
Moving Past Anti-VEGF: Novel Therapies for Treating Diabetic Retinopathy

Abstract
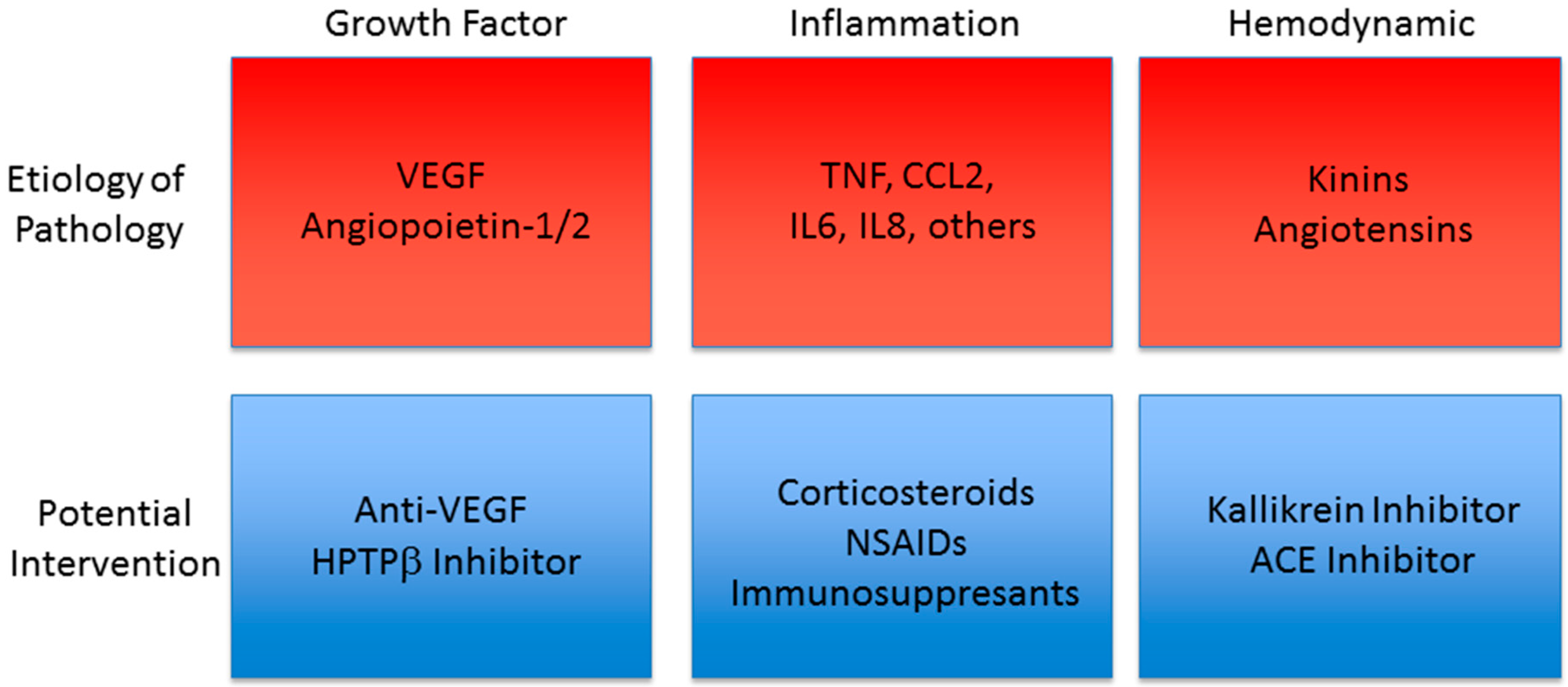
:1. Introduction
2. Retinal Anatomy and the Blood Retinal Barrier
2.1. The Blood Retinal Barrier
2.2. Tight Junctions
3. Diabetes-Associated Ophthalmic Pathologies
3.1. Diabetic Retinopathy
3.2. Diabetic Macular Edema
4. Therapeutic Intervention
4.1. VEGF
4.2. TNF-α
4.3. Corticosteroids
4.4. Kinin-Kallikrein System Inhibitors
4.5. Renin-Angiotensin System Inhibitors
4.6. RAS, KKS, and ACE
4.7. Angiopoietin
4.8. Non-Steroidal Anti-Inflammatory Drugs
4.9. Antibiotics and Immunosuppressants
4.10. Antioxidants
4.11. Vitreomacular Adhesion and Vitriol Viscosity Inhibitors
5. Summary
Acknowledgments
Conflicts of Interest
Abbreviations
| ACE | Angiotensin Converting Enzyme |
| Ang1 | Angiopoietin-1 |
| Ang2 | Angiopoietin-2 |
| B1R | Bradykinin-1 Receptor |
| B2R | Bradykinin-2 Receptor |
| BRB | Blood Retinal Barrier |
| COX | Cyclo-oxygenase |
| Coxib | COX Inhibitor |
| DME | Diabetic Macular Edema |
| DR | Diabetic Retinopathy |
| HAE | Hereditary Angioedema |
| HMWK | High Molecular Weight Kininogen |
| HPTPβ | Human Protein Tyrosine Phosphatase β |
| ICAM-1 | Intracellular Adhesion Molecule-1 |
| IOP | Intraocular Pressure |
| KKS | Kinin-Kallikrein System |
| LMWK | Low Molecular Weight Kininogen |
| NF-κB | Nuclear Factor Kappa B |
| NSAID | Non-Steroidal Anti-Inflammatory Drug |
| PVD | Posterior Vitreous Detachment |
| RAS | Renin-Angiotensin System |
| RPE | Retinal Pigmented Epithelium |
| TJ | Tight Junction |
| TNF-α | Tumor Necrosis Factor-Alpha |
| VEGF | Vascular Endothelial Growth Factor |
| VMA | Vitreomacular Adhesion |
| ZO-1, 2 | Zonula Occludens-1, 2 |
References
- Fong, D.S.; Aiello, L.; Gardner, T.W.; King, G.L.; Blankenship, G.; Cavallerano, J.D.; Ferris, F.L., 3rd; Klein, R. Retinopathy in Diabetes. Diabetes Care 2003, 26, s84–s87. [Google Scholar] [CrossRef]
- Aiello, L.P.; Gardner, T.W.; King, G.L.; Blankenship, G.; Cavallerano, J.D.; Ferris, F.L., 3rd; Klein, R. Diabetic retinopathy. Diabetes Care 1998, 21, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Prevention, Blindness caused by diabetes—Massachusetts, 1987–1994. Morb. Mortal. Wkly. Rep. 1996, 45, 937–941. [Google Scholar]
- Javitt, J.C.; Aiello, L.P.; Chiang, Y.; Ferris, F.L., 3rd; Canner, J.K.; Greenfield, S. Preventive eye care in people with diabetes is cost-saving to the federal government. Implications for health-care reform. Diabetes Care 1994, 17, 909–917. [Google Scholar] [CrossRef] [PubMed]
- NCD Risk Factor Collaboration (NCD-RisC). Worldwide trends in diabetes since 1980: A pooled analysis of 751 population-based studies with 4.4 million participants. Lancet 2016, 387, 1513–1530. [Google Scholar]
- Yau, J.W.; Rogers, S.L.; Kawasaki, R.; Lamoureux, E.L.; Kowalski, J.W.; Bek, T.; Chen, S.J.; Dekker, J.M.; Fletcher, A.; Grauslund, J.; et al. Global prevalence and major risk factors of diabetic retinopathy. Diabetes Care 2012, 35, 556–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyle, J.P.; Thompson, T.J.; Gregg, E.W.; Barker, L.E.; Williamson, D.F. Projection of the year 2050 burden of diabetes in the US adult population: Dynamic modeling of incidence, mortality, and prediabetes prevalence. Popul. Health Metr. 2010, 8. [Google Scholar] [CrossRef] [PubMed]
- Frey, T.; Antonetti, D.A. Alterations to the blood-retinal barrier in diabetes: Cytokines and reactive oxygen species. Antioxid. Redox Signal. 2011, 15, 1271–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winton-Brown, T.T.; Ting, A.; Mocellin, R.; Velakoulis, D.; Gaillard, F. Distinguishing neuroimaging features in patients presenting with visual hallucinations. Am. J. Neuroradiol. 2016, 37, 774–781. [Google Scholar] [CrossRef] [PubMed]
- Provis, J.M. Development of the primate retinal vasculature. Progress Retin. Eye Res. 2001, 20, 799–821. [Google Scholar] [CrossRef]
- Zhou, R.; Caspi, R.R. Ocular immune privilege. F1000 Biol. Rep. 2010, 2. [Google Scholar] [CrossRef] [PubMed]
- Antonetti, D.A.; Klein, R.; Gardner, T.W. Diabetic retinopathy. N. Engl. J. Med. 2012, 366, 1227–1239. [Google Scholar] [CrossRef] [PubMed]
- Maines, L.W.; Antonetti, D.A.; Wolpert, E.B.; Smith, C.D. Evaluation of the role of P-glycoprotein in the uptake of paroxetine, clozapine, phenytoin and carbamazapine by bovine retinal endothelial cells. Neuropharmacology 2005, 49, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, Y.; Tischfield, M.; Williams, J.; Smallwood, P.M.; Rattner, A.; Taketo, M.M.; Nathans, J. Canonical WNT signaling components in vascular development and barrier formation. J. Clin. Investig. 2014, 124, 3825–3846. [Google Scholar] [CrossRef] [PubMed]
- Armulik, A.; Genove, G.; Mae, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes regulate the blood-brain barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Antonetti, D.A.; Van Guilder, H.D.; Lin, C.M. Vascular permeability in diabetic retinopahty. In Diabetic Retinopathy; Duh, E.J., Ed.; Humana Press: Totwa, NJ, USA, 2008; pp. 333–352. [Google Scholar]
- Abcouwer, S.F.; Gardner, T.W. Diabetic retinopathy: Loss of neuroretinal adaptation to the diabetic metabolic environment. Ann. N. Y. Acad. Sci. 2014, 1311, 174–190. [Google Scholar] [CrossRef] [PubMed]
- Stem, M.S.; Gardner, T.W. Neurodegeneration in the pathogenesis of diabetic retinopathy: Molecular mechanisms and therapeutic implications. Curr. Med. Chem. 2013, 20, 3241–3250. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Cleghorn, W.; Contreras, L.; Linton, J.D.; Chan, G.C.; Chertov, A.O.; Saheki, T.; Govindaraju, V.; Sadilek, M.; Satrustegui, J.; et al. Cytosolic reducing power preserves glutamate in retina. Proc. Natl. Acad. Sci. USA 2013, 110, 18501–18506. [Google Scholar] [CrossRef] [PubMed]
- Buttery, R.G.; Hinrichsen, C.F.; Weller, W.L.; Haight, J.R. How thick should a retina be? A comparative study of mammalian species with and without intraretinal vasculature. Vis. Res. 1991, 31, 169–187. [Google Scholar] [CrossRef]
- Schneeberger, E.E.; Lynch, R.D. The tight junction: A multifunctional complex. Am. J. Physiol. Cell Physiol. 2004, 286, C1213–C1228. [Google Scholar] [CrossRef] [PubMed]
- Fanning, A.S.; Jameson, B.J.; Jesaitis, L.A.; Anderson, J.M. The tight junction protein ZO-1 establishes a link between the transmembrane protein occludin and the actin cytoskeleton. J. Biol. Chem. 1998, 273, 29745–29753. [Google Scholar] [CrossRef] [PubMed]
