Survivin-Based Treatment Strategies for Squamous Cell Carcinoma

 , , , ,
, , , , 
Abstract
:1. Introduction
1.1. Squamous Cell Carcinoma
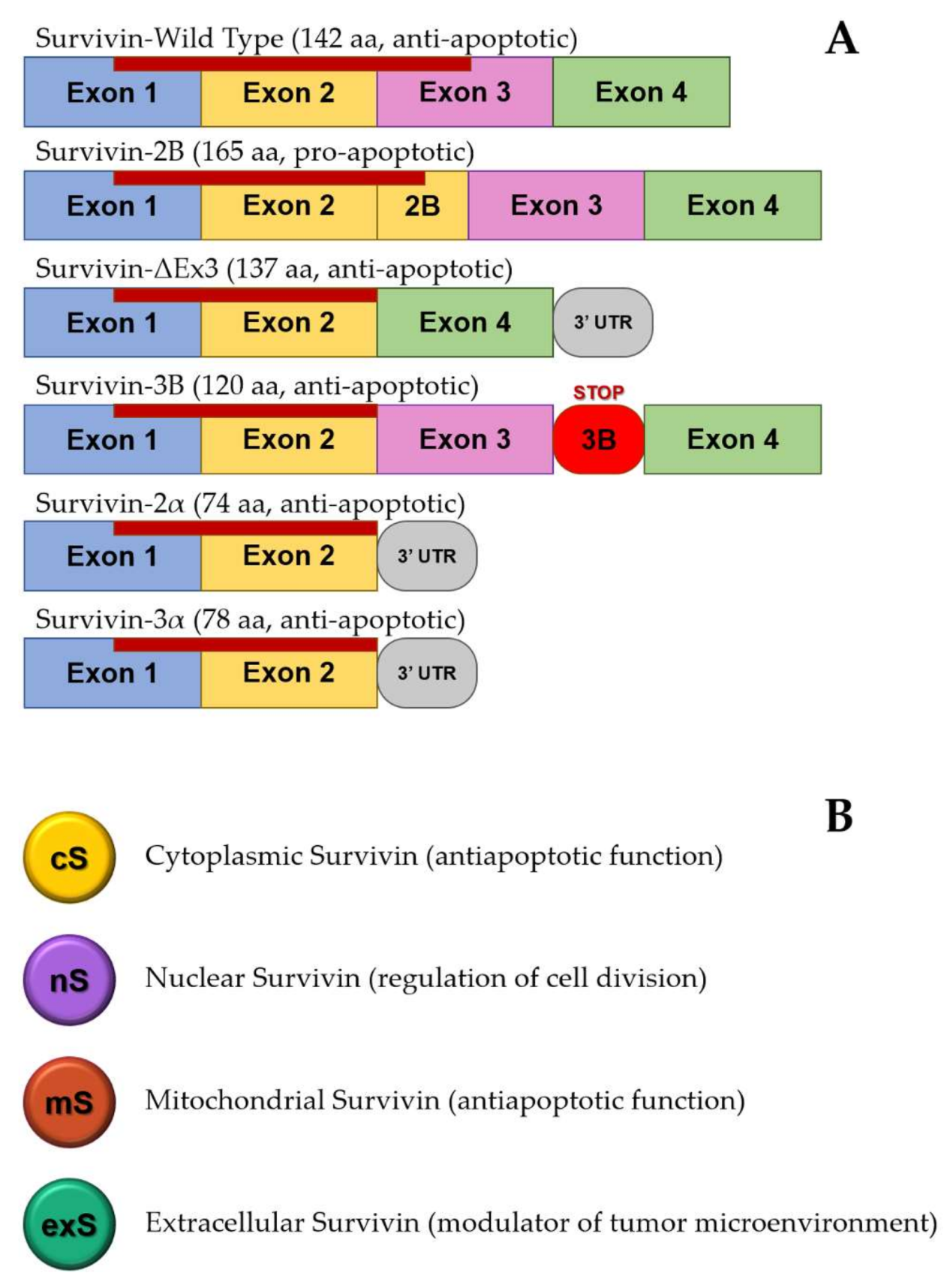
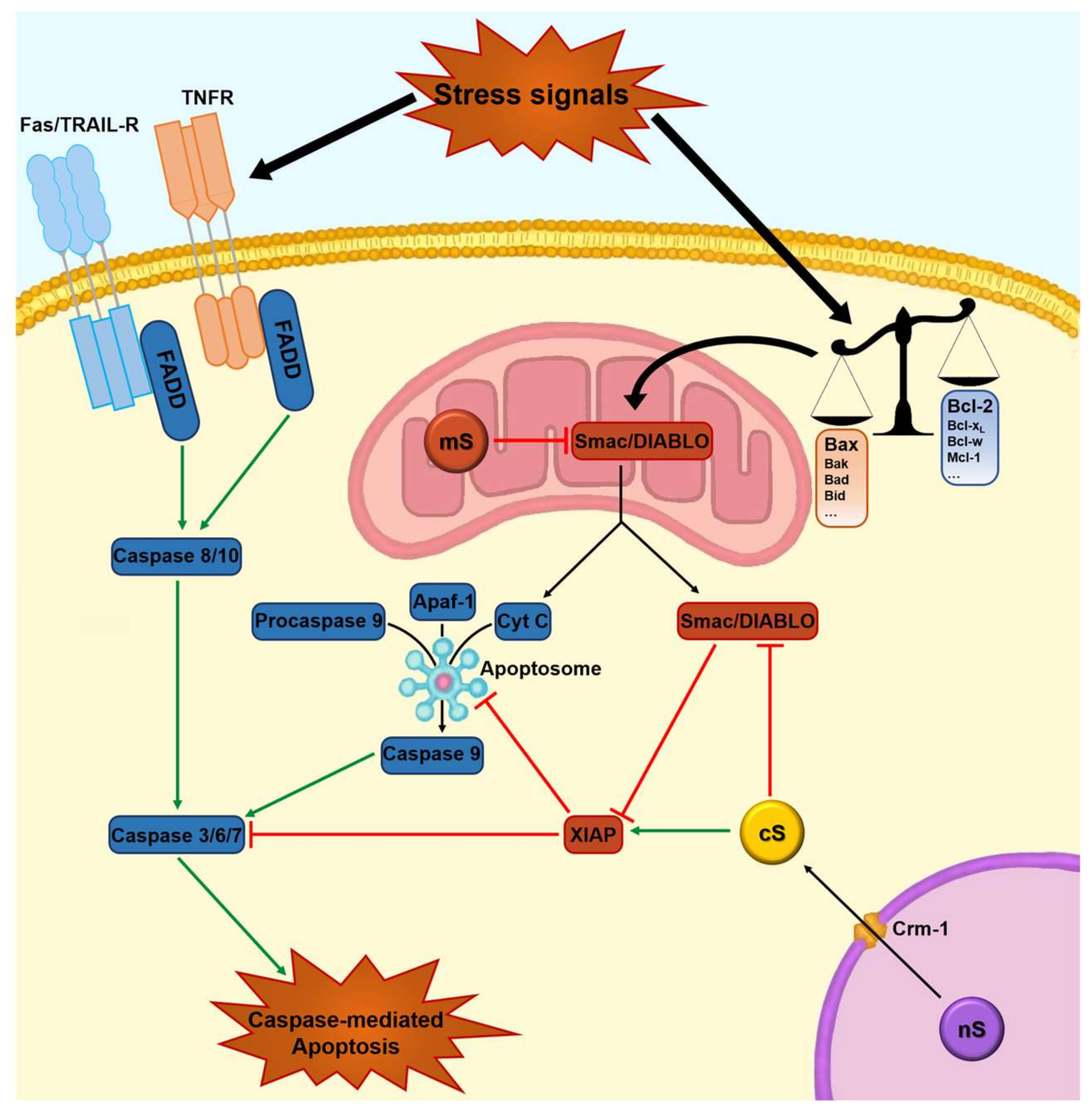
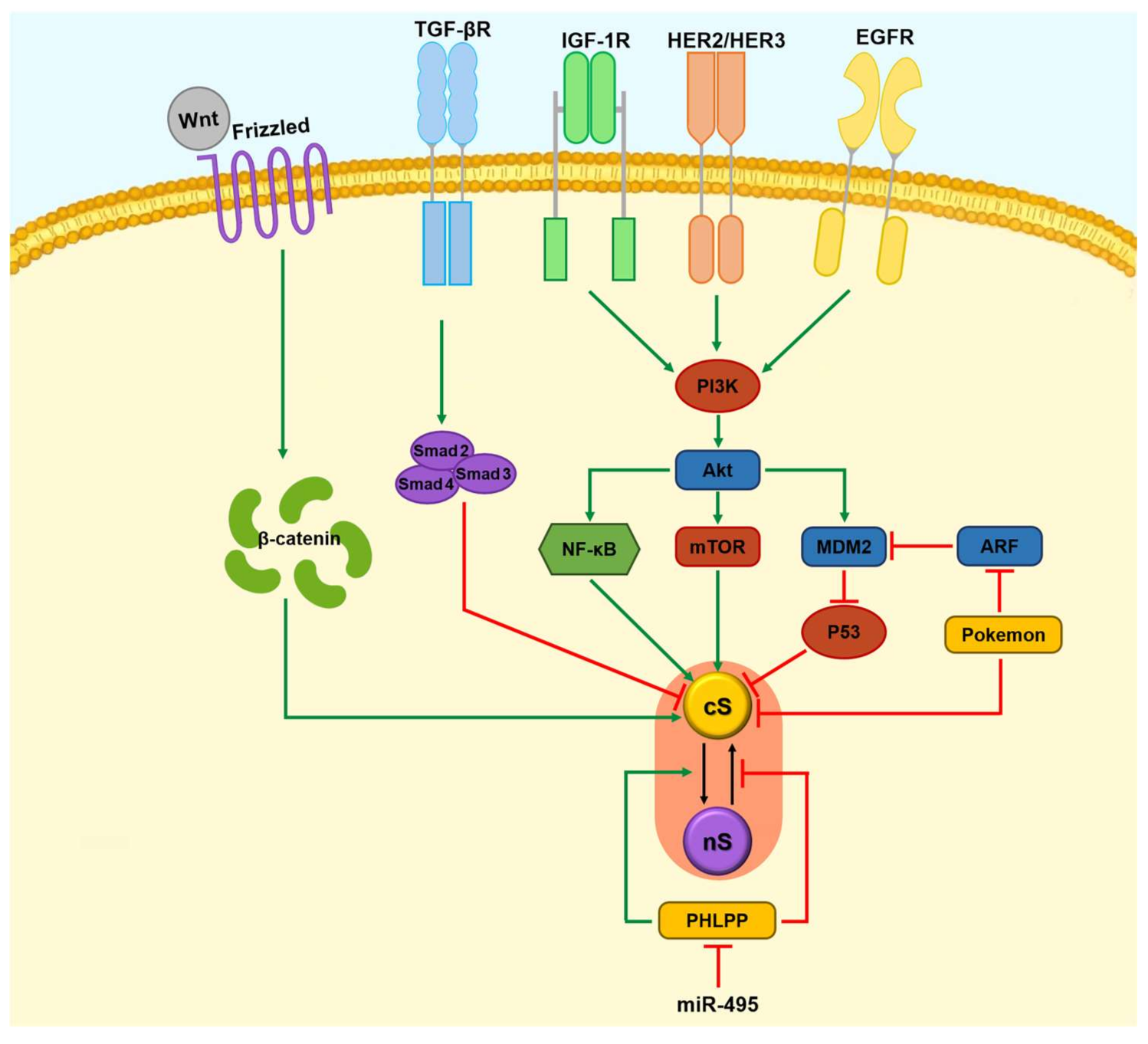
1.2. Apoptosis, Survivin, and Tumorigenesis
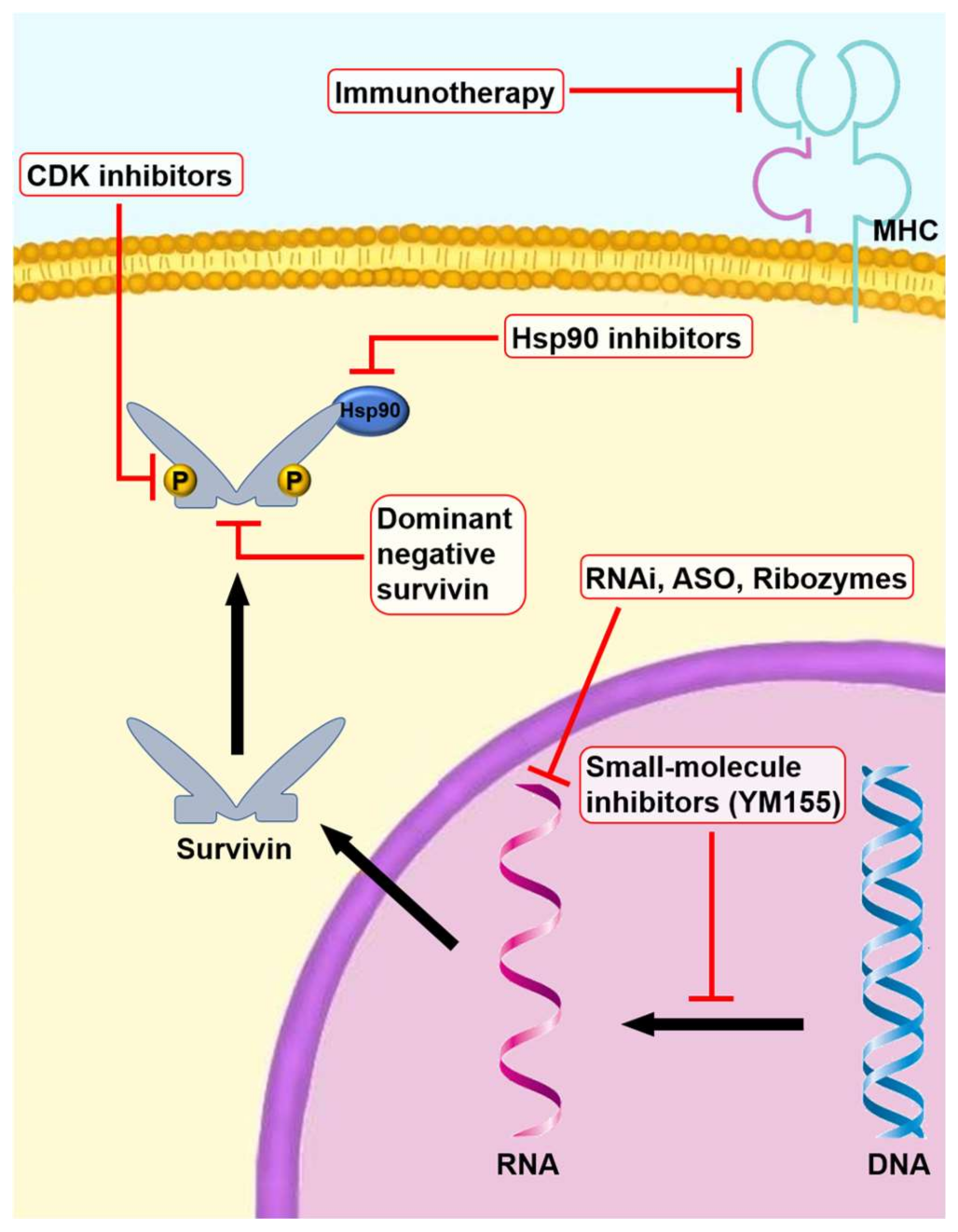
2. Gene Therapy
2.1. Dominant-Negative Survivin Mutants
2.2. RNAi (Small Interfering RNAs, Short Hairpin RNAs, and MicroRNAs)
2.3. Anti-Sense Oligonucleotides
3. Pharmacologic Therapy
Small-Molecule Inhibitors
4. Anti-Tumor Immunotherapy
Adoptive Cell Therapy
5. Side Effects
6. Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Khan, Z.; Khan, A.A.; Yadav, H.; Prasad, G.; Bisen, P.S. Survivin, a molecular target for therapeutic interventions in squamous cell carcinoma. Cell. Mol. Biol. Lett. 2017, 22, 8. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Wistuba, II; Emmert-Buck, M.R.; Erickson, H.S. Squamous cell carcinoma-similarities and differences among anatomical sites. Am. J. Cancer Res. 2011, 1, 275–300. [Google Scholar] [PubMed]
- Marur, S.; Forastiere, A.A. Head and neck squamous cell carcinoma: Update on epidemiology, diagnosis, and treatment. Mayo Clin. Proc. 2016, 91, 386–396. [Google Scholar] [CrossRef] [PubMed]
- Kallini, J.R.; Hamed, N.; Khachemoune, A. Squamous cell carcinoma of the skin: Epidemiology, classification, management, and novel trends. Int. J. Dermatol. 2015, 54, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Jennings, L.; Schmults, C.D. Management of high-risk cutaneous squamous cell carcinoma. J. Clin. Aesthet. Dermatol. 2010, 3, 39–48. [Google Scholar] [PubMed]
- Fahradyan, A.; Howell, A.C.; Wolfswinkel, E.M.; Tsuha, M.; Sheth, P.; Wong, A.K. Updates on the management of non-melanoma skin cancer (NMSC). Healthcare 2017, 5, 82. [Google Scholar] [CrossRef] [PubMed]
- Zhi, X.; Lamperska, K.; Golusinski, P.; Schork, N.J.; Luczewski, L.; Kolenda, T.; Golusinski, W.; Masternak, M.M. Gene expression analysis of head and neck squamous cell carcinoma survival and recurrence. Oncotarget 2015, 6, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Silveira, N.J.; Varuzza, L.; Machado-Lima, A.; Lauretto, M.S.; Pinheiro, D.G.; Rodrigues, R.V.; Severino, P.; Nobrega, F.G.; Silva, W.A.; de B Pereira, C.A.; et al. Searching for molecular markers in head and neck squamous cell carcinomas (HNSCC) by statistical and bioinformatic analysis of larynx-derived sage libraries. BMC Med. Genom. 2008, 1, 56. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xiong, Z.; Beasley, A.; D’Amico, T.; Chen, X.L. Personalized and targeted therapy of esophageal squamous cell carcinoma: An update. Ann. N. Y. Acad. Sci. 2016, 1381, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Kaifi, J.T.; Gusani, N.J.; Jiang, Y.; Mackley, H.B.; Dye, C.E.; Mathew, A.; Kimchi, E.T.; Reed, M.F.; Staveley-O’Carroll, K.F. Multidisciplinary management of early and locally advanced esophageal cancer. J. Clin. Gastroenterol. 2011, 45, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.; Belani, C. Systemic chemotherapy for advanced non-small cell lung cancer: Recent advances and future directions. Oncologist 2008, 13, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Tian, J.; Song, X.; Wu, B.; Liu, L. Causes of death and competing risk analysis of the associated factors for non-small cell lung cancer using the surveillance, epidemiology, and end results database. J. Cancer Res. Clin. Oncol. 2018, 144, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Agada, F.O.; Patmore, H.; Alhamarneh, O.; Stafford, N.D.; Greenman, J. Genetic profile of head and neck squamous cell carcinoma: Clinical implications. J. Laryngol. Otol. 2009, 123, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Lehrbach, D.M.; Nita, M.E.; Cecconello, I. Molecular aspects of esophageal squamous cell carcinoma carcinogenesis. Arq. Gastroenterol. 2003, 40, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 2014, 14, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Rodust, P.M.; Stockfleth, E.; Ulrich, C.; Leverkus, M.; Eberle, J. UV-induced squamous cell carcinoma—A role for antiapoptotic signalling pathways. Br. J. Dermatol. 2009, 161, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Bonner, J.A.; Harari, P.M.; Giralt, J.; Azarnia, N.; Shin, D.M.; Cohen, R.B.; Jones, C.U.; Sur, R.; Raben, D.; Jassem, J.; et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2006, 354, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Reid, B.J.; Blount, P.L.; Rabinovitch, P.S. Biomarkers in barrett’s esophagus. Gastrointest. Endosc. Clin. N. Am. 2003, 13, 369–397. [Google Scholar] [CrossRef]
- Brattstrom, D.; Bergqvist, M.; Lamberg, K.; Kraaz, W.; Scheibenflug, L.; Gustafsson, G.; Inganas, M.; Wagenius, G.; Brodin, O. Complete sequence of p53 gene in 20 patients with lung cancer: Comparison with chemosensitivity and immunohistochemistry. Med. Oncol. 1998, 15, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Chakravarti, D.; Flores, E.R. P63 steps into the limelight: Crucial roles in the suppression of tumorigenesis and metastasis. Nat. Rev. Cancer 2013, 13, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Molinolo, A.A.; Hewitt, S.M.; Amornphimoltham, P.; Keelawat, S.; Rangdaeng, S.; Meneses Garcia, A.; Raimondi, A.R.; Jufe, R.; Itoiz, M.; Gao, Y.; et al. Dissecting the Akt/mammalian target of rapamycin signaling network: Emerging results from the head and neck cancer tissue array initiative. Clin. Cancer Res. 2007, 13, 4964–4973. [Google Scholar] [CrossRef] [PubMed]
- Crook, N.E.; Clem, R.J.; Miller, L.K. An apoptosis-inhibiting baculovirus gene with a zinc finger-like motif. J. Virol. 1993, 67, 2168–2174. [Google Scholar] [PubMed]
- Ambrosini, G.; Adida, C.; Altieri, D.C. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat. Med. 1997, 3, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Altieri, D.C. Validating survivin as a cancer therapeutic target. Nat. Rev. Cancer 2003, 3, 46–54. [Google Scholar] [CrossRef] [PubMed]
- LaCasse, E.C.; Baird, S.; Korneluk, R.G.; MacKenzie, A.E. The inhibitors of apoptosis (IAPS) and their emerging role in cancer. Oncogene 1998, 17, 3247–3259. [Google Scholar] [CrossRef] [PubMed]
- Salvesen, G.S.; Duckett, C.S. Iap proteins: Blocking the road to death’s door. Nat. Rev. Mol. Cell Biol. 2002, 3, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Deveraux, Q.L.; Reed, J.C. Iap family proteins—Suppressors of apoptosis. Genes Dev 1999, 13, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef] [PubMed]
- Deveraux, Q.L.; Takahashi, R.; Salvesen, G.S.; Reed, J.C. X-linked iap is a direct inhibitor of cell-death proteases. Nature 1997, 388, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Tamm, I.; Wang, Y.; Sausville, E.; Scudiero, D.A.; Vigna, N.; Oltersdorf, T.; Reed, J.C. IAP-family protein survivin inhibits caspase activity and apoptosis induced by fas (CD95), bax, caspases, and anticancer drugs. Cancer Res. 1998, 58, 5315–5320. [Google Scholar] [PubMed]
- Yang, Y.; Fang, S.; Jensen, J.P.; Weissman, A.M.; Ashwell, J.D. Ubiquitin protein ligase activity of iaps and their degradation in proteasomes in response to apoptotic stimuli. Science 2000, 288, 874–877. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, S.; Pelus, L.M. Survivin, a cancer target with an emerging role in normal adult tissues. Mol. Cancer Ther. 2006, 5, 1087–1098. [Google Scholar] [CrossRef] [PubMed]
- Gurbuxani, S.; Xu, Y.; Keerthivasan, G.; Wickrema, A.; Crispino, J.D. Differential requirements for survivin in hematopoietic cell development. Proc. Natl. Acad. Sci. USA 2005, 102, 11480–11485. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, S.; Pelus, L.M. Activated H-RAS regulates hematopoietic cell survival by modulating survivin. Biochem. Biophys. Res. Commun. 2004, 323, 636–644. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Bakal, C.; Shahinian, A.; Elia, A.; Wakeham, A.; Suh, W.K.; Duncan, G.S.; Ciofani, M.; Rottapel, R.; Zuniga-Pflucker, J.C.; et al. Survivin loss in thymocytes triggers p53-mediated growth arrest and p53-independent cell death. J. Exp. Med. 2004, 199, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Altznauer, F.; Martinelli, S.; Yousefi, S.; Thurig, C.; Schmid, I.; Conway, E.M.; Schoni, M.H.; Vogt, P.; Mueller, C.; Fey, M.F.; et al. Inflammation-associated cell cycle-independent block of apoptosis by survivin in terminally differentiated neutrophils. J. Exp. Med. 2004, 199, 1343–1354. [Google Scholar] [CrossRef] [PubMed]
- Mesri, M.; Morales-Ruiz, M.; Ackermann, E.J.; Bennett, C.F.; Pober, J.S.; Sessa, W.C.; Altieri, D.C. Suppression of vascular endothelial growth factor-mediated endothelial cell protection by survivin targeting. Am. J. Pathol. 2001, 158, 1757–1765. [Google Scholar] [CrossRef]
- Blanc-Brude, O.P.; Mesri, M.; Wall, N.R.; Plescia, J.; Dohi, T.; Altieri, D.C. Therapeutic targeting of the survivin pathway in cancer: Initiation of mitochondrial apoptosis and suppression of tumor-associated angiogenesis. Clin. Cancer Res. 2003, 9, 2683–2692. [Google Scholar] [PubMed]
- Chiodino, C.; Cesinaro, A.M.; Ottani, D.; Fantini, F.; Giannetti, A.; Trentini, G.P.; Pincelli, C. Communication: Expression of the novel inhibitor of apoptosis survivin in normal and neoplastic skin. J. Investig. Dermatol. 1999, 113, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Suominen, J.S.; Hakovirta, H.; Parvinen, M.; Martinand-Mari, C.; Toppari, J.; Robbins, I. Survivin expression in rat testis is upregulated by stem-cell factor. Mol. Cell. Endocrinol. 2004, 218, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Kumazawa, Y.; Kawamura, K.; Sato, T.; Sato, N.; Konishi, Y.; Shimizu, Y.; Fukuda, J.; Kodama, H.; Tanaka, T. HCG up-regulates survivin mRNA in human granulosa cells. Mol. Hum. Reprod. 2005, 11, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Harfouche, R.; Hassessian, H.M.; Guo, Y.; Faivre, V.; Srikant, C.B.; Yancopoulos, G.D.; Hussain, S.N. Mechanisms which mediate the antiapoptotic effects of angiopoietin-1 on endothelial cells. Microvasc. Res. 2002, 64, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Mahotka, C.; Wenzel, M.; Springer, E.; Gabbert, H.E.; Gerharz, C.D. Survivin-deltaex3 and survivin-2b: Two novel splice variants of the apoptosis inhibitor survivin with different antiapoptotic properties. Cancer Res. 1999, 59, 6097–6102. [Google Scholar] [PubMed]
- Mahotka, C.; Liebmann, J.; Wenzel, M.; Suschek, C.V.; Schmitt, M.; Gabbert, H.E.; Gerharz, C.D. Differential subcellular localization of functionally divergent survivin splice variants. Cell Death Differ. 2002, 9, 1334–1342. [Google Scholar] [CrossRef] [PubMed]
- Mahotka, C.; Krieg, T.; Krieg, A.; Wenzel, M.; Suschek, C.V.; Heydthausen, M.; Gabbert, H.E.; Gerharz, C.D. Distinct in vivo expression patterns of survivin splice variants in renal cell carcinomas. Int. J. Cancer 2002, 100, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Badran, A.; Yoshida, A.; Ishikawa, K.; Goi, T.; Yamaguchi, A.; Ueda, T.; Inuzuka, M. Identification of a novel splice variant of the human anti-apoptopsis gene survivin. Biochem. Biophys. Res. Commun. 2004, 314, 902–907. [Google Scholar] [CrossRef] [PubMed]
- Caldas, H.; Honsey, L.E.; Altura, R.A. Survivin 2α: A novel survivin splice variant expressed in human malignancies. Mol. Cancer 2005, 4, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caldas, H.; Jiang, Y.; Holloway, M.P.; Fangusaro, J.; Mahotka, C.; Conway, E.M.; Altura, R.A. Survivin splice variants regulate the balance between proliferation and cell death. Oncogene 2005, 24, 1994–2007. [Google Scholar] [CrossRef] [PubMed]
- Garg, H.; Suri, P.; Gupta, J.C.; Talwar, G.P.; Dubey, S. Survivin: A unique target for tumor therapy. Cancer Cell Int. 2016, 16, 49. [Google Scholar] [CrossRef] [PubMed]
