Cellular Receptors of Amyloid β Oligomers (AβOs) in Alzheimer’s Disease
by
 , , and
, , and
Barbara Mroczko
1,2,3,* ,
,
Magdalena Groblewska
3,
Ala Litman-Zawadzka
1,
Johannes Kornhuber
4 and
Piotr Lewczuk
1,4 1
Department of Neurodegeneration Diagnostics, Medical University of Białystok, 15-089 Białystok, Poland
2
Department of Biochemical Diagnostics, Medical University of Białystok, 15-089 Białystok, Poland
3
Department of Biochemical Diagnostics, University Hospital in Białystok, 15-276 Białystok, Poland
4
Department of Psychiatry and Psychotherapy, Universitätsklinikum Erlangen, and Friedrich-Alexander Universität Erlangen-Nürnberg, 91054 Erlangen, Germany
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2018, 19(7), 1884; https://doi.org/10.3390/ijms19071884
Submission received: 23 May 2018
/
Revised: 19 June 2018
/
Accepted: 22 June 2018
/
Published: 27 June 2018
(This article belongs to the Special Issue Molecular Mechanism of Alzheimer's Disease)
Abstract
:It is estimated that Alzheimer’s disease (AD) affects tens of millions of people, comprising not only suffering patients, but also their relatives and caregivers. AD is one of age-related neurodegenerative diseases (NDs) characterized by progressive synaptic damage and neuronal loss, which result in gradual cognitive impairment leading to dementia. The cause of AD remains still unresolved, despite being studied for more than a century. The hallmark pathological features of this disease are senile plaques within patients’ brain composed of amyloid beta (Aβ) and neurofibrillary tangles (NFTs) of Tau protein. However, the roles of Aβ and Tau in AD pathology are being questioned and other causes of AD are postulated. One of the most interesting theories proposed is the causative role of amyloid β oligomers (AβOs) aggregation in the pathogenesis of AD. Moreover, binding of AβOs to cell membranes is probably mediated by certain proteins on the neuronal cell surface acting as AβO receptors. The aim of our paper is to describe alternative hypotheses of AD etiology, including genetic alterations and the role of misfolded proteins, especially Aβ oligomers, in Alzheimer’s disease. Furthermore, in this review we present various putative cellular AβO receptors related to toxic activity of oligomers.
1. Introduction
Alzheimer’s disease (AD) affects tens of millions of people worldwide and estimated number of AD patients would increase to over 130 million by 2050 [1]. AD is a big socio-economical problem because of comprising not only suffering patients, but also their relatives and caregivers. Additionally, current therapeutic strategies provide only palliative, not disease-modifying, agents.
AD belongs to a large group of neurodegenerative diseases (NDs), which include also Parkinson’s disease (PD) and PD-related disorders, prion disease, motor neuron diseases (MND), Huntington’s disease (HD), spinocerebellar ataxia (SCA), spinal muscular atrophy (SMA), and others [2]. Characterized features of NDs are the progressive degeneration and/or death of neuron cells. In AD, this neuronal loss is accompanied by progressive synaptic damage, which results in gradual cognitive impairment and, finally, dementia.
The main histopathological hallmarks of AD are the extracellular plaques within brain tissue consisted of variant forms of amyloid β (Aβ) and neurofibrillary tangles (NFTs) of many forms of phosphorylated Tau proteins (pTau), localized intraneuronally [3]. Primarily, these pathological alterations are seen within medial temporal lobe, whereas in later stages of AD they progress subsequently to brain regions associated with neocortex [4,5]. Formation of Aβ plaques and neuronal cell damage are preceded by reduced synaptic transmission and loss of dendritic spines, which lead to synaptic dysfunction in AD brain. It is estimated that these changes may anticipate the first cognitive decline symptoms even for two decades [6]. Furthermore, declined levels of Aβ42 in cerebrospinal fluid (CSF) and the presence of Aβ plaques in neuroimaging may head other AD-related alterations by many years [7].
2. Postulated Hypotheses of AD Etiology
2.1. Risk Factors of AD
The risk factors of AD include increasing age, genetic, and vascular factors, smoking, obesity and diabetes [8]. The presence of genetic mutations as the etiological factors of AD has been identified in 1–5% cases [8]. Although most of sporadic AD cases are unrelated to any autosomal-dominant inheritance, certain genetic changes may be linked with a significant risk of AD development. The mutations in presenilin1 (PS1), presenilin2 (PS2), and amyloid precursor protein (APP) genes are associated with the familial Alzheimer’s disease (FAD) (reviewed by Hardy [9]), while the presence of apolipoprotein E (APOE) ε4 genotype links to sporadic form of AD [10,11].
2.2. Amyloid Hypothesis
The precise etiology of AD remains unknown, despite over century passing from the first report of its symptoms by Alois Alzheimer [12]. Scientific efforts to elucidate AD etiopathogenesis lead to several different, partly complementary hypotheses. The relevant role of Aβ42 aggregation in AD pathogenesis has been disputed for over 25 years. Originally, the imbalance between production and clearance of Aβ42 in the very early stages of AD have been assumed as a causative and initiating factor of this disease. This dyshomeostasis may be the result of the mutations either in APP genes or in genes encoding presenilin, the substrate and enzyme of the reaction that generates Aβ42, respectively. It leads to the presence of Aβ deposits and the damage of the nerve tissue (reviewed by Selkoe [13]). Amyloid hypothesis may by supported by the observation that progressive Aβ deposition is present already in early, preclinical stages of AD and, finally, in all AD patients.
2.3. Isoform APOE4
The relationship between impaired amyloid deposition/clearance and genetic risk factors in AD were highlighted. As it was mentioned above, the best known genetic risk factor of AD is the ε4 allele of the APOE [14,15], which is associated with sporadic, late-onset AD. APOE is a polymorphic lipoprotein, with three major gene alleles: APOE-ε2, APOE-ε3, and APOE-ε4. It was shown that these three APOE isoforms bind Aβ differentially and modulate its fibrillogenesis [16,17,18]. The isoform APOE4 is unable to stimulate degradation of Aβ effectively, which results in a decreased brain clearance of Aβ and leads to the accumulation of amyloid deposits in the brain [19]. Moreover, there are more vascular and plaque deposits of Aβ observed in APOE4 carriers than in humans expressing only APOE3 [20]. This observation was also confirmed in genetically engineered mice [10]. Additionally, a quantitative evaluation in transgenic mice bearing human APP and APOE genes has shown decreased Aβ clearance in APOE4 carriers in comparison with E3 and E2 mice, which was paralleled by the degree of Aβ deposition [21].
2.4. Mutations in Presenilins PS1 and PS2 or APP Genes
In the familial form of AD, most cases are related to mutations in genes encoding one of three proteins: presenilins PS1 and PS2 or APP (i.e., the proteases and their substrate for generation of Aβ, respectively) [22]. APP is an essential membrane protein expressed mainly by the synapses and involved in their formation [23] as well as in neural plasticity [24] and iron export [25]. This protein is the precursor molecule that proteolytic processing generates various peptide fragments, including polypeptides of Aβ with 37 to 49 amino acid residues and molecular weight of approximately 4 kDa [26]. Proteolytic cleavage of APP is completed by enzymes of secretase family: α-, β-, and γ-secretase. Whereas APP processing by α- and β-secretase leads to removal of almost entire extracellular domain and produces membrane-anchored C-terminal fragments (reviewed by Zheng [27]), γ-secretase processing of APP results in generation of Aβ fragment [28]. γ-Secretase is a large, multi-subunit enzyme whose catalytic subunit is presenilin, a multi-pass transmembrane protein. The amyloidogenic processing of APP [29] and γ-secretase activity [30] have been associated with lipid rafts within cellular membrane. The role of cholesterol in lipid raft maintenance has been cited as a likely explanation for observations that high cholesterol and APOE-ε4 genotype are the major risk factors for Alzheimer’s disease [31].
Most mutations in the presenilin and APP genes enhance the production of Aβ42 [32] and early-onset deposition of this peptide [33,34,35]. Especially, the mutations in the region of APP molecule corresponding to the Aβ sequence lead to the production of more self-aggregating forms of amyloid [13]. Similarly, different presenilin mutations result in decreased ability of processing of APP by γ-secretase, and consequently increase the relative production of longer, more hydrophobic and more self-aggregating peptides of Aβ [13]. Peptides Aβ42, Aβ43, and longer express high potential of self-aggregation, whereas Aβ40 may rather be anti-amyloidogenic [36]. However, some of the pathogenic presenilin mutations only alter the ratio between Aβ42 and the other peptides of Aβ, especially Aβ40, but do not increase Aβ42 levels [37,38].
2.5. Down’s Syndrome
A gene for APP is located on chromosome 21. In subjects affected with Down’s syndrome due to the trisomy of this chromosome and possessing three copies of APP gene, AD is most likely to develop within the first 40 years of life [39,40]. This duplication of the wild-type APP gene leads to early-onset Aβ deposition, which occurs already in the teenagers, is then followed by microgliosis, astrocytosis, and accumulation of NFTs typical for AD. On the contrary, inheritance of a missense mutation in APP, that decreases the production and aggregation of Aβ, protects against AD and age-related cognitive decline [41].
2.6. Deposition of Misfolded Tau Protein
It was proposed that amyloid cascade is not the only pathway to AD (discussed by [42,43,44]). Although accumulation of Aβ in AD brain is followed by progressive deposition Tau protein, another hypothesis assumes that the abnormalities in the Tau protein initiate the cascade of events in AD [45]. In normal conditions, Tau is a soluble protein that is responsible for the association and stabilization of microtubules. In nerve cells, Tau is typically found in axons, but in the tauopathies, such as AD, progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), or inherited frontotemporal dementia and Parkinsonism linked to chromosome 17 (FTDP-17), this protein is present in an abnormal filamentous form and redistributed to the cell body and neurites [46]. Hyperphosphorylated forms of Tau aggregate in NFTs within the neurons [47]. This results in the disintegration of the microtubule and the destruction of neuronal transport [48], leading to impaired communication between neurons and, finally, their death [49]. This hypothesis may be partially confirmed in a model of AD created by Jack et al. [7], where Tau pathology in brain precedes Aβ deposition in time, but only on at a sub-threshold biomarker detection level. Although some human neuropathological studies suggest that NFTs may occur prior to presence of amyloid plaques (for review see: [13]), it is possible, that such studies might not have searched systematically for diffuse plaques or soluble, oligomeric forms of Aβ in the brain. Moreover, genetic studies prove that Aβ-elevating APP mutations lead to downstream alteration and aggregation of wild-type Tau, whereas Tau mutations do not lead to Aβ deposition and amyloid-related dementia. Some researchers suggest that Aβ can trigger AD-type Tau alterations, whereas Tau expression seems to permit certain downstream neuronal consequences of progressive Aβ build-up to arise [50]. This triggering feature is particularly addressed to soluble AβO [51].
2.7. Neuroinflammation
The immune system participates also in AD pathogenesis. It was demonstrated that AD patients express decreased levels of naturally-produced antibodies against Aβ when compared with healthy individuals [52]. The inflammatory reaction, oxidative stress and dyshomeostasis of metals metabolism also play an important role in AD pathogenesis [53,54]. It appears that insoluble Aβ deposits are recognized as foreign material and trigger activation of inflammatory response cascade [55,56]. Additionally, inflammation within AD brain may be partially linked with APOE4’s role as an aberrant immunomodulatory factor. The function of macrophages and microglia is regulated by APOE and may vary depending on isoform of this lipoprotein. Especially, APOε4 is associated with an enhanced inflammatory response compared to macrophages not expressing this allele [57]. It was shown that microglia derived from homozygotic mice possessing both alleles APOε4 demonstrated a pro-inflammatory phenotype, altered cell morphology, increased NOS2 mRNA levels and NO production, as well as higher pro-inflammatory cytokine production compared to microglia derived from APOε3 mice [58]. This effect was gene dose-dependent and increased with the number of APOε4 gene alleles. Although the immune aspects of AD draw increasing attention of researchers, the aim of this paper was to concentrate on other aspects of AD, such as soluble Aβ oligomers (AβOs) toxic activity and their putative cellular receptors.
2.8. Soluble AβOs Toxicity
Although initiated by Aβ, progression of AD is subsequently complicated and accelerated by other pathological processes, such as Tau pathology or inflammation. It is known that Aβ peptide may be present in various distinct states, including oligomeric forms of Aβ. These oligomeric species are antigenically distinct from monomeric and fibrillar conformations of Aβ peptide [59,60]. Currently, it is supposed that soluble AβOs, but not fibrillar Aβ42 within neuritic plaques, may be the toxic factors acting on a very early stage of AD, perhaps even initiating pathological cascade.
Mechanisms of AβO toxicity include synapse loss, the strongest pathological correlate of cognitive deficits in AD (Figure 1). AβO-induced decrease in synapse density is observed already in the earliest stages of AD [61], and the degree of synapse loss is greatest in close proximity of amyloid plaques [62]. The evidence for toxic AβO activity comes from observation that the loss of synapses in AD transgenic animals is correlated with the degree of colocalization of Aβ soluble oligomers with synaptic puncta [63].
Another mechanism of AβO toxicity is oligomers-induced disruption of synaptic transmission. It was demonstrated that soluble AβO could inhibit long-term potentiation (LTP) in mouse hippocampal tissue samples, suggesting that this form of Aβ might be the species triggering loss of synapses and memory impairment in AD [64]. It was also shown in mice model of AD that transgenic animals overexpressing mutant form of human APP exhibited lower density of presynaptic terminals, as well as severe impairments in synaptic transmission in the hippocampus for months before the presence of amyloid plaques [65]. This toxic activity of oligomers was confirmed in the study of Shankar et al., who demonstrated that soluble AβOs isolated from AD patients’ brains decreased number of synapses in animal models of AD, leading to enhanced long-term synaptic depression (LTD) and LTP in regions of brain which are responsible for memory [51].
What is interesting, it seems that intracellular soluble AβOs may be transmitted between neurons using synaptic connections, reaching even distant areas of the brain [66]. It was confirmed in AD mice models, where intracerebral injections of brain extracts from AD or Aβ aggregates induced amyloidogenesis [67]. Although various forms of AβOs may disseminate between neurons in this way, they do not spread between glial cells [68]. It is thought that the self-replication of Aβ and Tau aggregates and their spreading in a prion-like manner may contribute to the progressive nature of AD. Moreover, AβOs can be transferred not only within the brain, from one region to another, but it is supposed that oligomers might be transmitted between people [69].
A limitation of AβOs’ hypothesis may be the fact that oligomers are not homogenous species. Implications for the phenotypic diversity of Aβ in AD include clinical and neuropathological heterogeneity of AD, various distribution and significant differences in the expression of Aβ species in brain tissue, as well as different concentrations of Aβ peptides in CSF of AD patients [69]. AβOs’ molecular weight, morphology and conformation are highly variegated. There are different Aβ forms, from small, dimeric molecules, through trimers, and low molecular weight (LMW) and high molecular weight (HMW) oligomers up to protofibrils and fibrils, which range from relatively small molecules about 4 kDa to assemblies of 100 kDa. Therefore, the precise role of different oligomer species still remains to be elucidated [70,71].
3. Cellular Receptors Related to AβOs Activity
Although it is known that extracellular AβOs are able to bind to the surface of neurons, resulting in synaptic dysfunction and neurodegeneration, precise mechanism remains uncertain. It was suggested that AβOs may damage neuronal membranes directly, forming pores, which leads to the ionic dyshomeostasis, especially an increase in intracellular Ca2+ levels [72]. It was also proposed that AβOs at high concentration may interact directly with negatively charged phospholipid bilayers, leading to changes in their conductance in non-specific manner [73]. However, it is unclear how such membrane-disrupting activity could explain the selectivity of AβOs for the central nervous system (CNS), especially for the synapses, in AD.
Some other potential mechanisms of AβOs and their targets, including the abnormal activation of signaling pathways, are under extensive investigation [74]. As it was mentioned above, extracellular AβOs accrue generally at synapses, especially at synaptic spines, but detailed specificity of AβOs to particular cells was uncertain. It was shown that in hippocampal cultures Aβ oligomers were bound mostly to neurons, whereas in cortical and cerebellar cultures this binding occurred in a lesser degree [75,76]. It was also demonstrated that both AβOs prepared in vitro and those extracted from AD brain were the ligands targeting cultured mouse hippocampal cells. Soluble AβO isolated from AD patients bound to hippocampal dendrites in cultured mouse neurons with high, “ligand-like” specificity [77]. This specific targeting of neuronal cells is in line with rapid disruption of hippocampal LTP and LTD induced by oligomers [78,79] and with selective neuronal degeneration induced by soluble AβOs seen in brain slice preparations [80].
