
Transcriptional Factors Mediating Retinoic Acid Signals in the Control of Energy Metabolism



Abstract
:1. Introduction
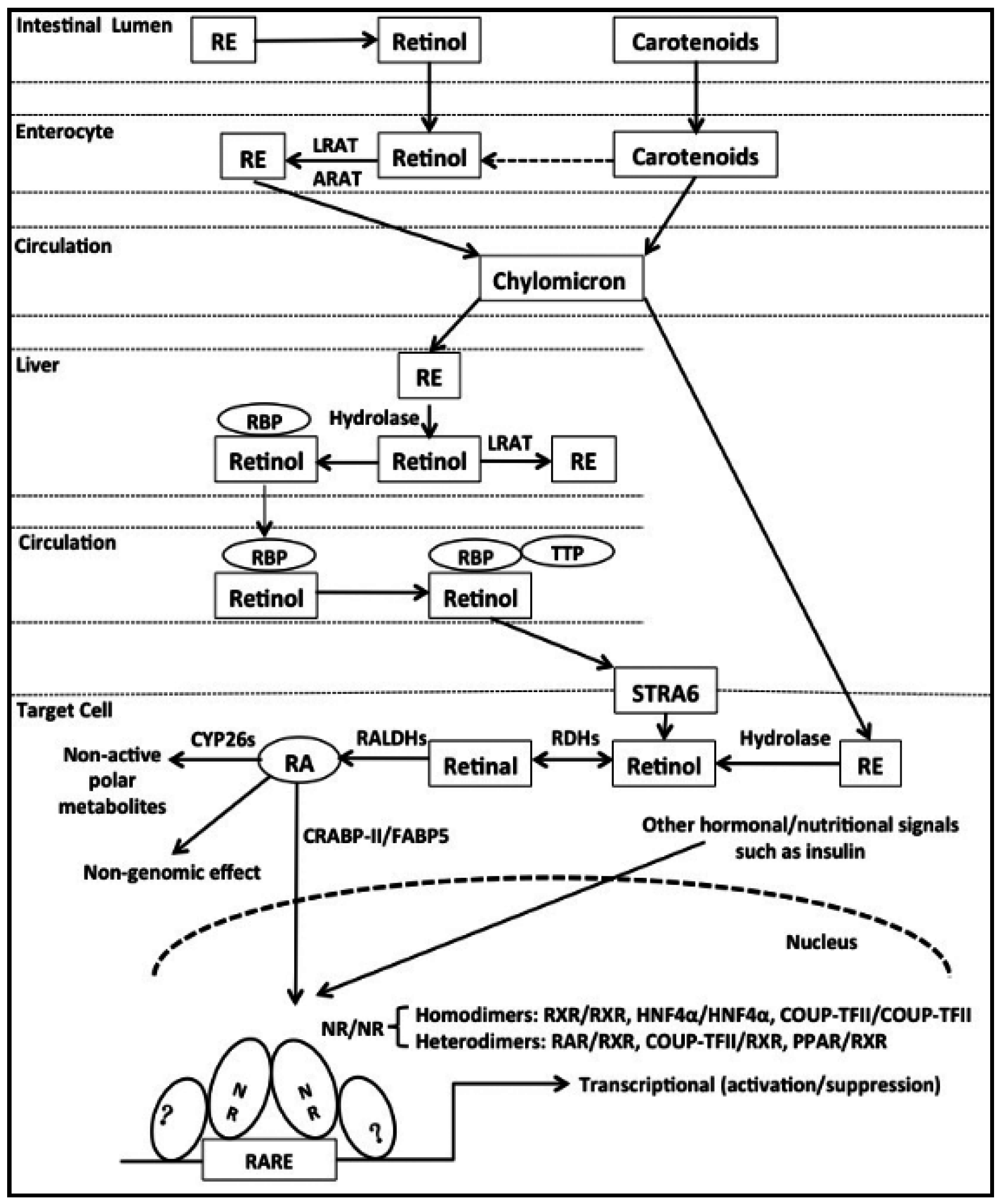
2. Uptake and Metabolism of Vitamin A (VA)

3. Characteristics of a Retinoic Acid Response Element (RARE)

{kind=link}
{kind=link}
| Type | Gene | RARE Sequences | References |
|---|---|---|---|
| DR0 | Socs3 | AGTTCA AGGTCA | Moutier et al. [48] |
| Msi2 | GGGTCA AGGTCA | ||
| DR1 | Crbp2 | AGGTCA c AGTTCA | Bastien et al. [55] |
| Crabp2 | TGACCT c TGCCCT | Durand et al. [56] | |
| DR2 | Gck | TGACCT tg TGACAC | Li et al. [54] |
| Epo | GGGTCA ag AGGTCA | Brade et al. [57] | |
| DR5 | Cyp26a1 | AGTTCA cccaa AGTTCA | Loudig et al. [58] |
| Rarb2 | GTTCAC cgaaa GTTCAC | Sucov et al. [43] | |
| Simple DR8 | Rqcd1 | GGGTCA gaggtgag AGGTCA | Moutier et al. [48] |
| Dedd | AGGTCA cgatctgg AGTTCA | ||
| Composite DR8 | Mafa | AGGTCA ga AGTTCA AGGTCA | |
| Nrp1 | GGATCA aa AGTTCA AGGTCA | ||
| IR0 | Trim16 | GGGTCA TGACCC | |
| Tr2-11 | GGGTCA CGAACT | Lee and Wei [59] |
4. Nuclear Receptors (NRs) Mediating Retinoic Acid (RA) Effects
4.1. Introduction of Nuclear Receptors (NRs)
4.2. RARs Are Primary Mediators of RA Signaling
4.3. The Role of RXRs in RA Signaling
4.4. HNF4α and Its Potential Association with RA Signaling Pathway
4.5. The Role of COUP-TFII in RA Signaling
4.6. The Role of PPARβ/δ in RA Signaling
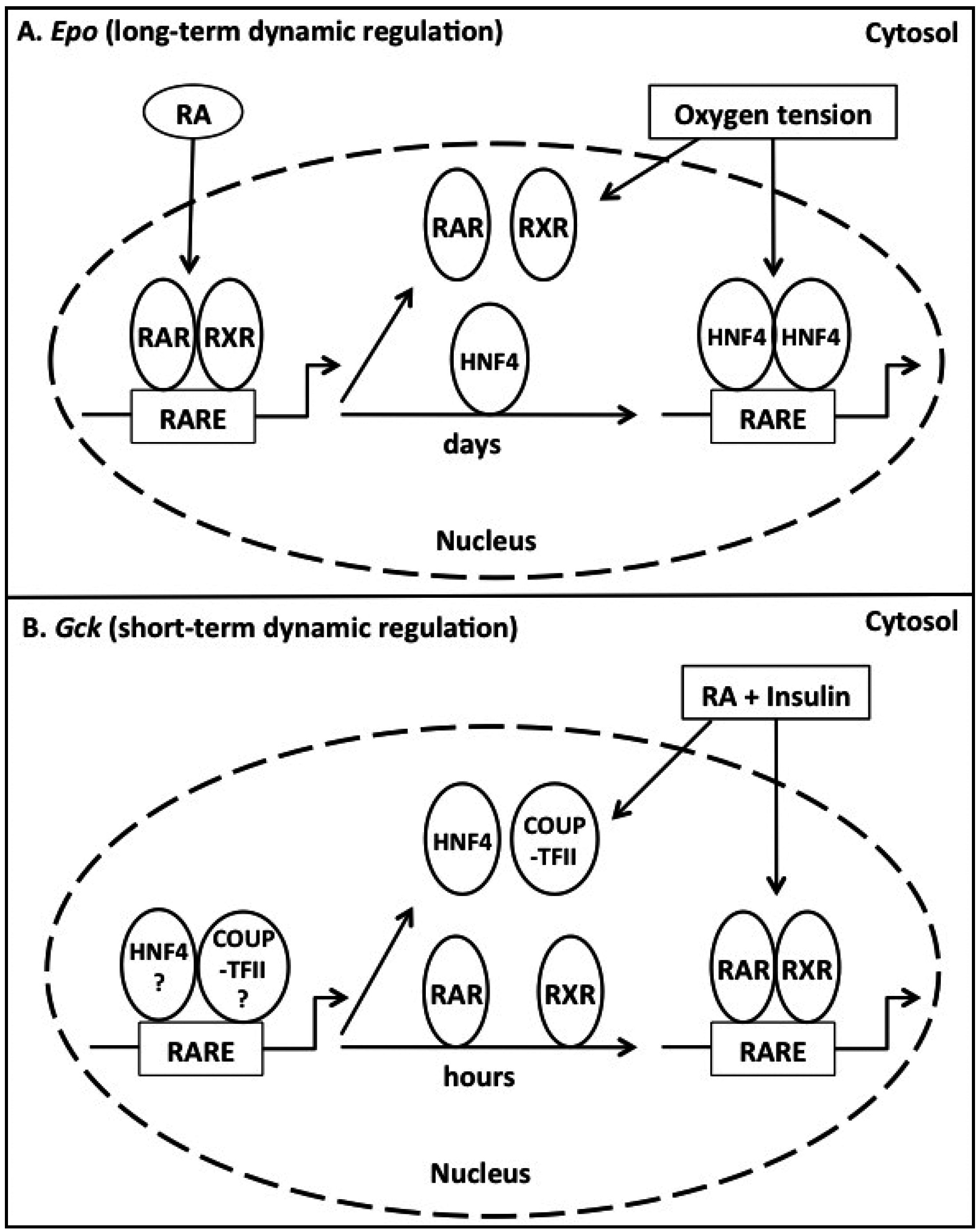
4.7. Promiscuous Binding of RARs, RXRs, HNF4, and COUP-TFII to RAREs
5. Conclusions and Future Perspectives

Acknowledgments
Author Contributions
Conflicts of Interests
References
- Zhao, S.; Li, R.; Li, Y.; Chen, W.; Zhang, Y.; Chen, G. Roles of vitamin A status and retinoids in glucose and fatty acid metabolism. Biochem. Cell Biol. 2012, 90, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.C.; Levi, L.; Noy, N. Holo-retinol-binding protein and its receptor STRA6 drive oncogenic transformation. Cancer Res. 2014, 74, 6341–6351. [Google Scholar] [CrossRef] [PubMed]
- Di Masi, A.; Leboffe, L.; de Marinis, E.; Pagano, F.; Cicconi, L.; Rochette-Egly, C.; Lo-Coco, F.; Ascenzi, P.; Nervi, C. Retinoic acid receptors: From molecular mechanisms to cancer therapy. Mol. Asp. Med. 2015, 41, 1–115. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Howell, M.L.; Li, Y.; Li, R.; Chen, G. Vitamin A and feeding statuses modulate the insulin-regulated gene expression in Zucker lean and fatty primary rat hepatocytes. PLoS ONE 2014, 9, e100868. [Google Scholar] [CrossRef] [PubMed]
- Olson, J.A. Needs and sources of carotenoids and vitamin A. Nutr. Rev. 1994, 52, S67–S73. [Google Scholar] [CrossRef] [PubMed]
- Harrison, E.H. Enzymes catalyzing the hydrolysis of retinyl esters. Biochim. Biophys. Acta 1993, 1170, 99–108. [Google Scholar] [CrossRef]
- Devery, J.; Milborrow, B.V. β-Carotene-15,15′-dioxygenase (EC 1.13.11.21) isolation reaction mechanism and an improved assay procedure. Br. J. Nutr. 1994, 72, 397–414. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.D.; Krinsky, N.I.; Tang, G.W.; Russell, R.M. Retinoic acid can be produced from excentric cleavage of β-carotene in human intestinal mucosa. Arch. Biochem. Biophys. 1992, 293, 298–304. [Google Scholar] [CrossRef]
- Helgerud, P.; Petersen, L.B.; Norum, K.R. Retinol esterification by microsomes from the mucosa of human small intestine. Evidence for acyl-Coenzyme A retinol acyltransferase activity. J. Clin. Investig. 1983, 71, 747–753. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, P.N.; Ong, D.E. Evidence for a lecithin-retinol acyltransferase activity in the rat small intestine. J. Biol. Chem. 1988, 263, 12478–12482. [Google Scholar] [PubMed]
- Blomhoff, R.; Green, M.H.; Green, J.B.; Berg, T.; Norum, K.R. Vitamin A metabolism: New perspectives on absorption, transport, and storage. Physiol. Rev. 1991, 71, 951–990. [Google Scholar] [PubMed]
- Wongsiriroj, N.; Piantedosi, R.; Palczewski, K.; Goldberg, I.J.; Johnston, T.P.; Li, E.; Blaner, W.S. The molecular basis of retinoid absorption: A genetic dissection. J. Biol. Chem. 2008, 283, 13510–13519. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, A.; Winston, A.; Lim, Y.H.; Gilbert, B.A.; Rando, R.R.; Bok, D. Molecular and biochemical characterization of lecithin retinol acyltransferase. J. Biol. Chem. 1999, 274, 3834–3841. [Google Scholar] [CrossRef] [PubMed]
- Shih, M.Y.; Kane, M.A.; Zhou, P.; Yen, C.L.; Streeper, R.S.; Napoli, J.L.; Farese, R.V., Jr. Retinol esterification by DGAT1 is essential for retinoid homeostasis in murine skin. J. Biol. Chem. 2009, 284, 4292–4299. [Google Scholar] [CrossRef] [PubMed]
- Orland, M.D.; Anwar, K.; Cromley, D.; Chu, C.H.; Chen, L.; Billheimer, J.T.; Hussain, M.M.; Cheng, D. Acyl coenzyme A dependent retinol esterification by acyl coenzyme A: Diacylglycerol acyltransferase 1. Biochim. Biophys. Acta 2005, 1737, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Harrison, E.H. Mechanisms of digestion and absorption of dietary vitamin A. Annu. Rev. Nutr. 2005, 25, 87–103. [Google Scholar] [CrossRef] [PubMed]
- During, A.; Harrison, E.H. Mechanisms of provitamin A (carotenoid) and vitamin A (retinol) transport into and out of intestinal Caco-2 cells. J. Lipid Res. 2007, 48, 2283–2294. [Google Scholar] [CrossRef] [PubMed]
- Goodman, D.W.; Huang, H.S.; Shiratori, T. Tissue distribution and metabolism of newly absorbed vitamin A in the rat. J. Lipid Res. 1965, 6, 390–396. [Google Scholar] [PubMed]
- Lobo, G.P.; Amengual, J.; Palczewski, G.; Babino, D.; von Lintig, J. Mammalian carotenoid-oxygenases: Key players for carotenoid function and homeostasis. Biochim. Biophys. Acta 2012, 1821, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Harrison, E.H.; Gad, M.Z.; Ross, A.C. Hepatic uptake and metabolism of chylomicron retinyl esters: Probable role of plasma membrane/endosomal retinyl ester hydrolases. J. Lipid Res. 1995, 36, 1498–1506. [Google Scholar] [PubMed]
- Raghu, P.; Sivakumar, B. Interactions amongst plasma retinol-binding protein, transthyretin and their ligands: Implications in vitamin A homeostasis and transthyretin amyloidosis. Biochim. Biophys. Acta 2004, 1703, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Noy, N. Retinoid-binding proteins: Mediators of retinoid action. Biochem. J. 2000, 348, 481–495. [Google Scholar] [CrossRef] [PubMed]
- Blomhoff, R.; Blomhoff, H.K. Overview of retinoid metabolism and function. J. Neurobiol. 2006, 66, 606–630. [Google Scholar] [CrossRef] [PubMed]
- Blomhoff, R. Transport and metabolism of vitamin A. Nutr. Rev. 1994, 52, S13–S23. [Google Scholar] [CrossRef] [PubMed]
- Batten, M.L.; Imanishi, Y.; Maeda, T.; Tu, D.C.; Moise, A.R.; Bronson, D.; Possin, D.; van Gelder, R.N.; Baehr, W.; Palczewski, K. Lecithin-retinol acyltransferase is essential for accumulation of all-trans-retinyl esters in the eye and in the liver. J. Biol. Chem. 2004, 279, 10422–10432. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, R.; Yu, J.; Honda, J.; Hu, J.; Whitelegge, J.; Ping, P.; Wiita, P.; Bok, D.; Sun, H. A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science 2007, 315, 820–825. [Google Scholar] [CrossRef] [PubMed]
- Blaner, W.S. STRA6, a cell-surface receptor for retinol-binding protein: The plot thickens. Cell Metab. 2007, 5, 164–166. [Google Scholar] [CrossRef] [PubMed]
- Isken, A.; Golczak, M.; Oberhauser, V.; Hunzelmann, S.; Driever, W.; Imanishi, Y.; Palczewski, K.; von Lintig, J. RBP4 disrupts vitamin A uptake homeostasis in a STRA6-deficient animal model for Matthew–Wood syndrome. Cell Metab. 2008, 7, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.C.; Noy, N. Signaling by vitamin A and retinol-binding protein in regulation of insulin responses and lipid homeostasis. Biochim. Biophys. Acta 2012, 1821, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.C.; Jin, H.; Majumdar, A.; Noy, N. Signaling by vitamin A and retinol-binding protein regulates gene expression to inhibit insulin responses. Proc. Natl. Acad. Sci. USA 2011, 108, 4340–4345. [Google Scholar] [CrossRef] [PubMed]
- Zemany, L.; Kraus, B.J.; Norseen, J.; Saito, T.; Peroni, O.D.; Johnson, R.L.; Kahn, B.B. Downregulation of STRA6 in adipocytes and adipose stromovascular fraction in obesity and effects of adipocyte-specific STRA6 knockdown in vivo. Mol. Cell. Biol. 2014, 34, 1170–1186. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.C.; Jacobs, H.; Marwarha, G.; Gely-Pernot, A.; O’Byrne, S.M.; DeSantis, D.; Klopfenstein, M.; Feret, B.; Dennefeld, C.; Blaner, W.S.; et al. The STRA6 receptor is essential for retinol-binding protein-induced insulin resistance but not for maintaining vitamin A homeostasis in tissues other than the eye. J. Biol. Chem. 2013, 288, 24528–24539. [Google Scholar] [CrossRef] [PubMed]
- Pares, X.; Farres, J.; Kedishvili, N.; Duester, G. Medium- and short-chain dehydrogenase/reductase gene and protein families: Medium-chain and short-chain dehydrogenases/reductases in retinoid metabolism. Cell. Mol. Life Sci. 2008, 65, 3936–3949. [Google Scholar] [CrossRef] [PubMed]
- Persson, B.; Hedlund, J.; Jornvall, H. Medium- and short-chain dehydrogenase/reductase gene and protein families: The MDR superfamily. Cell. Mol. Life Sci. 2008, 65, 3879–3894. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh, K.L.; Jornvall, H.; Persson, B.; Oppermann, U. Medium- and short-chain dehydrogenase/reductase gene and protein families: The SDR superfamily: Functional and structural diversity within a family of metabolic and regulatory enzymes. Cell. Mol. Life Sci. 2008, 65, 3895–3906. [Google Scholar] [CrossRef] [PubMed]
- Duester, G. Retinoic acid synthesis and signaling during early organogenesis. Cell 2008, 134, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Molotkov, A.; Duester, G. Genetic evidence that retinaldehyde dehydrogenase Raldh1 (Aldh1a1) functions downstream of alcohol dehydrogenase Adh1 in metabolism of retinol to retinoic acid. J. Biol. Chem. 2003, 278, 36085–36090. [Google Scholar] [CrossRef] [PubMed]
- Elizondo, G.; Corchero, J.; Sterneck, E.; Gonzalez, F.J. Feedback inhibition of the retinaldehyde dehydrogenase gene ALDH1 by retinoic acid through retinoic acid receptor α and CCAAT/enhancer-binding protein β. J. Biol. Chem. 2000, 275, 39747–39753. [Google Scholar] [CrossRef] [PubMed]
- Elizondo, G.; Medina-Diaz, I.M.; Cruz, R.; Gonzalez, F.J.; Vega, L. Retinoic acid modulates retinaldehyde dehydrogenase 1 gene expression through the induction of GADD153-C/EBPβ interaction. Biochem. Pharmacol. 2009, 77, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Sandell, L.L.; Lynn, M.L.; Inman, K.E.; McDowell, W.; Trainor, P.A. RDH10 oxidation of vitamin A is a critical control step in synthesis of retinoic acid during mouse embryogenesis. PLoS ONE 2012, 7, e30698. [Google Scholar] [CrossRef] [PubMed]
- Reijntjes, S.; Gale, E.; Maden, M. Generating gradients of retinoic acid in the chick embryo: Cyp26C1 expression and a comparative analysis of the Cyp26 enzymes. Dev. Dyn. 2004, 230, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Noy, N. The one-two punch: Retinoic acid suppresses obesity both by promoting energy expenditure and by inhibiting adipogenesis. Adipocyte 2013, 2, 184–187. [Google Scholar] [CrossRef] [PubMed]
- Sucov, H.M.; Murakami, K.K.; Evans, R.M. Characterization of an autoregulated response element in the mouse retinoic acid receptor type β gene. Proc. Natl. Acad. Sci. USA 1990, 87, 5392–5396. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, B.; Lehmann, J.M.; Zhang, X.K.; Hermann, T.; Husmann, M.; Graupner, G.; Pfahl, M. A retinoic acid receptor-specific element controls the retinoic acid receptor-β promoter. Mol. Endocrinol. 1990, 4, 1727–1736. [Google Scholar] [CrossRef] [PubMed]
- De The, H.; Vivanco-Ruiz, M.M.; Tiollais, P.; Stunnenberg, H.; Dejean, A. Identification of a retinoic acid responsive element in the retinoic acid receptor β gene. Nature 1990, 343, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Mahony, S.; Mazzoni, E.O.; McCuine, S.; Young, R.A.; Wichterle, H.; Gifford, D.K. Ligand-dependent dynamics of retinoic acid receptor binding during early neurogenesis. Genome Biol. 2011, 12, R2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balmer, J.E.; Blomhoff, R. A robust characterization of retinoic acid response elements based on a comparison of sites in three species. J. Steroid Biochem. Mol. Biol. 2005, 96, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Moutier, E.; Ye, T.; Choukrallah, M.A.; Urban, S.; Osz, J.; Chatagnon, A.; Delacroix, L.; Langer, D.; Rochel, N.; Moras, D.; et al. Retinoic acid receptors recognize the mouse genome through binding elements with diverse spacing and topology. J. Biol. Chem. 2012, 287, 26328–26341. [Google Scholar] [CrossRef] [PubMed]
- Mader, S.; Chen, J.Y.; Chen, Z.; White, J.; Chambon, P.; Gronemeyer, H. The patterns of binding of RAR, RXR and TR homo- and heterodimers to direct repeats are dictated by the binding specificites of the DNA binding domains. EMBO J. 1993, 12, 5029–5041. [Google Scholar] [PubMed]
- Perlmann, T.; Rangarajan, P.N.; Umesono, K.; Evans, R.M. Determinants for selective RAR and TR recognition of direct repeat HREs. Genes Dev. 1993, 7, 1411–1422. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, R.; DiRenzo, J.; Boehm, M.; Sugarman, J.; Gloss, B.; Rosenfeld, M.G.; Heyman, R.A.; Glass, C.K. Regulation of retinoid signalling by receptor polarity and allosteric control of ligand binding. Nature 1994, 371, 528–531. [Google Scholar] [CrossRef] [PubMed]
- Vasios, G.; Mader, S.; Gold, J.D.; Leid, M.; Lutz, Y.; Gaub, M.P.; Chambon, P.; Gudas, L. The late retinoic acid induction of laminin B1 gene transcription involves RAR binding to the responsive element. EMBO J. 1991, 10, 1149–1158. [Google Scholar] [PubMed]
- Vasios, G.W.; Gold, J.D.; Petkovich, M.; Chambon, P.; Gudas, L.J. A retinoic acid-responsive element is present in the 5′ flanking region of the laminin B1 gene. Proc. Natl. Acad. Sci. USA 1989, 86, 9099–9103. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zhang, R.; Li, Y.; Zhu, B.; Chen, W.; Zhang, Y.; Chen, G. A RARE of hepatic Gck promoter interacts with RARα, HNF4α and COUP-TFII that affect retinoic acid- and insulin-induced Gck expression. J. Nutr. Biochem. 2014, 25, 964–976. [Google Scholar] [CrossRef] [PubMed]
- Bastien, J.; Rochette-Egly, C. Nuclear retinoid receptors and the transcription of retinoid-target genes. Gene 2004, 328, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Durand, B.; Saunders, M.; Leroy, P.; Leid, M.; Chambon, P. All-trans and 9-cis retinoic acid induction of CRABPII transcription is mediated by RAR-RXR heterodimers bound to DR1 and DR2 repeated motifs. Cell 1992, 71, 73–85. [Google Scholar] [CrossRef]
- Brade, T.; Kumar, S.; Cunningham, T.J.; Chatzi, C.; Zhao, X.; Cavallero, S.; Li, P.; Sucov, H.M.; Ruiz-Lozano, P.; Duester, G. Retinoic acid stimulates myocardial expansion by induction of hepatic erythropoietin which activates epicardial Igf2. Development 2011, 138, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Loudig, O.; Babichuk, C.; White, J.; Abu-Abed, S.; Mueller, C.; Petkovich, M. Cytochrome P450RAI(CYP26) promoter: A distinct composite retinoic acid response element underlies the complex regulation of retinoic acid metabolism. Mol. Endocrinol. 2000, 14, 1483–1497. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Wei, L.N. Characterization of an inverted repeat with a zero spacer (IR0)-type retinoic acid response element from the mouse nuclear orphan receptor TR2–11 gene. Biochemistry 1999, 38, 8820–8825. [Google Scholar] [CrossRef] [PubMed]
- Chen, G. Roles of vitamin A metabolism in the development of hepatic insulin resistance. ISRN Hepatol. 2013, 2013, 1–21. [Google Scholar] [CrossRef]
- Germain, P.; Chambon, P.; Eichele, G.; Evans, R.M.; Lazar, M.A.; Leid, M.; de Lera, A.R.; Lotan, R.; Mangelsdorf, D.J.; Gronemeyer, H. International union of pharmacology. LX. Retinoic acid receptors. Pharmacol. Rev. 2006, 58, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Schug, T.T.; Berry, D.C.; Shaw, N.S.; Travis, S.N.; Noy, N. Opposing effects of retinoic acid on cell growth result from alternate activation of two different nuclear receptors. Cell 2007, 129, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Chambon, P. A decade of molecular biology of retinoic acid receptors. FASEB J. 1996, 10, 940–954. [Google Scholar] [PubMed]
- Berry, D.C.; Noy, N. Is PPARβ/δ a retinoid receptor? PPAR Res. 2007, 2007, 73256. [Google Scholar] [CrossRef] [PubMed]
- Stehlin-Gaon, C.; Willmann, D.; Zeyer, D.; Sanglier, S.; Van Dorsselaer, A.; Renaud, J.P.; Moras, D.; Schule, R. All-trans retinoic acid is a ligand for the orphan nuclear receptor ROR β. Nat. Struct. Biol. 2003, 10, 820–825. [Google Scholar] [CrossRef] [PubMed]
- Kruse, S.W.; Suino-Powell, K.; Zhou, X.E.; Kretschman, J.E.; Reynolds, R.; Vonrhein, C.; Xu, Y.; Wang, L.; Tsai, S.Y.; Tsai, M.J.; et al. Identification of COUP-TFII orphan nuclear receptor as a retinoic acid-activated receptor. PLoS Biol. 2008, 6, e227. [Google Scholar] [CrossRef] [PubMed]
- Bookout, A.L.; Jeong, Y.; Downes, M.; Yu, R.T.; Evans, R.M.; Mangelsdorf, D.J. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell 2006, 126, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Delfosse, V.; Maire, A.L.; Balaguer, P.; Bourguet, W. A structural perspective on nuclear receptors as targets of environmental compounds. Acta Pharmacol. Sin. 2014. [Google Scholar] [CrossRef]
- Gronemeyer, H.; Gustafsson, J.A.; Laudet, V. Principles for modulation of the nuclear receptor superfamily. Nat. Rev. Drug Discov. 2004, 3, 950–964. [Google Scholar] [CrossRef] [PubMed]
- Chawla, A.; Repa, J.J.; Evans, R.M.; Mangelsdorf, D.J. Nuclear receptors and lipid physiology: Opening the X-files. Science 2001, 294, 1866–1870. [Google Scholar] [CrossRef] [PubMed]
- Mangelsdorf, D.J.; Evans, R.M. The RXR heterodimers and orphan receptors. Cell 1995, 83, 841–850. [Google Scholar] [CrossRef]
- Glass, C.K.; Rosenfeld, M.G. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 2000, 14, 121–141. [Google Scholar] [PubMed]
- Durand, B.; Saunders, M.; Gaudon, C.; Roy, B.; Losson, R.; Chambon, P. Activation function 2 (AF-2) of retinoic acid receptor and 9-cis retinoic acid receptor: Presence of a conserved autonomous constitutive activating domain and influence of the nature of the response element on AF-2 activity. EMBO J. 1994, 13, 5370–5382. [Google Scholar] [PubMed]
- Mangelsdorf, D.J.; Thummel, C.; Beato, M.; Herrlich, P.; Schutz, G.; Umesono, K.; Blumberg, B.; Kastner, P.; Mark, M.; Chambon, P.; et al. The nuclear receptor superfamily: The second decade. Cell 1995, 83, 835–839. [Google Scholar] [CrossRef]
- Moras, D.; Gronemeyer, H. The nuclear receptor ligand-binding domain: Structure and function. Curr. Opin. Cell Biol. 1998, 10, 384–391. [Google Scholar] [CrossRef]
- Aranda, A.; Pascual, A. Nuclear hormone receptors and gene expression. Physiol. Rev. 2001, 81, 1269–1304. [Google Scholar] [PubMed]
- Gadaleta, R.M.; Magnani, L. Nuclear receptors and chromatin: An inducible couple. J. Mol. Endocrinol. 2014, 52, R137–R149. [Google Scholar] [CrossRef] [PubMed]
- Papacleovoulou, G.; Abu-Hayyeh, S.; Williamson, C. Nuclear receptor-driven alterations in bile acid and lipid metabolic pathways during gestation. Biochim. Biophys. Acta 2011, 1812, 879–887. [Google Scholar] [CrossRef] [PubMed]
- McKenna, N.J.; Xu, J.; Nawaz, Z.; Tsai, S.Y.; Tsai, M.J.; O’Malley, B.W. Nuclear receptor coactivators: Multiple enzymes, multiple complexes, multiple functions. J. Steroid Biochem. Mol. Biol. 1999, 69, 3–12. [Google Scholar] [CrossRef]
- Hermanson, O.; Glass, C.K.; Rosenfeld, M.G. Nuclear receptor coregulators: Multiple modes of modification. Trends Endocrinol. Metab. 2002, 13, 55–60. [Google Scholar] [CrossRef]
- Evans, R.M. The nuclear receptor superfamily: A rosetta stone for physiology. Mol. Endocrinol. 2005, 19, 1429–1438. [Google Scholar] [CrossRef] [PubMed]
- Heyman, R.A.; Mangelsdorf, D.J.; Dyck, J.A.; Stein, R.B.; Eichele, G.; Evans, R.M.; Thaller, C. 9-cis retinoic acid is a high affinity ligand for the retinoid X receptor. Cell 1992, 68, 397–406. [Google Scholar] [CrossRef]
- Levin, A.A.; Sturzenbecker, L.J.; Kazmer, S.; Bosakowski, T.; Huselton, C.; Allenby, G.; Speck, J.; Kratzeisen, C.; Rosenberger, M.; Lovey, A.; et al. 9-Cis retinoic acid stereoisomer binds and activates the nuclear receptor RXR α. Nature 1992, 355, 359–361. [Google Scholar] [CrossRef] [PubMed]
- Balmer, J.E.; Blomhoff, R. Gene expression regulation by retinoic acid. J. Lipid Res. 2002, 43, 1773–1808. [Google Scholar] [CrossRef] [PubMed]
- Kambhampati, S.; Li, Y.; Verma, A.; Sassano, A.; Majchrzak, B.; Deb, D.K.; Parmar, S.; Giafis, N.; Kalvakolanu, D.V.; Rahman, A.; et al. Activation of protein kinase C δ by all-trans-retinoic acid. J. Biol. Chem. 2003, 278, 32544–32551. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Han, M.; Shu, Y.N.; Li, Y.J.; Miao, S.B.; Zhang, X.H.; Shi, H.J.; Zhang, T.; Wen, J.K. HDAC2 phosphorylation-dependent Klf5 deacetylation and RARα acetylation induced by RAR agonist switch the transcription regulatory programs of p21 in VSMCs. Cell Res. 2011, 21, 1487–1508. [Google Scholar] [CrossRef] [PubMed]
- Petkovich, M.; Brand, N.J.; Krust, A.; Chambon, P. A human retinoic acid receptor which belongs to the family of nuclear receptors. Nature 1987, 330, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Giguere, V.; Ong, E.S.; Segui, P.; Evans, R.M. Identification of a receptor for the morphogen retinoic acid. Nature 1987, 330, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Evans, R. A transcriptional basis for physiology. Nat. Med. 2004, 10, 1022–1026. [Google Scholar] [CrossRef] [PubMed]
- Chambon, P. How I became one of the fathers of a superfamily. Nat. Med. 2004, 10, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Leroy, P.; Krust, A.; Zelent, A.; Mendelsohn, C.; Garnier, J.M.; Kastner, P.; Dierich, A.; Chambon, P. Multiple isoforms of the mouse retinoic acid receptor α are generated by alternative splicing and differential induction by retinoic acid. EMBO J. 1991, 10, 59–69. [Google Scholar] [PubMed]
- Brocard, J.; Kastner, P.; Chambon, P. Two novel RXR α isoforms from mouse testis. Biochem. Biophys. Res. Commun. 1996, 229, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Maruo, T.; Cao, Y.; Punj, V.; Mehta, R.; Das Gupta, T.K.; Christov, K. A novel RARβ isoform directed by a distinct promoter P3 and mediated by retinoic acid in breast cancer cells. Cancer Res. 2004, 64, 8911–8918. [Google Scholar] [CrossRef] [PubMed]
- Zelent, A.; Mendelsohn, C.; Kastner, P.; Krust, A.; Garnier, J.M.; Ruffenach, F.; Leroy, P.; Chambon, P. Differentially expressed isoforms of the mouse retinoic acid receptor β generated by usage of two promoters and alternative splicing. EMBO J. 1991, 10, 71–81. [Google Scholar] [PubMed]
- Kastner, P.; Krust, A.; Mendelsohn, C.; Garnier, J.M.; Zelent, A.; Leroy, P.; Staub, A.; Chambon, P. Murine isoforms of retinoic acid receptor γ with specific patterns of expression. Proc. Natl. Acad. Sci. USA 1990, 87, 2700–2704. [Google Scholar] [CrossRef] [PubMed]
- Dolle, P.; Ruberte, E.; Kastner, P.; Petkovich, M.; Stoner, C.M.; Gudas, L.J.; Chambon, P. Differential expression of genes encoding α, β and γ retinoic acid receptors and CRABP in the developing limbs of the mouse. Nature 1989, 342, 702–705. [Google Scholar] [CrossRef] [PubMed]
- Dolle, P.; Ruberte, E.; Leroy, P.; Morriss-Kay, G.; Chambon, P. Retinoic acid receptors and cellular retinoid binding proteins. I. A systematic study of their differential pattern of transcription during mouse organogenesis. Development 1990, 110, 1133–1151. [Google Scholar] [PubMed]
- Ruberte, E.; Dolle, P.; Chambon, P.; Morriss-Kay, G. Retinoic acid receptors and cellular retinoid binding proteins. II. Their differential pattern of transcription during early morphogenesis in mouse embryos. Development 1991, 111, 45–60. [Google Scholar] [PubMed]
- Ruberte, E.; Friederich, V.; Chambon, P.; Morriss-Kay, G. Retinoic acid receptors and cellular retinoid binding proteins. III. Their differential transcript distribution during mouse nervous system development. Development 1993, 118, 267–282. [Google Scholar] [PubMed]
- Mark, M.; Ghyselinck, N.B.; Chambon, P. Function of retinoic acid receptors during embryonic development. Nucl. Recept. Signal. 2009, 7, e002. [Google Scholar] [CrossRef] [PubMed]
- Theodosiou, M.; Laudet, V.; Schubert, M. From carrot to clinic: An overview of the retinoic acid signaling pathway. Cell. Mol. Life Sci. 2010, 67, 1423–1445. [Google Scholar] [CrossRef] [PubMed]
- Krezel, W.; Ghyselinck, N.; Samad, T.A.; Dupe, V.; Kastner, P.; Borrelli, E.; Chambon, P. Impaired locomotion and dopamine signaling in retinoid receptor mutant mice. Science 1998, 279, 863–867. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.L.; Tsai, H.C.; Wang, H.F.; Chang, J.; Lu, K.M.; Wu, H.L.; Lee, Y.C.; Tsai, T.F.; Takahashi, H.; Wagner, M.; et al. Modular patterning of structure and function of the striatum by retinoid receptor signaling. Proc. Natl. Acad. Sci. USA 2008, 105, 6765–6770. [Google Scholar] [CrossRef] [PubMed]
- Lohnes, D.; Kastner, P.; Dierich, A.; Mark, M.; LeMeur, M.; Chambon, P. Function of retinoic acid receptor γ in the mouse. Cell 1993, 73, 643–658. [Google Scholar] [CrossRef]
- Brun, P.J.; Grijalva, A.; Rausch, R.; Watson, E.; Yuen, J.J.; Das, B.C.; Shudo, K.; Kagechika, H.; Leibel, R.L.; Blaner, W.S. Retinoic acid receptor signaling is required to maintain glucose-stimulated insulin secretion and β-cell mass. FASEB J. 2015, 29, 671–683. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.C.; Noy, N. All-trans-retinoic acid represses obesity and insulin resistance by activating both peroxisome proliferation-activated receptor β/δ and retinoic acid receptor. Mol. Cell. Biol. 2009, 29, 3286–3296. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.C.; DeSantis, D.; Soltanian, H.; Croniger, C.M.; Noy, N. Retinoic acid upregulates preadipocyte genes to block adipogenesis and suppress diet-induced obesity. Diabetes 2012, 61, 1112–1121. [Google Scholar] [CrossRef] [PubMed]
- Scribner, K.B.; Odom, D.P.; McGrane, M.M. Nuclear receptor binding to the retinoic acid response elements of the phosphoenolpyruvate carboxykinase gene in vivo: Effects of vitamin A deficiency. J. Nutr. Biochem. 2007, 18, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, R.; Chen, W.; Li, Y.; Chen, G. Retinoids induced Pck1 expression and attenuated insulin-mediated suppression of its expression via activation of retinoic acid receptor in primary rat hepatocytes. Mol. Cell. Biochem. 2011, 355, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kane, M.A.; Chen, N.; Sparks, S.; Napoli, J.L. Quantification of endogenous retinoic acid in limited biological samples by LC/MS/MS. Biochem. J. 2005, 388, 363–369. [Google Scholar] [PubMed]
- Kane, M.A.; Folias, A.E.; Pingitore, A.; Perri, M.; Obrochta, K.M.; Krois, C.R.; Cione, E.; Ryu, J.Y.; Napoli, J.L. Identification of 9-cis-retinoic acid as a pancreas-specific autacoid that attenuates glucose-stimulated insulin secretion. Proc. Natl. Acad. Sci. USA 2010, 107, 21884–21889. [Google Scholar] [CrossRef] [PubMed]
- De Urquiza, A.M.; Liu, S.; Sjoberg, M.; Zetterstrom, R.H.; Griffiths, W.; Sjovall, J.; Perlmann, T. Docosahexaenoic acid, a ligand for the retinoid X receptor in mouse brain. Science 2000, 290, 2140–2144. [Google Scholar] [CrossRef] [PubMed]
- Ziouzenkova, O.; Orasanu, G.; Sukhova, G.; Lau, E.; Berger, J.P.; Tang, G.; Krinsky, N.I.; Dolnikowski, G.G.; Plutzky, J. Asymmetric cleavage of β-carotene yields a transcriptional repressor of retinoid X receptor and peroxisome proliferator-activated receptor responses. Mol. Endocrinol. 2007, 21, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, P.; Benomar, Y.; Staels, B. Retinoid X receptors: Common heterodimerization partners with distinct functions. Trends Endocrinol. Metab. 2010, 21, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Mangelsdorf, D.J.; Ong, E.S.; Dyck, J.A.; Evans, R.M. Nuclear receptor that identifies a novel retinoic acid response pathway. Nature 1990, 345, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Mangelsdorf, D.J.; Borgmeyer, U.; Heyman, R.A.; Zhou, J.Y.; Ong, E.S.; Oro, A.E.; Kakizuka, A.; Evans, R.M. Characterization of three RXR genes that mediate the action of 9-cis retinoic acid. Genes Dev. 1992, 6, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Dolle, P.; Fraulob, V.; Kastner, P.; Chambon, P. Developmental expression of murine retinoid X receptor (RXR) genes. Mech. Dev. 1994, 45, 91–104. [Google Scholar] [CrossRef]
- Mark, M.; Ghyselinck, N.B.; Chambon, P. Function of retinoid nuclear receptors: Lessons from genetic and pharmacological dissections of the retinoic acid signaling pathway during mouse embryogenesis. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 451–480. [Google Scholar] [CrossRef] [PubMed]
- Krezel, W.; Dupe, V.; Mark, M.; Dierich, A.; Kastner, P.; Chambon, P. RXR γ null mice are apparently normal and compound RXRα+/−/RXRβ−/−/RXRγ−/− mutant mice are viable. Proc. Natl. Acad. Sci. USA 1996, 93, 9010–9014. [Google Scholar] [CrossRef] [PubMed]
- Cooney, A.J.; Leng, X.; Tsai, S.Y.; O’Malley, B.W.; Tsai, M.J. Multiple mechanisms of chicken ovalbumin upstream promoter transcription factor-dependent repression of transactivation by the vitamin D, thyroid hormone, and retinoic acid receptors. J. Biol. Chem. 1993, 268, 4152–4160. [Google Scholar] [PubMed]
- Blumberg, B.; Evans, R.M. Orphan nuclear receptors—New ligands and new possibilities. Genes Dev. 1998, 12, 3149–3155. [Google Scholar] [CrossRef] [PubMed]
- Mascrez, B.; Mark, M.; Krezel, W.; Dupe, V.; LeMeur, M.; Ghyselinck, N.B.; Chambon, P. Differential contributions of AF-1 and AF-2 activities to the developmental functions of RXR α. Development 2001, 128, 2049–2062. [Google Scholar] [PubMed]
- Li, D.; Li, T.; Wang, F.; Tian, H.; Samuels, H.H. Functional evidence for retinoid X receptor (RXR) as a nonsilent partner in the thyroid hormone receptor/RXR heterodimer. Mol. Cell. Biol. 2002, 22, 5782–5792. [Google Scholar] [CrossRef] [PubMed]
- Singh Ahuja, H.; Liu, S.; Crombie, D.L.; Boehm, M.; Leibowitz, M.D.; Heyman, R.A.; Depre, C.; Nagy, L.; Tontonoz, P.; Davies, P.J. Differential effects of rexinoids and thiazolidinediones on metabolic gene expression in diabetic rodents. Mol. Pharmacol. 2001, 59, 765–773. [Google Scholar] [PubMed]
- Li, R.; Chen, W.; Li, Y.; Zhang, Y.; Chen, G. Retinoids synergized with insulin to induce Srebp-1c expression and activated its promoter via the two liver X receptor binding sites that mediate insulin action. Biochem. Biophys. Res. Commun. 2011, 406, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, R.; Davies, P.J.; Crombie, D.L.; Bischoff, E.D.; Cesario, R.M.; Jow, L.; Hamann, L.G.; Boehm, M.F.; Mondon, C.E.; Nadzan, A.M.; et al. Sensitization of diabetic and obese mice to insulin by retinoid X receptor agonists. Nature 1997, 386, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.J.; Han, G.; Cai, Y.; Dai, T.; Konishi, T.; Leng, A.S. Hepatocyte retinoid X receptor-α-deficient mice have reduced food intake, increased body weight, and improved glucose tolerance. Endocrinology 2003, 144, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Metzger, D.; Imai, T.; Jiang, M.; Takukawa, R.; Desvergne, B.; Wahli, W.; Chambon, P. Functional role of RXRs and PPARγ in mature adipocytes. Prostaglandins Leukot. Essent. Fat. Acids 2005, 73, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Ravnskjaer, K.; Boergesen, M.; Rubi, B.; Larsen, J.K.; Nielsen, T.; Fridriksson, J.; Maechler, P.; Mandrup, S. Peroxisome proliferator-activated receptor α (PPARα) potentiates, whereas PPARγ attenuates, glucose-stimulated insulin secretion in pancreatic β cells. Endocrinology 2005, 146, 3266–3276. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, S.; Taniguchi, H.; Moritoh, Y.; Tashiro, F.; Yamamoto, T.; Yamato, E.; Ikegami, H.; Ozato, K.; Miyazaki, J. Nuclear hormone retinoid X receptor (RXR) negatively regulates the glucose-stimulated insulin secretion of pancreatic β cells. Diabetes 2010, 59, 2854–2861. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.H.; Grayson, D.R.; Darnell, J.E., Jr. Multiple hepatocyte-enriched nuclear factors function in the regulation of transthyretin and α 1-antitrypsin genes. Mol. Cell. Biol. 1989, 9, 1415–1425. [Google Scholar] [PubMed]
- Sladek, F.M.; Zhong, W.M.; Lai, E.; Darnell, J.E., Jr. Liver-enriched transcription factor HNF4 is a novel member of the steroid hormone receptor superfamily. Genes Dev. 1990, 4, 23532365. [Google Scholar] [CrossRef]
- Drewes, T.; Senkel, S.; Holewa, B.; Ryffel, G.U. Human hepatocyte nuclear factor 4 isoforms are encoded by distinct and differentially expressed genes. Mol. Cell. Biol. 1996, 16, 925–931. [Google Scholar] [PubMed]
- Ellard, S.; Colclough, K. Mutations in the genes encoding the transcription factors hepatocyte nuclear factor 1α (HNF1A) and 4α (HNF4A) in maturity-onset diabetes of the young. Hum. Mutat. 2006, 27, 854–869. [Google Scholar] [CrossRef] [PubMed]
- Thomas, H.; Jaschkowitz, K.; Bulman, M.; Frayling, T.M.; Mitchell, S.M.; Roosen, S.; Lingott-Frieg, A.; Tack, C.J.; Ellard, S.; Ryffel, G.U.; et al. A distant upstream promoter of the HNF4α gene connects the transcription factors involved in maturity-onset diabetes of the young. Hum. Mol. Genet. 2001, 10, 2089–2097. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Liao, H.; Dang, H.; Pang, W.; Guan, Y.; Wang, X.; Shyy, J.Y.; Zhu, Y.; Sladek, F.M. Down-regulation of hepatic HNF4α gene expression during hyperinsulinemia via SREBPs. Mol. Endocrinol. 2009, 23, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Eeckhoute, J.; Moerman, E.; Bouckenooghe, T.; Lukoviak, B.; Pattou, F.; Formstecher, P.; Kerr-Conte, J.; Vandewalle, B.; Laine, B. Hepatocyte nuclear factor 4 α isoforms originated from the P1 promoter are expressed in human pancreatic β cells and exhibit stronger transcriptional potentials than P2 promoter-driven isoforms. Endocrinology 2003, 144, 1686–1694. [Google Scholar] [CrossRef] [PubMed]
- Stoffel, M.; Duncan, S.A. The maturity-onset diabetes of the young (MODY1) transcription factor HNF4α regulates expression of genes required for glucose transport and metabolism. Proc. Natl. Acad. Sci. USA 1997, 94, 13209–13214. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Nepomuceno, L.; Hopkins, K.; Sladek, F.M. Exclusive homodimerization of the orphan receptor hepatocyte nuclear factor 4 defines a new subclass of nuclear receptors. Mol. Cell. Biol. 1995, 15, 5131–5143. [Google Scholar] [PubMed]
- Bolotin, E.; Liao, H.; Ta, T.C.; Yang, C.; Hwang-Verslues, W.; Evans, J.R.; Jiang, T.; Sladek, F.M. Integrated approach for the identification of human hepatocyte nuclear factor 4α target genes using protein binding microarrays. Hepatology 2010, 51, 642–653. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Nepomuceno, L.; Yang, Q.; Sladek, F.M. Serine/threonine phosphorylation of orphan receptor hepatocyte nuclear factor 4. Arch. Biochem. Biophys. 1997, 340, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Viollet, B.; Kahn, A.; Raymondjean, M. Protein kinase A-dependent phosphorylation modulates DNA-binding activity of hepatocyte nuclear factor 4. Mol. Cell. Biol. 1997, 17, 4208–4219. [Google Scholar] [PubMed]
- Duncan, S.A.; Manova, K.; Chen, W.S.; Hoodless, P.; Weinstein, D.C.; Bachvarova, R.F.; Darnell, J.E., Jr. Expression of transcription factor HNF4 in the extraembryonic endoderm, gut, and nephrogenic tissue of the developing mouse embryo: HNF-4 is a marker for primary endoderm in the implanting blastocyst. Proc. Natl. Acad. Sci. USA 1994, 91, 7598–7602. [Google Scholar] [CrossRef] [PubMed]
- Taraviras, S.; Monaghan, A.P.; Schutz, G.; Kelsey, G. Characterization of the mouse HNF4 gene and its expression during mouse embryogenesis. Mech. Dev. 1994, 48, 67–79. [Google Scholar] [CrossRef]
- Chen, W.S.; Manova, K.; Weinstein, D.C.; Duncan, S.A.; Plump, A.S.; Prezioso, V.R.; Bachvarova, R.F.; Darnell, J.E., Jr. Disruption of the HNF4 gene, expressed in visceral endoderm, leads to cell death in embryonic ectoderm and impaired gastrulation of mouse embryos. Genes Dev. 1994, 8, 2466–2477. [Google Scholar] [CrossRef] [PubMed]
- Duncan, S.A.; Nagy, A.; Chan, W. Murine gastrulation requires HNF-4 regulated gene expression in the visceral endoderm: Tetraploid rescue of HNF4−/− embryos. Development 1997, 124, 279–287. [Google Scholar] [PubMed]
- Kiselyuk, A.; Lee, S.H.; Farber-Katz, S.; Zhang, M.; Athavankar, S.; Cohen, T.; Pinkerton, A.B.; Ye, M.; Bushway, P.; Richardson, A.D.; et al. HNF4α antagonists discovered by a high-throughput screen for modulators of the human insulin promoter. Chem. Biol. 2012, 19, 806–818. [Google Scholar] [CrossRef] [PubMed]
- Hayhurst, G.P.; Lee, Y.H.; Lambert, G.; Ward, J.M.; Gonzalez, F.J. Hepatocyte nuclear factor 4α (nuclear receptor 2A1) is essential for maintenance of hepatic gene expression and lipid homeostasis. Mol. Cell. Biol. 2001, 21, 1393–1403. [Google Scholar] [CrossRef] [PubMed]
- Palanker, L.; Tennessen, J.M.; Lam, G.; Thummel, C.S. Drosophila HNF4 regulates lipid mobilization and β-oxidation. Cell Metab. 2009, 9, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Ganjam, G.K.; Dimova, E.Y.; Unterman, T.G.; Kietzmann, T. FoxO1 and HNF4 are involved in regulation of hepatic glucokinase gene expression by resveratrol. J. Biol. Chem. 2009, 284, 30783–30797. [Google Scholar] [CrossRef] [PubMed]
- Roth, U.; Jungermann, K.; Kietzmann, T. Activation of glucokinase gene expression by hepatic nuclear factor 4α in primary hepatocytes. Biochem. J. 2002, 365, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Hirota, K.; Sakamaki, J.; Ishida, J.; Shimamoto, Y.; Nishihara, S.; Kodama, N.; Ohta, K.; Yamamoto, M.; Tanimoto, K.; Fukamizu, A. A combination of HNF4 and Foxo1 is required for reciprocal transcriptional regulation of glucokinase and glucose-6-phosphatase genes in response to fasting and feeding. J. Biol. Chem. 2008, 283, 32432–32441. [Google Scholar] [CrossRef] [PubMed]
- Miura, A.; Yamagata, K.; Kakei, M.; Hatakeyama, H.; Takahashi, N.; Fukui, K.; Nammo, T.; Yoneda, K.; Inoue, Y.; Sladek, F.M.; et al. Hepatocyte nuclear factor 4α is essential for glucose-stimulated insulin secretion by pancreatic β cells. J. Biol. Chem. 2006, 281, 5246–5257. [Google Scholar] [CrossRef] [PubMed]
- Bartoov-Shifman, R.; Hertz, R.; Wang, H.; Wollheim, C.B.; Bar-Tana, J.; Walker, M.D. Activation of the insulin gene promoter through a direct effect of hepatocyte nuclear factor 4α. J. Biol. Chem. 2002, 277, 25914–25919. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Maechler, P.; Antinozzi, P.A.; Hagenfeldt, K.A.; Wollheim, C.B. Hepatocyte nuclear factor 4α regulates the expression of pancreatic β cell genes implicated in glucose metabolism and nutrient-induced insulin secretion. J. Biol. Chem. 2000, 275, 35953–35959. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, K.; Furuta, H.; Oda, N.; Kaisaki, P.J.; Menzel, S.; Cox, N.J.; Fajans, S.S.; Signorini, S.; Stoffel, M.; Bell, G.I. Mutations in the hepatocyte nuclear factor 4α gene in maturity-onset diabetes of the young (MODY1). Nature 1996, 384, 458–460. [Google Scholar] [CrossRef] [PubMed]
- Moller, A.M.; Dalgaard, L.T.; Ambye, L.; Hansen, L.; Schmitz, O.; Hansen, T.; Pedersen, O. A novel Phe75fsdelT mutation in the hepatocyte nuclear factor 4α gene in a Danish pedigree with maturity-onset diabetes of the young. J. Clin. Endocrinol. Metab. 1999, 84, 367–369. [Google Scholar] [PubMed]
- Lehto, M.; Bitzen, P.O.; Isomaa, B.; Wipemo, C.; Wessman, Y.; Forsblom, C.; Tuomi, T.; Taskinen, M.R.; Groop, L. Mutation in the HNF4α gene affects insulin secretion and triglyceride metabolism. Diabetes 1999, 48, 423–425. [Google Scholar] [CrossRef] [PubMed]
- Lindner, T.; Gragnoli, C.; Furuta, H.; Cockburn, B.N.; Petzold, C.; Rietzsch, H.; Weiss, U.; Schulze, J.; Bell, G.I. Hepatic function in a family with a nonsense mutation (R154X) in the hepatocyte nuclear factor 4α/MODY1 gene. J. Clin. Investig. 1997, 100, 1400–1405. [Google Scholar] [CrossRef] [PubMed]
- Malecki, M.T.; Yang, Y.; Antonellis, A.; Curtis, S.; Warram, J.H.; Krolewski, A.S. Identification of new mutations in the hepatocyte nuclear factor 4α gene among families with early onset type 2 diabetes mellitus. Diabet. Med. 1999, 16, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Furuta, H.; Iwasaki, N.; Oda, N.; Hinokio, Y.; Horikawa, Y.; Yamagata, K.; Yano, N.; Sugahiro, J.; Ogata, M.; Ohgawara, H.; et al. Organization and partial sequence of the hepatocyte nuclear factor 4α/MODY1 gene and identification of a missense mutation, R127W, in a Japanese family with MODY. Diabetes 1997, 46, 1652–1657. [Google Scholar] [CrossRef] [PubMed]
- Moller, A.M.; Urhammer, S.A.; Dalgaard, L.T.; Reneland, R.; Berglund, L.; Hansen, T.; Clausen, J.O.; Lithell, H.; Pedersen, O. Studies of the genetic variability of the coding region of the hepatocyte nuclear factor 4α in Caucasians with maturity onset NIDDM. Diabetologia 1997, 40, 980–983. [Google Scholar] [PubMed]
- Bulman, M.P.; Dronsfield, M.J.; Frayling, T.; Appleton, M.; Bain, S.C.; Ellard, S.; Hattersley, A.T. A missense mutation in the hepatocyte nuclear factor 4α gene in a UK pedigree with maturity-onset diabetes of the young. Diabetologia 1997, 40, 859–862. [Google Scholar] [CrossRef] [PubMed]
- Hani, E.H.; Suaud, L.; Boutin, P.; Chevre, J.C.; Durand, E.; Philippi, A.; Demenais, F.; Vionnet, N.; Furuta, H.; Velho, G.; et al. A missense mutation in hepatocyte nuclear factor 4α, resulting in a reduced transactivation activity, in human late-onset non-insulin-dependent diabetes mellitus. J. Clin. Investig. 1998, 101, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Love-Gregory, L.D.; Wasson, J.; Ma, J.; Jin, C.H.; Glaser, B.; Suarez, B.K.; Permutt, M.A. A common polymorphism in the upstream promoter region of the hepatocyte nuclear factor 4α gene on chromosome 20q is associated with type 2 diabetes and appears to contribute to the evidence for linkage in an Ashkenazi Jewish population. Diabetes 2004, 53, 1134–1140. [Google Scholar] [CrossRef] [PubMed]
- Silander, K.; Mohlke, K.L.; Scott, L.J.; Peck, E.C.; Hollstein, P.; Skol, A.D.; Jackson, A.U.; Deloukas, P.; Hunt, S.; Stavrides, G.; et al. Genetic variation near the hepatocyte nuclear factor 4 alpha gene predicts susceptibility to type 2 diabetes. Diabetes 2004, 53, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Weedon, M.N.; Owen, K.R.; Shields, B.; Hitman, G.; Walker, M.; McCarthy, M.I.; Love-Gregory, L.D.; Permutt, M.A.; Hattersley, A.T.; Frayling, T.M. Common variants of the hepatocyte nuclear factor-4α P2 promoter are associated with type 2 diabetes in the UK. population. Diabetes 2004, 53, 3002–3006. [Google Scholar] [CrossRef] [PubMed]
- Hertz, R.; Magenheim, J.; Berman, I.; Bar-Tana, J. Fatty acyl-CoA thioesters are ligands of hepatic nuclear factor 4α. Nature 1998, 392, 512–516. [Google Scholar] [PubMed]
- Wisely, G.B.; Miller, A.B.; Davis, R.G.; Thornquest, A.D., Jr.; Johnson, R.; Spitzer, T.; Sefler, A.; Shearer, B.; Moore, J.T.; Miller, A.B.; et al. Hepatocyte nuclear factor 4 is a transcription factor that constitutively binds fatty acids. Structure 2002, 10, 1225–1234. [Google Scholar] [CrossRef]
- Dhe-Paganon, S.; Duda, K.; Iwamoto, M.; Chi, Y.I.; Shoelson, S.E. Crystal structure of the HNF4α ligand binding domain in complex with endogenous fatty acid ligand. J. Biol. Chem. 2002, 277, 37973–37976. [Google Scholar] [CrossRef] [PubMed]
- Divine, J.K.; McCaul, S.P.; Simon, T.C. HNF-1α and endodermal transcription factors cooperatively activate Fabpl: MODY3 mutations abrogate cooperativity. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 285, G62–G72. [Google Scholar] [CrossRef] [PubMed]
- Hatzis, P.; Talianidis, I. Regulatory mechanisms controlling human hepatocyte nuclear factor 4α gene expression. Mol. Cell. Biol. 2001, 21, 7320–7330. [Google Scholar] [CrossRef] [PubMed]
- Eeckhoute, J.; Formstecher, P.; Laine, B. Hepatocyte nuclear factor 4α enhances the hepatocyte nuclear factor 1α-mediated activation of transcription. Nucleic Acids Res. 2004, 32, 2586–2593. [Google Scholar] [CrossRef] [PubMed]
- Ktistaki, E.; Talianidis, I. Modulation of hepatic gene expression by hepatocyte nuclear factor 1. Science 1997, 277, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Rowley, C.W.; Staloch, L.J.; Divine, J.K.; McCaul, S.P.; Simon, T.C. Mechanisms of mutual functional interactions between HNF4α and HNF1α revealed by mutations that cause maturity onset diabetes of the young. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G466–G475. [Google Scholar] [CrossRef] [PubMed]
- Suaud, L.; Hemimou, Y.; Formstecher, P.; Laine, B. Functional study of the E276Q mutant hepatocyte nuclear factor 4α found in type 1 maturity-onset diabetes of the young: Impaired synergy with chicken ovalbumin upstream promoter transcription factor II on the hepatocyte nuclear factor-1 promoter. Diabetes 1999, 48, 1162–1167. [Google Scholar] [CrossRef] [PubMed]
- Hall, R.K.; Sladek, F.M.; Granner, D.K. The orphan receptors COUP-TF and HNF4 serve as accessory factors required for induction of phosphoenolpyruvate carboxykinase gene transcription by glucocorticoids. Proc. Natl. Acad. Sci. USA 1995, 92, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Stroup, D.; Chiang, J.Y. HNF4 and COUP-TFII interact to modulate transcription of the cholesterol 7α-hydroxylase gene (CYP7A1). J. Lipid Res. 2000, 41, 1–11. [Google Scholar] [PubMed]
- Chan, J.; Nakabayashi, H.; Wong, N.C. HNF4 increases activity of the rat Apo A1 gene. Nucleic Acids Res. 1993, 21, 1205–1211. [Google Scholar] [CrossRef] [PubMed]
- Ladias, J.A.; Hadzopoulou-Cladaras, M.; Kardassis, D.; Cardot, P.; Cheng, J.; Zannis, V.; Cladaras, C. Transcriptional regulation of human apolipoprotein genes ApoB, ApoCIII, and ApoAII by members of the steroid hormone receptor superfamily HNF4, ARP-1, EAR-2, and EAR-3. J. Biol. Chem. 1992, 267, 15849–15860. [Google Scholar] [PubMed]
- Mietus-Snyder, M.; Sladek, F.M.; Ginsburg, G.S.; Kuo, C.F.; Ladias, J.A.; Darnell, J.E., Jr.; Karathanasis, S.K. Antagonism between apolipoprotein AI regulatory protein 1, Ear3/COUP-TF, and hepatocyte nuclear factor 4 modulates apolipoprotein CIII gene expression in liver and intestinal cells. Mol. Cell. Biol. 1992, 12, 1708–1718. [Google Scholar] [PubMed]
- Ktistaki, E.; Talianidis, I. Chicken ovalbumin upstream promoter transcription factors act as auxiliary cofactors for hepatocyte nuclear factor 4 and enhance hepatic gene expression. Mol. Cell. Biol. 1997, 17, 2790–2797. [Google Scholar] [PubMed]
- Magee, T.R.; Cai, Y.; El-Houseini, M.E.; Locker, J.; Wan, Y.J. Retinoic acid mediates down-regulation of the α-fetoprotein gene through decreased expression of hepatocyte nuclear factors. J. Biol. Chem. 1998, 273, 30024–30032. [Google Scholar] [CrossRef] [PubMed]
- Nakshatri, H.; Chambon, P. The directly repeated RG(G/T)TCA motifs of the rat and mouse cellular retinol-binding protein II genes are promiscuous binding sites for RAR, RXR, HNF4, and ARP-1 homo- and heterodimers. J. Biol. Chem. 1994, 269, 890–902. [Google Scholar] [PubMed]
- Makita, T.; Hernandez-Hoyos, G.; Chen, T.H.; Wu, H.; Rothenberg, E.V.; Sucov, H.M. A developmental transition in definitive erythropoiesis: Erythropoietin expression is sequentially regulated by retinoic acid receptors and HNF4. Genes Dev. 2001, 15, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Pastorcic, M.; Wang, H.; Elbrecht, A.; Tsai, S.Y.; Tsai, M.J.; O’Malley, B.W. Control of transcription initiation in vitro requires binding of a transcription factor to the distal promoter of the ovalbumin gene. Mol. Cell. Biol. 1986, 6, 2784–2791. [Google Scholar] [PubMed]
- Sagami, I.; Tsai, S.Y.; Wang, H.; