L.M.; Galkin, A.P.; Nizhnikov, A.A. Proteomic analysis of Escherichia coli protein fractions resistant to solubilization by ionic detergents. Biochemistry 2016, 81, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Nizhnikov, A.A.; Ryzhova, T.A.; Volkov, K.V.; Zadorsky, S.P.; Sopova, J.V.; Inge-Vechtomov, S.G.; Galkin, A.P. Interaction of prions causes heritable traits in Saccharomyces cerevisiae. PLOS Genet. 2016, 12, e1006504. [Google Scholar] [CrossRef] [PubMed]
- Saifitdinova, A.F.; Nizhnikov, A.A.; Lada, A.G.; Rubel, A.A.; Magomedova, Z.M.; Ignatova, V.V.; Inge-Vechtomov, S.G.; Galkin, A.P. [NSI+]: A novel non-Mendelian nonsense suppressor determinant in Saccharomyces cerevisiae. Curr. Genet. 2010, 56, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Nizhnikov, A.A.; Magomedova, Z.M.; Rubel, A.A.; Kondrashkina, A.M.; Inge-Vechtomov, S.G.; Galkin, A.P. [NSI+] determinant has a pleiotropic phenotypic manifestation that is modulated by SUP35, SUP45, and VTS1 genes. Curr. Genet. 2012, 58, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Reiter, L.; Claassen, M.; Schrimpf, S.P.; Jovanovic, M.; Schmidt, A.; Buhmann, J.M.; Hengartner, M.O.; Aebersold, R. Protein identification false discovery rates for very large proteomics data sets generated by tandem mass spectrometry. Mol. Cell. Proteom. 2009, 8, 2405–2417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teschendorf, D.; Link, C.D. What have worm models told us about the mechanisms of neuronal dysfunction in human neurodegenerative diseases? Mol. Neurodegener. 2009, 4, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, A.G.; Marfil, V.; Li, C. Use of C. elegans as a model to study Alzheimer’s disease and other neurodegenerative diseases. Front. Genet. 2014, 5, 279. [Google Scholar] [CrossRef] [PubMed]
- Newman, M.; Ebrahimie, E.; Lardelli, M. Using the zebrafish model for Alzheimer’s disease research. Front. Genet. 2014, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Moussaud, S.; Jones, D.R.; Moussaud-Lamodière, E.L.; Delenclos, M.; Ross, O.A.; McLean, P.J. Alpha-synuclein and tau: Teammates in neurodegeneration? Mol. Neurodegener. 2014, 9, 43. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Funez, P.; de Mena, L.; Rincon-Limas, D.E. Modeling the complex pathology of Alzheimer’s disease in Drosophila. Exp. Neurol. 2015, 274, 58–71. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.; Götz, J. What we can learn from animal models about cerebral multi-morbidity. Alzheimers Res. Ther. 2015, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sasaguri, H.; Nilsson, P.; Hashimoto, S.; Nagata, K.; Saito, T.; De Strooper, B.; Hardy, J.; Vassar, R.; Winblad, B.; Saido, T.C. APP mouse models for Alzheimer’s disease preclinical studies. EMBO J. 2017, 36, 2473–2487. [Google Scholar] [CrossRef] [PubMed]
- Drummond, E.; Wisniewski, T. Alzheimer’s disease: Experimental models and reality. Acta Neuropathol. 2017, 133, 155–175. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Rockenstein, E.; Veinbergs, I.; Sagara, Y.; Mallory, M.; Hashimoto, M.; Mucke, L. β-amyloid peptides enhance α-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer’s disease and Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2001, 98, 12245–12250. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.L.; Covell, D.J.; Daniels, J.P.; Iba, M.; Stieber, A.; Zhang, B.; Riddle, D.M.; Kwong, L.K.; Xu, Y.; Trojanowski, J.Q.; et al. Distinct α-synuclein strains differentially promote Tau inclusions in neurons. Cell 2013, 154, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Mougenot, A.L.J.; Bencsik, A.; Nicot, S.; Vulin, J.; Morignat, E.; Verchère, J.; Bétemps, D.; Lakhdar, L.; Legastelois, S.; Baron, T.G. Transmission of prion strains in a transgenic mouse model overexpressing human A53T mutated α-synuclein. J. Neuropathol. Exp. Neurol. 2011, 70, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Rockenstein, E.; Inglis, C.; Adame, A.; Bett, C.; Lucero, M.; Sigurdson, C.J. Prion infection promotes extensive accumulation of α-synuclein in aged human α-synuclein transgenic mice. Prion 2012, 6, 184–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, J.A.; Metzler, M.; Georgiou, J.; Mage, M.; Roder, J.C.; Rose, A.M.; Hayden, M.R.; Neri, C. Huntingtin-interacting protein 1 influences worm and mouse presynaptic function and protects Caenorhabditis elegans neurons against mutant polyglutamine toxicity. J. Neurosci. 2007, 27, 11056–11064. [Google Scholar] [CrossRef] [PubMed]
- Roy, B.; Jackson, G.R. Interactions between Tau and α-synuclein augment neurotoxicity in a Drosophila model of Parkinson’s disease. Hum. Mol. Genet. 2014, 23, 3008–3023. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Xu, C.; Nagarajan, S.; Liu, Z.; Hemphill, W.O.; Shi, R.; Uversky, V.N.; Caldwell, G.A.; Caldwell, K.A.; Witt, S.N. Alpha-synuclein inhibits Snx3-retromer-mediated retrograde recycling of iron transporters in S. cerevisiae and C. elegans models of Parkinson’s disease. Hum. Mol. Genet. 2018. [Google Scholar] [CrossRef] [PubMed]
- Menezes, R.; Tenreiro, S.; Macedo, D.; Santos, C.; Outeiro, T. From the baker to the bedside: Yeast models of Parkinson’s disease. Microb. Cell 2015, 2, 262–279. [Google Scholar] [CrossRef] [PubMed]
- Heinisch, J.J.; Brandt, R. Signaling pathways and posttranslational modifications of tau in Alzheimer’s disease: The humanization of yeast cells. Microb. Cell 2016, 3, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Ciaccioli, G.; Martins, A.; Rodrigues, C.; Vieira, H.; Calado, P. A powerful yeast model to investigate the synergistic interaction of α-synuclein and Tau in neurodegeneration. PLoS ONE 2013, 8, e55848. [Google Scholar] [CrossRef] [PubMed]
- Rostovtseva, T.K.; Gurnev, P.A.; Protchenko, O.; Hoogerheide, D.P.; Yap, T.L.; Philpott, C.C.; Lee, J.C.; Bezrukov, S.M. α-synuclein shows high affinity interaction with voltage-dependent anion channel, suggesting mechanisms of mitochondrial regulation and toxicity in Parkinson disease. J. Biol. Chem. 2015, 290, 18467–18477. [Google Scholar] [CrossRef] [PubMed]
- Chandramowlishwaran, P.; Sun, M.; Casey, K.L.; Romanyuk, A.V.; Grizel, A.V.; Sopova, J.V.; Rubel, A.A.; Nussbaum-Krammer, C.; Vorberg, I.M.; Chernoff, Y.O. Mammalian amyloidogenic proteins promote prion nucleation in yeast. J. Biol. Chem. 2018, 293, 3436–3450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, M.; Gallagher, J.E.G. Systems biology from a yeast omics perspective. FEBS Lett. 2009, 583, 3895–3899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brückner, A.; Polge, C.; Lentze, N.; Auerbach, D.; Schlattner, U. Yeast two-hybrid, a powerful tool for systems biology. Int. J. Mol. Sci. 2009, 10, 2763–2788. [Google Scholar] [CrossRef] [PubMed]
- Stynen, B.; Tournu, H.; Tavernier, J.; Van Dijck, P. Diversity in genetic in vivo methods for protein-protein interaction studies: From the yeast two-hybrid system to the mammalian split-luciferase system. Microbiol. Mol. Biol. Rev. 2012, 76, 331–382. [Google Scholar] [CrossRef] [PubMed]
- Goehler, H.; Lalowski, M.; Stelzl, U.; Waelter, S.; Stroedicke, M.; Worm, U.; Droege, A.; Lindenberg, K.S.; Knoblich, M.; Haenig, C.; et al. A protein interaction network links GIT1, an enhancer of huntingtin aggregation, to Huntington’s disease. Mol. Cell 2004, 15, 853–865. [Google Scholar] [CrossRef] [PubMed]
- Kaltenbach, L.S.; Romero, E.; Becklin, R.R.; Chettier, R.; Bell, R.; Phansalkar, A.; Strand, A.; Torcassi, C.; Savage, J.; Hurlburt, A.; et al. Huntingtin interacting proteins are genetic modifiers of neurodegeneration. PLoS Genet. 2007, 3, 689–708. [Google Scholar] [CrossRef] [PubMed]
- Fiumara, F.; Fioriti, L.; Kandel, E.R.; Hendrickson, W.A. Essential role of coiled coils for aggregation and activity of Q/N-rich prions and polyQ proteins. Cell 2010, 143, 1121–1135. [Google Scholar] [CrossRef] [PubMed]
- Petrakis, S.; Schaefer, M.H.; Wanker, E.E.; Andrade-Navarro, M.A. Aggregation of polyQ-extended proteins is promoted by interaction with their natural coiled-coil partners. BioEssays 2013, 35, 503–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Totzeck, F.; Andrade-Navarro, M.A.; Mier, P. The protein structure context of polyQ regions. PLoS ONE 2017, 12, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Harbi, D.; Harrison, P.M. Interaction networks of prion, prionogenic and prion-like proteins in budding yeast, and their role in gene regulation. PLoS ONE 2014, 9, e100615. [Google Scholar] [CrossRef] [PubMed]
- Biza, K.V.; Nastou, K.C.; Tsiolaki, P.L.; Mastrokalou, C.V.; Hamodrakas, S.J.; Iconomidou, V.A. The amyloid interactome: Exploring protein aggregation. PLoS ONE 2017, 12, e0173163. [Google Scholar] [CrossRef] [PubMed]
- Kalathur, R.K.R.; Pedro Pinto, J.; Sahoo, B.; Chaurasia, G.; Futschik, M.E. HDNetDB: A molecular interaction database for network-oriented investigations into Huntington’s disease. Sci. Rep. 2017, 7, 5216. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, J.L.; Nettleton, E.J.; Bouchard, M.; Robinson, C.V.; Dobson, C.M.; Saibil, H.R. The protofilament structure of insulin amyloid fibrils. Proc. Natl. Acad. Sci. USA 2002, 99, 9196–9201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.-P.; Arai, T.; Miklossy, J.; McGeer, P.L. Aβ and tau form soluble complexes that may promote self aggregation of both into the insoluble forms observed in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 1953–1958. [Google Scholar] [CrossRef] [PubMed]