- Antonetti, D.A.; Barber, A.J.; Khin, S.; Lieth, E.; Tarbell, J.M.; Gardner, T.W. Vascular permeability in experimental diabetes is associated with reduced endothelial occludin content: Vascular endothelial growth factor decreases occludin in retinal endothelial cells. Penn State Retina Research Group. Diabetes 1998, 47, 1953–1959. [Google Scholar] [CrossRef] [PubMed]
- Hirase, T.; Staddon, J.M.; Saitou, M.; Ando-Akatsuka, Y.; Itoh, M.; Furuse, M.; Fujimoto, K.; Tsukita, S.; Rubin, L.L. Occludin as a possible determinant of tight junction permeability in endothelial cells. J. Cell Sci. 1997, 110 Pt 14, 1603–1613. [Google Scholar] [PubMed]
- Phillips, B.E.; Antonetti, D.A. Blood-retinal barrier, retinal vascular leakage and macular edema. In Retinal Vascular Diseases; Joussen, A.M., Gardner, T.W., Kirshhof, B., Ryan, S.J., Eds.; Springer-Verlag: Berlin, Germany, 2007; pp. 139–147. [Google Scholar]
- Katsuno, T.; Umeda, K.; Matsui, T.; Hata, M.; Tamura, A.; Itoh, M.; Takeuchi, K.; Fujimori, T.; Nabeshima, Y.; Noda, T.; et al. Deficiency of zonula occludens-1 causes embryonic lethal phenotype associated with defected yolk sac angiogenesis and apoptosis of embryonic cells. Mol. Biol. Cell 2008, 19, 2465–2475. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Kausalya, P.J.; Phua, D.C.; Ali, S.M.; Hossain, Z.; Hunziker, W. Early embryonic lethality of mice lacking ZO-2, but Not ZO-3, reveals critical and nonredundant roles for individual zonula occludens proteins in mammalian development. Mol. Cell. Biol. 2008, 28, 1669–1678. [Google Scholar] [CrossRef] [PubMed]
- Furuse, M.; Hata, M.; Furuse, K.; Yoshida, Y.; Haratake, A.; Sugitani, Y.; Noda, T.; Kubo, A.; Tsukita, S. Claudin-based tight junctions are crucial for the mammalian epidermal barrier: A lesson from claudin-1-deficient mice. J. Cell Biol. 2002, 156, 1099–1111. [Google Scholar] [CrossRef] [PubMed]
- Muto, S.; Hata, M.; Taniguchi, J.; Tsuruoka, S.; Moriwaki, K.; Saitou, M.; Furuse, K.; Sasaki, H.; Fujimura, A.; Imai, M.; et al. Claudin-2-deficient mice are defective in the leaky and cation-selective paracellular permeability properties of renal proximal tubules. Proc. Natl. Acad. Sci. USA. 2010, 107, 8011–8016. [Google Scholar] [CrossRef] [PubMed]
- Breiderhoff, T.; Himmerkus, N.; Stuiver, M.; Mutig, K.; Will, C.; Meij, I.C.; Bachmann, S.; Bleich, M.; Willnow, T.E.; Muller, D. Deletion of claudin-10 (Cldn10) in the thick ascending limb impairs paracellular sodium permeability and leads to hypermagnesemia and nephrocalcinosis. Proc. Natl. Acad. Sci. USA 2012, 109, 14241–14246. [Google Scholar] [CrossRef] [PubMed]
- Nitta, T.; Hata, M.; Gotoh, S.; Seo, Y.; Sasaki, H.; Hashimoto, N.; Furuse, M.; Tsukita, S. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J. Cell Biol. 2003, 161, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Flodby, P.; Luo, J.; Kage, H.; Sipos, A.; Gao, D.; Ji, Y.; Beard, L.L.; Marconett, C.N.; DeMaio, L.; et al. Knockout mice reveal key roles for claudin 18 in alveolar barrier properties and fluid homeostasis. Am. J. Respir. Cell Mol. Biol. 2014, 51, 210–222. [Google Scholar] [CrossRef] [PubMed]
- Saitou, M.; Furuse, M.; Sasaki, H.; Schulzke, J.D.; Fromm, M.; Takano, H.; Noda, T.; Tsukita, S. Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol. Biol. Cell 2000, 11, 4131–4142. [Google Scholar] [CrossRef] [PubMed]
- Schulzke, J.D.; Gitter, A.H.; Mankertz, J.; Spiegel, S.; Seidler, U.; Amasheh, S.; Saitou, M.; Tsukita, S.; Fromm, M. Epithelial transport and barrier function in occludin-deficient mice. Biochim. Biophys. Acta 2005, 1669, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Felinski, E.A.; Antonetti, D.A. Occludin phosphorylation and ubiquitination regulate tight junction trafficking and vascular endothelial growth factor-induced permeability. J. Biol. Chem. 2009, 284, 21036–21046. [Google Scholar] [CrossRef] [PubMed]
- Wessel, F.; Winderlich, M.; Holm, M.; Frye, M.; Rivera-Galdos, R.; Vockel, M.; Linnepe, R.; Ipe, U.; Stadtmann, A.; Zarbock, A.; et al. Leukocyte extravasation and vascular permeabilitity are each controlled in vivo by different tyrosine residues of VE-cadherin. Nat. Immunol. 2014, 15, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.; Airey, M.; Baxter, H.; Forrester, J.; Kennedy-Martin, T.; Girach, A. Epidemiology of diabetic retinopathy and macular oedema: A systematic review. Eye 2004, 18, 963–983. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, T.; Jalil, A.; Antoniou, Y.; Bishop, P.N.; Vallejo-Garcia, J.L.; Patton, N. Vitrectomy for primary symptomatic vitreous opacities: An evidence-based review. Eye 2016, 30, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Ali, F.A. A review of diabetic macular edema. Dig. J. Ophthalmol. 1997, 3. [Google Scholar]
- Klein, R.; Knudtson, M.D.; Lee, K.E.; Gangnon, R.; Klein, B.E. The Wisconsin Epidemiologic Study of Diabetic Retinopathy XXIII: The twenty-five-year incidence of macular edema in persons with type 1 diabetes. Ophthalmology 2009, 116, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.; Moss, S. A comparison of the study populations in the Diabetes Control and Complications Trial and the Wisconsin Epidemiologic Study of Diabetic Retinopathy. Arch. Intern. Med. 1995, 155, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Romero-Aroca, P.; Baget-Bernaldiz, M.; Fernandez-Ballart, J.; Plana-Gil, N.; Soler-Lluis, N.; Mendez-Marin, I.; Bautista-Perez, A. Ten-year incidence of diabetic retinopathy and macular edema. Risk factors in a sample of people with type 1 diabetes. Diabetes Res. Clin. Pract. 2011, 94, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Diabetic Retinopathy Clinical Research Network; Browning, D.J.; Glassman, A.R.; Aiello, L.P.; Beck, R.W.; Brown, D.W.; Fong, D.S.; Bressler, N.M.; Danis, R.P.; Kinyoun, J.L.; et al. Relationship between optical coherence tomography-measured central retinal thickness and visual acuity in diabetic macular edema. Ophthalmology 2007, 114, 525–536. [Google Scholar] [PubMed]
- Patelli, F.; Fasolino, G.; Radice, P.; Russo, S.; Zumbo, G.; di Tizio, F.M.; Frisone, G.; Marchi, S. Time course of changes in retinal thickness and visual acuity after intravitreal triamcinolone acetonide for diffuse diabetic macular edema with and without previous macular laser treatment. Retina 2005, 25, 840–845. [Google Scholar] [CrossRef] [PubMed]
- Gardner, T.W.; Larsen, M.; Girach, A.; Zhi, X. Diabetic macular oedema and visual loss: Relationship to location, severity and duration. Acta Ophthalmol. 2009, 87, 709–713. [Google Scholar] [CrossRef] [PubMed]
- Sander, B.; Thornit, D.N.; Colmorn, L.; Strom, C.; Girach, A.; Hubbard, L.D.; Lund-Andersen, H.; Larsen, M. Progression of diabetic macular edema: Correlation with blood retinal barrier permeability, retinal thickness, and retinal vessel diameter. Investig. Ophthalmol. Vis. Sci. 2007, 48, 3983–3987. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.; Wong, T.Y.; Sabanayagam, C. Epidemiology of diabetic retinopathy, diabetic macular edema and related vision loss. Eye Vis. 2015, 2. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Schwickerath, G. Light coagulation; a method for treatment and prevention of the retinal detachment. Albrecht Graefes Arch. Ophthalmol. 1954, 156, 2–34. [Google Scholar] [CrossRef]
- The Early Treatment Diabetic Retinopathy Study Research Group. Techniques for scatter and local photocoagulation treatment of diabetic retinopathy: Early Treatment Diabetic Retinopathy Study Report No. 3. Int. Ophthalmol. Clin. 1987, 27, 254–264. [Google Scholar]
- The Diabetic Retinopathy Study Research Group. Photocoagulation treatment of proliferative diabetic retinopathy. Clinical application of Diabetic Retinopathy Study (DRS) findings, DRS Report Number 8. Ophthalmology 1981, 88, 583–600. [Google Scholar]
- Fong, D.S.; Girach, A.; Boney, A. Visual side effects of successful scatter laser photocoagulation surgery for proliferative diabetic retinopathy: A literature review. Retina 2007, 27, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Action to Control Cardiovascular Risk in Diabetes Study Group; Gerstein, H.C.; Miller, M.E.; Byington, R.P.; Goff, D.C., Jr.; Bigger, J.T.; Buse, J.B.; Cushman, W.C.; Genuth, S.; Ismail-Beigi, F.; et al. Effects of intensive glucose lowering in type 2 diabetes. N. Engl. J. Med. 2008, 358, 2545–2559. [Google Scholar] [PubMed]
- Chew, E.Y.; Davis, M.D.; Danis, R.P.; Lovato, J.F.; Perdue, L.H.; Greven, C.; Genuth, S.; Goff, D.C.; Leiter, L.A.; Ismail-Beigi, F.; et al. The effects of medical management on the progression of diabetic retinopathy in persons with type 2 diabetes: The Action to Control Cardiovascular Risk in Diabetes (ACCORD) Eye Study. Ophthalmology 2014, 121, 2443–2451. [Google Scholar] [CrossRef] [PubMed]
- UK Prospective Diabetes Study Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes. Lancet 1998, 352, 837–853. [Google Scholar]
- Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N. Engl. J. Med. 1993, 329, 978–986. [Google Scholar]
- Nathan, D.M.; Cleary, P.A.; Backlund, J.Y.; Genuth, S.M.; Lachin, J.M.; Orchard, T.J.; Raskin, P.; Zinman, B. Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) Study Research Group. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N. Engl. J. Med. 2005, 353, 2643–2653. [Google Scholar] [PubMed]
- Keck, P.J.; Hauser, S.D.; Krivi, G.; Sanzo, K.; Warren, T.; Feder, J.; Connolly, D.T. Vascular permeability factor, an endothelial cell mitogen related to PDGF. Science 1989, 246, 1309–1312. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.W.; Cachianes, G.; Kuang, W.J.; Goeddel, D.V.; Ferrara, N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 1989, 246, 1306–1309. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.W.; Adamis, A.P.; Shima, D.T.; D’Amore, P.A.; Moulton, R.S.; O’Reilly, M.S.; Folkman, J.; Dvorak, H.F.; Brown, L.F.; Berse, B.; et al. Vascular endothelial growth factor/vascular permeability factor is temporally and spatially correlated with ocular angiogenesis in a primate model. Am. J. Pathol. 1994, 145, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Aiello, L.P.; Avery, R.L.; Arrigg, P.G.; Keyt, B.A.; Jampel, H.D.; Shah, S.T.; Pasquale, L.R.; Thieme, H.; Iwamoto, M.A.; Park, J.E. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N. Engl. J. Med. 1994, 331, 1480–1487. [Google Scholar] [CrossRef] [PubMed]
- Argaw, A.T.; Gurfein, B.T.; Zhang, Y.; Zameer, A.; John, G.R. VEGF-mediated disruption of endothelial CLN-5 promotes blood-brain barrier breakdown. Proc. Natl. Acad. Sci. USA 2009, 106, 1977–1982. [Google Scholar] [CrossRef] [PubMed]
- Simo, R.; Sundstrom, J.M.; Antonetti, D.A. Ocular Anti-VEGF therapy for diabetic retinopathy: The role of VEGF in the pathogenesis of diabetic retinopathy. Diabetes Care 2014, 37, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Stefanini, F.R.; Badaro, E.; Falabella, P.; Koss, M.; Farah, M.E.; Maia, M. Anti-VEGF for the management of diabetic macular edema. J. Immunol. Res. 2014, 2014, 632307. [Google Scholar] [CrossRef] [PubMed]
- Osaadon, P.; Fagan, X.J.; Lifshitz, T.; Levy, J. A review of anti-VEGF agents for proliferative diabetic retinopathy. Eye 2014, 28, 510–520. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, Q.D.; Brown, D.M.; Marcus, D.M.; Boyer, D.S.; Patel, S.; Feiner, L.; Gibson, A.; Sy, J.; Rundle, A.C.; Hopkins, J.J.; et al. Ranibizumab for diabetic macular edema; results from 2 phase III randomized trials; RISE and RIDE. Ophthalmology 2012, 119, 789–801. [Google Scholar] [CrossRef] [PubMed]
- Writing Committee for the Diabetic Retinopathy Clinical Research Network; Gross, J.G.; Glassman, A.R.; Jampol, L.M.; Inusah, S.; Aiello, L.P.; Antoszyk, A.N.; Baker, C.W.; Berger, B.B.; Bressler, N.M.; et al. Panretinal photocoagulation vs. intravitreous ranibizumab for proliferative diabetic retinopathy: A randomized clinical trial. JAMA 2015, 314, 2137–2146. [Google Scholar] [PubMed]
- Group, C.R.; Martin, D.F.; Maguire, M.G.; Ying, G.S.; Grunwald, J.E.; Fine, S.L.; Jaffe, G.J. Ranibizumab and bevacizumab for neovascular age-related macular degeneration. N. Engl. J. Med. 2011, 364, 1897–1908. [Google Scholar]
- Dabir, S.S.; Das, D.; Nallathambi, J.; Mangalesh, S.; Yadav, N.K.; Schouten, J.S. Differential systemic gene expression profile in patients with diabetic macular edema: Responders versus nonresponders to standard treatment. Indian J. Ophthalmol. 2014, 62, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Simo-Servat, O.; Hernandez, C.; Simo, R. Usefulness of the vitreous fluid analysis in the translational research of diabetic retinopathy. Mediat. Inflamm. 2012, 2012, 872978. [Google Scholar] [CrossRef] [PubMed]
- Semeraro, F.; Cancarini, A.; dell’Omo, R.; Rezzola, S.; Romano, M.R.; Costagliola, C. Diabetic Retinopathy: Vascular and Inflammatory Disease. J. Diabetes Res. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Kern, T.S. Contributions of inflammatory processes to the development of the early stages of diabetic retinopathy. Exp. Diabetes Res. 2007, 2007. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Odenbach, S. Role of interleukin-1beta in the pathogenesis of diabetic retinopathy. Br. J. Ophthalmol. 2004, 88, 1343–1347. [Google Scholar] [CrossRef] [PubMed]
- Kahn, S.E.; Hull, R.L.; Utzschneider, K.M. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 2006, 444, 840–846. [Google Scholar] [CrossRef] [PubMed]
- Doganay, S.; Evereklioglu, C.; Er, H.; Turkoz, Y.; Sevinc, A.; Mehmet, N.; Savli, H. Comparison of serum NO, TNF-α, IL-β, sIL-2R, IL-6 and IL-8 levels with grades of retinopathy in patients with diabetes mellitus. Eye 2002, 16, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Bamforth, S.D.; Lightman, S.; Greenwood, J. The effect of TNF-α and IL-6 on the permeability of the rat blood-retinal barrier in vivo. Acta Neuropathol. 1996, 91, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Aveleira, C.A.; Lin, C.M.; Abcouwer, S.F.; Ambrosio, A.F.; Antonetti, D.A. TNF-alpha signals through PKCζ/NF-κB to alter the tight junction complex and increase retinal endothelial cell permeability. Diabetes 2010, 59, 2872–2882. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zhao, N.; Xu, X.; Xu, Y.; Li, S.; Zhang, J.; Yang, P. Dose-specific effects of tumor necrosis factor alpha on osteogenic differentiation of mesenchymal stem cells. Cell Prolif. 2011, 44, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Sfikakis, P.P.; Grigoropoulos, V.; Emfietzoglou, I.; Theodossiadis, G.; Tentolouris, N.; Delicha, E.; Katsiari, C.; Alexiadou, K.; Hatziagelaki, E.; Theodossiadis, P.G. Infliximab for diabetic macular edema refractory to laser photocoagulation: A randomized, double-blind, placebo-controlled, crossover, 32-week study. Diabetes Care 2010, 33, 1523–1528. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Hernandez-Bogantes, E.; Roca, J.A.; Arevalo, J.F.; Barraza, K.; Lasave, A.F. Intravitreal tumor necrosis factor inhibitors in the treatment of refractory diabetic macular edema: A pilot study from the Pan-American Collaborative Retina Study Group. Retina 2011, 31, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Giganti, M.; Beer, P.M.; Lemanski, N.; Hartman, C.; Schartman, J.; Falk, N. Adverse events after intravitreal infliximab (Remicade). Retina 2010, 30, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Levy-Clarke, G.; Jabs, D.A.; Read, R.W.; Rosenbaum, J.T.; Vitale, A.; van Gelder, R.N. Expert panel recommendations for the use of anti-tumor necrosis factor biologic agents in patients with ocular inflammatory disorders. Ophthalmology 2014, 121, 785–796. [Google Scholar] [CrossRef]
- McLean, J.M. Use of ACTH and cortisone. Trans. Am. Ophthalmol. Soc. 1950, 48, 293–296. [Google Scholar]
- Jonas, J.B.; Jonas, R.A.; Neumaier, M.; Findeisen, P. Cytokine concentration in aqueous humor of eyes with diabetic macular edema. Retina 2012, 32, 2150–2157. [Google Scholar] [CrossRef]
- Caceres-del-Carpio, J.; Costa, R.D.; Haider, A.; Narayanan, R.; Kuppermann, B.D. Corticosteroids: Triamcinolone, Dexamethasone and Fluocinolone. Dev. Ophthalmol. 2016, 55, 221–231. [Google Scholar]
- Pagano, G.; Bruno, A.; Cavallo-Perin, P.; Cesco, L.; Imbimbo, B. Glucose intolerance after short-term administration of corticosteroids in healthy subjects. Prednisone, deflazacort, and betamethasone. Arch. Intern. Med. 1989, 149, 1098–1101. [Google Scholar] [CrossRef] [PubMed]
- Saag, K.G. Glucocorticoid-induced osteoporosis. Endocrinol. Metab. Clin. N. Am. 2003, 32, 135–157. [Google Scholar] [CrossRef]
- Francois, J. Cortisone et tension oculaire (in French). Ann. d’Oculist 1954, 187, 805. [Google Scholar]
- Diabetic Retinopathy Clinical Research Network; Elman, M.J.; Aiello, L.P.; Beck, R.W.; Bressler, N.M.; Bressler, S.B.; Edwards, A.R.; Ferris, F.L., 3rd; Friedman, S.M.; Glassman, A.R.; et al. Randomized trial evaluating ranibizumab plus prompt or deferred laser or triamcinolone plus prompt laser for diabetic macular edema. Ophthalmology 2010, 117, 1064–1077. [Google Scholar] [CrossRef] [PubMed]
- Jonas, J.B.; Kreissig, I.; Degenring, R. Intraocular pressure after intravitreal injection of triamcinolone acetonide. Br. J. Ophthalmol. 2003, 87, 24–27. [Google Scholar] [CrossRef] [PubMed]
- Gillies, M.C.; Kuzniarz, M.; Craig, J.; Ball, M.; Luo, W.; Simpson, J.M. Intravitreal triamcinolone-induced elevated intraocular pressure is associated with the development of posterior subcapsular cataract. Ophthalmology 2005, 112, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Abcouwer, S.F. Angiogenic factors and cytokines in diabetic retinopathy. J. Clin. Cell. Immunol. 2013, S1, 1–12. [Google Scholar] [CrossRef]
- Chang-Lin, J.E.; Attar, M.; Acheampong, A.A.; Robinson, M.R.; Whitcup, S.M.; Kuppermann, B.D.; Welty, D. Pharmacokinetics and pharmacodynamics of a sustained-release dexamethasone intravitreal implant. Investig. Ophthalmol. Vis. Sci. 2011, 52, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Boyer, D.S.; Yoon, Y.H.; Belfort, R., Jr.; Bandello, F.; Maturi, R.K.; Augustin, A.J.; Li, X.Y.; Cui, H.; Hashad, Y.; Whitcup, S.M.; et al. Three-year, randomized, sham-controlled trial of dexamethasone intravitreal implant in patients with diabetic macular edema. Ophthalmology 2014, 121, 1904–1914. [Google Scholar] [CrossRef] [PubMed]
- Zhioua, I.; Semoun, O.; Lalloum, F.; Souied, E.H. Intravitreal Dexamethasone Implant in Patients with Ranibizumab Persistent Diabetic Macular Edema. Retina 2015, 35, 1429–1435. [Google Scholar] [CrossRef] [PubMed]
- Lazic, R.; Lukic, M.; Boras, I.; Draca, N.; Vlasic, M.; Gabric, N.; Tomic, Z. Treatment of anti-vascular endothelial growth factor-resistant diabetic macular edema with dexamethasone intravitreal implant. Retina 2014, 34, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Arcinue, C.A.; Ceron, O.M.; Foster, C.S. A comparison between the fluocinolone acetonide (Retisert) and dexamethasone (Ozurdex) intravitreal implants in uveitis. J. Ocul. Pharmacol. Ther. 2013, 29, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.G.; Flynn, H.W., Jr. Fluocinolone acetonide implantable device for diabetic retinopathy. Curr. Pharm. Biotechnol. 2011, 12, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Pearson, P.A.; Comstock, T.L.; Ip, M.; Callanan, D.; Morse, L.S.; Ashton, P.; Levy, B.; Mann, E.S.; Eliott, D. Fluocinolone acetonide intravitreal implant for diabetic macular edema: A 3-year multicenter, randomized, controlled clinical trial. Ophthalmology 2011, 118, 1580–1587. [Google Scholar] [CrossRef] [PubMed]