Several studies postulate various possible receptors involved in the toxicity of AβOs. Binding of AβOs to cell membranes is probably mediated by particular cell surface proteins that act as toxin receptors. This hypothesis could explain various mechanisms of AβOs’ activity, resulting in synaptic dysfunction and neurodegeneration [64].
It is possible that these receptor proteins might be expressed only on certain cells, converting action of AβOs into harmful responses. Moreover, such receptors for extracellular AβO are rather localized at neuronal synapses and should have a high affinity for AβO. These receptors should be also more selective for AβOs than for monomeric or fibrillar Aβ, because monomers of Aβ are present ubiquitously in all individuals, while their levels do not substantially change with disease. Furthermore, putative AβO receptors should have the ability to transduce extracellular triggering factors into certain intracellular changes. It may be achieved either directly or by connection with other active molecules.
There are over 20 candidates for AβO receptors, including glutamate, adrenergic, acetylcholine receptors and others. Unfortunately, no single candidate receptor protein has been shown yet to be responsible for all features of AβO activity. Moreover, the heterogeneity of AβOs results in their diverse affinity as ligands when binding to various putative oligomers’ receptors [81].
3.1. Glutamate Receptor NMDAR
Synaptotoxic activity of AβOs includes inappropriate increase of extracellular glutamate concentration and activation of glutamate receptors, which results in rapid impairment of synaptic plasticity [82]. The N-methyl-d-aspartate (NMDA) receptor is a glutamate receptor with ion channel activity. It plays a role in controlling of synaptic plasticity and synapse formation, which are responsible for memory function, learning and formation of neuronal networks in CNS [83]. It was suggested that oligomeric Aβ toxicity may involve NMDAR activation, although it remains controversial whether AβOs trigger loss or gain of its function.
Some studies indicate that AβOs initiate impairment of NMDAR activity by removal from the cell surface and triggering of synaptic depression signaling pathways [84,85,86]. By modulation of NMDAR-dependent signaling pathway, AβOs induce reversible synapse loss causing the decrease in spine density [85,87]. Both in vivo and in vitro studies demonstrated that Aβ can disrupt induction of LTP depending on this type receptor [88]. Moreover, activity of this receptor is required for AβOs-induced synaptic depression [87].
On the contrary, other authors demonstrated that AβOs cause an increase of NMDAR receptor function. AβOs induce neuronal oxidative stress through an NMDAR-dependent mechanism. This activity of AβOs is blocked by memantine, an uncompetitive NMDAR antagonist and the drug used to relief AD symptoms [89]. Moreover, it was reported in animal models of AD, that chronic treatment with memantine reduced Aβ deposition in the brain, both insoluble Aβ fibrils and soluble AβOs. Memantine not only inhibited the formation of different types of Aβ aggregates in a concentration-dependent manner, but also led to disaggregation of Aβ42 fibrils [90]. Interestingly, specific antibody to the extracellular domain of the NR1 subunit of NMDARs led also to reduction of AβOs binding to neurons and completely blocked the formation of reactive oxygen species (ROS) [89].
Dysregulation of Ca2+ signaling and membrane disturbance, which is thought as a ubiquitous mechanism of soluble AβOs neurotoxicity [91], may also be mediated by activation of NMDAR [92]. AβOs disrupt NMDAR-mediated postsynaptic Ca2+ signaling in response to presynaptic stimulation by enhancing the accessibility of extracellular glutamate as well as directly disturbing the NMDARs [93]. This excessive activation of NMDAR leads to disproportionate inflow of Ca2+ to neurons and may cause excitotoxicity, a pathological mechanism recognized in some NDs, including AD. It is thought that this aberrant regulation of intracellular Ca2+ signaling is an early event in AD, prior to the presence of clinical symptoms. Dysregulation of Ca2+ signaling is also believed to be a crucial factor contributing to AD pathogenesis (for review see: [94]).
Moreover, AβOs interfere specifically with several proteins involved in calcium-related signaling pathways, such as calcineurin, which is Ca2+-dependent phosphatase, and Ca2+/calmodulin-dependent kinase II (CaMKII) [88]. The dynamic balance between these enzymes is presumed to be important for synaptic plasticity. It was demonstrated that LMW AβOs may inhibit CaMKII activity and thus disrupt the equilibrium between above mentioned enzymes [95]. In addition, activation of NMDAR by soluble AβOs involves Ca2+-mediated mitochondrial dysfunction as well as decreased CaMKII levels at synapses. This results in dramatic loss of synaptic proteins such as postsynaptic density-95 (PSD-95), dynamin-1 and synaptophysin [96].
NMDAR may also mediate the toxic impact of AβOs on glucose metabolism in neurons. AMP-activated kinase (AMPK) is a key enzyme in energy sensing and metabolic reprogramming under cellular energy restriction, which is associated with some peripheral metabolic diseases, including diabetes. An impaired AMPK function has been linked recently to certain neurological disorders, such as AD [97]. The intracellular ATP levels and AMPK activity were decreased in cultured hippocampal neurons already after short-term exposure to AβOs. This AβO-dependent reduction in AMPK activity is also mediated by glutamate receptors NMDARs, which results in removal of glucose transporters (GLUTs) from the surfaces of hippocampal neurons [97].
3.2. Glutamate Receptor AMPAR
The α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) is also a glutamate ionotropic transmembrane receptor that mediates synaptic transmission in CNS. AMPAR is classified as a non-NMDA-type receptor. AMPARs are tetrameric receptors composed of four subunits, labelled as GluA1, GluA2, GluA3, and GluA4. Subunits GluA1 and GluA2 play an important role in synaptic plasticity and LTP [98]. The permeability of AMPAR to Ca2+ is related to the GluA2 subunit [99]. Phosphorylation of AMPARs influences ion channel localization and its conductance. Subunit GluA1 has four known phosphorylation sites, but serine 845 (S845) is a residue that plays an essential role in the trafficking of AMPARs toward extrasynaptic sites [100].
Aβ oligomers may cause synaptic dysfunction also by inducing calcineurin-dependent internalization of AMPAR [101]. It was shown in cortical cultures that soluble oligomers of Aβ, but not monomers, mediate the internalization of the AMPAR subunits GluA1/GluA2 by endocytosis [102]. Short-term exposure of hippocampal neurons to AβOs led to noticeable removal of AMPARs from postsynaptic surface and to impaired insertion of this receptor during synaptic potentiation [103]. It is in accordance with the finding that acute exposure of cultured neurons to soluble AβOs induced AMPAR ubiquitination associated with the removal of this receptor from the plasma membrane [104]. Aβ oligomers reduce basal levels of S845 phosphorylation and surface expression of AMPARs affecting AMPAR subunit composition contributing to early synapse dysfunction in a transgenic mouse model of AD [105].
Binding of AβOs to neurons occurs in dendritic spines expressing AMPARs, preferentially GluA2, which is calcium impermeable [106]. Furthermore, pharmacological inhibition of AMPARs leads to reduced AβOs binding. It was demonstrated that the process of rapid internalization of AβOs with surface AMPAR subunits is mediated by calcineurin, whereas inhibition of this phosphatase and AMPARs prevents AβOs-induced synaptic disruption and spine loss [106].
Whereas the role of AMPARs in hippocampal pyramidal neurons containing GluA1 and GluA2 subunits (GluA1/2) has been extensively examined, the importance of AMPAR type having GluA2 and GluA3 (GluA2/3) for synapse physiology was not clear. It was recently revealed that activation of GluA3 AMPARs may constitute novel type of plasticity at synapses [107]. Animal studies shown that in basal conditions GluA2/3 AMPARs are in low-conductance state, shifting to a high-conductance GluA2/3 channels with increased intracellular cyclic AMP (cAMP) levels, which led to synaptic potentiation [107]. It was also indicated that some forms of LTP, such as vestibulo-cerebellar motor learning, may rather require GluA3-AMPARs activation by increasing single-channel conductance mediated by cAMP signaling [108].
Furthermore, the presence of GluA3-containing AMPARs may be also relevant for synaptic and cognitive deficits mediated by AβOs. It was shown in experiments in AD mouse models that all the effects on synapses and memory mediated by soluble oligomeric clusters of Aβ required presence of AMPA receptor subunit GluA3 [109]. Moreover, AβOs blocked synaptic LTP only in neurons expressing this subunit, whereas GluA3-deficient hippocampal neurons were resistant to toxic AβO activity, such as synaptic depression and spine loss. What is important, mice lacking GluA3 subunit did not express memory impairments [109].
Interestingly, abnormal Tau phosphorylation may contribute to AβOs-induced signaling deficits of AMPAR [110]. AβOs led to abnormal Tau distribution in dendritic spines in cultured rodent hippocampal neurons. Aberrant Tau localization was dependent on the phosphorylation of this protein and resulted in early cognitive deficits and synaptic loss [110].
3.3. Metabotropic Glutamate Receptor 5 mGluR5
Glutamate is one of the main excitatory neurotransmitters in human CNS and glutamatergic neurotransmission is involved in most aspects of normal human brain function [111]. This neurotransmitter signals through ligand-gated ion channels, such as AMPAR, or through metabotropic glutamate receptors (mGluRs), a family of several G protein-coupled receptors. Two principal signal transduction pathways involving mGluRs are known: cAMP and phosphatidylinositol signal pathways [112].
Metabotropic glutamate receptor 5 (mGluR5) belongs to group I of metabotropic glutamate receptors and activates phospholipase C. This type of receptor has been implicated in a diverse variety of physiological neuronal functions. Moreover, mGluR5 acts postsynaptically as a co-receptor for AβO [113]. Soluble extracellular AβOs bind to lipid-anchored cellular prion protein (PrPC) with high affinity and specificity [114,115]. The coexpression of mGluR5 allows PrPC-bound AβO for activation of intracellular Fyn kinase, what results in the disruption of synapses [113]. Complexes of AβO with PrPC generate mGluR5-mediated influx of Ca2+ in neurons. This influx may be also driven by human AD brain extracts. Aβ peptides also disturb intracellular Ca2+ homeostasis. It was demonstrated that Aβ42 oligomers, but not monomers, significantly altered Ca2+ release from intracellular stores, which involved mGluR5 and required network activity [116]. In addition, dendritic spine loss is also mediated by AβO-PrPC-mGluR5 complexes signaling pathway [113].
3.4. Cellular Prion Protein PrPC
It seems that significant part of AβO toxicity in AD may be mediated after initial interaction with PrPC on the neuronal surface. In the normal brain, the expression of PrPC is controlled by a feedback loop with amyloid intracellular domain (AICD). PrPC inhibits the activity of β-secretase (β-site APP cleaving enzyme-1, BACE1) [117] as well as AICD production [118], whereas AICD upregulates PrPC expression, which maintains the inhibitory effect of PrPC on BACE1.
This reaction is disrupted in AD, resulting in the binding of increased levels of AβOs to PrPC and disturbed regulation of BACE1 activity. Moreover, PrPC inhibits formation of fibrillar aggregates of Aβ, trapping this peptide in an oligomeric state [119]. Only recently it was demonstrated that PrPC specifically inhibits elongation of Aβ fibrils by binding to the ends of growing polymers [120]. It was shown that this inhibitory effect requires the globular C-terminal domain of PrPC, which suggests that PrPC might recognize specific structure that is common to the ends of both oligomeric and fibrillar form of Aβ [120]. This interaction could probably contribute to the neurotoxicity of AβOs.
As it was mentioned above, cellular prion protein PrPC was identified as AβO co-receptor, although the infectious form PrPSc conformation is not required [115]. PrPC binds Aβ42-oligomers with high affinity and high selectivity. Purified recombinant PrPC interacted directly with AβOs, whereas the binding of synthetic AβOs to neurons decreased in PrPC-null mice.
Moreover, PrPC mediates impairment of synaptic plasticity by AβOs [115]. The effects of interaction between PrPC and AβOs on LTP were compared between wild-type and PrPC-null mice [115]. It was shown that soluble AβOs reduced LTP in the wild-type mice, but not in the PrPC-null mice. It may indicate that PrPC is required to mediate these toxic effects of AβOs. It was also demonstrated that binding onto PrPC induces intracellular Ca2+ increase in neurons via the complex PrPC-mGluR5, with harmful effects on synaptic transmission [121].
Although additional receptors may contribute to mediation of AβO action, recent investigations indicate that PrPC supposedly plays a primary role (reviewed by Del Rio [122]). PrPC is a glycosylphosphatidylinositol (GPI)-anchored protein. Thus, the mediation of the signal transduction requires the formation of complexes between PrPC and certain transmembrane proteins, such as acetylcholine and glutamate receptors [113,123,124,125,126]. The complex PrPC-mGluR5 plays an important role in AβO binding and activity of oligomers in neurons. The signal transduction downstream of AβO-PrPC complexes involves mGluR5, as well as kinases Fyn and Pyk2 [113]. Additionally, after AβOs to binding PrPC and activation of Fyn tyrosine kinase, NMDARs are phosphorylated, which in turn results in altered surface expression, dysregulation of receptor function, excitotoxicity, and dendritic spine retraction [113]. This mechanism is consistent with previous discovery that Fyn is essential for AβO-induced synaptotoxicity [64,127].
Interestingly, it was shown that PrPc also appears to be relevant in α-synucleopathies, such as PD, participating in α-synuclein binding and brain spreading [122].
3.5. β2-Adrenergic Receptors
The β2-adrenergic receptors (β2ARs) are expressed in the brain, especially in regions involved in AD pathogenesis, i.e. hippocampus and cortex [128]. β2ARs play an important role in cognitive functioning. The activation of β2ARs is essential for normal learning and memory [129,130]. Stimulation of β2ARs promotes synaptic LTP in dentate gyrus and hippocampus [131,132,133,134,135,136]. The role of β2AR in memory formation may be confirmed by enhanced expression of β2ARs in dendritic spines [137,138]. β2AR roles in brain are associated with the AMPA-type glutamate receptor [137].
It was demonstrated that β2ARs activation enhances neurogenesis in APP/PS1 mice, a mouse model of AD [139]. Stimulation of these receptors attenuated memory deficits and reduced Aβ accumulation in mouse brain. Moreover, activation of β2ARs enabled the recovery of memory deficits in APP/PS1 mice, enhanced neurogenesis in the dentate gyrus, restored dendritic branches, and spine density in the hippocampus as well as increased the levels of synapse-associated proteins such as synaptophysin, synapsin 1, and PSD-95 [139]. These findings suggest that activation of β2AR protects synapses in this animal model of AD.
Alterations in β2ARs function have been linked to AD, although the results were not consistent. Decreased levels of β2ARs in certain regions of post-mortem human AD brain, such as locus coeruleus and hippocampus, were demonstrated [140,141,142]. Activation of β2ARs resulted in enhanced γ-secretase activity and intensified amyloid plaque formation [143], whereas use of β2AR antagonists conversely attenuated production of Aβ induced by acute stress [144].
On the other side, the administration of ICI, a selective β2AR antagonist, enhanced neuropathological changes, such as increased Aβ plaque burden, as well as accumulation of phosphorylated Tau in a mouse model of AD [145]. Moreover, blockade of β2AR led to cognitive deficits in mice. These results suggest that selective pharmacologic inhibition of β2ARs may have negative effects on AD-like pathology in this animal model of AD. It should be highlighted that the link between β2ARs and AD is likely highly complex.
It was shown that human AβOs, when applied to slices of rodent brain, are able to induce the degradation of β2ARs [146,147]. β2AR levels in hippocampal slices were decreased significantly after exposition to AβOs. Although HMW soluble oligomers of Aβ extracted from AD brain had faint or none cytotoxic activity, they dissociated in alkaline environment to smaller, LMW oligomers (approximately 8–70 kDa). Postincubation LMW were much more bioactive. They induced impaired hippocampal LTP, activated brain microglia and led to decrease in the neuronal levels of β2ARs in mice in vivo [148].
3.6. Acetylcholine Receptor α7nAChR
It was shown that Aβ42 binds with high, picomolar affinity to α7 nicotinic acetylcholine receptor (α7nAChR), a neuronal pentameric cation channel [149]. This binding is accompanied with the loss of cholinergic neurons in the brain, resulting in receptor internalization and intracellular accumulation of Aβ [150]. Furthermore, formation of the α7nAChR-Aβ42 complex was suppressed by shorter chains of Aβ (12–28), indicating that this sequence region contains the binding epitope of amyloid [149].
It was also demonstrated in immunohistochemical studies on human sporadic AD brains that α7nAChR is present in neuritic plaques and co-localizes with Aβ in individual cortical neurons [149]. Moreover, the presence of intracellular AβOs was shown in human cholinergic basal forebrain neurons, suggesting the role of amyloid oligomers in cholinergic deficiency [151]. It was confirmed in a triple-transgenic mouse model of AD, where loss of the α7nAChRs was restricted to brain regions that accumulate Aβ intraneuronally [152].
The loss of α7nAChR enhances AβOs accumulation in a mouse model of AD, exacerbating early-stage cognitive decline and septo-hippocampal pathology [153]. In α7nAChR-null mice crossed with those transgenic for mutant human APP, a neurodegeneration in hippocampus and cognitive decline were found already in early, pre-plaque stage of AD. These changes were associated with the appearance of a small, dodecameric form of AβO [153]. Presented findings suggest that α7nAChR plays a protective role for AβOs toxicity. What is more, restoring LTP impaired by AβOs is possible by using a selective neuronal nicotinic receptor partial agonist SSR180711, which completely rescued both early and late LTP impaired by Aβ42 oligomers [154].
3.7. Insulin Receptor
A pathophysiological connection between AD and diabetes was confirmed in numerous studies (reviewed by de Felice [155]). An increasing body of evidence indicates that AD may be called a “brain-specific form of diabetes” or “type 3 diabetes” [156,157]. Both diseases are characterized by key pathological features such as insulin resistance, inflammation, and altered metabolism. Diabetic pathophysiology includes reduction in brain insulin signaling, decreased levels of brain insulin, and elevated levels of glucose.
Toxic activity of AβOs may be also linked with impaired insulin signaling and brain insulin resistance, which lead to elevated Aβ production and reduced AβO clearance, resulting in oligomers’ deposits in the brain and neuronal damage [158]. Furthermore, it was shown that signal transduction by neuronal insulin receptors (IRs) is highly sensitive to soluble AβO. AβOs themselves can influence IRs and decrease brain insulin signaling [158]. In addition, AβOs bind to neuronal IRs and affect its insulin-induced autophosphorylation, preventing activation of specific kinases required for LTP [159]. In cultures of mature hippocampal neurons, soluble oligomers caused a rapid, substantial loss of surface IRs, especially on dendrites bound by AβOs [106].
3.8. p75 Neurotrophin Receptor p75NTR
It was suggested that AβOs may induce neuronal death via nerve growth factor (NGF) receptor by alteration of NGF-mediated signaling in cultured cells [75,160]. NGF mediates cell loss through low-affinity receptor for nerve growth factor, also called p75 neurotrophin receptor (p75NTR), which belongs to the tumor necrosis factor (TNF) receptor superfamily [74]. Precisely, toxic effects of Aβ mediated by p75NTR depend on a death domain in the cytoplasmic part of this receptor molecule [161,162]. Synapse targeting of AβOs involves activation of p75NTR. AβOs, together with PrPC, bind at the membrane receptors, forming annular amyloid pores and ion channels to induce aberrant cytoskeletal changes in dendritic spines [96].
In the mouse hippocampus, the expression of p75NTR induced by AβOs involves insulin-like growth factor 1 receptor (IGF-1R) signaling [163]. Significantly elevated hippocampal expression of membrane-associated p75NTR protein was shown in transgenic AD mice and was associated with the age-dependent increase of Aβ42 levels. Moreover, it was demonstrated that microinjections of AβOs induced p75NTR expression in the hippocampus through phosphorylation of IGF-1R, whereas co-administration of IGF-1R inhibitor blocked AβOs-induced overexpression of p75NTR [163].
Conflicting evidence exists regarding the role of p75NTR in AD, especially against toxicity of AβOs. Although an important role of p75NTR in Aβ metabolism and Aβ-mediated neurodegeneration in AD brains was shown, this protein also promotes the differentiation and survival of vertebrate neurons [164]. Furthermore, a conflicting role of p75NTR in the cytotoxic function of Aβ depends on the different state of this peptide. In fact, the neurotoxicity of the two forms of Aβ, insoluble fibrillar or soluble oligomeric form, occurs with different mechanisms. Primarily, it was proved that the expression of p75NTR is required for cell death by fibrillar form of Aβ [165]. Interestingly, the toxicity of fibrillar Aβ species is strictly dependent on p75NTR, whereas neurotoxicity of soluble AβOs is independent of p75NTR and is even decreased by the presence of this receptor. Moreover, the expression of p75NTR protects against the neurotoxicity of oligomers [166]. This protective effect results from an active function of the juxtamembrane sequence of the cytoplasmic region of p75NTR and is mediated by phosphatidylinositide 3-kinase (PI3K) activity [166]. These results suggest that p75NTR might have diverse functions in cell death and survival.
3.9. Immunoglobulin and Immunoglobulin-Like Receptors
Human leukocyte immunoglobulin-like receptor B2 (LilrB2) belongs to the subfamily B class of leukocyte immunoglobulin-like receptors (LIR) expressed on immune cells. LilrB2 inhibits stimulation of an immune response, controls inflammatory responses and cytotoxicity, and limits autoreactivity of immune system. LilrB2 binds to major histocompatibility complex (MHC) class I molecules on antigen-presenting cells. It was indicated that MHC class I molecules have additional functions in CNS [167]. Furthermore, numerous MHC class I antigens and their binding partners are found to be expressed in CNS neurons and might be involved in activity-dependent synaptic plasticity [167]. LilrB2 also participates in the process of synaptic plasticity and neurite growth in CNS [168].
Murine homolog of LilrB2, paired immunoglobulin-like receptor B (PirB), is an immune inhibitory receptor, primarily identified in mouse immune cells [169]. Expression of PirB is also observed in subsets of neurons throughout mouse brain. In addition, PirB participates in the inhibition of axonal regeneration [170,171]. It was also suggested that PirB plays an important role in age-related hippocampal aging, synaptic loss and neurotransmitter release, which causes cognitive dysfunction associated with AD [172].
Importantly, murine PirB and its human orthologue LilrB2 are thought to be nanomolar affinity receptors for Aβ oligomers [168]. The interaction between AβOs and PirB/LilrB2 are mediated by the first two extracellular immunoglobulin domains of the receptors [168]. PirB regulates synaptic plasticity, affecting hippocampal LTP, which contributes to Aβ-induced deficits of memory in a mouse model of AD [168]. Moreover, high PirB expression is required for the harmful effect of AβOs on hippocampal formation [168]. In double transgenic APP/PS1 mice, ocular dominance plasticity (ODP) was defective during the very early period of synaptic plasticity development [173]. This observation is in contrast with enhanced ODP during the critical period and in adult mice lacking PirB [174]. It suggests that impaired ODP is one of the earliest Aβ-induced deficits in a mouse model of AD. While Aβ42 oligomers robustly bound to LilrB2-expressing heterologous cells, only a minimal binding of monomeric Aβ42 to LilrB2 was observed [168], which suggests selectivity of AβOs reaction with this receptor. Although similar levels of LilrB2 were detected either in human AD brains or in specimens from non-demented adults, downstream signaling was altered in AD specimens.
It was suggested that FcγRIIb (Fragment crystallizable gamma receptor II b) may also play a role as a AβOs receptor, mediating neurodegeneration and toxic activity of oligomers [175]. FcγRIIb belongs to family of Fc-gamma receptors (FcγR) which have a binding specificity for the Fc (Fragment, crystallizable) region of immunoglobulin gamma (IgG) [176]. They are present on the surface of B lymphocytes, dendritic cells, natural killer cells, macrophages, granulocytes, mast cells, and other cells of the immune system. Additionally, all of the Fcγ receptors (FcγR) belong to the immunoglobulin superfamily and differ in their affinities for IgG due to variegated molecular structure of different IgG subclasses.
It was demonstrated that FcγRIIb is an important factor contributing to the AβOs’ neurotoxicity and memory impairment. This protein was significantly upregulated in the hippocampus of AD brains and neuronal cells exposed to synthetic Aβ [175]. Soluble Aβ oligomers interacted with FcγRIIb both in vitro and in AD brains, whereas inhibition of that interaction blocked neurotoxicity of synthetic AβO. Moreover, in mouse model of AD, genetic depletion of FcγRIIb rescued memory impairments and prevented AβO-induced inhibition of LTP, which supports an idea that this receptor could play an essential role in Aβ-mediated neuronal dysfunction [175].
3.10. Triggering Receptor Expressed on Myeloid Cells 2 TREM2
Triggering receptor expressed on myeloid cells 2 (TREM2) is a transmembrane-glycoprotein receptor that is present on the surface of immune cells of myeloid origin [177]. As a lipid-sensing activating receptor, TREM2 binds to phospholipids, apolipoproteins, and lipoproteins through its immunoglobulin-like domain [178]. Moreover, TREM2 interacts with TYRO protein tyrosine kinase-binding protein, also known as DNAX-activating protein of 12 kDa (DAP12), which is an adapter protein for this receptor. In the brain, this interaction triggers the phagocytosis of apoptotic neurons and Aβ peptide in microglia with no inflammatory effects [179].
It was shown that certain coding variants in TREM2 gene are associated with increased risk for AD [180], which suggests that immune cell dysfunction may also play a role in AD pathogenesis. Normal proteolytic maturation of full-length TREM2 at the plasma membrane is disturbed in mutations of TREM2 gene, resulting in impaired phagocytosis, which may contribute to the pathogenesis of AD [179].
In normal conditions, TREM2 directly binds to AβOs with nanomolar affinity. Only recently, it was demonstrated that in AD-associated TREM2 mutations this binding is reduced [181]. Moreover, the degradation of Aβ in primary microglial culture and mouse brain was impaired in TREM2 deficiency, resulting in microglial depolarization, induction of K+ current into cells as well as increased cytokine expression and secretion, cells migration, proliferation, apoptosis, and morphological changes are dependent on TREM2 [182]. Additionally, TREM2-DAP12 interaction was enhanced by AβOs, which demonstrates that TREM2 may act as a microglial AβO receptor that mediates physiological and AD-related pathological effects [181].
3.11. Tyrosine Kinase Ephrin Receptors Eph4A and EphB2
It was suggested that the tyrosine kinase Eph receptors may also play a role in AβOs-induced synaptotoxicity [183]. Eph receptors were named after the cell line from which the cDNA was first isolated, erythropoietin-producing hepatocellular carcinoma. Based on the affinities for binding ligands and similarity of extracellular domain sequences, they are divided into two functionally different groups: EphA and EphB. There are nine EphA receptors (1–9), which bind to ephrin-A ligands (ephrin-A 1–5), proteins anchored to the cell membrane by GPI motif, whereas five EphB receptors (1–5) bind to ephrin-B ligands (ephrin-B 1–3) with a transmembrane domain and a short cytoplasmic region [184].
Eph receptors and their ligands play a key role in the physiological functioning, development and maturation of nervous system [183]. Since Eph ligands and receptors are both membrane-bound proteins, the Eph/ephrin binding and activation of their intracellular signalling pathways may occur via direct cell-to-cell interactions only. In particular, their presence both in pre- and postsynaptic regions is necessary for the development and stabilization of synapses, although EphB and EphA play opposite roles [185].
It was shown, that EphB promotes morphogenesis and growth of dendritic spines, whereas their development is aberrant in the absence of these receptors in hippocampal neurons [186]. Moreover, the formation of synapses is induced by activation of EphB2 receptor via interaction with NMDAR [187].
EphB receptors are also important factors in the pathophysiology of AD and other neuropathologies [188]. It was demonstrated that EphB2 levels in the membrane of hippocampal neurons were decreased after short term treatment of AβOs [189], which could be a result of NMDAR activation [190]. It was also shown that AβOs binding to the fibronectin (FN) type III repeat domain of EphB2 triggers to endocytosis of this receptor and its degradation in the proteasome [191]. The results of EphB2 degradation are the impairment of NMDAR functioning and cognitive deficits. What is interesting, these interaction sites of the EphB2 FN domain with AβOs may be blocked by a small, 10 amino acids length peptide Pep63, which rescued memory deficits in mouse model of AD [192]. These results suggest that inhibition of EphB2-AβOs interactions may be a promising strategy for AD treatment. Furthermore, it was shown induction of the EphB2 expression in the dentate gyrus prevented the cognitive deficits and LTP impairments in mice model of AD [106]. On the other hand, the decrease of AMPAR and NMDAR levels induced by AβOs may be prevented by overexpression of EphB2. This protective effect could be directly related to PDZ-binding motif of EphB2 [191,193,194].
EphA receptors bind membrane-bound ephrinA family ligands residing on adjacent cells, leading to contact-dependent bidirectional signalling [195]. EphA4 receptor plays also an important role in the regulation of synapses functioning in the nervous system and in the repairment after injury, preventing axonal regeneration as well as in the angiogenesis and formation of vessels within central nervous system [183].
EphA4 mediates dendritic spine remodeling and contributes to homeostatic plasticity through the regulation of AMPAR levels [195,196,197]. Activation of EphA4 receptor induces a decrease in the strength of the excitatory synapse as well as reduction of spine length and density in hippocampal slices [195]. This receptor is also associated with the loss of dendritic spines, their retraction and growth cone collapse [195]. It was confirmed in EphA4-knockout mice, which expressed disorganized, longer, and more numerous spines than wild-type mice [183].
Moreover, EphA receptors seems to be key player in the pathophysiology of AD and other neuropathologies, such as motor neuron degeneration in amyotrophic lateral sclerosis (ALS) [198,199]. It was shown in AD brains, that hippocampal distribution of EphA4 was co-localized with neuritic plaques already at early stages (Braak stage II), which suggests that EphA4 may contribute to synaptic dysfunction [200]. Additionally, it was demonstrated that levels of EphA4 mRNA in synaptoneurosomes from AD patients were twofold higher than in non-demented controls [201].
Furthermore, AβOs aberrantly activate EphA4 leading to dendritic spine elimination, whereas blockade or absence of this receptor in hippocampal neurons prevents synaptic loss [202,203]. This AβOs-EphA4 axis involves c-Abl tyrosine kinase activation by AβOs in dendritic spines of cultured hippocampal neurons, which is required for AβOs-induced synaptic loss [183,203].
3.12. Receptor for Advanced Glycation Endproducts RAGE
RAGE is a small, 35 kDa transmembrane protein, which belongs to the immunoglobulin superfamily and plays a role in innate immunity. RAGE is composed of three extracellular Ig-like domains (Vd, C1d, C2d), with a single transmembrane domain, and a short cytoplasmic tail [204]. This receptor is described as a “pattern recognition” receptor because of its ability to recognize common structural motifs. RAGE is able to bind multiple ligands, such as advanced glycation endproducts (AGE), glycans and glycoproteins, as well as chromatin protein high mobility group box 1 protein (HMGB-1), calprotectin and S100B [205]. After stimulation, RAGE activates certain pro-inflammatory genes, which mediate Aβ-induced oxidative stress.
Moreover, activation of RAGE results in continuous instigation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) [206,207]. On the other hand, RAGE itself is upregulated by NF-κB, thus creating RAGE/NF-κB axis. This forms a positive feed-back loop leading to chronic inflammation, altered micro- and macrovasculature, and tissue damage, pathological events observed also in AD.
Expression of RAGE is increased in the AD brain. Moreover, enhanced levels of RAGE ligands were observed in a range of inflammatory diseases, atherosclerosis, diabetes and cancer as well as in AD, which suggests a causative role of this receptor in inflammatory chronic state [207]. RAGE was also identified as one of the cell-surface binding sites for Aβ peptide at the plasma membrane of neurons, microglial cells, and endothelial cells of the vessel wall [206].
Furthermore, RAGE participates in the clearance of Aβ. The level of Aβ peptides as well as other substances in the brain is not only a result of specific equilibrium between their synthesis and degradation, but also depends on the transport into the brain from blood and efflux from the brain into blood through blood-brain barrier (BBB). Both in normal aging and in AD, the rate of CSF reabsorption into the blood, known as bulk flow, is also impaired. The main receptors for the transport of Aβ across BBB are RAGE and low-density lipoprotein receptor related protein-1 (LRP1). RAGE is responsible for Aβ influx, whereas LRP1 is the main receptor controlling the efflux across the BBB to the plasma [208]. Soluble form of LRP1 sequesters 70–90% of plasma Aβ peptides in normal conditions. In AD, this Aβ clearance is disturbed [206]. Both in AD mouse models and in AD patients, the brain endothelial expression of RAGE is elevated, whereas plasma levels of sLRP1 and its Aβ-binding capacity are decreased, leading to increase in free Aβ fraction in plasma [209].