- Oláh, J.; Vincze, O.; Virók, D.; Simon, D.; Bozsó, Z.; Tokési, N.; Horváth, I.; Hlavanda, E.; Kovács, J.; Magyar, A.; et al. Interactions of pathological hallmark proteins: Tubulin polymerization promoting protein/p25,β-amyloid, and α-synuclein. J. Biol. Chem. 2011, 286, 34088–34100. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Smith, D.; Leong, B.J.; Brannstrom, K.; Almqvist, F.; Chapman, M.R. Promiscuous cross-seeding between bacterial amyloids promotes interspecies biofilms. J. Biol. Chem. 2012, 287, 35092–35103. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.J.; Hubauer-Brenner, M.; Gruber, H.J.; Cui, Y.; Traxler, L.; Siligan, C.; Park, S.; Hinterdorfer, P. Curli mediate bacterial adhesion to fibronectin via tensile multiple bonds. Sci. Rep. 2016, 6, 33909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal phosphorylation of the microtubule-associated protein τ (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef] [PubMed]
- Sepulcre, J.; Schultz, A.P.; Sabuncu, M.; Gomez-Isla, T.; Chhatwal, J.; Becker, A.; Sperling, R.; Johnson, K.A. In vivo Tau, amyloid, and gray matter profiles in the aging brain. J. Neurosci. 2016, 36, 7364–7374. [Google Scholar] [CrossRef] [PubMed]
- Rank, K.B.; Pauley, A.M.; Bhattacharya, K.; Wang, Z.; Evans, D.B.; Fleck, T.J.; Johnston, J.A.; Sharma, S.K. Direct interaction of soluble human recombinant tau protein with Aβ 1–42 results in tau aggregation and hyperphosphorylation by tau protein kinase II. FEBS Lett. 2002, 514, 263–268. [Google Scholar] [CrossRef]
- Kotzbauer, P.T.; Giasson, B.I.; Kravitz, A.V.; Golbe, L.I.; Mark, M.H.; Trojanowski, J.Q.; Lee, V.M.Y. Fibrillization of α-synuclein and tau in familial Parkinson’s disease caused by the A53T α-synuclein mutation. Exp. Neurol. 2004, 187, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Irwin, D.J.; Grossman, M.; Weintraub, D.; Hurtig, H.I.; Duda, J.E.; Xie, S.X.; Lee, E.B.; Van Deerlin, V.M.; Lopez, O.L.; Kofler, J.K.; et al. Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: A retrospective analysis. Lancet Neurol. 2017, 16, 55–65. [Google Scholar] [CrossRef]
- Kessels, H.W.; Nguyen, L.N.; Nabavi, S.; Malinow, R. The prion protein as a receptor for amyloid-β. Nature 2010, 466, E3–E4. [Google Scholar] [CrossRef] [PubMed]
- Laurén, J.; Gimbel, D.A.; Nygaard, H.B.; Gilbert, J.W.; Strittmatter, S.M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 2009, 457, 1128–1132. [Google Scholar] [CrossRef] [PubMed]
- Um, J.W.; Kaufman, A.C.; Kostylev, M.; Heiss, J.K.; Stagi, M.; Takahashi, H.; Kerrisk, M.E.; Vortmeyer, A.; Wisniewski, T.; Koleske, A.J.; et al. Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer Aβ oligomer bound to cellular prion protein. Neuron 2013, 79, 887–902. [Google Scholar] [CrossRef] [PubMed]
- Larson, M.; Sherman, M.A.; Amar, F.; Nuvolone, M.; Schneider, J.A.; Bennett, D.A.; Aguzzi, A.; Lesne, S.E. The complex PrPc-Fyn couples human oligomeric Aβ with pathological Tau changes in Alzheimer’s disease. J. Neurosci. 2012, 32, 16857–16871. [Google Scholar] [CrossRef] [PubMed]
- Debatin, L.; Streffer, J.; Geissen, M.; Matschke, J.; Aguzzi, A.; Glatzel, M. Association between deposition of beta-amyloid and pathological prion protein in sporadic Creutzfeldt-Jakob disease. Neurodegener. Dis. 2008, 5, 347–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghoshal, N.; Cali, I.; Perrin, R.J.; Josephson, S.A.; Sun, N.; Gambetti, P.; Morris, J.C. Codistribution of amyloid β plaques and spongiform degeneration in familial Creutzfeldt-Jakob disease with the E200K-129M haplotype. Arch. Neurol. 2009, 66, 1240–1246. [Google Scholar] [CrossRef] [PubMed]
- Scherzinger, E.; Lurz, R.; Turmaine, M.; Mangiarini, L.; Hollenbach, B.; Hasenbank, R.; Bates, G.; Davies, S.; Lehrach, H.; Wanker, E. Huntingtin encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell 1997, 90, 549–558. [Google Scholar] [CrossRef]
- Huang, C.C.; Faber, P.W.; Persichetti, F.; Mittal, V.; Vonsattel, J.P.; MacDonald, M.E.; Gusella, J.F. Amyloid formation by mutant huntingtin: Threshold, progressivity and recruitment of normal polyglutamine proteins. Somat. Cell Mol. Genet. 1998, 24, 217–233. [Google Scholar] [CrossRef] [PubMed]
- Davies, S.W.; Turmaine, M.; Cozens, B.A.; DiFiglia, M.; Sharp, A.H.; Ross, C.A.; Scherzinger, E.; Wanker, E.E.; Mangiarini, L.; Bates, G.P. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 1997, 90, 537–548. [Google Scholar] [CrossRef]
- Nucifora, F.C., Jr. Interference by Huntingtin and Atrophin-1 with CBP-mediated transcription leading to cellular toxicity. Science 2001, 291, 2423–2428. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-H.; Cheng, A.L.; Zhou, H.; Lam, S.; Rao, M.; Li, H.; Li, X.-J. Interaction of Huntington disease protein with transcriptional activator Sp1. Mol. Cell. Biol. 2002, 22, 1277–1287. [Google Scholar] [CrossRef] [PubMed]
- Steffan, J.S.; Kazantsev, A.; Spasic-Boskovic, O.; Greenwald, M.; Zhu, Y.Z.; Gohler, H.; Wanker, E.E.; Bates, G.P.; Housman, D.E.; Thompson, L.M. The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc. Natl. Acad. Sci. USA 2000, 97, 6763–6768. [Google Scholar] [CrossRef] [PubMed]
- Boutell, J.M.; Thomas, P.; Neal, J.W.; Weston, V.J.; Duce, J.; Harper, P.S.; Jones, A.L. Aberrant interactions of transcriptional repressor proteins with the Huntington’s disease gene product, huntingtin. Hum. Mol. Genet. 1999, 8, 1647–1655. [Google Scholar] [CrossRef] [PubMed]
- Shimohata, T.; Nakajima, T.; Yamada, M.; Uchida, C.; Onodera, O.; Naruse, S.; Kimura, T.; Koide, R.; Nozaki, K.; Sano, Y.; et al. Expanded polyglutamine stretches interact with TAF(II)130, interfering with CREB-dependent transcription. Nat. Genet. 2000, 26, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Doi, H.; Koyano, S.; Suzuki, Y.; Nukina, N.; Kuroiwa, Y. The RNA-binding protein FUS/TLS is a common aggregate-interacting protein in polyglutamine diseases. Neurosci. Res. 2010, 66, 131–133. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.-X.; Li, S.-H.; Nguyen, H.-P.; Li, X.-J. Huntingtin inclusions do not deplete polyglutamine-containing transcription factors in HD mice. Hum. Mol. Genet. 2002, 11, 905–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benn, C.L.; Sun, T.; Sadri-Vakili, G.; McFarland, K.N.; DiRocco, D.P.; Yohrling, G.J.; Clark, T.W.; Bouzou, B.; Cha, J.-H.J. Huntingtin modulates transcription, occupies gene promoters in vivo, and binds directly to DNA in a polyglutamine-dependent manner. J. Neurosci. 2008, 28, 10720–10733. [Google Scholar] [CrossRef] [PubMed]
- Duennwald, M.L.; Jagadish, S.; Giorgini, F.; Muchowski, P.J.; Lindquist, S. A network of protein interactions determines polyglutamine toxicity. Proc. Natl. Acad. Sci. USA 2006, 103, 11051–11056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urakov, V.N.; Vishnevskaya, A.B.; Alexandrov, I.M.; Kushnirov, V.V.; Smirnov, V.N.; Ter-Avanesyan, M.D. Interdependence of amyloid formation in yeast: Implications for polyglutamine disorders and biological functions. Prion 2010, 4, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Serpionov, G.V.; Alexandrov, A.I.; Ter-Avanesyan, M.D. Distinct mechanisms of mutant huntingtin toxicity in different yeast strains. FEMS Yeast Res. 2017, 17, fow102. [Google Scholar] [CrossRef] [PubMed]
- Nizhnikov, A.A.; Antonets, K.S.; Inge-Vechtomov, S.G.; Derkatch, I.L. Modulation of efficiency of translation termination in Saccharomyces cerevisiae. Prion 2014, 8, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Chakrabortee, S.; Byers, J.S.; Jones, S.; Garcia, D.M.; Bhullar, B.; Chang, A.; She, R.; Lee, L.; Fremin, B.; Lindquist, S.; et al. Intrinsically disordered proteins drive emergence and inheritance of biological traits. Cell 2016, 167, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Culver, B.P.; Savas, J.N.; Park, S.K.; Choi, J.H.; Zheng, S.; Zeitlin, S.O.; Yates, J.R.; Tanese, N. Proteomic analysis of wild-type and mutant huntingtin-associated proteins in mouse brains identifies unique interactions and involvement in protein synthesis. J. Biol. Chem. 2012, 287, 21599–21614. [Google Scholar] [CrossRef] [PubMed]
- Shirasaki, D.I.; Greiner, E.R.; Al-Ramahi, I.; Gray, M.; Boontheung, P.; Geschwind, D.H.; Botas, J.; Coppola, G.; Horvath, S.; Loo, J.A.; et al. Network organization of the huntingtin proteomic interactome in mammalian brain. Neuron 2012, 75, 41–57. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Cantle, J.P.; Chatzopoulou, D.; Wang, N.; Gao, F.; Al-Ramahi, I.; Lu, X.H.; Ramos, E.M.; El-Zein, K.; Zhao, Y.; et al. Integrated genomics and proteomics define huntingtin CAG length-dependent networks in mice. Nat. Neurosci. 2016, 19, 623–633. [Google Scholar] [CrossRef]
- Söderberg, L.; Bogdanovic, N.; Axelsson, B.; Winblad, B.; Näslund, J.; Tjernberg, L.O. Analysis of single Alzheimer solid plaque cores by laser capture microscopy and nanoelectrospray/tandem mass spectrometry. Biochemistry 2006, 45, 9849–9856. [Google Scholar] [CrossRef] [PubMed]
- Gozal, Y.M.; Duong, D.M.; Gearing, M.; Cheng, D.; Hanfelt, J.J.; Funderburk, C.; Peng, J.; Lah, J.J.; Levey, A.I. Proteomics analysis reveals novel components in the detergent-insoluble subproteome in Alzheimer’s disease. J. Proteome Res. 2009, 8, 5069–5079. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein e and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Rostagno, A.; Lashley, T.; Ng, D.; Meyerson, J.; Braendgaard, H.; Plant, G.; Bojsen-Møller, M.; Holton, J.; Frangione, B.; Revesz, T.; et al. Preferential association of serum amyloid P component with fibrillar deposits in familial British and Danish dementias: Similarities with Alzheimer’s disease. J. Neurol. Sci. 2007, 257, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Yasojima, K.; Schwab, C.; McGeer, E.G.; McGeer, P.L. Up-regulated production and activation of the complement system in Alzheimer’s disease brain. Am. J. Pathol. 1999, 154, 927–936. [Google Scholar] [CrossRef]
- Sondheimer, N.; Lindquist, S. Rnq1: An epigenetic modifier of protein function in yeast. Mol. Cell 2000, 5, 163–172. [Google Scholar] [CrossRef]
- Derkatch, I.L.; Bradley, M.E.; Hong, J.Y.; Liebman, S.W. Prions affect the appearance of other prions: The story of [PIN+]. Cell 2001, 106, 171–182. [Google Scholar] [CrossRef]
- Bradley, M.E.; Edskes, H.K.; Hong, J.Y.; Wickner, R.B.; Liebman, S.W. Interactions among prions and prion “strains” in yeast. Proc. Natl. Acad. Sci. USA 2002, 99, 16392–16399. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, G.; Shimazu, N.; Tanaka, M. A yeast prion, Mod5, promotes acquired drug resistance and cell survival under environmental stress. Science 2012, 336, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Derkatch, I.L.; Bradley, M.E.; Masse, S.V.; Zadorsky, S.P.; Polozkov, G.V.; Inge-Vechtomov, S.G.; Liebman, S.W. Dependence and independence of [PSI+] and [PIN+]: A two-prion system in yeast? EMBO J. 2000, 19, 1942–1952. [Google Scholar] [CrossRef] [PubMed]
- Bagriantsev, S.; Liebman, S.W. Specificity of prion assembly in vivo: [PSI+] and [PIN+] form separate structures in yeast. J. Biol. Chem. 2004, 279, 51042–51048. [Google Scholar] [CrossRef] [PubMed]
- Schwimmer, C.; Masison, D.C. Antagonistic interactions between yeast [PSI+] and [URE3] prions and curing of [URE3] by Hsp70 protein chaperone Ssa1p but not by Ssa2p. Mol. Cell. Biol. 2002, 22, 3590–3598. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Park, K.-W.; Yu, H.; Fan, Q.; Li, L. Newly identified prion linked to the chromatin-remodeling factor Swi1 in Saccharomyces cerevisiae. Nat. Genet. 2008, 40, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Li, L. Investigating the Interactions of Yeast Prions: [SWI+], [PSI+], and [PIN+]. Genetics 2014, 197, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Halfmann, R.; Wright, J.R.; Alberti, S.; Lindquist, S.; Rexach, M. Prion formation by a yeast GLFG nucleoporin. Prion 2012, 6, 391–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Z.; Zhang, Y.; Li, L. The yeast prion [SWI+] abolishes multicellular growth by triggering conformational changes of multiple regulators required for flocculin gene expression. Cell Rep. 2015, 13, 2865–2878. [Google Scholar] [CrossRef] [PubMed]
- Chernova, T.A.; Wilkinson, K.D.; Chernoff, Y.O. Prions, chaperones, and proteostasis in yeast. Cold Spring Harb. Perspect. Biol. 2017, 9, a023663. [Google Scholar] [CrossRef] [PubMed]
- Allen, K.D.; Wegrzyn, R.D.; Chernova, T.A.; Müller, S.; Newnam, G.P.; Winslett, P.A.; Wittich, K.B.; Wilkinson, K.D.; Chernoff, Y.O. Hsp70 chaperones as modulators of prion life cycle. Genetics 2005, 169, 1227–1242. [Google Scholar] [CrossRef] [PubMed]
- Helsen, C.W.; Glover, J.R. Insight into molecular basis of curing of [PSI+] prion by overexpression of 104-kDa heat shock protein (Hsp104). J. Biol. Chem. 2012, 287, 542–556. [Google Scholar] [CrossRef] [PubMed]
- Ohta, S.; Kawai-Noma, S.; Kitamura, A.; Pack, C.G.; Kinjo, M.; Taguchi, H. The interaction of Hsp104 with yeast prion Sup35 as analyzed by fluorescence cross-correlation spectroscopy. Biochem. Biophys. Res. Commun. 2013, 442, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Doyle, S.M.; Genest, O.; Wickner, S. Protein rescue from aggregates by powerful molecular chaperone machines. Nat. Rev. Mol. Cell Biol. 2013, 14, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Winkler, J.; Tyedmers, J.; Bukau, B.; Mogk, A. Hsp70 targets Hsp100 chaperones to substrates for protein disaggregation and prion fragmentation. J. Cell Biol. 2012, 198, 387–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiktev, D.A.; Patterson, J.C.; Muller, S.; Bariar, B.; Pan, T.; Chernoff, Y.O. Regulation of Chaperone Effects on a Yeast Prion by Cochaperone Sgt2. Mol. Cell. Biol. 2012, 32, 4960–4970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; McQuade, T.; Siemer, A.B.; Napetschnig, J.; Moriwaki, K.; Hsiao, Y.-S.; Damko, E.; Moquin, D.; Walz, T.; McDermott, A.; et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 2012, 150, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.N.; Yang, Z.H.; Wang, X.K.; Zhang, Y.; Wan, H.; Song, Y.; Chen, X.; Shao, J.; Han, J. Distinct roles of RIP1-RIP3 hetero-and RIP3-RIP3 homo-interaction in mediating necroptosis. Cell Death Differ. 2014, 21, 1709–1720. [Google Scholar] [CrossRef] [PubMed]
- Kajava, A.V.; Klopffleisch, K.; Chen, S.; Hofmann, K. Evolutionary link between metazoan RHIM motif and prion-forming domain of fungal heterokaryon incompatibility factor HET-s/HET-s. Sci. Rep. 2014, 4, 7436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turcq, B.; Deleu, C.; Denayrolles, M.; Begueret, J. Two allelic genes responsible for vegetative incompatibility in the fungus Podospora anserina are not essential for cell viability. MGG Mol. Gen. Genet. 1991, 228, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Seuring, C.; Greenwald, J.; Wasmer, C.; Wepf, R.; Saupe, S.J.; Meier, B.H.; Riek, R. The mechanism of toxicity in HET-S/HET-s prion incompatibility. PLoS Biol. 2012, 10, e1001451. [Google Scholar] [CrossRef] [PubMed]
- Maddelein, M.-L.; Dos Reis, S.; Duvezin-Caubet, S.; Coulary-Salin, B.; Saupe, S.J. Amyloid aggregates of the HET-s prion protein are infectious. Proc. Natl. Acad. Sci. USA 2002, 99, 7402–7407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritter, C.; Maddelein, M.L.; Siemer, A.B.; Lührs, T.; Ernst, M.; Meier, B.H.; Saupe, S.J.; Riek, R. Correlation of structural elements and infectivity of the HET-s prion. Nature 2005, 435, 844–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daskalov, A.; Habenstein, B.; Martinez, D.; Debets, A.J.M.; Sabaté, R.; Loquet, A.; Saupe, S.J. Signal transduction by a fungal NOD-like receptor based on propagation of a prion amyloid fold. PLOS Biol. 2015, 13, e1002059. [Google Scholar] [CrossRef] [PubMed]
- Loquet, A.; Saupe, S. Diversity of amyloid motifs in NLR signaling in fungi. Biomolecules 2017, 7, 38. [Google Scholar] [CrossRef] [PubMed]
- Mompeán, M.; Li, W.; Li, J.; Laage, S.; Siemer, A.B.; Bozkurt, G.; Wu, H.; McDermott, A.E. The structure of the necrosome RIPK1-RIPK3 core, a human hetero-amyloid signaling complex. Cell 2018, 173, 1244–1253. [Google Scholar] [CrossRef] [PubMed]
- Baxa, U.; Cassese, T.; Kajava, A.V.; Steven, A.C. Structure, function, and amyloidogenesis of fungal prions: Filament polymorphism and prion variants. Adv. Protein Chem. 2006, 73, 125–180. [Google Scholar] [CrossRef] [PubMed]
- Azizyan, R.A.; Garro, A.; Radkova, Z.; Anikeenko, A.; Bakulina, A.; Dumas, C.; Kajava, A.V. Establishment of constraints on amyloid formation imposed by steric exclusion of globular domains. J. Mol. Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Bondarev, S.A.; Bondareva, O.V.; Zhouravleva, G.A.; Kajava, A.V. BetaSerpentine: A bioinformatics tool for reconstruction of amyloid structures. Bioinformatics 2018, 34, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Kleino, A.; Ramia, N.F.; Bozkurt, G.; Shen, Y.; Nailwal, H.; Huang, J.; Napetschnig, J.; Gangloff, M.; Chan, F.K.M.; Wu, H.; et al. Peptidoglycan-sensing receptors trigger the formation of functional amyloids of the adaptor protein Imd to initiate Drosophila NF-κB signaling. Immunity 2017, 47, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, W.J.; Offermann, M.K. Apoptosis induced by the Toll-like receptor adaptor TRIF is dependent on its receptor interacting protein homotypic interaction motif. J. Immunol. 2005, 174, 4942–4952. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, W.J.; Upton, J.W.; Mocarski, E.S. Receptor-interacting protein homotypic interaction motif-dependent control of NF-κB activation via the DNA-dependent activator of IFN regulatory factors. J. Immunol. 2008, 181, 6427–6434. [Google Scholar] [CrossRef] [PubMed]
- Rebsamen, M.; Heinz, L.X.; Meylan, E.; Michallet, M.-C.; Schroder, K.; Hofmann, K.; Vazquez, J.; Benedict, C.A.; Tschopp, J. DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-κB. EMBO Rep. 2009, 10, 916–922. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Omoto, S.; Harris, P.A.; Finger, J.N.; Bertin, J.; Gough, P.J.; Kaiser, W.J.; Mocarski, E.S. Herpes simplex virus suppresses necroptosis in human cells. Cell Host Microbe 2015, 17, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Wu, S.-Q.; Liang, Y.; Zhou, X.; Chen, W.; Li, L.; Wu, J.; Zhuang, Q.; Chen, C.; Li, J.; et al. RIP1/RIP3 binding to HSV-1 ICP6 initiates necroptosis to restrict virus propagation in mice. Cell Host Microbe 2015, 17, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, Y.; Liu, S.; Yu, X.; Li, L.; Shi, C.; He, W.; Li, J.; Xu, L.; Hu, Z.; et al. Direct activation of RIP3/MLKL-dependent necrosis by herpes simplex virus 1 (HSV-1) protein ICP6 triggers host antiviral defense. Proc. Natl. Acad. Sci. USA 2014, 111, 15438–15443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daskalov, A. On the evolutionary trajectories of signal-transducing amyloids in fungi and beyond. Prion 2016, 10, 362–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, M.R. Role of Escherichia coli Curli operons in directing amyloid fiber formation. Science 2002, 295, 851–855. [Google Scholar] [CrossRef] [PubMed]
- White, A.P.; Collinson, S.K.; Banser, P.A.; Gibson, D.L.; Paetzel, M.; Strynadka, N.C.; Kay, W.W. Structure and characterization of AgfB from Salmonella enteritidis thin aggregative fimbriae. J. Mol. Biol. 2001, 311, 735–749. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Smith, D.R.; Jones, J.W.; Chapman, M.R. In vitro polymerization of a functional Escherichia coli amyloid protein. J. Biol. Chem. 2007, 282, 3713–3719. [Google Scholar] [CrossRef] [PubMed]
- Hammer, N.D.; Schmidt, J.C.; Chapman, M.R. The curli nucleator protein, CsgB, contains an amyloidogenic domain that directs CsgA polymerization. Proc. Natl. Acad. Sci. USA 2007, 104, 12494–12499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, Q.; Crick, S.L.; Pinkner, J.S.; Ford, B.; Hultgren, S.J.; Frieden, C. The E. coli CsgB nucleator of curli assembles to b-sheet oligomers that alter the CsgA fibrillization mechanism. Proc. Natl. Acad. Sci. USA 2012, 109, 6502–6507. [Google Scholar] [CrossRef] [PubMed]
- Blanco, L.P.; Evans, M.L.; Smith, D.R.; Badtke, M.P.; Chapman, M.R. Diversity, biogenesis and function of microbial amyloids. Trends Microbiol. 2012, 20, 66–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberti, S. The wisdom of crowds: Regulating cell function through condensed states of living matter. J. Cell Sci. 2017, 130, 2789–2796. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Han, T.W.; Xie, S.; Shi, K.; Du, X.; Wu, L.C.; Mirzaei, H.; Goldsmith, E.J.; Longgood, J.; Pei, J.; et al. Cell-free formation of RNA granules: Low complexity sequence domains form dynamic fibers within hydrogels. Cell 2012, 149, 753–767. [Google Scholar] [CrossRef] [PubMed]
- Kedersha, N.L.; Gupta, M.; Li, W.; Miller, I.; Anderson, P. RNA-binding proteins Tia-1 and Tiar link the phosphorylation of Eif-2α to the assembly of mammalian stress granules. J. Cell Biol. 1999, 147, 1431–1442. [Google Scholar] [CrossRef] [PubMed]
- Gilks, N.; Kedersha, N.; Ayodele, M.; Shen, L.; Stoecklin, G.; Dember, L.M.; Anderson, P. Stress granule assembly Is mediated by prion-like aggregation of TIA-1. Mol. Biol. Cell 2004, 15, 5383–5398. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Rayman, J.B.; Kandel, E.R.; Derkatch, I.L. Functional role of Tia1/Pub1 and Sup35 prion domains: Directing protein synthesis machinery to the tubulin cytoskeleton. Mol. Cell 2014, 55, 305–318. [Google Scholar] [CrossRef] [PubMed]
- Kroschwald, S.; Maharana, S.; Mateju, D.; Malinovska, L.; Nüske, E.; Poser, I.; Richter, D.; Alberti, S. Promiscuous interactions and protein disaggregases determine the material state of stress-inducible RNP granules. eLife 2015, 4, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Bourgade, K.; Dupuis, G.; Frost, E.H.; Fülöp, T. Anti-Viral Properties of Amyloid-β Peptides. J. Alzheimers Dis. 2016, 54, 859–878. [Google Scholar] [CrossRef] [PubMed]
- Torrent, M.; Pulido, D.; Nogués, M.V.; Boix, E. Exploring New Biological Functions of Amyloids: Bacteria Cell Agglutination Mediated by Host Protein Aggregation. PLoS Pathog. 2012, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paushkin, S.V.; Kushnirov, V.V.; Smirnov, V.N.; Ter-Avanesyan, M.D. Interaction between yeast Sup45p (eRF1) and Sup35p (eRF3) polypeptide chain release factors: Implications for prion-dependent regulation. Mol. Cell. Biol. 1997, 17, 2798–2805. [Google Scholar] [CrossRef] [PubMed]
- Stansfield, I.; Jones, K.M.; Ter-Avanesyan, M.D.; Tuite, M.F. The products of the SUP45 (eRF1) and SUP35 genes interact to mediate translation termination in Saccharomyces cerevisiae. EMBO J. 1995, 14, 4365–4373. [Google Scholar] [CrossRef] [PubMed]
- Zhouravleva, G.; Frolova, L.; Le Goff, X.; Le Guellec, R.; Inge-Vechtomov, S.; Kisselev, L.; Philippe, M. Termination of translation in eukaryotes is governed by two interacting polypeptide chain release factors, eRF1 and eRF3. EMBO J. 1995, 14, 4065–4072. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amyloid-Forming Protein | Interacting Proteins | Class of Co-Aggregation | Experiments | References |
|---|---|---|---|---|
| Sup35 | Ssa1, Ssa2, Hsp104, Sse1, Ssb1, Ssb2, Ydj1, Sis1 | sequestration | differential centrifugation | [35,36] |
| Sis1, Hsp104 | sequestration | colocalization, FCCS | [49] | |
| Ssa1, Ssa2, Sis1, Hsp104, Hsp110 (Sse) | sequestration | colocalization | [39] | |
| Sgt2 | sequestration | colocalization, differential centrifugation | [189] | |
| Ure2 1, New1 1 | co-aggregation | colocalization, FCCS | [49] | |
| Rnq1 1 | axial co-aggregation | affinity chromatography, colocalization, seeding, crosslinking, FRET | [31,32,37,38,49,67,68] | |
| Sla2 | titration | co-IP, differential centrifugation | [35,36] | |
| Sup45 | titration | differential centrifugation | [225] | |
| Pub11 | co-aggregation | SDD-AGE | [53] | |
| Rnq1 | Pub11 | co-aggregation | SDD-AGE | [53] |
| Swi1 | Mss1 1, Sap30 1, Msn1 1 | co-aggregation | colocalization | [182] |
| csgA | csgB 1 | axial co-aggregation | seeding, SPR, structure modelling | [214,215,216] |
| fibronectin | sequestration | single molecular force spectroscopy measurments | [136] | |
| α-Syn | Tau 1 | co-aggregation | seeding, colocalization, affinity chromatography, FRET | [25,28,41,50,69,110,118] |
| Aβ 1 | co-aggregation | seeding, co-IP | [26,66] | |
| IAPP | axial co-aggregation | seeding | [60] | |
| Aβ | PrP 1 | co-aggregation | co-IP, colocalization, seeding | [24,40,48,146,147] |
| Tau 1 | Lateral co-aggregation | seeding, colocalization, molecular dynamics simulations | [44,138,139] | |
| AApoAII | AA 1 | co-aggregation | seeding, colocalization | [43] |
| Htt, atrophin-1 | CBP | sequestration | colocalization, co-IP | [151] |
| Htt | p53 | sequestration | diffential centrifugation, colocalization | [153] |
| mSin3a | sequestration | diffential centrifugation, colocalization | [154] | |
| TAFII130 | sequestration | yeast two hybrid, co-IP | [155] | |
| TBP | sequestration | diffential centrifugation | [149] | |
| FUS | sequestration | colocalization | [156] | |
| Def1 1, Pub1 1, Rpn10 1, Ent2 1, Bmh2 1 | co-aggregation | PSIA | [94,162] | |
| TIA-1 | sequestration | colocalization | [42] | |
| Rip1/Rip3 | Rip1 1/Rip3 1 | axial co-aggregation | seeding, gel filtration, co-IP | [190] |
| MLKL | titration | co-IP | [191] | |
| HET-s | NWD2 1 | co-aggregation | seeding, colocalization | [197] |
| PGRP-LE | Imd 1 | co-aggregation | seeding | [203] |
| Titration | Sequestration | Axial Co-Aggregation | Lateral Co-Aggregation |
|---|---|---|---|
| The Interaction between Soluble Protein and Amyloid | The Interaction of Two Proteins in the Amyloid Conformation | ||
| Soluble protein interacts with amyloid via specific binding site (s) | Soluble protein interacts with amyloid non-specifically | Molecules of different proteins stack along the fibril axis and form common amyloid fibril | Different proteins form separate amyloid fibrils which interact with lateral surfaces of each other |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bondarev, S.A.; Antonets, K.S.; Kajava, A.V.; Nizhnikov, A.A.; Zhouravleva, G.A. Protein Co-Aggregation Related to Amyloids: Methods of Investigation, Diversity, and Classification. Int. J. Mol. Sci. 2018, 19, 2292. https://doi.org/10.3390/ijms19082292
Bondarev SA, Antonets KS, Kajava AV, Nizhnikov AA, Zhouravleva GA. Protein Co-Aggregation Related to Amyloids: Methods of Investigation, Diversity, and Classification. International Journal of Molecular Sciences. 2018; 19(8):2292. https://doi.org/10.3390/ijms19082292
Chicago/Turabian StyleBondarev, Stanislav A., Kirill S. Antonets, Andrey V. Kajava, Anton A. Nizhnikov, and Galina A. Zhouravleva. 2018. "Protein Co-Aggregation Related to Amyloids: Methods of Investigation, Diversity, and Classification" International Journal of Molecular Sciences 19, no. 8: 2292. https://doi.org/10.3390/ijms19082292