- Altieri, D.C. Survivin, cancer networks and pathway-directed drug discovery. Nat. Rev. Cancer 2008, 8, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Mak, T.W. Pathways of apoptotic and non-apoptotic death in tumour cells. Nat. Rev. Cancer 2004, 4, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Welm, A.; Bishop, J.M. Cell division and cell survival in the absence of survivin. Proc. Natl. Acad. Sci. USA 2004, 101, 15100–15105. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Altieri, D.C. The cancer antiapoptosis mouse survivin gene: Characterization of locus and transcriptional requirements of basal and cell cycle-dependent expression. Cancer Res. 1999, 59, 3143–3151. [Google Scholar] [PubMed]
- Vader, G.; Kauw, J.J.; Medema, R.H.; Lens, S.M. Survivin mediates targeting of the chromosomal passenger complex to the centromere and midbody. EMBO Rep. 2006, 7, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Ruchaud, S.; Carmena, M.; Earnshaw, W.C. Chromosomal passengers: Conducting cell division. Nat. Rev. Mol. Cell Biol. 2007, 8, 798–812. [Google Scholar] [CrossRef] [PubMed]
- Sampath, S.C.; Ohi, R.; Leismann, O.; Salic, A.; Pozniakovski, A.; Funabiki, H. The chromosomal passenger complex is required for chromatin-induced microtubule stabilization and spindle assembly. Cell 2004, 118, 187–202. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Aspe, J.R.; Asumen, M.G.; Almaguel, F.; Odumosu, O.; Acevedo-Martinez, S.; de Leon, M.; Langridge, W.H.; Wall, N.R. Extracellular, cell-permeable survivin inhibits apoptosis while promoting proliferative and metastatic potential. Br. J. Cancer 2009, 100, 1073–1086. [Google Scholar] [CrossRef] [PubMed]
- Greening, D.W.; Gopal, S.K.; Mathias, R.A.; Liu, L.; Sheng, J.; Zhu, H.J.; Simpson, R.J. Emerging roles of exosomes during epithelial-mesenchymal transition and cancer progression. Semin. Cell Dev. Biol. 2015, 40, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Ferguson Bennit, H.; Asuncion Valenzuela, M.M.; Turay, D.; Diaz Osterman, C.J.; Moyron, R.B.; Esebanmen, G.E.; Ashok, A.; Wall, N.R. Localization and upregulation of survivin in cancer health disparities: A clinical perspective. Biologics 2015, 9, 57–67. [Google Scholar] [PubMed]
- Galbo, P.M., Jr.; Ciesielski, M.J.; Figel, S.; Maguire, O.; Qiu, J.; Wiltsie, L.; Minderman, H.; Fenstermaker, R.A. Circulating CD9+/GFAP+/survivin+ exosomes in malignant glioma patients following survivin vaccination. Oncotarget 2017, 8, 114722–114735. [Google Scholar] [CrossRef] [PubMed]
- Lo Muzio, L.; Pannone, G.; Santarelli, A.; Bambini, F.; Mascitti, M.; Rubini, C.; Testa, N.F.; Dioguardi, M.; Leuci, S.; Bascones, A.; et al. Is expression of p120ctn in oral squamous cell carcinomas a prognostic factor? Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2013, 115, 789–798. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Jutzy, J.M.; Valenzuela, M.M.; Turay, D.; Aspe, J.R.; Ashok, A.; Mirshahidi, S.; Mercola, D.; Lilly, M.B.; Wall, N.R. Plasma-derived exosomal survivin, a plausible biomarker for early detection of prostate cancer. PLoS ONE 2012, 7, e46737. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Duan, N.; Zhang, C.; Zhang, W. Survivin and tumorigenesis: Molecular mechanisms and therapeutic strategies. J. Cancer 2016, 7, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.J.; Lopez-Chavez, A.; Citrin, D.; Janik, J.E.; Morris, J.C. Impacting tumor cell-fate by targeting the inhibitor of apoptosis protein survivin. Mol. Cancer 2011, 10, 35. [Google Scholar] [CrossRef] [PubMed]
- Ausserlechner, M.J.; Hagenbuchner, J. Mitochondrial survivin—An achilles’ heel in cancer chemoresistance. Mol. Cell. Oncol. 2016, 3, e1076589. [Google Scholar] [CrossRef] [PubMed]
- Ceballos-Cancino, G.; Espinosa, M.; Maldonado, V.; Melendez-Zajgla, J. Regulation of mitochondrial smac/diablo-selective release by survivin. Oncogene 2007, 26, 7569–7575. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.J.; Plescia, J.; Clevers, H.; Fearon, E.R.; Altieri, D.C. Survivin and molecular pathogenesis of colorectal cancer. Lancet 2003, 362, 205–209. [Google Scholar] [CrossRef]
- Pise-Masison, C.A.; Radonovich, M.; Dohoney, K.; Morris, J.C.; O’Mahony, D.; Lee, M.J.; Trepel, J.; Waldmann, T.A.; Janik, J.E.; Brady, J.N. Gene expression profiling of atl patients: Compilation of disease-related genes and evidence for tcf4 involvement in BIRC5 gene expression and cell viability. Blood 2009, 113, 4016–4026. [Google Scholar] [CrossRef] [PubMed]
- Carter, B.Z.; Milella, M.; Altieri, D.C.; Andreeff, M. Cytokine-regulated expression of survivin in myeloid leukemia. Blood 2001, 97, 2784–2790. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, J. Stat proteins and oncogenesis. J. Clin. Investig. 2002, 109, 1139–1142. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, M.; Kieff, E.; Izumi, K.M. The epstein-barr virus latent membrane protein 1 putative janus kinase 3 (JAK3) binding domain does not mediate JAK3 association or activation in B-lymphoma or lymphoblastoid cell lines. J. Virol. 2002, 76, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Fortugno, P.; Beltrami, E.; Plescia, J.; Fontana, J.; Pradhan, D.; Marchisio, P.C.; Sessa, W.C.; Altieri, D.C. Regulation of survivin function by HSP90. Proc. Natl. Acad. Sci. USA 2003, 100, 13791–13796. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Fang, M.; Li, Y.; Li, L.; Wang, X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating iap inhibition. Cell 2000, 102, 33–42. [Google Scholar] [CrossRef]
- Verhagen, A.M.; Ekert, P.G.; Pakusch, M.; Silke, J.; Connolly, L.M.; Reid, G.E.; Moritz, R.L.; Simpson, R.J.; Vaux, D.L. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 2000, 102, 43–53. [Google Scholar] [CrossRef]