Interestingly, distinct regions of RAGE are induced by different Aβ conformations in AD-related apoptosis [204]. It was demonstrated that anti-RAGE antibodies significantly improved survival of cortical rat neurons and RAGE-expressing cells exposed to either AβOs or aggregated Aβ. Moreover, the use of site-specific antibodies against domain Vd of this receptor prevented AβOs-induced neurotoxicity, whereas blockade of the apoptosis induced by aggregated Aβ required neutralization of C1d domain of RAGE [204].
3.13. Megalin Receptor
Megalin, also known as glycoprotein 330 (gp330) or low-density lipoprotein-related protein 2 (LRP2), is a large, approximately 600 kDa protein, which is a multiligand binding receptor expressed in the plasma membrane of epithelial cells [210]. In CNS, megalin is present in choroid plexus epithelium and ependymal cells covering the brain ventricles. LRP2 is a member of a family of receptors with structural similarities to the low-density lipoprotein receptor (LDLR). Megalin mediates endocytosis of its ligands, which results in the degradation in lysosomes or transcytosis [211]. This receptor has been shown to interact with various ligands, vitamin-binding proteins, carrier proteins, lipoproteins, hormones and hormone precursors, as well as drugs and toxins [212]. Moreover, this receptor has also functions in cellular communication and signal transduction, with PSD-95 as an interaction partner [213].
LRP2 is also an endocytic receptor for apolipoprotein J (ApoJ)/clusterin involved in rapid receptor-mediated uptake or bidirectional exchanges of soluble Aβ across BBB [214]. ApoJ has been revealed to be the major protein binding Aβ in CSF. Megalin mediates cellular uptake and transport of ApoJ alone and ApoJ complexed with Aβ-40, the most abundant amyloid isoform found in Aβ deposits of the blood vessels, from the periphery into the brain at the cerebral vascular endothelium and choroid epithelium [215]. This interaction of ApoJ-Aβ complex with megalin is thought to be another, besides RAGE and LRP1, mechanism preventing pathological accumulation of Aβ [216]. It was shown that Aβ alone did not bind directly to LRP-2, whereas complexes of Aβ-40 with apoJ were able to react with megalin. Moreover, ApoJ/Aβ binding interaction was blocked polyclonal anti-megalin antibodies, which supports the role of LRP-2 as a mediator of the clearance of ApoJ/Aβ complex from CSF and in the regulation of Aβ accumulation [216].
3.14. Nuclear Receptors
Nuclear receptors (NRs) constitute a class of proteins that mediate certain, relatively small, molecules pathways, thus controlling the development, homeostasis, and various metabolic processes. There are currently 48 nuclear receptors known in the human genome, most of them have identified specific ligands [217]. NRs are involved in the synthesis and metabolism of steroid and thyroid hormones as well as and various other lipid-soluble signals, including retinoic acid, oxysterols, vitamin D, cholesterol, lipids, and bile acids or thyroid hormone [217].
Moreover, many of NRs, but not all, directly bind to signalling molecules. These molecules are small and have lipophilic character, therefore they can easily enter the target cell. Thus, unlike described above membrane-bound receptors, NRs are intracellular proteins which are capable of direct binding to DNA, thus controlling the expression of adjacent genes, which is their unique property that differentiates them from other classes of receptors. Because of this ability, NRs are classified as transcription factors [218].
The nuclear receptor superfamily may be divided according to their amino acid sequence similarities in six subfamilies, which are thyroid hormone receptor-like, retinoid x receptor-like, estrogen receptor-like, nerve growth factor IB-like, steroidogenic factor-like and germ cell nuclear factor-like [217]. Moreover, some NRs require heterodimerization with retinoid X receptor (RXR) [217]. Furthermore, NRs-ligands interactions are characterized by certain redundancy: ligands are nonselective for particular receptors, which also share their transcriptional targets, serving as transcriptional inducers of one another. However, several NRs remain with unknown ligands and are described as “orphan receptors” [219].
All NRs are conservative and similar in their general structure, which includes a ligand-binding/dimerization domain (LBD) and DNA-binding/weak dimerization domain (DBD) as well as at least one N-terminal ligand-independent transactivation region, referred to as AF-1 for activation function 1 (or the A/B domain), and a ligand-dependent transcription region AF-2. To bind DNA, AF-2 may form complexes with co-regulatory proteins that can act as co-activators or co-repressors. AF-2 co-activators regulate histone acetyltransferase activity, whereas its co-repressors control histone deacetylase activity [218].
Certain NRs are also linked with AD pathology as well as with AβOs toxicity. One of these receptors is vitamin D receptor (VDR) that is broadly expressed in brain and regulates many genes. VDR mediates action of Vitamin D (1,25-(OH)2D3), an important neurosteroid, which plays key role in the brain functioning, such as calcium signaling, cell proliferation and differentiation. Vitamin D is also a neurotrophic factor that regulates neurotransmission and synaptic plasticity. It was revealed recently, that Vitamin D treatment results in significant increase of LRP1 expression both in-vivo and in-vitro studies [220]. Moreover, it was suggested that VDR deficiency/inhibition can be a potential risk factor for AD [221]. It was shown that Vitamin D may be also involved in Aβ clearance. 1,25-(OH)2D3 increases transport of Aβ across the BBB by regulating expression of amyloid transporters, such as LRP-1, via its nuclear receptor VDR only, or by binding heterodimeric complexes of VDR with RXR [222].
3.15. Sirtuin
Sirtuin 1 (SIRT 1), one of NRs that has recently emerged as a crucial protein that may play protective roles in AD and other NDs, including PD and MND (for review see [223]). SIRT1 belongs to the family of sirtuins (Sir2, silent information regulator 2 protein) that was shown to regulate lifespan in lower organisms and affect diseases of aging in mammals. SIRT1 is a nicotinamide adenine dinucleotide (NAD+)-dependent histone deacetylase involved in calorie restriction (CR) (reviewed in [224]). Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. As it was mentioned above, it was proposed that AD may be described as new form of diabetes or “type 3 diabetes”. The resistance to insulin and insulin-like growth factor are thought to be crucial for the progression of AD [225]. SIRT1 deficiency is also ascribed to be responsible for the increased risk of insulin resistance, obesity and diabetes, including type 3 diabetes, whereas low-calorie diet and nutrition reverse type 3 diabetes and accelerated aging linked to global chronic diseases [226].
SIRT1 is involved in neurodevelopment, including axon elongation, neurite outgrowth and dendritic branching [223]. Furthermore, this NR is also essential for normal cognitive function and synaptic plasticity. It was demonstrated that SIRT1 attenuates amyloidogenic processing of APP by increasing α-secretase activity via SIRT1-coupled retinoic acid receptor-β (RARβ) activation [227]. Upregulation of α-secretase shifts APP processing to non-amyloidogenic cleavage of APP and reduces the pathological accumulation of the toxic Aβ species that results from β- and γ-secretase activity. It may be confirmed by the fact that a significant decrease in SIRT1 level, both mRNA and protein, was observed in the cortex of AD patients [228]. SIRT1 reduction paralleled tau accumulation in the AD brain and may be closely associated with deposition of Aβ in the cerebral cortex of patients with AD.
Although it is difficult to determine when exactly SIRT1 loss occurs in AD, it was suggested that it may be rather a relatively late event. A significant correlation was observed between SIRT1 and the duration of AD symptoms, accumulation of tau, as well as Aβ42 deposition [228]. Furthermore, it seems that AβOs toxicity and their binding to AβO receptors are rather primary toxic effects, than secondary to NRs disturbances with consecutive AβOs receptor interactions.
4. Conclusions
Since its first description over hundred years ago, Alzheimer’s disease is one of the diseases in modern biomedicine that have garnered most scientific attention. Within these 100 years there have emerged various hypotheses in order to explain underlying pathology. The dominant model of AD pathogenesis is amyloid hypothesis, although its details were changing over this time, indicating increasing role of oligomeric amyloid beta species as the main toxic factors leading to damage of neurons and loss of synapses. Recent studies have identified that soluble Aβ oligomers interact with certain receptor proteins.
In conclusion, a variety of specific receptors could be responsible for mediating the synaptotoxicity caused by AβOs in AD (Table 1). The AβO-associated receptors include ionotropic and metabotropic glutamate receptors NMDAR, AMPAR, and mGluR, their co-receptor—cellular prion protein PrPc, ephrin receptors EphB2 and EphA4, RAGE, immunoglobulin and immunoglobulin-like receptors FcγRIIB and PirB/LiL2R, neurotrophin receptor p75NTR, β-adrenergic as well as acetylcholine receptors a7nAChRs. Despite over twenty various protein receptors proposed within over twenty years of amyloid hypothesis, no single candidate receptor has been revealed to be necessary and sufficient to account for all features of AβO toxic activity. Taken together, it seems that among this abundancy glutamate and Eph receptors could explain most of the pathophysiological defects and structural changes observed in central nervous system. However, further studies are needed to determine the relevance and contribution of each of these molecules to the pathogenesis of this disease.
Funding
This research was funded by grants for neurodegenerative diseases, Medical University of Białystok, Poland. This research received no external funding.
Acknowledgments
B.M. has received consultation and/or lecture honoraria from Roche, Cormay and Biameditek. P.L. received research support from the Innovative Medicines Initiative Joint Undertaking under grant agreement n° 115372, resources of which are composed of financial contribution from the European Union’s Seventh Framework Programme (FP7/2007–2013) and EFPIA companies’ in kind contribution, and he received consultation and lectures honoraria from Innogenetics/Fujirebio Europe, IBL International, AJ Roboscreen, and Roche.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AβOs | Amyloid β oligomers |
| AD | Alzheimer’s disease |
| NDs | Neurodegenerative diseases |
| Aβ | Amyloid beta |
| NFTs | Neurofibrillary tangles |
| PD | Parkinson’s disease |
| MND | Motor neuron diseases |
| HD | Huntington’s disease |
| SCA | Spinocerebellar ataxia |
| SMA | Spinal muscular atrophy |
| pTau | Tau protein |
| CSF | Cerebrospinal fluid |
| PS | Presenilin |
| APP | Amyloid precursor protein |
| FAD | Familial Alzheimer’s disease |
| APOE | Apolipoprotein E |
| PSP | Progressive supranuclear palsy |
| CBD | Corticobasal degeneration |
| FTDP-17 | Frontotemporal dementia and Parkinsonism linked to chromosome 17 |
| LTP | Long-term potentiation |
| LTD | Long-term synaptic depression |
| LMW | Low molecular weight |
| HMW | High molecular weight |
| CNS | Central nervous system |
| NMDAR | N-methyl-d-aspartate receptor |
| ROS | reactive oxygen species |
| CaMKII | Ca2+/calmodulin-dependent kinase II |
| PSD-95 | Postsynaptic density-95 |
| AMPK | AMP-activated kinase |
| GLUTs | Glucose transporters |
| AMPAR | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor |
| mGluRs | Metabotropic glutamate receptors |
| PrPC | Cellular prion protein |
| AICD | Amyloid intracellular domain |
| BACE1 | β-site APP cleaving enzyme-1 |
| GPI | Glycosylphosphatidylinositol |
| β2ARs | β2-adrenergic receptors |
| α7nAChR | α7 nicotinic acetylcholine receptor |
| IR | Insulin receptor |
| NGF | Nerve growth factor |
| p75NTR | p75 neurotrophin receptor |
| TNF | Tumor necrosis factor |
| IGF-1R | Insulin-like growth factor 1 receptor |
| PI3K | Phosphatidylinositide 3-kinase |
| LilrB2 | Leukocyte immunoglobulin-like receptor B2 |
| LIR | Leukocyte immunoglobulin-like receptors |
| MHC | Major histocompatibility complex |
| PirB | Paired immunoglobulin-like receptor B |
| ODP | Ocular dominance plasticity |
| FcγR | Fc-gamma receptors |
| IgG | Immunoglobulin gamma |
| TREM2 | Triggering receptor expressed on myeloid cells 2 |
| DAP12 | DNAX-activating protein of 12 kDa |
| Eph | Erythropoietin-producing hepatocellular carcinoma |
| FN | Fibronectin |
| RAGE | Receptor for advanced glycation endproducts |
| LRP | Low density lipoprotein-related protein |
| RARβ | Retinoic acid receptor-β |
| HMGB-1 | High mobility group box 1 protein |
| AGE | Advanced glycation endproducts |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| gp330 | Glycoprotein 330 |
| LDLR | Low density lipoprotein receptor |
| NR | Nuclear receptor |
| VDR | Vitamin D receptor |
| LBD | Ligand-binding/dimerization domain |
| DBD | DNA-binding/weak dimerization domain |
| AF-1 | Activation function 1 |
| RXR | Retinoid X receptor |
| SIRT 1 | Sirtuin 1 |
References
- World Alzheimer Report 2015. Available online: https://www.alz.co.uk/research/WorldAlzheimerReport2015.pdf (accessed on 12 May 2018).
- JPND Research. Available online: http://www.neurodegenerationresearch.eu/about/what/ (accessed on 12 May 2018).