- Campochiaro, P.A.; Nguyen, Q.D.; Hafiz, G.; Bloom, S.; Brown, D.M.; Busquets, M.; Ciulla, T.; Feiner, L.; Sabates, N.; Billman, K.; et al. Aqueous levels of fluocinolone acetonide after administration of fluocinolone acetonide inserts or fluocinolone acetonide implants. Ophthalmology 2013, 120, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Campochiaro, P.A.; Hafiz, G.; Shah, S.M.; Bloom, S.; Brown, D.M.; Busquets, M.; Ciulla, T.; Feiner, L.; Sabates, N.; Billman, K.; et al. Sustained ocular delivery of fluocinolone acetonide by an intravitreal insert. Ophthalmology 2010, 117, 1393–1399. [Google Scholar] [CrossRef] [PubMed]
- Kita, T.; Clermont, A.C.; Murugesan, N.; Zhou, Q.; Fujisawa, K.; Ishibashi, T.; Aiello, L.P.; Feener, E.P. Plasma Kallikrein-Kinin System as a VEGF-Independent Mediator of Diabetic Macular Edema. Diabetes 2015, 64, 3588–3599. [Google Scholar] [CrossRef] [PubMed]
- Stone, O.A.; Richer, C.; Emanueli, C.; van Weel, V.; Quax, P.H.; Katare, R.; Kraenkel, N.; Campagnolo, P.; Barcelos, L.S.; Siragusa, M.; et al. Critical role of tissue kallikrein in vessel formation and maturation: Implications for therapeutic revascularization. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Maas, C.; Govers-Riemslag, J.W.; Bouma, B.; Schiks, B.; Hazenberg, B.P.; Lokhorst, H.M.; Hammarstrom, P.; ten Cate, H.; de Groot, P.G.; Bouma, B.N.; et al. Misfolded proteins activate factor XII in humans, leading to kallikrein formation without initiating coagulation. J. Clin. Investig. 2008, 118, 3208–3218. [Google Scholar] [CrossRef] [PubMed]
- Catanzaro, O.L.; Dziubecki, D.; Obregon, P.; Rodriguez, R.R.; Sirois, P. Antidiabetic efficacy of bradykinin antagonist R-954 on glucose tolerance test in diabetic type 1 mice. Neuropeptides 2010, 44, 187–189. [Google Scholar] [CrossRef] [PubMed]
- Leeb-Lundberg, L.M.; Marceau, F.; Muller-Esterl, W.; Pettibone, D.J.; Zuraw, B.L. International union of pharmacology. XLV. Classification of the kinin receptor family: From molecular mechanisms to pathophysiological consequences. Pharmacol. Rev. 2005, 57, 27–77. [Google Scholar] [CrossRef] [PubMed]
- Chao, J.; Yin, H.; Gao, L.; Hagiwara, M.; Shen, B.; Yang, Z.R.; Chao, L. Tissue kallikrein elicits cardioprotection by direct kinin b2 receptor activation independent of kinin formation. Hypertension 2008, 52, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Abdouh, M.; Talbot, S.; Couture, R.; Hassessian, H.M. Retinal plasma extravasation in streptozotocin-diabetic rats mediated by kinin B1 and B2 receptors. Br. J. Pharmacol. 2008, 154, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Mejia, A.J.; Matus, C.E.; Pavicic, F.; Concha, M.; Ehrenfeld, P.; Figueroa, C.D. Intracellular signaling pathways involved in the release of IL-4 and VEGF from human keratinocytes by activation of kinin B1 receptor: Functional relevance to angiogenesis. Arch. Dermatol. Res. 2015, 307, 803–817. [Google Scholar] [CrossRef] [PubMed]
- Bhoola, K.D.; Figueroa, C.D.; Worthy, K. Bioregulation of kinins: Kallikreins, kininogens, and kininases. Pharmacol. Rev. 1992, 44, 1–80. [Google Scholar] [PubMed]
- Bhat, M.; Pouliot, M.; Couture, R.; Vaucher, E. The kallikrein-kinin system in diabetic retinopathy. Prog. Drug Res. 2014, 69, 111–143. [Google Scholar] [PubMed]
- Abdouh, M.; Khanjari, A.; Abdelazziz, N.; Ongali, B.; Couture, R.; Hassessian, H.M. Early upregulation of kinin B1 receptors in retinal microvessels of the streptozotocin-diabetic rat. Br. J. Pharmacol. 2003, 140, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Ni, A.; Yin, H.; Agata, J.; Yang, Z.; Chao, L.; Chao, J. Overexpression of kinin B1 receptors induces hypertensive response to des-Arg9-bradykinin and susceptibility to inflammation. J. Biol. Chem. 2003, 278, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Pesquero, J.B.; Araujo, R.C.; Heppenstall, P.A.; Stucky, C.L.; Silva, J.A., Jr.; Walther, T.; Oliveira, S.M.; Pesquero, J.L.; Paiva, A.C.; Calixto, J.B.; et al. Hypoalgesia and altered inflammatory responses in mice lacking kinin B1 receptors. Proc. Natl. Acad. Sci. USA 2000, 97, 8140–8145. [Google Scholar] [CrossRef] [PubMed]
- Kedzierska, K.; Ciechanowski, K.; Golembiewska, E.; Safranow, K.; Ciechanowicz, A.; Domanski, L.; Myslak, M.; Rozanski, J. Plasma prekallikrein as a risk factor for diabetic retinopathy. Arch. Med. Res. 2005, 36, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Sharma, J.N.; Al-Shoumer, K.A.; Matar, K.M.; Al-Gharee, H.Y.; Madathil, N.V. Bradykinin-forming components in Kuwaiti patients with type 2 diabetes. Int. J. Immunopathol. Pharmacol. 2013, 26, 699–705. [Google Scholar] [PubMed]
- Phipps, J.A.; Clermont, A.C.; Sinha, S.; Chilcote, T.J.; Bursell, S.E.; Feener, E.P. Plasma kallikrein mediates angiotensin II type 1 receptor-stimulated retinal vascular permeability. Hypertension 2009, 53, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Clermont, A.; Rook, S.; Fonda, S.J.; Srinivasan, V.J.; Wojtkowski, M.; Fujimoto, J.G.; Avery, R.L.; Arrigg, P.G.; Bursell, S.; et al. Extracellular carbonic anhydrase mediates hemorrhagic retinal and cerebral vascular permeability through prekallikrein activation. Nat. Med. 2007, 13, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Zuraw, B.L.; Banerji, A.; Bernstein, J.A.; Busse, P.J.; Christiansen, S.C.; Davis-Lorton, M.; Frank, M.M.; Li, H.H.; Lumry, W.R.; Riedl, M.; et al. US Hereditary Angioedema Association Medical Advisory Board 2013 recommendations for the management of hereditary angioedema due to C1 inhibitor deficiency. J. Allergy Clin. Immunol. Pract. 2013, 1, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Chyung, Y.; Vince, B.; Iarrobino, R.; Sexton, D.; Kenniston, J.; Faucette, R.; TenHoor, C.; Stolz, L.E.; Stevens, C.; Biedenkapp, J.; et al. A phase 1 study investigating DX-2930 in healthy subjects. Ann. Allergy Asthma Immunol. 2014, 113, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Wegman-Ostrosky, T.; Soto-Reyes, E.; Vidal-Millan, S.; Sanchez-Corona, J. The renin-angiotensin system meets the hallmarks of cancer. J. Renin Angiotensin Aldosterone Syst. 2015, 16, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Vincent, F.; Bonnin, P.; Clemessy, M.; Contreres, J.O.; Lamande, N.; Gasc, J.M.; Vilar, J.; Hainaud, P.; Tobelem, G.; Corvol, P.; et al. Angiotensinogen delays angiogenesis and tumor growth of hepatocarcinoma in transgenic mice. Cancer Res. 2009, 69, 2853–2860. [Google Scholar] [CrossRef] [PubMed]
- Sjolie, A.K.; Klein, R.; Porta, M.; Orchard, T.; Fuller, J.; Parving, H.H.; Bilous, R.; Aldington, S.; Chaturvedi, N. Retinal microaneurysm count predicts progression and regression of diabetic retinopathy. Post-hoc results from the DIRECT Programme. Diabet. Med. 2011, 28, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Mauer, M.; Zinman, B.; Gardiner, R.; Suissa, S.; Sinaiko, A.; Strand, T.; Drummond, K.; Donnelly, S.; Goodyer, P.; Gubler, M.C.; et al. Renal and retinal effects of enalapril and losartan in type 1 diabetes. N. Engl. J. Med. 2009, 361, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Bader, M. Kallikrein-kinin system in neovascularization. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 617–619. [Google Scholar] [CrossRef] [PubMed]
- Ismael, M.A.; Talbot, S.; Carbonneau, C.L.; Beausejour, C.M.; Couture, R. Blockade of sensory abnormalities and kinin B(1) receptor expression by N-acetyl-l-cysteine and ramipril in a rat model of insulin resistance. Eur. J. Pharmacol. 2008, 589, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T. Kallikrein-Kinin System: An Emerging Competitor or Collaborator for VEGF in Diabetic Macular Edema? Diabetes 2015, 64, 3350–3352. [Google Scholar] [CrossRef] [PubMed]
- Bas, M.; Greve, J.; Stelter, K.; Havel, M.; Strassen, U.; Rotter, N.; Veit, J.; Schossow, B.; Hapfelmeier, A.; Kehl, V.; et al. A randomized trial of icatibant in ACE-inhibitor-induced angioedema. N. Engl. J. Med. 2015, 372, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Joseph, K.; Tholanikunnel, T.E.; Kaplan, A.P. In vitro comparison of bradykinin degradation by aliskiren, a renin inhibitor, and an inhibitor of angiotensin-converting enzyme. J. Renin Angiotensin Aldosterone Syst. 2015, 16, 321–327. [Google Scholar] [CrossRef] [PubMed]
- UK Prospective Diabetes Study Group. Tight blood pressure control and risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 38. BMJ 1998, 317, 703–713. [Google Scholar]
- Chaturvedi, N.; Sjolie, A.K.; Stephenson, J.M.; Abrahamian, H.; Keipes, M.; Castellarin, A.; Rogulja-Pepeonik, Z.; Fuller, J.H. Effect of lisinopril on progression of retinopathy in normotensive people with type 1 diabetes. The EUCLID Study Group. EURODIAB Controlled Trial of Lisinopril in Insulin-Dependent Diabetes Mellitus. Lancet 1998, 351, 28–31. [Google Scholar] [CrossRef]
- Wang, B.; Wang, F.; Zhang, Y.; Zhao, S.H.; Zhao, W.J.; Yan, S.L.; Wang, Y.G. Effects of RAS inhibitors on diabetic retinopathy: A systematic review and meta-analysis. Lancet Diabetes Endocrinol. 2015, 3, 263–274. [Google Scholar] [CrossRef]
- Yacyshyn, O.K.; Lai, P.F.; Forse, K.; Teichert-Kuliszewska, K.; Jurasz, P.; Stewart, D.J. Tyrosine phosphatase beta regulates angiopoietin-Tie2 signaling in human endothelial cells. Angiogenesis 2009, 12, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.; Aldrich, T.H.; Jones, P.F.; Acheson, A.; Compton, D.L.; Jain, V.; Ryan, T.E.; Bruno, J.; Radziejewski, C.; Maisonpierre, P.C.; et al. Isolation of angiopoietin-1, a ligand for the TIE2 receptor, by secretion-trap expression cloning. Cell 1996, 87, 1161–1169. [Google Scholar] [CrossRef]
- Gamble, J.R.; Drew, J.; Trezise, L.; Underwood, A.; Parsons, M.; Kasminkas, L.; Rudge, J.; Yancopoulos, G.; Vadas, M.A. Angiopoietin-1 is an antipermeability and anti-inflammatory agent in vitro and targets cell junctions. Circ. Res. 2000, 87, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Joussen, A.M.; Poulaki, V.; Tsujikawa, A.; Qin, W.; Qaum, T.; Xu, Q.; Moromizato, Y.; Bursell, S.E.; Wiegand, S.J.; Rudge, J.; et al. Suppression of diabetic retinopathy with angiopoietin-1. Am. J. Pathol. 2002, 160, 1683–1693. [Google Scholar] [CrossRef]
- Holash, J.; Maisonpierre, P.C.; Compton, D.; Boland, P.; Alexander, C.R.; Zagzag, D.; Yancopoulos, G.D.; Wiegand, S.J. Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science 1999, 284, 1994–1998. [Google Scholar] [CrossRef] [PubMed]
- Scharpfenecker, M.; Fiedler, U.; Reiss, Y.; Augustin, H.G. The Tie-2 ligand angiopoietin-2 destabilizes quiescent endothelium through an internal autocrine loop mechanism. J. Cell Sci. 2005, 118, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, D.; Suzuma, K.; Suzuma, I.; Ohashi, H.; Ojima, T.; Kurimoto, M.; Murakami, T.; Kimura, T.; Takagi, H. Vitreous levels of angiopoietin 2 and vascular endothelial growth factor in patients with proliferative diabetic retinopathy. Am. J. Ophthalmol. 2005, 139, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Tuuminen, R.; Haukka, J.; Loukovaara, S. Poor glycemic control associates with high intravitreal angiopoietin-2 levels in patients with diabetic retinopathy. Acta Ophthalmol. 2015, 93, e515–e516. [Google Scholar] [CrossRef] [PubMed]
- Rangasamy, S.; McGuire, P.G.; Das, A. Diabetic retinopathy and inflammation: Novel therapeutic targets. Middle East Afr. J. Ophthalmol. 2012, 19, 52–59. [Google Scholar] [PubMed]
- Tang, J.; Kern, T.S. Inflammation in diabetic retinopathy. Prog. Retin. Eye Res. 2011, 30, 343–358. [Google Scholar] [CrossRef] [PubMed]
- Early Treatment Diabetic Retinopathy Study Research Group. Effects of Aspirin Treatment on Diabetic Retinopathy. ETDRS report number 8. Ophthalmology 1991, 98, 757–765. [Google Scholar]
- Esgin, H.; Samut, H.S. Topical ketorolac 0.5% for ocular pain relief during scatter laser photocoagulation with 532 nm green laser. J. Ocul. Pharmacol. Ther. 2006, 22, 460–464. [Google Scholar] [CrossRef] [PubMed]
- Ngo, L.L.; Ward, K.K.; Mody, S.K. Ketorolac for Pain Control With Intrauterine Device Placement: A Randomized Controlled Trial. Obstet. Gynecol. 2015, 126, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Siribumrungwong, K.; Cheewakidakarn, J.; Tangtrakulwanich, B.; Nimmaanrat, S. Comparing parecoxib and ketorolac as preemptive analgesia in patients undergoing posterior lumbar spinal fusion: A prospective randomized double-blinded placebo-controlled trial. BMC Musculoskelet. Disord. 2015, 16, 59. [Google Scholar] [CrossRef] [PubMed]
- Patricio, J.P.; Barbosa, J.P.; Ramos, R.M.; Antunes, N.F.; de Melo, P.C. Relative cardiovascular and gastrointestinal safety of non-selective non-steroidal anti-inflammatory drugs versus cyclo-oxygenase-2 inhibitors: Implications for clinical practice. Clin. Drug Investig. 2013, 33, 167–183. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, R.M.; Vianna, R.N.; Cardoso, G.P.; de Magalhaes, A.V.; Burnier, M.N., Jr. Intravitreal injection of commercially available ketorolac tromethamine in eyes with diabetic macular edema refractory to laser photocoagulation. Curr. Eye Res. 2011, 36, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Schoenberger, S.D.; Kim, S.J.; Shah, R.; Sheng, J.; Cherney, E. Reduction of interleukin 8 and platelet-derived growth factor levels by topical ketorolac, 0.45%, in patients with diabetic retinopathy. JAMA Ophthalmol. 2014, 132, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.M.; Almukhtar, T.H.; Baker, C.W.; Glassman, A.R.; Elman, M.J.; Bressler, N.M.; Maker, M.P.; Jampol, L.M.; Melia, M.; Diabetic retinopathy clinical research network. Topical nepafenec in eyes with noncentral diabetic macular edema. Retina 2015, 35, 944–956. [Google Scholar] [CrossRef] [PubMed]
- Kulik, A.; Bykov, K.; Choudhry, N.K.; Bateman, B.T. Non-steroidal anti-inflammatory drug administration after coronary artery bypass surgery: Utilization persists despite the boxed warning. Pharmacoepidemiol. Drug Saf. 2015, 24, 647–653. [Google Scholar] [CrossRef] [PubMed]
- McCormack, P.L. Ketorolac 0.45% ophthalmic solution. Drugs Aging 2011, 28, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Solomon, K.D.; Cheetham, J.K.; DeGryse, R.; Brint, S.F.; Rosenthal, A. Topical ketorolac tromethamine 0.5% ophthalmic solution in ocular inflammation after cataract surgery. Ophthalmology 2001, 108, 331–337. [Google Scholar] [CrossRef]
- Krady, J.K.; Basu, A.; Allen, C.M.; Xu, Y.; LaNoue, K.F.; Gardner, T.W.; Levison, S.W. Minocycline reduces proinflammatory cytokine expression, microglial activation, and caspase-3 activation in a rodent model of diabetic retinopathy. Diabetes 2005, 54, 1559–1565. [Google Scholar] [CrossRef] [PubMed]
- Cukras, C.A.; Petrou, P.; Chew, E.Y.; Meyerle, C.B.; Wong, W.T. Oral minocycline for the treatment of diabetic macular edema (DME): Results of a phase I/II clinical study. Investig. Ophthalmol. Vis. Sci. 2012, 53, 3865–3874. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Chen, Y.; Wu, Q.; Jia, L.; Du, X. Minocycline inhibits PARP1 expression and decreases apoptosis in diabetic retinopathy. Mol. Med. Rep. 2015, 12, 4887–4894. [Google Scholar] [PubMed]
- Jordan, J.; Fernandez-Gomez, F.J.; Ramos, M.; Ikuta, I.; Aguirre, N.; Galindo, M.F. Minocycline and cytoprotection: Shedding new light on a shadowy controversy. Curr. Drug Deliv. 2007, 4, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.S.; Wehrli, S.; Roder, H.; Rogers, M.; Forrest, J.N., Jr.; McCrimmon, D.; Zasloff, M. Squalamine: An aminosterol antibiotic from the shark. Proc. Natl. Acad. Sci. USA 1993, 90, 1354–1358. [Google Scholar] [CrossRef] [PubMed]
- Ciulla, T.A.; Criswell, M.H.; Danis, R.P.; Williams, J.I.; McLane, M.P.; Holroyd, K.J. Squalamine lactate reduces choroidal neovascularization in a laser-injury model in the rat. Retina 2003, 23, 808–814. [Google Scholar] [CrossRef] [PubMed]
- Higgins, R.D.; Yan, Y.; Geng, Y.; Zasloff, M.; Williams, J.I. Regression of retinopathy by squalamine in a mouse model. Pediatr. Res. 2004, 56, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Webster, A.C.; Lee, V.W.; Chapman, J.R.; Craig, J.C. Target of rapamycin inhibitors (sirolimus and everolimus) for primary immunosuppression of kidney transplant recipients: A systematic review and meta-analysis of randomized trials. Transplantation 2006, 81, 1234–1248. [Google Scholar] [CrossRef] [PubMed]
- Liegl, R.; Koenig, S.; Siedlecki, J.; Haritoglou, C.; Kampik, A.; Kernt, M. Temsirolimus inhibits proliferation and migration in retinal pigment epithelial and endothelial cells via mTOR inhibition and decreases VEGF and PDGF expression. PLoS ONE 2014, 9, e88203. [Google Scholar] [CrossRef] [PubMed]
- Ozdemir, G.; Kilinc, M.; Ergun, Y.; Sahin, E. Rapamycin inhibits oxidative and angiogenic mediators in diabetic retinopathy. Can. J. Ophthalmol. J. Can. D’ophtalmol. 2014, 49, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Yagasaki, R.; Nakahara, T.; Ushikubo, H.; Mori, A.; Sakamoto, K.; Ishii, K. Anti-angiogenic effects of mammalian target of rapamycin inhibitors in a mouse model of oxygen-induced retinopathy. Biol. Pharm. Bull. 2014, 37, 1838–1842. [Google Scholar] [CrossRef] [PubMed]
- Dugel, P.U.; Blumenkranz, M.S.; Haller, J.A.; Williams, G.A.; Solley, W.A.; Kleinman, D.M.; Naor, J. A randomized, dose-escalation study of subconjunctival and intravitreal injections of sirolimus in patients with diabetic macular edema. Ophthalmology 2012, 119, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Seto, B. Rapamycin and mTOR: A serendipitous discovery and implications for breast cancer. Clin. Transl. Med. 2012, 1. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Veenstra, A.; Palczewski, K.; Kern, T.S. Photoreceptor cells are major contributors to diabetes-induced oxidative stress and local inflammation in the retina. Proc. Natl. Acad. Sci. USA 2013, 110, 16586–16591. [Google Scholar] [CrossRef] [PubMed]
- Millen, A.E.; Klein, R.; Folsom, A.R.; Stevens, J.; Palta, M.; Mares, J.A. Relation between intake of vitamins C and E and risk of diabetic retinopathy in the atherosclerosis risk in communities study. Am. J. Clin. Nutr. 2004, 79, 865–873. [Google Scholar] [PubMed]
- Lee, C.T.; Gayton, E.L.; Beulens, J.W.; Flanagan, D.W.; Adler, A.I. Micronutrients and diabetic retinopathy a systematic review. Opthalmology 2010, 117, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Chous, A.P.; Richer, S.P.; Gerson, J.D.; Kowluru, R.A. The diabetes visual function supplement study (DiVFuSS). Br. J. Ophthalmol. 2016, 100, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Akiba, J.; Arzabe, C.W.; Trempe, C.L. Posterior vitreous detachment and neovascularization in diabetic retinopathy. Ophthalmology 1990, 97, 889–891. [Google Scholar] [CrossRef]
- Sebag, J. Vitreous: In Health and Disease, 1st ed.; Springer: New York, NY, USA, 2014. [Google Scholar]