- Qiu, Y.; Li, X.; Yi, B.; Zheng, J.; Peng, Z.; Zhang, Z.; Wu, M.; Shen, F.; Su, C. Protein phosphatase phlpp induces cell apoptosis and exerts anticancer activity by inhibiting survivin phosphorylation and nuclear export in gallbladder cancer. Oncotarget 2015, 6, 19148–19162. [Google Scholar] [CrossRef] [PubMed]
- Zu, X.; Ma, J.; Liu, H.; Liu, F.; Tan, C.; Yu, L.; Wang, J.; Xie, Z.; Cao, D.; Jiang, Y. Pro-oncogene pokemon promotes breast cancer progression by upregulating survivin expression. Breast Cancer Res. 2011, 13, R26. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Ackermann, E.J.; Bennett, C.F.; Rothermel, A.L.; Plescia, J.; Tognin, S.; Villa, A.; Marchisio, P.C.; Altieri, D.C. Pleiotropic cell-division defects and apoptosis induced by interference with survivin function. Nat. Cell Biol. 1999, 1, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Swana, H.S.; Grossman, D.; Anthony, J.N.; Weiss, R.M.; Altieri, D.C. Tumor content of the antiapoptosis molecule survivin and recurrence of bladder cancer. N. Engl. J. Med. 1999, 341, 452–453. [Google Scholar] [CrossRef] [PubMed]
- Sarela, A.I.; Macadam, R.C.; Farmery, S.M.; Markham, A.F.; Guillou, P.J. Expression of the antiapoptosis gene, survivin, predicts death from recurrent colorectal carcinoma. Gut 2000, 46, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Iwamoto, S.; Gon, G.; Nohara, T.; Iwamoto, M.; Tanigawa, N. Expression of survivin and its relationship to loss of apoptosis in breast carcinomas. Clin. Cancer Res. 2000, 6, 127–134. [Google Scholar] [PubMed]
- Kawasaki, H.; Altieri, D.C.; Lu, C.D.; Toyoda, M.; Tenjo, T.; Tanigawa, N. Inhibition of apoptosis by survivin predicts shorter survival rates in colorectal cancer. Cancer Res. 1998, 58, 5071–5074. [Google Scholar] [PubMed]
- Santarelli, A.; Mascitti, M.; Rubini, C.; Bambini, F.; Giannatempo, G.; lo Russo, L.; Sartini, D.; Emanuelli, M.; Procaccini, M.; lo Muzio, L. Nuclear survivin as a prognostic factor in squamous-cell carcinoma of the oral cavity. Appl. Immunohistochem. Mol. Morphol. 2017, 25, 566–570. [Google Scholar] [CrossRef] [PubMed]
- Adida, C.; Berrebi, D.; Peuchmaur, M.; Reyes-Mugica, M.; Altieri, D.C. Anti-apoptosis gene, survivin, and prognosis of neuroblastoma. Lancet 1998, 351, 882–883. [Google Scholar] [CrossRef]
- Lo Muzio, L.; Staibano, S.; Pannone, G.; Mignogna, M.D.; Mariggio, A.; Salvatore, G.; Chieffi, P.; Tramontano, D.; de Rosa, G.; Altieri, D.C. Expression of the apoptosis inhibitor survivin in aggressive squamous cell carcinoma. Exp. Mol. Pathol. 2001, 70, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Yao, H.; Wang, S.; Hong, M.; He, J.; Cao, S.; Min, H.; Song, E.; Guo, X. Prognostic value of survivin and livin in nasopharyngeal carcinoma. Laryngoscope 2006, 116, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, H.; Toyoda, M.; Shinohara, H.; Okuda, J.; Watanabe, I.; Yamamoto, T.; Tanaka, K.; Tenjo, T.; Tanigawa, N. Expression of survivin correlates with apoptosis, proliferation, and angiogenesis during human colorectal tumorigenesis. Cancer 2001, 91, 2026–2032. [Google Scholar] [CrossRef]
- Kato, J.; Kuwabara, Y.; Mitani, M.; Shinoda, N.; Sato, A.; Toyama, T.; Mitsui, A.; Nishiwaki, T.; Moriyama, S.; Kudo, J.; et al. Expression of survivin in esophageal cancer: Correlation with the prognosis and response to chemotherapy. Int. J. Cancer 2001, 95, 92–95. [Google Scholar] [CrossRef]
- Ding, Y.; Prieto, V.G.; Zhang, P.S.; Rosenthal, S.; Smith, K.J.; Skelton, H.G.; Diwan, A.H. Nuclear expression of the antiapoptotic protein survivin in malignant melanoma. Cancer 2006, 106, 1123–1129. [Google Scholar] [CrossRef] [PubMed]
- Span, P.N.; Tjan-Heijnen, V.C.; Manders, P.; van Tienoven, D.; Lehr, J.; Sweep, F.C. High survivin predicts a poor response to endocrine therapy, but a good response to chemotherapy in advanced breast cancer. Breast Cancer Res. Treat. 2006, 98, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Vaira, V.; Lee, C.W.; Goel, H.L.; Bosari, S.; Languino, L.R.; Altieri, D.C. Regulation of survivin expression by IGF-1/MTOR signaling. Oncogene 2007, 26, 2678–2684. [Google Scholar] [CrossRef] [PubMed]
- Lo Muzio, L.; Pannone, G.; Staibano, S.; Mignogna, M.D.; Rubini, C.; Mariggio, M.A.; Procaccini, M.; Ferrari, F.; De Rosa, G.; Altieri, D.C. Survivin expression in oral squamous cell carcinoma. Br. J. Cancer 2003, 89, 2244–2248. [Google Scholar] [CrossRef] [PubMed]
- Lodi, G.; Franchini, R.; Bez, C.; Sardella, A.; Moneghini, L.; Pellegrini, C.; Bosari, S.; Manfredi, M.; Vescovi, P.; Carrassi, A. Detection of survivin mRNA in healthy oral mucosa, oral leucoplakia and oral cancer. Oral Dis. 2010, 16, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Lo Muzio, L.; Farina, A.; Rubini, C.; Pezzetti, F.; Stabellini, G.; Laino, G.; Santarelli, A.; Pannone, G.; Bufo, P.; de Lillo, A.; et al. Survivin as prognostic factor in squamous cell carcinoma of the oral cavity. Cancer Lett. 2005, 225, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Lo Muzio, L.; Pannone, G.; Leonardi, R.; Staibano, S.; Mignogna, M.D.; De Rosa, G.; Kudo, Y.; Takata, T.; Altieri, D.C. Survivin, a potential early predictor of tumor progression in the oral mucosa. J. Dent. Res. 2003, 82, 923–928. [Google Scholar] [CrossRef] [PubMed]
- Velculescu, V.E.; Madden, S.L.; Zhang, L.; Lash, A.E.; Yu, J.; Rago, C.; Lal, A.; Wang, C.J.; Beaudry, G.A.; Ciriello, K.M.; et al. Analysis of human transcriptomes. Nat. Genet. 1999, 23, 387–388. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.H.; Svane, I.M.; Becker, J.C.; Straten, P.T. The universal character of the tumor-associated antigen survivin. Clin. Cancer Res. 2007, 13, 5991–5994. [Google Scholar] [CrossRef] [PubMed]
- Altieri, D.C. Targeted therapy by disabling crossroad signaling networks: The survivin paradigm. Mol. Cancer Ther. 2006, 5, 478–482. [Google Scholar] [CrossRef] [PubMed]