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological Alterations in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- De Leon, M.J.; Golomb, J.; George, A.E.; Convit, A.; Tarshish, C.Y.; McRae, T.; de Santi, S.; Smith, G.; Ferris, S.H.; Noz, M.; et al. The radiologic prediction of Alzheimer disease: The atrophic hippocampal formation. AJNR Am. J. Neuroradiol. 1993, 14, 897–906. [Google Scholar] [PubMed]
- Braak, H.; Braak, E. Evolution of the neuropathology of Alzheimer’s disease. Acta Neurol. Scand. Suppl. 1996, 165, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Beason-Held, L.L.; Goh, J.O.; An, Y.; Kraut, M.A.; O’Brien, R.J.; Ferrucci, L.; Resnick, S.M. Changes in brain function occur years before the onset of cognitive impairment. J. Neurosci. 2013, 33, 18008–18014. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R.; Knopman, D.S.; Jagust, W.J.; Petersen, R.C.; Weiner, M.W.; Aisen, P.S.; Shaw, L.M.; Vemuri, P.; Wiste, H.J.; Weigand, S.D.; et al. Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013, 12, 207–216. [Google Scholar] [CrossRef]
- Reitz, C.; Mayeux, R. Alzheimer disease: Epidemiology, Diagnostic Criteria, Risk Factors and Biomarkers. Biochem. Pharmacol. 2014, 88, 640–651. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Gwinn-Hardy, K. Genetic classification of primary neurodegenerative disease. Science 1998, 282, 1075–1079. [Google Scholar] [CrossRef] [PubMed]
- Holtzman, D.M.; Herz, J.; Bu, G. Apolipoprotein E and apolipoprotein E receptors: Normal biology and roles in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006312. [Google Scholar] [CrossRef] [PubMed]
- Spinney, L. Alzheimer’s disease: The forgetting gene. Nature 2014, 510, 26–28. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer, A. Über eine eigenartige Erkrankung der Hirnrinde. Allgemeine Zeitschrift fur Psychiatrie und Psychisch-Gerichtliche Medizin 1907, 64, 146–148. [Google Scholar]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Mahley, R.W.; Weisgraber, K.H.; Huang, Y. Apolipoprotein E4: A causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5644–5651. [Google Scholar] [CrossRef] [PubMed]
- Strittmatter, W.J.; Saunders, A.M.; Schmechel, D.; Pericak-Vance, M.; Enghild, J.; Salvesen, G.S.; Roses, A.D. Apolipoprotein E: High-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 1977–1981. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Yee, A.; Brewer, H.B.; Das, S.; Potter, H. The amyloid-associated proteins a1-antichymotrypsin and apolipoprotein E promote the assembly of the Alzheimer b-protein into filaments. Nature 1994, 372, 92–94. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, T.; Castano, A.M.; Golabek, A.; Vogel, T.; Frangione, B. Acceleration of Alzheimer’s fibril formation by apolipoprotein E in vitro. Am. J. Pathol. 1994, 145, 1030–1035. [Google Scholar] [PubMed]
- Evans, K.C.; Berger, E.P.; Cho, C.G.; Weisgraber, K.H.; Lansbury, P.T. Apolipoprotein E is a kinetic but not a thermodynamic inhibitor of amyloid formation: Implications for the pathogenesis and treatment of Alzheimer disease. Proc. Natl. Acad. Sci. USA 1995, 92, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Polvikoski, T.; Sulkava, R.; Haltia, M.; Kainulainen, K.; Vuorio, A.; Verkkoniemi, A.; Niinistö, L.; Halonen, P.; Kontula, K. Apolipoprotein E, dementia, and cortical deposition of beta-amyloid protein. N. Engl. J. Med. 1995, 333, 1242–1247. [Google Scholar] [CrossRef] [PubMed]
- Rebeck, G.W.; Reiter, J.S.; Strickland, D.K.; Hyman, B.T. Apolipoprotein E in sporadic Alzheimer’s disease: Allelic variation and receptor interactions. Neuron 1993, 11, 575–580. [Google Scholar] [CrossRef]
- Castellano, J.M.; Kim, J.; Stewart, F.R.; Jiang, H.; DeMattos, R.B.; Patterson, B.W.; Fagan, A.M.; Morris, J.C.; Mawuenyega, K.G.; Cruchaga, C.; et al. Human apoE isoforms differentially regulate brain amyloid-b peptide clearance. Sci. Transl. Med. 2011, 3, 89ra57. [Google Scholar] [CrossRef] [PubMed]
- Waring, S.C.; Rosenberg, R.N. Genome-Wide Association Studies in Alzheimer Disease. Arch. Neurol. 2008, 65, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Priller, C.; Bauer, T.; Mitteregger, G.; Krebs, B.; Kretzschmar, H.A.; Herms, J. Synapse formation and function is modulated by the amyloid precursor protein. J. Neurosci. 2006, 26, 7212–7221. [Google Scholar] [CrossRef] [PubMed]
- Turner, P.R.; O’Connor, K.; Tate, W.P.; Abraham, W.C. Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory. Prog. Neurobiol. 2003, 70, 1–32. [Google Scholar] [CrossRef]
- Duce, J.A.; Tsatsanis, A.; Cater, M.A.; James, S.A.; Robb, E.; Wikhe, K.; Leong, S.L.; Perez, K.; Johanssen, T.; Greenough, M.A.; et al. Iron-export ferroxidase activity of β-amyloid precursor protein is inhibited by zinc in Alzheimer’s disease. Cell 2010, 142, 857–867. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B.; Annaert, W. Proteolytic processing and cell biological functions of the amyloid precursor protein. J. Cell Sci. 2000, 113, 1857–1870. [Google Scholar] [PubMed]
- Zheng, H.; Koo, E.H. The amyloid precursor protein: Beyond amyloid. Mol. Neurodegener. 2006, 1, 5. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Hasegawa, H.; Schmitt-Ulms, G.; Kawarai, T.; Bohm, C.; Katayama, T.; Gu, Y.; Sanjo, N.; Glista, M.; Rogaeva, E.; et al. TMP21 is a presenilin complex component that modulates gamma-secretase but not epsilon-secretase activity. Nature 2006, 440, 1208–1212. [Google Scholar] [CrossRef] [PubMed]
- Ehehalt, R.; Keller, P.; Haass, C.; Thiele, C.; Simons, K. Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J. Cell Biol. 2003, 160, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Vetrivel, K.S.; Cheng, H.; Lin, W.; Sakurai, T.; Li, T.; Nukina, N.; Wong, P.C.; Xu, H.; Thinakaran, G. Association of gamma-secretase with lipid rafts in post-Golgi and endosome membranes. J. Biol. Chem. 2004, 279, 44945–44954. [Google Scholar] [CrossRef] [PubMed]
- Riddell, D.R.; Christie, G.; Hussain, I.; Dingwall, C. Compartmentalization of beta-secretase (Asp2) into low-buoyant density, noncaveolar lipid rafts. Curr. Biol. 2001, 11, 1288–1293. [Google Scholar] [CrossRef]
- Selkoe, D.J. Translating cell biology into therapeutic advances in Alzheimer’s disease. Nature 1999, 399, A23–A31. [Google Scholar] [CrossRef] [PubMed]
- Lemere, C.A.; Blustzjan, J.K.; Yamaguchi, H.; Wisniewski, T.; Saido, T.C.; Selkoe, D.J. Sequence of deposition of heterogeneous amyloid b-peptides and Apo E in Down syndrome: Implications for initial events in amyloid plaque formation. Neurobiol. Dis. 1996, 3, 16–32. [Google Scholar] [CrossRef] [PubMed]
- Lemere, C.A.; Lopera, F.; Kosik, K.S.; Lendon, C.L.; Ossa, J.; Saido, T.C.; Yamaguchi, H.; Ruiz, A.; Martinez, A.; Madrigal, L.; et al. The E280A presenilin 1 Alzheimer mutation produces increased Ab42 deposition and severe cerebellar pathology. Nat. Med. 1996, 2, 1146–1150. [Google Scholar] [CrossRef] [PubMed]
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 2012, 367, 795–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Onstead, L.; Randle, S.; Price, R.; Smithson, L.; Zwizinski, C.; Dickson, D.W.; Golde, T.; McGowan, E. Abeta40 inhibits amyloid deposition in vivo. J. Neurosci. 2007, 27, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Borchelt, D.R.; Thinakaran, G.; Eckman, C.B.; Lee, M.K.; Davenport, F.; Ratovitsky, T.; Prada, C.M.; Kim, G.; Seekins, S.; Yager, D.; et al. Familial Alzheimer’s Disease–Linked Presenilin 1 Variants Elevate Aβ1–42/1–40 Ratio In Vitro and In Vivo. Neuron 1996, 17, 1005–1013. [Google Scholar] [CrossRef]
- Shioi, J.; Georgakopoulos, A.; Mehta, P.; Kouchi, Z.; Litterst, C.M.; Baki, L.; Robakis, N.K. FAD mutants unable to increase neurotoxic Abeta 42 suggest that mutation effects on neurodegeneration may be independent of effects on Abeta. J. Neurochem. 2007, 101, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Nistor, M.; Don, M.; Parekh, M.; Sarsoza, F.; Goodus, M.; Lopez, G.E.; Kawas, C.; Leverenz, J.; Doran, E.; Lott, I.T.; Hill, M.; Head, E. Alpha- and beta-secretase activity as a function of age and beta-amyloid in Down syndrome and normal brain. Neurobiol. Aging 2007, 28, 1493–1506. [Google Scholar] [CrossRef] [PubMed]
- Lott, I.T.; Head, E. Alzheimer disease and Down syndrome: Factors in pathogenesis. Neurobiol. Aging 2005, 26, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, T.; Atwal, J.K.; Steinberg, S.; Snaedal, J.; Jonsson, P.V.; Bjornsson, S.; Stefansson, H.; Sulem, P.; Gudbjartsson, D.; Maloney, J.; et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 2012, 488, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Chételat, G. Alzheimer disease: Aβ-independent processes-rethinking preclinical AD. Nat. Rev. Neurol. 2013, 9, 123–124. [Google Scholar] [CrossRef] [PubMed]
- Chételat, G. Reply: The amyloid cascade is not the only pathway to AD. Nat. Rev. Neurol. 2013, 9, 356. [Google Scholar] [CrossRef] [PubMed]
- Vishnu, V.Y. Can tauopathy shake the amyloid cascade hypothesis? Nat. Rev. Neurol. 2013, 9, 356. [Google Scholar] [CrossRef] [PubMed]
- Mudher, A.; Lovestone, S. Alzheimer’s disease-do tauists and baptists finally shake hands? Trends Neurosci. 2002, 25, 22–26. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G.; Jakes, R.; Rutherford, D.; Crowther, R.A. Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles in Alzheimer’s disease. Neuron 1989, 3, 519–526. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G.; Crowther, R.A. Tau proteins and neurofibrillary degeneration. Brain Pathol. 1991, 1, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; del C Alonso, A.; Chen, S.; Chohan, M.O.; El-Akkad, E.; Gong, C.X.; Khatoon, S.; Li, B.; Liu, F.; Rahman, A.; et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim. Biophys. Acta 2005, 1739, 198–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun, W.; Johnson, G.V. The role of tau phosphorylation and cleavage in neuronal cell death. Front. Biosci. 2007, 12, 733–756. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, M.; Shimada, H.; Suhara, T.; Shinotoh, H.; Ji, B.; Maeda, J.; Zhang, M.R.; Trojanowski, J.Q.; Lee, V.M.; Ono, M.; et al. Imaging of tau pathology in a tauopathy mouse model and in Alzheimer patients compared to normal controls. Neuron 2013, 79, 1094–1108. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Weksler, M.E.; Relkin, N.; Turkenich, R.; LaRusse, S.; Zhou, L.; Szabo, P. Patients with Alzheimer disease have lower levels of serum anti-amyloid peptide antibodies than healthy elderly individuals. Exp. Gerontol. 2002, 37, 943–948. [Google Scholar] [CrossRef]
- Su, B.; Wang, X.; Nunomura, A.; Moreira, P.I.; Lee, H.G.; Perry, G.; Smith, M.A.; Zhu, X. Oxidative stress signaling in Alzheimer’s disease. Curr. Alzheimer Res. 2008, 5, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Kastenholz, B.; Garfin, D.E.; Horst, J.; Nagel, K.A. Plant metal chaperones: A novel perspective in dementia therapy. Amyloid 2009, 16, 81–83. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.V.; Leitner, W.P.; Rivest, A.J.; Staples, M.K.; Radeke, M.J.; Anderson, D.H. The Alzheimer’s Aß-peptide is deposited at sites of complement activation in pathologic deposits associated with aging and age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2002, 99, 11830–11835. [Google Scholar] [CrossRef] [PubMed]
- Tuppo, E.E.; Arias, H.R. The role of inflammation in Alzheimer’s disease. Int. J. Biochem. Cell Biol. 2005, 37, 289–305. [Google Scholar] [CrossRef] [PubMed]
- Rebeck, G.W. The role of APOE on lipid homeostasis and inflammation in normal brains. J. Lipid Res. 2017, 58, 1493–1499. [Google Scholar] [CrossRef] [PubMed]
- Vitek, M.P.; Brown, C.M.; Colton, C.A. APOE genotype-specific differences in the innate immune response. Neurobiol. Aging 2009, 30, 1350–1360. [Google Scholar] [CrossRef] [PubMed]
- Lansbury, P.T. Evolution of amyloid: What normal protein folding may tell us about fibrillogenesis and disease. Proc. Natl. Acad. Sci. USA 1999, 96, 3342–3344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003, 300, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Scheff, S.W.; Price, D.A.; Schmitt, F.A.; DeKosky, S.T.; Mufson, E.J. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology 2007, 68, 1501–1508. [Google Scholar] [CrossRef] [PubMed]
- Lanz, T.A.; Carter, D.B.; Merchant, K.M. Dendritic spine loss in the hippocampus of young PDAPP and Tg2576 mice and its prevention by the ApoE2 genotype. Neurobiol. Dis. 2003, 13, 246–253. [Google Scholar] [CrossRef]
- Koffie, R.M.; Meyer-Luehmann, M.; Hashimoto, T.; Adams, K.W.; Mielke, M.L.; Garcia-Alloza, M.; Micheva, K.D.; Smith, S.J.; Kim, M.L.; Lee, V.M.; et al. Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc. Natl. Acad. Sci. USA 2009, 106, 4012–4017. [Google Scholar] [CrossRef] [PubMed]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, nonfibrillar ligands derived from A 1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA 1998, 95, 6448–6453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsia, A.Y.; Masliah, E.; McConlogue, L.; Yu, G.Q.; Tatsuno, G.; Hu, K.; Kholodenko, D.; Malenka, R.C.; Nicoll, R.A.; Mucke, L. Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc. Natl. Acad. Sci. USA 1999, 96, 3228–3233. [Google Scholar] [CrossRef] [PubMed]
- Nath, S.; Agholme, L.; Kurudenkandy, F.R.; Granseth, B.; Marcusson, J.; Hallbeck, M. Spreading of neurodegenerative pathology via neuron-to-neuron transmission of beta-amyloid. J. Neurosci. 2012, 32, 8767–8777. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Fritschi, S.K.; Schelle, J.; Obermuller, U.; Degenhardt, K.; Kaeser, S.A.; Eisele, Y.S.; Walker, L.C.; Baumann, F.; Staufenbiel, M.; et al. Persistence of Abeta seeds in APP null mouse brain. Nat. Neurosci. 2015, 18, 1559–1561. [Google Scholar] [CrossRef] [PubMed]
- Domert, J.; Rao, S.B.; Agholme, L.; Brorsson, A.C.; Marcusson, J.; Hallbeck, M.; Nath, S. Spreading of amyloid-beta peptides via neuritic cell-to-cell transfer is dependent on insufficient cellular clearance. Neurobiol. Dis. 2014, 65, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Condello, C.; Stöehr, J. Aβ propagation and strains: Implications for the phenotypic diversity in Alzheimer’s disease. Neurobiol. Dis. 2018, 109, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Benilova, I.; Karran, E.; De Strooper, B. The toxic Abeta oligomer and Alzheimer’s disease: An emperor in need of clothes. Nat. Neurosci. 2012, 15, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Kostylev, M.A.; Kaufman, A.C.; Nygaard, H.B.; Patel, P.; Haas, L.T.; Gunther, E.C.; Vortmeyer, A.; Strittmatter, S.M. Prion-protein-interacting amyloid-beta oligomers of high molecular weight are tightly correlated with memory impairment in multiple Alzheimer mouse models. J. Biol. Chem. 2015, 290, 17415–17438. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda, F.J.; Parodi, J.; Peoples, R.W.; Opazo, C.; Aguayo, L.G. Synaptotoxicity of Alzheimer Beta Amyloid Can Be Explained by Its Membrane Perforating Property. PLoS ONE 2010, 5, e11820. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, K.C.; Marunde, M.R.; Das, A.; Velasco, P.T.; Kuhns, B.D.; Marty, M.T.; Klein, W.L. Nanoscale Synaptic Membrane Mimetic Allows Unbiased High Throughput Screen That Targets Binding Sites for Alzheimer’s-Associated Aβ Oligomers. PLoS ONE 2015, 10, e0125263. [Google Scholar] [CrossRef] [PubMed]
- Kayed, R.; Lasagna-Reeves, C.A. Molecular mechanisms of amyloid oligomers toxicity. J. Alzheimers Dis. 2013, 33, S67–S78. [Google Scholar] [CrossRef] [PubMed]
- Chromy, B.A.; Nowak, R.J.; Lambert, M.P.; Viola, K.L.; Chang, L.; Velasco, P.T.; Jones, B.W.; Fernandez, S.J.; Lacor, P.N.; Horowitz, P.; et al. Self-assembly of Abeta(1-42) into globular neurotoxins. Biochemistry 2003, 42, 12749–12760. [Google Scholar] [CrossRef] [PubMed]
- Lambert, M.P.; Viola, K.L.; Chromy, B.A.; Chang, L.; Morgan, T.E.; Yu, J.; Venton, D.L.; Krafft, G.A.; Finch, C.E.; Klein, W.L. Vaccination with soluble Abeta oligomers generates toxicity-neutralizing antibodies. J. Neurochem. 2001, 79, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Chang, L.; Viola, K.L.; Lacor, P.N.; Lambert, M.P.; Finch, C.E.; Krafft, G.A.; Klein, W.L. Alzheimer’s disease-affected brain: Presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc. Natl. Acad. Sci. USA 2003, 100, 10417–10422. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.W.; Pasternak, J.F.; Kuo, H.; Ristic, H.; Lambert, M.P.; Chromy, B.; Viola, K.L.; Klein, W.L.; Stine, W.B.; Krafft, G.A. Soluble oligomers of beta amyloid (1-42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain Res. 2002, 924, 133–140. [Google Scholar] [CrossRef]
- Kim, H.J.; Chae, S.C.; Lee, D.K. Selective neuronal degeneration induced by soluble oligomeric amyloid beta protein. FASEB J. 2003, 17, 118–120. [Google Scholar] [CrossRef] [PubMed]
- Jarosz-Griffiths, H.H.; Noble, E.; Rushworth, J.V.; Hooper, N.M. Amyloid-β Receptors: The Good, the Bad, and the Prion Protein. J. Biol. Chem. 2016, 291, 3174–3183. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Qi, Y.; Klyubin, I.; Ondrejcak, T.; Sarell, C.J.; Cuello, A.C.; Collinge, J.; Rowan, M.J. Targeting glutamatergic and cellular prion protein mechanisms of amyloid β-mediated persistent synaptic plasticity disruption: Longitudinal studies. Neuropharmacology 2017, 15, 231–246. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Tsien, J.Z. Memory and the NMDA Receptors. N. Engl. J. Med. 2009, 361, 302–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, E.M.; Nong, Y.; Almeida, C.G.; Paul, S.; Moran, T.; Choi, E.Y.; Nairn, A.C.; Salter, M.W.; Lombroso, P.J.; Gouras, G.K.; et al. Regulation of NMDA receptor trafficking by amyloid-beta. Nat. Neurosci. 2005, 8, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Bloodgood, B.L.; Townsend, M.; Walsh, D.M.; Selkoe, D.J.; Sabatini, B.L. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptordependent signaling pathway. J. Neurosci. 2007, 27, 2866–2875. [Google Scholar] [CrossRef] [PubMed]
- Tamburri, A.; Dudilot, A.; Licea, S.; Bourgeois, C.; Boehm, J. NMDA-receptor activation but not ion flux is required for amyloid-beta induced synaptic depression. PLoS ONE 2013, 8, e65350. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Nguyen, L.N.; Kessels, H.W.; Hagiwara, H.; Sisodia, S.; Malinow, R. Amyloid beta from axons and dendrites reduces local spine number and plasticity. Nat. Neurosci. 2010, 13, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Yamin, G. NMDA receptor-dependent signaling pathways that underlie amyloid beta-protein disruption of LTP in the hippocampus. J. Neurosci. Res. 2009, 87, 1729–1736. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G.; Velasco, P.T.; Lambert, M.P.; Viola, K.; Fernandez, S.J.; Ferreira, S.T.; Klein, W.L. Abeta oligomers induce neuronal oxidative stress through an N-methyl-d-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J. Biol. Chem. 2007, 282, 11590–11601. [Google Scholar] [CrossRef] [PubMed]
- Takahashi-Ito, K.; Makino, M.; Okado, K.; Tomita, T. Memantine inhibits β-amyloid aggregation and disassembles preformed β-amyloid aggregates. Biochem. Biophys. Res. Commun. 2017, 493, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Demuro, A.; Mina, E.; Kayed, R.; Milton, S.C.; Parker, I.; Glabe, C.G. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J. Biol. Chem. 2005, 280, 17294–17300. [Google Scholar] [CrossRef] [PubMed]
- Alberdi, E.; Sánchez-Gómez, M.V.; Cavaliere, F.; Pérez-Samartín, A.; Zugaza, J.L.; Trullas, R.; Domercq, M.; Matute, C. Amyloid beta oligomers induce Ca2+ dysregulation and neuronal death through activation of ionotropic glutamate receptors. Cell Calcium 2010, 47, 264–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, J.; Kulasiri, D.; Samarasinghe, S. Computational investigation of Amyloid-β-induced location- and subunit-specific disturbances of NMDAR at hippocampal dendritic spine in Alzheimer’s disease. PLoS ONE 2017, 12, e0182743. [Google Scholar] [CrossRef] [PubMed]
- Popugaeva, E.; Pchitskaya, E.; Bezprozvanny, I. Dysregulation of neuronal calcium homeostasis in Alzheimer’s disease—A therapeutic opportunity? Biochem. Biophys. Res. Commun. 2017, 483, 998–1004. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Watson, J.B.; Xie, C.W. Amyloid beta prevents activation of calcium/calmodulin-dependent protein kinase II and AMPA receptor phosphorylation during hippocampal long-term potentiation. J. Neurophysiol. 2004, 92, 2853–2858. [Google Scholar] [CrossRef] [PubMed]
- Sivanesan, S.; Tan, A.; Rajadas, J. Pathogenesis of Abeta oligomers in synaptic failure. Curr. Alzheimer Res. 2013, 10, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Seixas da Silva, G.S.; Melo, H.M.; Lourenco, M.V.; Lyra, E.; Silva, N.M.; de Carvalho, M.B.; Alves-Leon, S.V.; de Souza, J.M.; Klein, W.L.; da-Silva, W.S.; et al. Amyloid-β oligomers transiently inhibit AMP-activated kinase and cause metabolic defects in hippocampal neurons. J. Biol. Chem. 2017, 292, 7395–7406. [Google Scholar] [CrossRef] [PubMed]
- Boehm, J.; Kang, M.G.; Johnson, R.C.; Esteban, J.; Huganir, R.L.; Malinow, R. Synaptic incorporation of AMPA receptors during LTP is controlled by a PKC phosphorylation site on GluR1. Neuron 2006, 51, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Maren, S.; Tocco, G.; Standley, S.; Baudry, M.; Thompson, R.F. Postsynaptic factors in the expression of long-term potentiation (LTP): Increased glutamate receptor binding following LTP induction in vivo. Proc. Natl. Acad. Sci. USA 1993, 90, 9654–9658. [Google Scholar] [CrossRef] [PubMed]
- Banke, T.G.; Bowie, D.; Lee, H.; Huganir, R.L.; Schousboe, A.; Traynelis, S.F. Control of GluR1 AMPA receptor function by cAMP-dependent protein kinase. J. Neurosci. 2000, 20, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, H.; Boehm, J.; Sato, C.; Iwatsubo, T.; Tomita, T.; Sisodia, S.; Malinow, R. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron 2006, 52, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kurup, P.; Xu, J.; Anderson, G.M.; Greengard, P.; Nairn, A.C.; Lombroso, P.J. Reduced levels of the tyrosine phosphatase STEP block β amyloid-mediated GluA1/GluA2 receptor internalization. J. Neurochem. 2011, 119, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Rui, Y.; Gu, J.; Yu, K.; Hartzell, H.C.; Zheng, J.Q. Inhibition of AMPA receptor trafficking at hippocampal synapses by beta-amyloid oligomers: The mitochondrial contribution. Mol. Brain 2010, 3, 10. [Google Scholar] [CrossRef] [PubMed]
- Guntupalli, S.; Jang, S.E.; Zhu, T.; Huganir, R.L.; Widagdo, J.; Anggono, V. GluA1 subunit ubiquitination mediates amyloid-β-induced loss of surface α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors. J. Biol. Chem. 2017, 292, 8186–8194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miñano-Molina, A.J.; España, J.; Martín, E. Soluble oligomers of amyloid-β peptide disrupt membrane trafficking of α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor contributing to early synapse dysfunction. J. Biol. Chem. 2011, 286, 27311–27321. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.Q.; Santini, F.; Breese, R.; Ross, D.; Zhang, X.D.; Stone, D.J.; Ferrer, M.; Townsend, M.; Wolfe, A.L.; Seager, M.A.; et al. Inhibition of calcineurin-mediated endocytosis and alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors prevents amyloid beta oligomer-induced synaptic disruption. J. Biol. Chem. 2010, 285, 7619–7632. [Google Scholar] [CrossRef] [PubMed]
- Renner, M.C.; Albers, E.H.; Gutierrez-Castellanos, N.; Reinders, N.R.; van Huijstee, A.N.; Xiong, H.; Lodder, T.R.; Kessels, H.W. Synaptic plasticity through activation of GluA3-containing AMPA-receptors. eLife 2017, 6, e25462. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Castellanos, N.; Da Silva-Matos, C.M.; Zhou, K.; Canto, C.B.; Renner, M.C.; Koene, L.M.C.; Ozyildirim, O.; Sprengel, R.; Kessels, H.W.; De Zeeuw, C.I. Motor Learning Requires Purkinje Cell Synaptic Potentiation through Activation of AMPA-Receptor Subunit GluA3. Neuron 2017, 93, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Reinders, N.R.; Pao, Y.; Renner, M.C.; da Silva-Matos, C.M.; Lodder, T.R.; Malinow, R.; Kessels, H.W. Amyloid-β effects on synapses and memory require AMPA receptor subunit GluA3. Proc. Natl. Acad. Sci. USA 2016, 113, E6526–E6534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, E.C.; Teravskis, P.J.; Dummer, B.W.; Zhao, X.; Huganir, R.L.; Liao, D. Tau phosphorylation and tau mislocalization mediate soluble Aβ oligomer-induced AMPA glutamate receptor signaling deficits. Eur. J. Neurosci. 2014, 39, 1214–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minakami, R.; Katsuki, F.; Yamamoto, T.; Nakamura, K.; Sugiyama, H. Molecular cloning and the functional expression of two isoforms of human metabotropic glutamate receptor subtype 5. Biochem. Biophys. Res. Commun. 1994, 199, 1136–1143. [Google Scholar] [CrossRef] [PubMed]
- Gilman, A.G. G proteins: Transducers of receptor-generated signals. Annu. Rev. Biochem. 1987, 56, 615–649. [Google Scholar] [CrossRef] [PubMed]
- Um, J.W.; Kaufman, A.C.; Kostylev, M.; Heiss, J.K.; Stagi, M.; Takahashi, H.; Kerrisk, M.E.; Vortmeyer, A.; Wisniewski, T.; Koleske, A.J.; et al. Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer aβ oligomer bound to cellular prion protein. Neuron 2013, 79, 887–902. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Yadav, S.P.; Surewicz, W.K. Interaction between human prion protein and amyloid-beta (Abeta) oligomers: Role OF N-terminal residues. J. Biol. Chem. 2010, 285, 26377–26383. [Google Scholar] [CrossRef] [PubMed]
- Lauren, J.; Gimbel, D.A.; Nygaard, H.B.; Gilbert, J.W.; Strittmatter, S.M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 2009, 457, 1128–1132. [Google Scholar] [CrossRef] [PubMed]
- Lazzari, C.; Kipanyula, M.J.; Agostini, M.; Pozzan, T.; Fasolato, C. Aβ42 oligomers selectively disrupt neuronal calcium release. Neurobiol. Aging 2015, 36, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Parkin, E.T.; Watt, N.T.; Hussain, I.; Eckman, E.A.; Eckman, C.B.; Manson, J.C.; Baybutt, H.N.; Turner, A.J.; Hooper, N.M. Cellular prion protein regulates beta-secretase cleavage of the Alzheimer’s amyloid precursor protein. Proc. Natl. Acad. Sci. USA 2007, 104, 11062–11067. [Google Scholar] [CrossRef] [PubMed]
- Vincent, B.; Sunyach, C.; Orzechowski, H.D.; St George-Hyslop, P.; Checler, F. p53-Dependent transcriptional control of cellular prion by presenilins. J. Neurosci. 2009, 29, 6752–6760. [Google Scholar] [CrossRef] [PubMed]
- Younan, N.D.; Sarell, C.J.; Davies, P.; Brown, D.R.; Viles, J.H. The cellular prion protein traps Alzheimer’s Abeta in an oligomeric form and disassembles amyloid fibers. FASEB J. 2013, 27, 1847–1858. [Google Scholar] [CrossRef] [PubMed]
- Bove-Fenderson, E.; Urano, R.; Straub, J.E.; Harris, D.A. Cellular prion protein targets amyloid-β fibril ends via its C-terminal domain to prevent elongation. J. Biol. Chem. 2017, 292, 16858–16871. [Google Scholar] [CrossRef] [PubMed]
- Beraldo, F.H.; Ostapchenko, V.G.; Caetano, F.A.; Guimaraes, A.L.; Ferretti, G.D.; Daude, N.; Bertram, L.; Nogueira, K.O.; Silva, J.L.; Westaway, D.; et al. Regulation of Amyloid β Oligomer Binding to Neurons and Neurotoxicity by the Prion Protein-mGluR5 Complex. J. Biol. Chem. 2016, 291, 21945–21955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Río, J.A.; Ferrer, I.; Gavín, R. Role of cellular prion protein in interneuronal amyloidtransmission. Prog. Neurobiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Beraldo, F.H.; Arantes, C.P.; Santos, T.G.; Queiroz, N.G.; Young, K.; Rylett, R.J.; Markus, R.P.; Prado, M.A.; Martins, V.R. Role of alpha7 nicotinic acetylcholine receptor in calcium signaling induced by prion protein interaction with stress-inducible protein 1. J. Biol. Chem. 2010, 285, 36542–36550. [Google Scholar] [CrossRef] [PubMed]
- Beraldo, F.H.; Arantes, C.P.; Santos, T.G.; Machado, C.F.; Roffe, M.; Hajj, G.N.; Lee, K.S.; Magalhães, A.C.; Caetano, F.A.; Mancini, G.L.; et al. Metabotropic glutamate receptors transduce signals for neurite outgrowth after binding of the prion protein to laminin gamma1 chain. FASEB J. 2011, 25, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Haas, L.T.; Salazar, S.V.; Kostylev, M.A.; Um, J.W.; Kaufman, A.C.; Strittmatter, S.M. Metabotropic glutamate receptor 5 couples cellular prion protein to intracellular signalling in Alzheimer’s disease. Brain 2016, 139, 526–546. [Google Scholar] [CrossRef] [PubMed]
- You, H.; Tsutsui, S.; Hameed, S.; Kannanayakal, T.J.; Chen, L.; Xia, P.; Engbers, J.D.; Lipton, S.A.; Stys, P.K.; Zamponi, G.W. Abeta neurotoxicity depends on interactions between copper ions, prion protein, and N-methyl-d-aspartate receptors. Proc. Natl. Acad. Sci. USA 2012, 109, 1737–1742. [Google Scholar] [CrossRef] [PubMed]
- Chin, J.; Palop, J.J.; Yu, G.Q.; Kojima, N.; Masliah, E.; Mucke, L. Fyn kinase modulates synaptotoxicity, but not aberrant sprouting, in human amyloid precursor protein transgenic mice. J. Neurosci. 2004, 24, 4692–4697. [Google Scholar] [CrossRef] [PubMed]
- Daly, C.J.; McGrath, J.C. Previously unsuspected widespread cellular and tissue distribution of beta-adrenoceptors and its relevance to drug action. Trends Pharmacol. Sci. 2011, 32, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, M.E.; Summers, R.J. Role of adrenoceptor subtypes in memory consolidation. Prog. Neurobiol. 2002, 67, 345–391. [Google Scholar] [CrossRef]
- McIntyre, C.K.; McGaugh, J.L.; Williams, C.L. Interacting Brain Systems Modulate Memory Consolidation. Neurosci. Biobehav. Rev. 2012, 36, 1750–1762. [Google Scholar] [CrossRef] [PubMed]
- Connor, S.A.; Wang, Y.T.; Nguyen, P.V. Activation of β-adrenergic receptors facilitates heterosynaptic translation-dependent long-term potentiation. J. Physiol. 2011, 589, 4321–4340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelinas, J.N.; Nguyen, P.V. Beta-adrenergic receptor activation facilitates induction of a protein synthesis-dependent late phase of long-term potentiation. J. Neurosci. 2005, 25, 3294–3303. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.W.; Min, M.Y.; Chiu, T.H.; Yang, H.W. Enhancement of associative long-term potentiation by activation of beta-adrenergic receptors at CA1 synapses in rat hippocampal slices. J. Neurosci. 2003, 23, 74173–74181. [Google Scholar] [CrossRef]
- Qian, H.; Matt, L.; Zhang, M.; Nguyen, M.; Patriarchi, T.; Koval, O.M.; Anderson, M.E.; He, K.; Lee, H.K.; Hell, J.W. β2-Adrenergic receptor supports prolonged theta tetanus-induced LTP. J. Neurophysiol. 2012, 107, 2703–2712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, M.J.; Moody, T.D.; Makhinson, M.; O’Dell, T.J. Activity-dependent beta-adrenergic modulation of low frequency stimulation induced LTP in the hippocampal CA1 region. Neuron 1996, 17, 475–482. [Google Scholar] [CrossRef]
- Walling, S.G.; Harley, C.W. Locus ceruleus activation initiates delayed synaptic potentiation of perforant path input to the dentate gyrus in awake rats: A novel beta-adrenergic- and protein synthesis-dependent mammalian plasticity mechanism. J. Neurosci. 2004, 24, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Joiner, M.L.; Lise, M.F.; Yuen, E.Y.; Kam, A.Y.; Zhang, M.; Hall, D.D.; Malik, Z.A.; Qian, H.; Chen, Y.; Ulrich, J.D.; et al. Assembly of a beta2-adrenergic receptor-GluR1 signalling complex for localized cAMP signalling. EMBO J. 2010, 29, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Govindaiah, G.; Liu, R.; De Arcangelis, V.; Cox, C.L.; Xiang, Y.K. Binding of amyloid beta peptide to beta2 adrenergic receptor induces PKA-dependent AMPA receptor hyperactivity. FASEB J. 2010, 24, 3511–3521. [Google Scholar] [CrossRef] [PubMed]
- Chai, G.; Wang, Y.; Yasheng, A.; Zhao, P. Beta 2-adrenergic receptor activation enhances neurogenesis in Alzheimer’s disease mice. Neural Regen. Res. 2016, 11, 1617–1624. [Google Scholar] [CrossRef] [PubMed]
- Marien, M.R.; Colpaert, F.C.; Rosenquist, A.C. Noradrenergic mechanisms in neurodegenerative diseases: A theory. Brain Res. Brain Res. Rev. 2004, 45, 38–78. [Google Scholar] [CrossRef] [PubMed]
- Szot, P.; White, S.S.; Greenup, J.L.; Leverenz, J.B.; Peskind, E.R.; Raskind, M.A. Compensatory changes in the noradrenergic nervous system in the locus ceruleus and hippocampus of postmortem subjects with Alzheimer’s disease and dementia with Lewy bodies. J. Neurosci. 2006, 26, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Manaye, K.F.; Mouton, P.R.; Xu, G.; Drew, A.; Lei, D.L.; Sharma, Y.; Rebeck, G.W.; Turner, S. Age-related loss of noradrenergic neurons in the brains of triple transgenic mice. Age 2013, 35, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Zhao, X.; Bao, G.; Zou, L.; Teng, L.; Wang, Z.; Song, M.; Xiong, J.; Bai, Y.; Pei, G. Activation of beta2-adrenergic receptor stimulates gamma-secretase activity and accelerates amyloid plaque formation. Nat. Med. 2006, 12, 1390–1396. [Google Scholar] [CrossRef] [PubMed]
- Yu, N.N.; Wang, X.X.; Yu, J.T.; Wang, ND.; Lu, RC.; Miao, D.; Tian, Y.; Tan, L. Blocking beta2-adrenergic receptor attenuates acute stress-induced amyloid beta peptides production. Brain Res. 2010, 1317, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Branca, C.; Wisely, E.V.; Hartman, L.K.; Caccamo, A.; Oddo, S. Administration of a selective β2 adrenergic receptor antagonist exacerbates neuropathology and cognitive deficits in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2014, 35, 2726–2735. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Yuen, E.Y.; Zhou, Y.; Yan, Z.; Xiang, Y.K. Amyloid beta peptide-(1–42) induces internalization and degradation of beta2 adrenergic receptors in prefrontal cortical neurons. J. Biol. Chem. 2011, 286, 31852–31863. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Jin, M.; Zhang, D.; Yang, T.; Koeglsperger, T.; Fu, H.; Selkoe, D.J. Environmental novelty activates beta2-adrenergic signaling to prevent the impairment of hippocampal LTP by Abeta oligomers. Neuron 2013, 77, 929–941. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Li, S.; Xu, H.; Walsh, D.M.; Selkoe, D.J. Large Soluble Oligomers of Amyloid β-Protein from Alzheimer Brain Are Far Less Neuroactive Than the Smaller Oligomers to Which They Dissociate. J. Neurosci. 2017, 37, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Lee, D.H.; D’Andrea, M.R.; Peterson, P.A.; Shank, R.P.; Reitz, A.B. beta-Amyloid(1-42) binds to alpha7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer’s disease pathology. J. Biol. Chem. 2000, 275, 5626–5632. [Google Scholar] [CrossRef] [PubMed]
- Nagele, R.G.; D’Andrea, M.R.; Anderson, W.J.; Wang, H.Y. Intracellular accumulation of beta-amyloid(1-42) in neurons is facilitated by the alpha 7 nicotinic acetylcholine receptor in Alzheimer’s disease. Neuroscience 2002, 110, 199–211. [Google Scholar] [CrossRef]
- Baker-Nigh, A.; Vahedi, S.; Davis, E.G.; Weintraub, S.; Bigio, E.H.; Klein, W.L.; Geula, C. Neuronal amyloid-β accumulation within cholinergic basal forebrain in ageing and Alzheimer’s disease. Brain 2015, 138, 1722–1737. [Google Scholar] [CrossRef] [PubMed]
- Oddo, S.; Caccamo, A.; Green, K.N.; Liang, K.; Tran, L.; Chen, Y.; Leslie, F.M.; LaFerla, F.M. Chronic nicotine administration exacerbates tau pathology in a transgenic model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2005, 102, 3046–3051. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, C.M.; Kayed, R.; Zheng, H.; Sweatt, J.D.; Dineley, K.T. Loss of alpha7 nicotinic receptors enhances beta-amyloid oligomer accumulation, exacerbating earlystage cognitive decline and septohippocampal pathology in a mouse model of Alzheimer’s disease. J. Neurosci. 2010, 30, 2442–2453. [Google Scholar] [CrossRef] [PubMed]
- Kroker, K.S.; Moreth, J.; Kussmaul, L.; Rast, G.; Rosenbrock, H. Restoring long-term potentiation impaired by amyloid-beta oligomers: Comparison of an acetylcholinesterase inhibitior and selective neuronal nicotinic receptor agonists. Brain Res. Bull. 2013, 96, 28–38. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G.; Lourenco, M.V.; Ferreira, S.T. How does brain insulin resistance develop in Alzheimer’s disease? Alzheimers Dement. J. Alzheimers Assoc. 2014, 10, S26–S32. [Google Scholar] [CrossRef] [PubMed]
- De la Monte, S.M.; Wands, J.R. Alzheimer’s disease is type 3 diabetes-evidence reviewed. J. Diabetes Sci. Technol. 2008, 2, 1101–1113. [Google Scholar] [CrossRef] [PubMed]
- De la Monte, S.M. Type 3 diabetes is sporadic Alzheimers disease: Mini-review. Eur. Neuropsychopharmacol. 2014, 24, 1954–1960. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G.; Vieira, M.N.; Bomfim, T.R.; Decker, H.; Velasco, P.T.; Lambert, M.P.; Viola, K.L.; Zhao, W.Q.; Ferreira, S.T.; Klein, W.L. Protection of synapses against Alzheimer’s-linked toxins: Insulin signaling prevents the pathogenic binding of Abeta oligomers. Proc. Natl. Acad. Sci. USA 2009, 106, 1971–1976. [Google Scholar] [CrossRef] [PubMed]
- Townsend, M.; Mehta, T.; Selkoe, D.J. Soluble Abeta inhibits specific signal transduction cascades common to the insulin receptor pathway. J. Biol. Chem. 2007, 282, 33305–33312. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, N.; Matsubara, E.; Maeda, S.; Minagawa, H.; Takashima, A.; Maruyama, W.; Michikawa, M.; Yanagisawa, K. A ganglioside-induced toxic soluble Abeta assembly. Its enhanced formation from Abeta bearing the Arctic mutation. J. Biol. Chem. 2007, 282, 2646–2655. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hong, Y.; Bounhar, Y.; Blacker, M.; Roucou, X.; Tounekti, O.; Vereker, E.; Bowers, W.J.; Federoff, H.J.; Goodyer, C.G.; et al. p75 neurotrophin receptor protects primary cultures of human neurons against extracellular amyloid beta peptide cytotoxicity. J. Neurosci. 2003, 23, 7385–7394. [Google Scholar] [CrossRef] [PubMed]
- Costantini, C.; Rossi, F.; Formaggio, E.; Bernardoni, R.; Cecconi, D.; Della-Bianca, V. Characterization of the signalling pathway downstream p75 neurotrophin receptor involved in beta-amyloid peptide-dependent cell death. J. Mol. Neurosci. 2005, 25, 141–156. [Google Scholar] [CrossRef]
- Ito, S.; Ménard, M.; Atkinson, T.; Gaudet, C.; Brown, L.; Whitfield, J.; Chakravarthy, B. Involvement of insulin-like growth factor 1 receptor signaling in the amyloid-β peptide oligomers-induced p75 neurotrophin receptor protein expression in mouse hippocampus. J. Alzheimers Dis. 2012, 31, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Dechant, G.; Barde, Y.A. The neurotrophin receptor p75(NTR): Novel functions and implications for diseases of the nervous system. Nat. Neurosci. 2002, 5, 1131–1136. [Google Scholar] [CrossRef] [PubMed]
- Perini, G.; Della-Bianca, V.; Politi, V.; Della Valle, G.; Dal-Pra, I.; Rossi, F.; Armato, U. Role of p75 neurotrophin receptor in the neurotoxicity by beta-amyloid peptides and synergistic effect of inflammatory cytokines. J. Exp. Med. 2002, 195, 907–918. [Google Scholar] [CrossRef] [PubMed]
- Costantini, C.; Della-Bianca, V.; Formaggio, E.; Chiamulera, C.; Montresor, A.; Rossi, F. The expression of p75 neurotrophin receptor protects against the neurotoxicity of soluble oligomers of beta-amyloid. Exp. Cell Res. 2005, 311, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, L.M.; Shatz, C.J. Immune signalling in neural development, synaptic plasticity and disease. Nat. Rev. Neurosci. 2004, 5, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Vidal, G.S.; Djurisic, M.; William, C.M.; Birnbaum, M.E.; Garcia, K.C.; Hyman, B.T.; Shatz, C.J. Human LilrB2 Is a β-Amyloid Receptor and Its Murine Homolog PirB Regulates Synaptic Plasticity in an Alzheimer’s Model. Science 2013, 341, 1399–1404. [Google Scholar] [CrossRef] [PubMed]
- Kubagawa, H.; Burrows, P.D.; Cooper, M.D. A novel pair of immunoglobulin-like receptors expressed by B cells and myeloid cells. Proc. Natl. Acad. Sci. USA 1997, 94, 5261–5266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Wang, Y.; Fu, W. Axon regeneration impediment: The role of paired immunoglobulin-like receptor B. Neural Regen. Res. 2015, 10, 1338–1342. [Google Scholar] [CrossRef] [PubMed]
- Atwal, J.K.; Pinkston-Gosse, J.; Syken, J.; Stawicki, S.; Wu, Y.; Shatz, C.; Tessier-Lavigne, M. PirB is a functional receptor for myelin inhibitors of axonal regeneration. Science 2008, 322, 967–970. [Google Scholar] [CrossRef] [PubMed]
- VanGuilder Starkey, H.D.; Van Kirk, C.A.; Bixler, G.V.; Imperio, C.G.; Kale, V.P.; Serfass, J.M.; Farley, J.A.; Yan, H.; Warrington, J.P.; Han, S.; et al. Neuroglial expression of the MHCI pathway and PirB receptor is upregulated in the hippocampus with advanced aging. J. Mol. Neurosci. 2012, 48, 111–126. [Google Scholar] [CrossRef] [PubMed]
- William, C.M.; Andermann, M.L.; Goldey, G.J.; Roumis, DK.; Reid, RC.; Shatz, CJ.; Albers, MW.; Frosch, MP.; Hyman, BT. Synaptic plasticity defect following visual deprivation in Alzheimer’s disease model transgenic mice. J. Neurosci. 2012, 32, 8004–8011. [Google Scholar] [CrossRef] [PubMed]
- Syken, J.; Grandpre, T.; Kanold, P.O.; Shatz, C.J. PirB restricts ocular dominance plasticity in visual cortex. Science 2006, 313, 1795–1800. [Google Scholar] [CrossRef] [PubMed]
- Kam, T.I.; Song, S.; Gwon, Y.; Park, H.; Yan, J.J.; Im, I.; Choi, J.W.; Choi, T.Y.; Kim, J.; Song, D.K.; et al. FcγRIIb mediates amyloid-β neurotoxicity and memory impairment in Alzheimer’s disease. J. Clin. Investig. 2013, 123, 2791–2802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maverakis, E.; Kim, K.; Shimoda, M.; Gershwin, M.E.; Patel, F.; Wilken, R.; Lebrilla, C.B. Glycans in the Immune system and The Altered Glycan Theory of Autoimmunity: A Critical Review. J. Autoimmun. 2015, 57, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Efthymiou, A.G.; Goate, A.M. Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol. Neurodegener. 2017, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Hooli, B.; Mullin, K.; Jin, S.C.; Cella, M.; Ulland, T.K.; Wang, Y.; Tanzi, R.E.; Colonna, M. Alzheimer’s disease-associated TREM2 variants exhibit either decreased or increased ligand-dependent activation. Alzheimers Dement. 2017, 4, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Lue, L.F.; Schmitz, C.; Walker, D.G. What happens to microglial TREM2 in Alzheimer’s disease: Immunoregulatory turned into immunopathogenic? Neuroscience 2015, 302, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.C.; Benitez, B.A.; Karch, C.M.; Cooper, B.; Skorupa, T.; Carrell, D.; Cruchaga, C. Coding variants in TREM2 increase risk for Alzheimer’s disease. Hum. Mol. Gen. 2014, 23, 5838–5846. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wu, X.; Li, X.; Jiang, L.L.; Gui, X.; Liu, Y.; Sun, Y.; Zhu, B.; Piña-Crespo, J.C.; Zhang, M.; et al. TREM2 Is a Receptor for β-Amyloid that Mediates Microglial Function. Neuron 2018, 97, 1023–1031.e7. [Google Scholar] [CrossRef] [PubMed]
- Yeh, F.L.; Hansen, D.V.; Sheng, M. TREM2, Microglia, and Neurodegenerative Diseases. Trends Mol. Med. 2017, 23, 512–533. [Google Scholar] [CrossRef] [PubMed]
- Vargas, L.M.; Cerpa, W.; Muñoz, F.J.; Zanlungo, S.; Alvarez, A.R. Amyloid-β oligomers synaptotoxicity: The emerging role of EphA4/c-Abl signaling in Alzheimer’s disease. Biochim. Biophys. Acta 2018, 1864, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Klein, R. Bidirectional modulation of synaptic functions by Eph/ephrin signaling. Nat. Neurosci. 2009, 12, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Hruska, M.; Dalva, M.B. Ephrin regulation of synapse formation, function and plasticity. Mol. Cell. Neurosci. 2012, 50, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, Y.; Pasquale, E.B. Eph receptors in the adult brain. Curr. Opin. Neurobiol. 2004, 14, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Henkemeyer, M.; Itkis, O.S.; Ngo, M.; Hickmott, P.W.; Ethell, I.M. Multiple EphB receptor tyrosine kinases shape dendritic spines in the hippocampus. J. Cell Biol. 2003, 163, 1313–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attwood, B.K.; Patel, S.; Pawlak, R. Ephs and ephrins: Emerging therapeutic targets in neuropathology. Int. J. Biochem. Cell Biol. 2012, 44, 578–581. [Google Scholar] [CrossRef] [PubMed]
- Lacor, P.N.; Buniel, M.C.; Furlow, P.W.; Clemente, AS.; Velasco, PT.; Wood, M.; Viola, KL.; Klein, WL. Aβ Oligomer-Induced Aberrations in Synapse Composition, Shape, and Density Provide a Molecular Basis for Loss of Connectivity in Alzheimer’s Disease. J. Neurosci. 2007, 27, 796–807. [Google Scholar] [CrossRef] [PubMed]
- Takasu, M.A.; Dalva, M.B.; Zigmond, R.E.; Greenberg, M.E. Modulation of NMDA receptor-dependent calcium influx and gene expression through EphB receptors. Science 2002, 295, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Cisse, M.; Halabisky, B.; Harris, J.; Devidze, N.; Dubal, D.B.; Sun, B.; Orr, A.; Lotz, G.; Kim, D.H.; Hamto, P.; et al. Reversing EphB2 depletion rescues cognitive functions in Alzheimer model. Nature 2011, 469, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.D.; Sun, K.; Hu, R.; Liu, X.Y.; Hu, Q.M.; Sun, X.Y.; Yao, B.; Sun, N.; Hao, J.R.; Wei, P.; et al. Blocking the Interaction between EphB2 and ADDLs by a Small Peptide Rescues Impaired Synaptic Plasticity and Memory Deficits in a Mouse Model of Alzheimer’s Disease. J. Neurosci. 2016, 36, 11959–11973. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, T.; Kim, D.; Knox, J.A.; Johnson, E.; Mucke, L. Increasing the receptor tyrosine kinase EphB2 prevents amyloid-beta-induced depletion of cell surface glutamate receptors by a mechanism that requires the PDZ-binding motif of EphB2 and neuronal activity. J. Biol. Chem. 2016, 291, 1719–1734. [Google Scholar] [CrossRef] [PubMed]
- Simon, A.M.; de Maturana, R.L.; Ricobaraza, A.; Escribano, L.; Schiapparelli, L.; Cuadrado-Tejedor, M.; Perez-Mediavilla, A.; Avila, J.; Del Rio, J.; Frechilla, D. Early changes in hippocampal Eph receptors precede the onset of memory decline in mouse models of Alzheimer’s disease. J. Alzheimers Dis. 2009, 17, 773–786. [Google Scholar] [CrossRef] [PubMed]
- Murai, K.K.; Nguyen, L.N.; Irie, F.; Yamaguchi, Y.; Pasquale, E.B. Control of hippocampal dendritic spine morphology through ephrin-A3/EphA4 signaling. Nat. Neurosci. 2003, 6, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Fu, A.K.; Hung, K.W.; Fu, W.Y.; Shen, C.; Chen, Y.; Xia, J.; Lai, K.O.; Ip, N.Y. APC(Cdh1) mediates EphA4-dependent downregulation of AMPA receptors in homeostatic plasticity. Nat. Neurosci. 2011, 14, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.R.; Hou, Z.H.; Yu, X. The kinase activity of EphA4 mediates homeostatic scaling-down of synaptic strength via activation of Cdk5. Neuropharmacology 2013, 65, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Van Hoecke, A.; Schoonaert, L.; Lemmens, R.; Timmers, M.; Staats, K.A.; Laird, A.S.; Peeters, E.; Philips, T.; Goris, A.; Dubois, B.; et al. EPHA4 is a disease modifier of amyotrophic lateral sclerosis in animal models and in humans. Nat. Med. 2012, 18, 1418–1422. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Boyd, A.W.; Bartlett, P.F. The identification of a novel isoform of EphA4 and its expression in SOD1(G93A) mice. Neuroscience 2017, 347, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Rosenberger, A.F.; Rozemuller, A.J.; van der Flier, W.M.; Scheltens, P.; van der Vies, S.M.; Hoozemans, J.J. Altered distribution of the EphA4 kinase in hippocampal brain tissue of patients with Alzheimer’s disease correlates with pathology. Acta Neuropathol. Commun. 2014, 2, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, C.; Mehrian Shai, R.; Wu, Y.; Hsu, Y.-H.; Sitzer, T.; Spann, B.; Miller, C.A. Transcriptome Analysis of Synaptoneurosomes Identifies Neuroplasticity Genes Overexpressed in Incipient Alzheimer’s Disease. PLoS ONE 2009, 4, e4936. [Google Scholar] [CrossRef] [PubMed]
- Fu, A.K.; Hung, K.W.; Huang, H.; Gu, S.; Shen, Y.; Cheng, E.Y.; Ip, F.C.; Huang, X.; Fu, W.Y.; Ip, N.Y. Blockade of EphA4 signaling ameliorates hippocampal synaptic dysfunctions in mouse models of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2014, 111, 9959–9964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vargas, L.M.; Leal, N.; Estrada, L.D.; Gonzalez, A.; Serrano, F.; Araya, K.; Gysling, K.; Inestrosa, N.C.; Pasquale, E.B.; Alvarez, A.R. EphA4 activation of c-Abl mediates synaptic loss and LTP blockade caused by amyloid-beta oligomers. PLoS ONE 2014, 9, e92309. [Google Scholar] [CrossRef] [PubMed]
- Sturchler, E.; Galichet, A.; Weibel, M.; Leclerc, E.; Heizmann, C.W. Site-Specific Blockade of RAGE-Vd Prevents Amyloid-β Oligomer Neurotoxicity. J. Neurosci. 2008, 28, 5149–5158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neeper, M.; Schmidt, A.M.; Brett, J.; Yan, SD.; Wang, F.; Pan, YC.; Elliston, K.; Stern, D.; Shaw, A. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J. Biol. Chem. 1992, 267, 14998–15004. [Google Scholar] [PubMed]
- Yan, S.D.; Chen, X.; Fu, J.; Chen, M.; Zhu, H.; Roher, A.; Slattery, T.; Zhao, L.; Nagashima, M.; Morser, J.; et al. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature 1996, 382, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Bierhaus, A.; Schiekofer, S.; Schwaninger, M.; Andrassy, M.; Humpert, PM.; Chen, J.; Hong, M.; Luther, T.; Henle, T.; Klöting, I.; et al. Diabetes-associated sustained activation of the transcription factor nuclear factor-kappaB. Diabetes 2001, 50, 2792–2808. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.; Wu, Z.; Zlokovic, B.V. RAGE (yin) versus LRP (yang) balance regulates alzheimer amyloid beta-peptide clearance through transport across the blood-brain barrier. Stroke 2004, 35, 2628–2631. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.; Bell, R.; Sagare, A.; Zlokovic, B. Clearance of amyloid-β peptide across the blood-brain barrier: Implication for therapies in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2009, 8, 16–30. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Pietromonaco, S.; Loo, A.K.-C.; Farquhar, M.G. Complete cloning and sequencing of rat gp330/‘megalin’, a distinctive member of the low density lipoprotein receptor gene family. Proc. Natl. Acad. Sci. USA 1994, 91, 9725–9729. [Google Scholar] [CrossRef] [PubMed]
- Marzolo, M.P.; Farfán, P. New insights into the roles of megalin/LRP2 and the regulation of its functional expression. Biol. Res. 2011, 44, 89–105. [Google Scholar] [CrossRef] [PubMed]
- Christensen, E.I.; Birn, H. Megalin and cubilin: Multifunctional endocytic receptors. Nat. Rev. Mol. Cell Biol. 2002, 3, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Larsson, M.; Hjälm, G.; Sakwe, A.M.; Engström, A.; Höglund, AS.; Larsson, E.; Robinson, RC.; Sundberg, C.; Rask, L. Selective interaction of megalin with postsynaptic density-95 (PSD-95)-like membrane-associated guanylate kinase (MAGUK) proteins. Biochem. J. 2003, 373, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Kounnas, M.Z.; Loukinova, E.B.; Stefansson, S.; Harmony, J.A.; Brewer, B.; Strickland, D.K.; Argraves, W.S. Identification of glycoprotein 330 as an endocytic receptor for apolipoprotein J/clusterin. J. Biol. Chem. 1995, 270, 13070–13075. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V.; Martel, C.L.; Matsubara, E.; McComb, JG.; Zheng, G.; McCluskey, RT.; Frangione, B.; Ghiso, J. Glycoprotein 330/megalin: Probable role in receptor-mediated transport of apolipoprotein J alone and in a complex with Alzheimer disease amyloid beta at the blood-brain and blood-cerebrospinal fluid barriers. Proc. Natl. Acad. Sci. USA 1996, 93, 4229–4234. [Google Scholar] [CrossRef] [PubMed]
- Hammad, S.M.; Ranganathan, S.; Loukinova, E.; Twal, W.O.; Argraves, W.S. Interaction of apolipoprotein J-amyloid beta-peptide complex with low density lipoprotein receptor-related protein-2/megalin. A mechanism to prevent pathological accumulation of amyloid beta-peptide. J. Biol. Chem. 1997, 272, 18644–18649. [Google Scholar] [CrossRef] [PubMed]
- Mangelsdorf, D.J.; Thummel, C.; Beato, M.; Herrlich, P.; Schütz, G.; Umesono, K.; Blumberg, B.; Kastner, P.; Mark, M.; Chambon, P.; et al. The nuclear receptor superfamily: The second decade. Cell 1995, 83, 835–839. [Google Scholar] [CrossRef] [Green Version]
- Woods, C.G.; Heuvel, J.P.; Rusyn, I. Genomic profiling in nuclear receptor-mediated toxicity. Toxicol. Pathol. 2007, 35, 474–494. [Google Scholar] [CrossRef] [PubMed]
- Burris, T.P.; Busby, S.A.; Griffin, P.R. Targeting orphan nuclear receptors for treatment of metabolic diseases and autoimmunity. Chem. Biol. 2012, 19, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.; Shah, J. Role of Vitamin D in Amyloid clearance via LRP-1 upregulation in Alzheimer’s disease: A potential therapeutic target? J. Chem. Neuroanat. 2017, 85, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Gezen-Ak, D.; Yılmazer, S.; Dursun, E. Why Vitamin D in Alzheimer’s disease? The hypothesis. J. Alzheimers Dis. 2014, 40, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Ohtsuki, S.; Nezu, Y.; Koitabashi, Y.; Murata, S.; Terasaki, T. 1α,25-Dihydroxyvitamin D3 enhances cerebral clearance of human amyloid-β peptide(1-40) from mouse brain across the blood-brain barrier. Fluids Barriers CNS 2011, 8, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herskovits, A.Z.; Guarente, L. SIRT1 in neurodevelopment and brain senescence. Neuron 2014, 81, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Martins, I.J.; Calderón, A.M. Diet and Nutrition reverse Type 3 Diabetes and Accelerated Aging linked to Global chronic diseases. J. Diabetes Res. Ther. 2016, 2. [Google Scholar] [CrossRef]
- Talbot, K. Brain insulin resistance in Alzheimer’s disease and its potential treatment with GLP-1 analogs. Neurodegener. Dis. Manag. 2014, 4, 31–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, F.; Kume, S.; Koya, D. SIRT1 and insulin resistance. Nat. Rev. Endocrinol. 2009, 5, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.R.; Shin, H.K.; Park, S.Y.; Kim, H.Y.; Lee, W.S.; Rhim, B.Y.; Hong, K.W.; Kim, C.D. Cilostazol suppresses β-amyloid production by activating a disintegrin and metalloproteinase 10 via the upregulation of SIRT1-coupled retinoic acid receptor-β. J. Neurosci. Res. 2014, 92, 1581–1590. [Google Scholar] [CrossRef] [PubMed]
- Julien, C.; Tremblay, C.; Emond, V.; Lebbadi, M.; Salem, N., Jr.; Bennett, D.A.; Calon, F. Sirtuin 1 reduction parallels the accumulation of tau in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2009, 68, 48–58. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Toxic activity of amyloid β oligomers (AβOs).


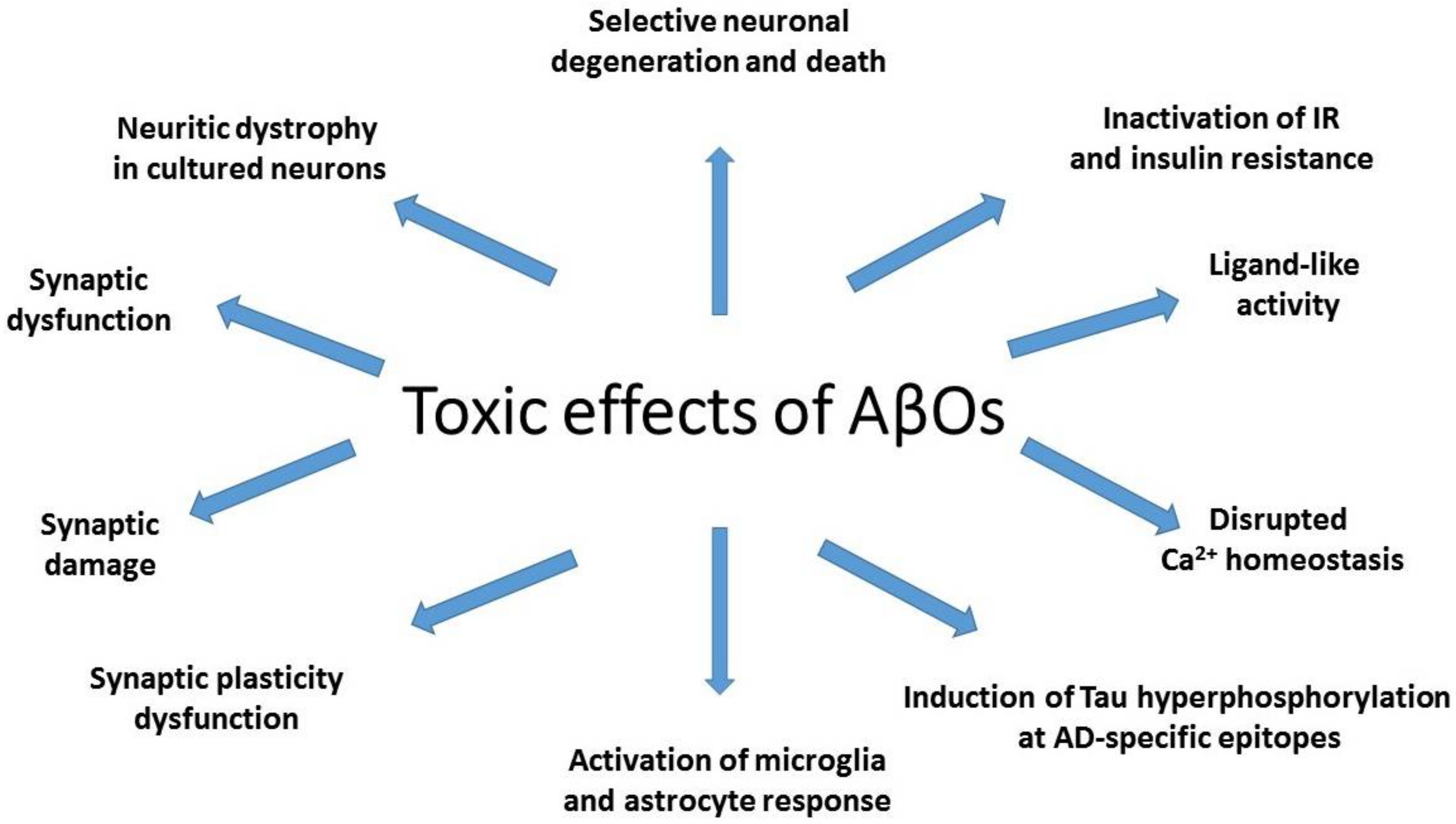{kind=link}
Table 1.
Cellular receptors related to amyloid β oligomer (AβO) activity.
| Name of the Receptor | Abbreviation | References |
|---|---|---|
| N-Methyl-d-aspartate receptor | NMDAR | [83,84,85,86,87,88,89,94,96,97] |
| α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor | AMPAR | [100,101,103,104,105,106,107,108,109,110] |
| Metabotropic glutamate receptor | mGluR | [112,113,116] |
| Cellular prion protein | PrPc | [113,115,120,121,122] |
| β2-Adrenergic receptor | β2AR | [128,140,141,142,143,144,145,146,147,148] |
| α7 nicotinic acetylcholine receptor | α7nAChR | [149,150,151,152,153,154] |
| Insulin receptor | IR | [158,159] |
| p75 neutrotrophin receptor | p75NTR | [96,163,164,165,166] |
| Human leukocyte immunoglobulin-like receptor B2 | LilrB2 | [167,168] |
| Paired immunoglobulin-like receptor B | PirB | [168,172] |
| Fragment crystallizable gamma receptor II b | FcγRIIb | [175,176] |
| Triggering receptor expressed on myeloid cells 2 | TREM2 | [181,182] |
| Tyrosine kinase ephrin type-A receptor 4 | Eph4A | [183,202,203] |
| Tyrosine kinase ephrin type-B receptor 2 | EphB2 | [183,188,189,190,191,192,193,194] |
| Receptor for advanced glycation endproducts | RAGE | [204,206,207,208,209] |
| Megalin (glycoprotein 330, low density lipoprotein-related protein 2) | gp330, LRP2 | [210,211,212,213,214,215,216] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mroczko, B.; Groblewska, M.; Litman-Zawadzka, A.; Kornhuber, J.; Lewczuk, P. Cellular Receptors of Amyloid β Oligomers (AβOs) in Alzheimer’s Disease. Int. J. Mol. Sci. 2018, 19, 1884. https://doi.org/10.3390/ijms19071884
AMA Style
Mroczko B, Groblewska M, Litman-Zawadzka A, Kornhuber J, Lewczuk P. Cellular Receptors of Amyloid β Oligomers (AβOs) in Alzheimer’s Disease. International Journal of Molecular Sciences. 2018; 19(7):1884. https://doi.org/10.3390/ijms19071884
Chicago/Turabian StyleMroczko, Barbara, Magdalena Groblewska, Ala Litman-Zawadzka, Johannes Kornhuber, and Piotr Lewczuk. 2018. "Cellular Receptors of Amyloid β Oligomers (AβOs) in Alzheimer’s Disease" International Journal of Molecular Sciences 19, no. 7: 1884. https://doi.org/10.3390/ijms19071884
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.