- Khoshnevis, M.; Sebag, J. Pharmacologic vitreolysis with ocriplasmin: Rationale for use and therapeutic potential in vitreo-retinal disorders. BioDrugs 2015, 29, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Kuppermann, B.D. Ocriplasmin for pharmacologic vitreolysis. Retina 2012, 32, S225–S228; discussion S228–S231. [Google Scholar] [CrossRef] [PubMed]
- Haller, J.A.; Stalmans, P.; Benz, M.S.; Gandorfer, A.; Pakola, S.J.; Girach, A.; Kampik, A.; Jaffe, G.J.; Toth, C.A.; Group, M.-T.S. Efficacy of intravitreal ocriplasmin for treatment of vitreomacular adhesion: Subgroup analyses from two randomized trials. Ophthalmology 2015, 122, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Varma, R.; Haller, J.A.; Kaiser, P.K. Improvement in Patient-Reported Visual Function after Ocriplasmin for Vitreomacular Adhesion: Results of the Microplasmin for Intravitreous Injection-Traction Release without Surgical Treatment (MIVI-TRUST) Trials. JAMA Ophthalmol. 2015, 133, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Soldi, R.; Mitola, S.; Strasly, M.; Defilippi, P.; Tarone, G.; Bussolino, F. Role of alphavbeta3 integrin in the activation of vascular endothelial growth factor receptor-2. EMBO J. 1999, 18, 882–892. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Li, X.H.; Wang, L.F.; Kuang, X.; Hang, Z.X.; Deng, Y.; Du, J.R. Therapeutic efficacy of a novel non-peptide alphavbeta3 integrin antagonist for pathological retinal angiogenesis in mice. Exp. Eye Res. 2014, 129, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Gong, J.; Xu, Z.; Wei, Y.; Duh, E.J. Inhibition of pathological retinal angiogenesis by the integrin alphavbeta3 antagonist tetraiodothyroacetic acid (tetrac). Exp. Eye Res. 2012, 94, 41–48. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Drug | Class | Company | Currently in Listed Clinical Trials for DR/DME? | FDA Approval for DR or DME? | FDA Approved for Other Condition? |
|---|---|---|---|---|---|
| Bevacizumab | Anti-VEGF | Genentech (South San Francisco, CA, USA) | Yes (NCT02462304) | No | Yes (Various forms of cancer) |
| Ranibizumab | Anti-VEGF | Genentech | Yes (NCT02462304) | Yes | Yes (Neovascular Age-Related Macular Degeneration) |
| Aflibrocept | Anti-VEGF (VEGF-Trap) | Regeneron (Tarrytown, NY, USA) | Yes | Yes | Yes (Neovascular Age-Related Macular Degeneration) |
| Pegaptanib | Anti-VEGF | Pfizer (New York, NY, USA) | No | No | Yes (Neovascular Age-Related Macular Degeneration) |
| PAN-90806 | Anti-VEGF | PanOptica (Bernardsville, NJ, USA) | Yes (NCT02475109) | No | No |
| Infliximab | TNF-α Inhibitor | Janssen Biotech (Horsham, PA, USA) | No | No | Yes (Crohn′s Disease, Ulcerative Colitis, and Various forms of Arthritis) |
| Adalimumab | TNF-α Inhibitor | AbbVie (North Chicago, IL, USA) | No | No | Yes (Various Autoimmune Disorders Including Arthritis) |
| Ozurdex | Corticosteroid Implant (dexamethasone) | Allergan (Parsippany-Troy Hills, NJ, USA) | No | Yes | Yes (Uveitis) |
| Retisert | Corticosteroid Implant (fluocinolone) | Bausch & Lomb (Rochester, NY, USA) | No | No | Yes (Uveitis) |
| Iluvien | Corticosteroid Implant (fluocinolone) | Alimera Sciences (Alpharetta, GA, USA) | Yes | Yes | No |
| Candasartin | Angiotensin Receptor Blocker | AstraZeneca (London, UK), Generics | No | No | Yes (Hypertension and Heart Failure) |
| Losartan | Angiotensin Receptor Blocker | Merck (Kenilworth, UK), Generics | No | No | Yes (Hypertension, Diabetic Nephropathy) |
| KVD001 | Kallikrein Inhibitor | KalVista Pharm. (Porton Down, UK) | No | No | No |
| DM199 | Recomenant human tissue kallikrein-1 | DiaMedica (Minneapolis, MN, USA) | No | No | No |
| Ecallantide/DX-88 | Kallikrein Inhibitor | Dyax (Lexington, MA, USA) | No | No | Yes (Herditary Angioedema) |
| DX-2930 | Human monoclonal anti-Kallikrein antibody | Shire (Dublin, Ireland) | No | No | No |
| BCX7353 | Kallikrein Inhibitor | BioCryst (Durham, UK) | No | No | No |
| Avoralstat/BCX4161 | Kallikrein Inhibitor | BioCryst | No | No | No |
| Icatibant | Bradykinin receptor-2 antagonist | Shire | No | No | Yes (Herditary Angioedema) |
| Enalapril | ACE Inhibitor | Multiple | No | No | Yes (Hypertension) |
| Lisinopril | ACE Inhibitor | Multiple | No | No | Yes (Hypertension, Heart Failure, Acute Myocardial Infarction) |
| AKB-9778 | Tie-2 Activator | Akebia Therapeutics (Cambridge, MA, USA) | No | No | No |
| AKB-9875 | Tie-2 Activator | Akebia Therapeutics | No | No | No |
| AKB-9089 | Tie-2 Activator | Akebia Therapeutics | No | No | No |
| HPTPβ Antibody | Tie-2 Activator | Akebia Therapeutics) | No | No | No |
| Trebananib | Angiopoietin Blocker | Amgen (Thousand Oaks, CA, USA) | No | No | No |
| Ketorolac | NSAID (coxib) | Roche (Basel, Switzerland) | Yes (NCT01609881) | No | Yes (Postoperative Ophthalmic Inflammation) |
| Nepafenic | NSAID (coxib) | Alcon (Hunenberg, Switzerland) | No | No | Yes (Postoperative Ophthalmic Inflammation) |
| Diclofenac | NSAID | Mutiple | Yes (NCT01694212) | No | Yes (Analgesic, Osteoarthritis) |
| Minocycline | Antibiotic | Multiple | No | No | Yes (Acne, Various Bacterial Infections) |
| Squalamine | Anti-microbial | Ohr Pharm. (New York, NY, USA) | Yes | No | No |
| Rapamycin (Sirolimus) | Immunosuppresant/mTOR Inhibitor | Pfizer | No | No | Yes (Organ Transplant Immunosuppresion, Lymphangioleiomyomatosis) |
| Everolimus | Immunosuppresant/mTOR Inhibitor | Novartis (Basel, Switzerland) | No | No | Yes (Organ Transplant Immunosuppresion, Various Neuroendocrine tumors) |
| Vitreosolve | Posterier Vitreous Detachment Agent | Vitreoretinal Tech. (Irvine, CA, USA) | No | No | No |
| Ocriplasmin | Posterier Vitreous Detachment Agent | ThromboGenics (Leuven, Belgium) | Yes | No | Yes (Symptomatic VMA) |
| Luminate | Anti-Integrin/Posterier Vitreous Detachment Agent | Allegro Ophthalmics (San Juan Capistrano, CA, USA) | Yes (NCT02435862) | No | No |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bolinger, M.T.; Antonetti, D.A. Moving Past Anti-VEGF: Novel Therapies for Treating Diabetic Retinopathy. Int. J. Mol. Sci. 2016, 17, 1498. https://doi.org/10.3390/ijms17091498
Bolinger MT, Antonetti DA. Moving Past Anti-VEGF: Novel Therapies for Treating Diabetic Retinopathy. International Journal of Molecular Sciences. 2016; 17(9):1498. https://doi.org/10.3390/ijms17091498
Chicago/Turabian StyleBolinger, Mark T., and David A. Antonetti. 2016. "Moving Past Anti-VEGF: Novel Therapies for Treating Diabetic Retinopathy" International Journal of Molecular Sciences 17, no. 9: 1498. https://doi.org/10.3390/ijms17091498