- Sawyers, C. Targeted cancer therapy. Nature 2004, 432, 294–297. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Longley, D.B.; Johnston, P.G. Molecular mechanisms of drug resistance. J. Pathol. 2005, 205, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Grossman, D.; McNiff, J.M.; Li, F.; Altieri, D.C. Expression of the apoptosis inhibitor, survivin, in nonmelanoma skin cancer and gene targeting in a keratinocyte cell line. Lab. Investig. 1999, 79, 1121–1126. [Google Scholar] [PubMed]
- Grossman, D.; McNiff, J.M.; Li, F.; Altieri, D.C. Expression and targeting of the apoptosis inhibitor, survivin, in human melanoma. J. Investig. Dermatol. 1999, 113, 1076–1081. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.P.; Cui, J.T.; Liston, P.; Huajiang, X.; Xu, R.; Lin, M.C.; Zhu, Y.B.; Zou, B.; Ng, S.S.; Jiang, S.H.; et al. Gene therapy for colon cancer by adeno-associated viral vector-mediated transfer of survivin CYS84ALA mutant. Gastroenterology 2005, 128, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.P.; Jiang, X.H.; Lin, M.C.; Cui, J.T.; Yang, Y.; Lum, C.T.; Zou, B.; Zhu, Y.B.; Jiang, S.H.; Wong, W.M.; et al. Suppression of survivin expression inhibits in vivo tumorigenicity and angiogenesis in gastric cancer. Cancer Res. 2003, 63, 7724–7732. [Google Scholar] [PubMed]
- O’Connor, D.S.; Grossman, D.; Plescia, J.; Li, F.; Zhang, H.; Villa, A.; Tognin, S.; Marchisio, P.C.; Altieri, D.C. Regulation of apoptosis at cell division by P34CDC2 phosphorylation of survivin. Proc. Natl. Acad. Sci. USA 2000, 97, 13103–13107. [Google Scholar] [CrossRef] [PubMed]
- Mesri, M.; Wall, N.R.; Li, J.; Kim, R.W.; Altieri, D.C. Cancer gene therapy using a survivin mutant adenovirus. J. Clin. Investig. 2001, 108, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Wall, N.R.; O’Connor, D.S.; Plescia, J.; Pommier, Y.; Altieri, D.C. Suppression of survivin phosphorylation on THR34 by flavopiridol enhances tumor cell apoptosis. Cancer Res. 2003, 63, 230–235. [Google Scholar] [PubMed]
- Aspe, J.R.; Wall, N.R. Survivin-t34a: Molecular mechanism and therapeutic potential. OncoTargets Ther. 2010, 3, 247–254. [Google Scholar]
- Barrett, R.M.; Osborne, T.P.; Wheatley, S.P. Phosphorylation of survivin at threonine 34 inhibits its mitotic function and enhances its cytoprotective activity. Cell Cycle 2009, 8, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.; Carmena, M.; Sambade, C.; Earnshaw, W.C.; Wheatley, S.P. Survivin is required for stable checkpoint activation in taxol-treated hela cells. J. Cell Sci. 2003, 116, 2987–2998. [Google Scholar] [CrossRef] [PubMed]
- Pennati, M.; Folini, M.; Zaffaroni, N. Targeting survivin in cancer therapy: Fulfilled promises and open questions. Carcinogenesis 2007, 28, 1133–1139. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Li, J.; Zeng, Z.; Xian, L. Lentivirus-mediated gene therapy by suppressing survivin in balb/c nude mice bearing oral squamous cell carcinoma. Cancer Biol. Ther. 2006, 5, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Wang, Y.; Xiao, M.; Lin, Y.; Yu, L. Up-regulation of survivin in oral squamous cell carcinoma correlates with poor prognosis and chemoresistance. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2010, 110, 484–491. [Google Scholar] [PubMed]
- Xu, J.H.; Wang, A.X.; Huang, H.Z.; Wang, J.G.; Pan, C.B.; Zhang, B. Survivin shRNA induces caspase-3-dependent apoptosis and enhances cisplatin sensitivity in squamous cell carcinoma of the tongue. Oncol. Res. 2010, 18, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.M.; Luan, X.Y.; Lei, D.P.; Ma, X.J.; Liu, X.X.; Liu, J.; Pan, X.L. Suppression of survivin expression by short hairpin rna induces apoptosis in human laryngeal carcinoma cells. ORL J. Otorhinolaryngol. Relat. Spec. 2008, 70, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Ngan, C.Y.; Yamamoto, H.; Takagi, A.; Fujie, Y.; Takemasa, I.; Ikeda, M.; Takahashi-Yanaga, F.; Sasaguri, T.; Sekimoto, M.; Matsuura, N.; et al. Oxaliplatin induces mitotic catastrophe and apoptosis in esophageal cancer cells. Cancer Sci. 2008, 99, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhu, H.; Quan, L.; Zhou, C.; Bai, J.; Zhang, G.; Zhan, Q.; Xu, N. Downregulation of survivin by RNAI inhibits the growth of esophageal carcinoma cells. Cancer Biol. Ther. 2005, 4, 974–978. [Google Scholar] [CrossRef] [PubMed]
- Vattemi, E.; Claudio, P.P. Adenoviral gene therapy in head and neck cancer. Drug News Perspect. 2006, 19, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Cartier, N.; Hacein-Bey-Abina, S.; Bartholomae, C.C.; Veres, G.; Schmidt, M.; Kutschera, I.; Vidaud, M.; Abel, U.; dal Cortivo, L.; Caccavelli, L.; et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science 2009, 326, 818–823. [Google Scholar] [CrossRef] [PubMed]
- Khan, Z.; Khan, N.; Varma, A.K.; Tiwari, R.P.; Mouhamad, S.; Prasad, G.B.; Bisen, P.S. Oxaliplatin-mediated inhibition of survivin increases sensitivity of head and neck squamous cell carcinoma cell lines to paclitaxel. Curr. Cancer Drug Targets 2010, 10, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Esquela-Kerscher, A.; Slack, F.J. OncomiRs-microRNAs with a role in cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Novina, C.D.; Sharp, P.A. The RNAI revolution. Nature 2004, 430, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Choi, Y.C.; Lee, S.; Jeong, Y.; Yoon, J.; Baek, K. Induction of growth arrest by miR-542-3p that targets survivin. FEBS Lett. 2010, 584, 4048–4052. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Fu, H.; Xing, R.; Tie, Y.; Zhu, J.; Sun, Z.; Zheng, X. Survivin knockdown combined with apoptin overexpression inhibits cell growth significantly. Cancer Biol. Ther. 2008, 7, 1053–1060. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Lin, R.; Dai, J.; Jin, D.; Wang, S.Q. Suppression of tumor growth using antisense oligonucleotide against survivin in an orthotopic transplant model of human hepatocellular carcinoma in nude mice. Oligonucleotides 2006, 16, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Rodel, F.; Frey, B.; Leitmann, W.; Capalbo, G.; Weiss, C.; Rodel, C. Survivin antisense oligonucleotides effectively radiosensitize colorectal cancer cells in both tissue culture and murine xenograft models. Int. J. Radiat. Oncol. Biol. Phys. 2008, 71, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Sah, N.K.; Munshi, A.; Hobbs, M.; Carter, B.Z.; Andreeff, M.; Meyn, R.E. Effect of downregulation of survivin expression on radiosensitivity of human epidermoid carcinoma cells. Int. J. Radiat. Oncol. Biol. Phys. 2006, 66, 852–859. [Google Scholar] [CrossRef] [PubMed]
- Sharma, H.; Sen, S.; Lo Muzio, L.; Mariggio, A.; Singh, N. Antisense-mediated downregulation of anti-apoptotic proteins induces apoptosis and sensitizes head and neck squamous cell carcinoma cells to chemotherapy. Cancer Biol. Ther. 2005, 4, 720–727. [Google Scholar] [CrossRef] [PubMed]
- Kojima, H.; Iida, M.; Yaguchi, Y.; Suzuki, R.; Hayashi, N.; Moriyama, H.; Manome, Y. Enhancement of cisplatin sensitivity in squamous cell carcinoma of the head and neck transfected with a survivin antisense gene. Arch. Otolaryngol. Head Neck Surg. 2006, 132, 682–685. [Google Scholar] [CrossRef] [PubMed]
- Talbot, D.C.; Ranson, M.; Davies, J.; Lahn, M.; Callies, S.; Andre, V.; Kadam, S.; Burgess, M.; Slapak, C.; Olsen, A.L.; et al. Tumor survivin is downregulated by the antisense oligonucleotide ly2181308: A proof-of-concept, first-in-human dose study. Clin. Cancer Res. 2010, 16, 6150–6158. [Google Scholar] [CrossRef] [PubMed]
- Tanioka, M.; Nokihara, H.; Yamamoto, N.; Yamada, Y.; Yamada, K.; Goto, Y.; Fujimoto, T.; Sekiguchi, R.; Uenaka, K.; Callies, S.; et al. Phase i study of ly2181308, an antisense oligonucleotide against survivin, in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2011, 68, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Carter, B.Z.; Wang, R.Y.; Schober, W.D.; Milella, M.; Chism, D.; Andreeff, M. Targeting survivin expression induces cell proliferation defect and subsequent cell death involving mitochondrial pathway in myeloid leukemic cells. Cell Cycle 2003, 2, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M.; Arendt, B.K.; Grote, D.M.; Jelinek, D.F.; Novak, A.J.; Wellik, L.E.; Remstein, E.D.; Bennett, C.F.; Fielding, A. Inhibition of survivin expression suppresses the growth of aggressive non-hodgkin’s lymphoma. Leukemia 2004, 18, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, T.; Takeuchi, M.; Kinoyama, I.; Minematsu, T.; Shirasuna, K.; Matsuhisa, A.; Kita, A.; Tominaga, F.; Yamanaka, K.; Kudoh, M.; et al. YM155, a novel small-molecule survivin suppressant, induces regression of established human hormone-refractory prostate tumor xenografts. Cancer Res. 2007, 67, 8014–8021. [Google Scholar] [CrossRef] [PubMed]
- Iwasa, T.; Okamoto, I.; Suzuki, M.; Nakahara, T.; Yamanaka, K.; Hatashita, E.; Yamada, Y.; Fukuoka, M.; Ono, K.; Nakagawa, K. Radiosensitizing effect of YM155, a novel small-molecule survivin suppressant, in non-small cell lung cancer cell lines. Clin. Cancer Res. 2008, 14, 6496–6504. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, T.; Kita, A.; Yamanaka, K.; Mori, M.; Amino, N.; Takeuchi, M.; Tominaga, F.; Kinoyama, I.; Matsuhisa, A.; Kudou, M.; et al. Broad spectrum and potent antitumor activities of YM155, a novel small-molecule survivin suppressant, in a wide variety of human cancer cell lines and xenograft models. Cancer Sci. 2011, 102, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Minematsu, T.; Iwai, M.; Umehara, K.; Usui, T.; Kamimura, H. Characterization of human organic cation transporter 1 (OCT1/SLC22A1)- and OCT2 (SLC22A2)-mediated transport of 1-(2-methoxyethyl)-2-methyl-4,9-dioxo-3-(pyrazin-2-ylmethyl)-4,9-dihydro-1h-naphtho[2,3-d]imidazolium bromide (YM155 monobromide), a novel small molecule survivin suppressant. Drug Metab. Dispos. 2010, 38, 1–4. [Google Scholar] [PubMed]
- Iwasa, T.; Okamoto, I.; Takezawa, K.; Yamanaka, K.; Nakahara, T.; Kita, A.; Koutoku, H.; Sasamata, M.; Hatashita, E.; Yamada, Y.; et al. Marked anti-tumour activity of the combination of YM155, a novel survivin suppressant, and platinum-based drugs. Br. J. Cancer 2010, 103, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.W.; Mita, A.; Lewis, L.D.; Garrett, C.R.; Till, E.; Daud, A.I.; Patnaik, A.; Papadopoulos, K.; Takimoto, C.; Bartels, P.; et al. Phase I and pharmacokinetic study of YM155, a small-molecule inhibitor of survivin. J. Clin. Oncol. 2008, 26, 5198–5203. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Okamoto, I.; Miyazaki, M.; Morinaga, R.; Tsuya, A.; Hasegawa, Y.; Terashima, M.; Ueda, S.; Fukuoka, M.; Ariyoshi, Y.; et al. Phase I study of YM155, a novel survivin suppressant, in patients with advanced solid tumors. Clin. Cancer Res. 2009, 15, 3872–3880. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K.D.; Samlowski, W.; Ward, J.; Catlett, J.; Cranmer, L.; Kirkwood, J.; Lawson, D.; Whitman, E.; Gonzalez, R. A multi-center phase II evaluation of the small molecule survivin suppressor YM155 in patients with unresectable stage III or IV melanoma. Investig. New Drugs 2011, 29, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Giaccone, G.; Zatloukal, P.; Roubec, J.; Floor, K.; Musil, J.; Kuta, M.; van Klaveren, R.J.; Chaudhary, S.; Gunther, A.; Shamsili, S. Multicenter phase II trial of YM155, a small-molecule suppressor of survivin, in patients with advanced, refractory, non-small-cell lung cancer. J. Clin. Oncol. 2009, 27, 4481–4486. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, D.S.; Wall, N.R.; Porter, A.C.; Altieri, D.C. A p34(CDC2) survival checkpoint in cancer. Cancer Cell 2002, 2, 43–54. [Google Scholar] [CrossRef]
- Pennati, M.; Campbell, A.J.; Curto, M.; Binda, M.; Cheng, Y.; Wang, L.Z.; Curtin, N.; Golding, B.T.; Griffin, R.J.; Hardcastle, I.R.; et al. Potentiation of paclitaxel-induced apoptosis by the novel cyclin-dependent kinase inhibitor NU6140: A possible role for survivin down-regulation. Mol. Cancer Ther. 2005, 4, 1328–1337. [Google Scholar] [CrossRef] [PubMed]
- Prystowsky, M.B.; Adomako, A.; Smith, R.V.; Kawachi, N.; McKimpson, W.; Atadja, P.; Chen, Q.; Schlecht, N.F.; Parish, J.L.; Childs, G.; et al. The histone deacetylase inhibitor lbh589 inhibits expression of mitotic genes causing G2/M arrest and cell death in head and neck squamous cell carcinoma cell lines. J. Pathol. 2009, 218, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Castelli, C.; Rivoltini, L.; Andreola, G.; Carrabba, M.; Renkvist, N.; Parmiani, G. T-cell recognition of melanoma-associated antigens. J. Cell. Physiol. 2000, 182, 323–331. [Google Scholar] [CrossRef]
- Rohayem, J.; Diestelkoetter, P.; Weigle, B.; Oehmichen, A.; Schmitz, M.; Mehlhorn, J.; Conrad, K.; Rieber, E.P. Antibody response to the tumor-associated inhibitor of apoptosis protein survivin in cancer patients. Cancer Res. 2000, 60, 1815–1817. [Google Scholar] [PubMed]
- Schmidt, S.M.; Schag, K.; Muller, M.R.; Weck, M.M.; Appel, S.; Kanz, L.; Grunebach, F.; Brossart, P. Survivin is a shared tumor-associated antigen expressed in a broad variety of malignancies and recognized by specific cytotoxic T cells. Blood 2003, 102, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, M.; Diestelkoetter, P.; Weigle, B.; Schmachtenberg, F.; Stevanovic, S.; Ockert, D.; Rammensee, H.G.; Rieber, E.P. Generation of survivin-specific CD8+ T effector cells by dendritic cells pulsed with protein or selected peptides. Cancer Res. 2000, 60, 4845–4849. [Google Scholar] [PubMed]
- Reker, S.; Meier, A.; Holten-Andersen, L.; Svane, I.M.; Becker, J.C.; thor Straten, P.; Andersen, M.H. Identification of novel survivin-derived CTL epitopes. Cancer Biol. Ther. 2004, 3, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.H.; thor, S.P. Survivin—A universal tumor antigen. Histol. Histopathol. 2002, 17, 669–675. [Google Scholar] [PubMed]
- Miyazaki, A.; Kobayashi, J.; Torigoe, T.; Hirohashi, Y.; Yamamoto, T.; Yamaguchi, A.; Asanuma, H.; Takahashi, A.; Michifuri, Y.; Nakamori, K.; et al. Phase i clinical trial of survivin-derived peptide vaccine therapy for patients with advanced or recurrent oral cancer. Cancer Sci. 2011, 102, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Hirohashi, Y.; Torigoe, T.; Maeda, A.; Nabeta, Y.; Kamiguchi, K.; Sato, T.; Yoda, J.; Ikeda, H.; Hirata, K.; Yamanaka, N.; et al. An HLA-A24-restricted cytotoxic T lymphocyte epitope of a tumor-associated protein, survivin. Clin. Cancer Res. 2002, 8, 1731–1739. [Google Scholar] [PubMed]
- Idenoue, S.; Hirohashi, Y.; Torigoe, T.; Sato, Y.; Tamura, Y.; Hariu, H.; Yamamoto, M.; Kurotaki, T.; Tsuruma, T.; Asanuma, H.; et al. A potent immunogenic general cancer vaccine that targets survivin, an inhibitor of apoptosis proteins. Clin. Cancer Res. 2005, 11, 1474–1482. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, J.; Torigoe, T.; Hirohashi, Y.; Idenoue, S.; Miyazaki, A.; Yamaguchi, A.; Hiratsuka, H.; Sato, N. Comparative study on the immunogenicity between an HLA-A24-restricted cytotoxic T-cell epitope derived from survivin and that from its splice variant survivin-2b in oral cancer patients. J. Transl. Med. 2009, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- Kandiloros, D.; Eleftheriadou, A.; Chalastras, T.; Kyriou, L.; Yiotakis, I.; Ferekidis, E. Prospective study of a panel of tumor markers as prognostic factors in patients with squamous cell carcinoma of head and neck. Med. Oncol. 2006, 23, 463–470. [Google Scholar] [CrossRef]
- Meyer, F.; Samson, E.; Douville, P.; Duchesne, T.; Liu, G.; Bairati, I. Serum prognostic markers in head and neck cancer. Clin. Cancer Res. 2010, 16, 1008–1015. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, M.; Yang, D.; Hu, X.; Liu, F.; Singh, N.; Browning, D.; Ganapathy, V.; Chandler, P.; Choubey, D.; Abrams, S.I.; et al. IFN-γ upregulates survivin and IFI202 expression to induce survival and proliferation of tumor-specific t cells. PLoS ONE 2010, 5, e14076. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Yannelli, J.R.; Yang, J.C.; Topalian, S.L.; Schwartzentruber, D.J.; Weber, J.S.; Parkinson, D.R.; Seipp, C.A.; Einhorn, J.H.; White, D.E. Treatment of patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and interleukin 2. J. Natl. Cancer Inst. 1994, 86, 1159–1166. [Google Scholar] [CrossRef] [PubMed]
- Dudley, M.E.; Wunderlich, J.R.; Robbins, P.F.; Yang, J.C.; Hwu, P.; Schwartzentruber, D.J.; Topalian, S.L.; Sherry, R.; Restifo, N.P.; Hubicki, A.M.; et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 2002, 298, 850–854. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Dudley, M.E.; Restifo, N.P. Cancer immunotherapy. N. Engl. J. Med. 2008, 359, 1072. [Google Scholar] [PubMed]
- Leung, C.G.; Xu, Y.; Mularski, B.; Liu, H.; Gurbuxani, S.; Crispino, J.D. Requirements for survivin in terminal differentiation of erythroid cells and maintenance of hematopoietic stem and progenitor cells. J. Exp. Med. 2007, 204, 1603–1611. [Google Scholar] [CrossRef] [PubMed]
- Sharief, M.K.; Semra, Y.K. Down-regulation of survivin expression in T lymphocytes after interferon beta-1a treatment in patients with multiple sclerosis. Arch. Neurol. 2002, 59, 1115–1121. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, W.H.; Biade, S.; Zilfou, J.T.; Chen, J.; Murphy, M. Transcriptional repression of the anti-apoptotic survivin gene by wild type p53. J. Biol. Chem. 2002, 277, 3247–3257. [Google Scholar] [CrossRef] [PubMed]
- Mirza, A.; McGuirk, M.; Hockenberry, T.N.; Wu, Q.; Ashar, H.; Black, S.; Wen, S.F.; Wang, L.; Kirschmeier, P.; Bishop, W.R.; et al. Human survivin is negatively regulated by wild-type p53 and participates in p53-dependent apoptotic pathway. Oncogene 2002, 21, 2613–2622. [Google Scholar] [CrossRef] [PubMed]
- Vile, R.G.; Russell, S.J.; Lemoine, N.R. Cancer gene therapy: Hard lessons and new courses. Gene Ther. 2000, 7, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.M.; Grandis, J.R. The current state of head and neck cancer gene therapy. Hum. Gene Ther. 2009, 20, 1565–1575. [Google Scholar] [CrossRef] [PubMed]
- Brenner, M.K.; Gottschalk, S.; Leen, A.M.; Vera, J.F. Is cancer gene therapy an empty suit? Lancet Oncol. 2013, 14, e447–e456. [Google Scholar] [CrossRef]
- Cross, D.; Burmester, J.K. Gene therapy for cancer treatment: Past, present and future. Clin Med Res 2006, 4, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Ventola, C.L. Cancer immunotherapy, part 3: Challenges and future trends. Pharm. Ther. 2017, 42, 514–521. [Google Scholar]
- Pardoll, D. Cancer and the immune system: Basic concepts and targets for intervention. Semin. Oncol. 2015, 42, 523–538. [Google Scholar] [CrossRef] [PubMed]
- Tsuruma, T.; Iwayama, Y.; Ohmura, T.; Katsuramaki, T.; Hata, F.; Furuhata, T.; Yamaguchi, K.; Kimura, Y.; Torigoe, T.; Toyota, N.; et al. Clinical and immunological evaluation of anti-apoptosis protein, survivin-derived peptide vaccine in phase i clinical study for patients with advanced or recurrent breast cancer. J. Transl. Med. 2008, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Fenstermaker, R.A.; Ciesielski, M.J.; Qiu, J.; Yang, N.; Frank, C.L.; Lee, K.P.; Mechtler, L.R.; Belal, A.; Ahluwalia, M.S.; Hutson, A.D. Clinical study of a survivin long peptide vaccine (survaxm) in patients with recurrent malignant glioma. Cancer Immunol. Immunother. 2016, 65, 1339–1352. [Google Scholar] [CrossRef] [PubMed]
- Clemens, M.R.; Gladkov, O.A.; Gartner, E.; Vladimirov, V.; Crown, J.; Steinberg, J.; Jie, F.; Keating, A. Phase II, multicenter, open-label, randomized study of YM155 plus docetaxel as first-line treatment in patients with HER2-negative metastatic breast cancer. Breast Cancer Res. Treat. 2015, 149, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.W.; Quinn, D.I.; Ferrari, A.; Ahmann, F.; Giaccone, G.; Drake, T.; Keating, A.; de Bono, J.S. A phase ii study of YM155, a novel small-molecule suppressor of survivin, in castration-resistant taxane-pretreated prostate cancer. Ann. Oncol. 2012, 23, 968–973. [Google Scholar] [CrossRef] [PubMed]
- Brany, D.; Dvorska, D.; Slavik, P.; Skolka, R.; Adamkov, M. Survivin and gynaecological tumours. Pathol. Res. Pract. 2017, 213, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Winograd, E.K.; Ciesielski, M.J.; Fenstermaker, R.A. Novel vaccines for glioblastoma: Clinical update and perspective. Immunotherapy 2016, 8, 1293–1308. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.J.; Thomas, A.; Rajan, A.; Chun, G.; Lopez-Chavez, A.; Szabo, E.; Spencer, S.; Carter, C.A.; Guha, U.; Khozin, S.; et al. A phase I/II study of sepantronium bromide (YM155, survivin suppressor) with paclitaxel and carboplatin in patients with advanced non-small-cell lung cancer. Ann. Oncol. 2013, 24, 2601–2606. [Google Scholar] [CrossRef] [PubMed]
- Honma, I.; Kitamura, H.; Torigoe, T.; Takahashi, A.; Tanaka, T.; Sato, E.; Hirohashi, Y.; Masumori, N.; Tsukamoto, T.; Sato, N. Phase I clinical study of anti-apoptosis protein survivin-derived peptide vaccination for patients with advanced or recurrent urothelial cancer. Cancer Immunol. Immunother. 2009, 58, 1801–1807. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Kitamura, H.; Inoue, R.; Nishida, S.; Takahashi-Takaya, A.; Kawami, S.; Torigoe, T.; Hirohashi, Y.; Tsukamoto, T.; Sato, N.; et al. Potential survival benefit of anti-apoptosis protein: Survivin-derived peptide vaccine with and without interferon alpha therapy for patients with advanced or recurrent urothelial cancer—Results from phase I clinical trials. Clin. Dev. Immunol. 2013, 2013, 262967. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author | Agent | Phase | Site of Cancer | Results | Ref./ID Code |
|---|---|---|---|---|---|
| Talbot et al. | LY2181308 | I | HNSCC, esophagus | Effective downregulation of survivin. Dose level of 750 mg. | [131] |
| Tanioka et al. | LY2181308 | I | Esophagus | Grade I/II toxicities. Dose level of 750 mg. No case of SD. | [132] |
| Tolcher et al. | YM155 | I | HNSCC, NSCLC | MTD of 4.8 mg/m2. 1 case of NSCLC had minor response. | [140] |
| Satoh et al. | YM155 | I | NSCLC, esophagus | MTD of 8.0 mg/m2/day. Grade I/II toxicities. | [141] |
| Kelly et al. | YM155 | I/II | NSCLC | Grade II hematological toxicities. Two cases of PR. | [179] NCT01100931 |
| Giaccone et al. | YM155 | II | NSCLC | Grade III toxicities 18.9%. Two cases of PR, 14 cases of SD. | [143] |
| Honma et al. | Peptide vaccine (2B80-88) | I | Urinary tract | Grade I toxicities in six patients. Two cases of SD. | [180] |
| Miyazaki et al. | Peptide vaccine (2B80-88) | I | Oral cavity | No cases of toxicity. One case of PR. | [153] UMIN000000976 |
| Tanaka et al. | Peptide vaccine (2B80-88) | I | Urinary tract | Grade I toxicities. Six cases of SD. | [181] UMIN00005859 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santarelli, A.; Mascitti, M.; Lo Russo, L.; Sartini, D.; Troiano, G.; Emanuelli, M.; Lo Muzio, L. Survivin-Based Treatment Strategies for Squamous Cell Carcinoma. Int. J. Mol. Sci. 2018, 19, 971. https://doi.org/10.3390/ijms19040971
Santarelli A, Mascitti M, Lo Russo L, Sartini D, Troiano G, Emanuelli M, Lo Muzio L. Survivin-Based Treatment Strategies for Squamous Cell Carcinoma. International Journal of Molecular Sciences. 2018; 19(4):971. https://doi.org/10.3390/ijms19040971
Chicago/Turabian StyleSantarelli, Andrea, Marco Mascitti, Lucio Lo Russo, Davide Sartini, Giuseppe Troiano, Monica Emanuelli, and Lorenzo Lo Muzio. 2018. "Survivin-Based Treatment Strategies for Squamous Cell Carcinoma" International Journal of Molecular Sciences 19, no. 4: 971. https://doi.org/10.3390/ijms19040971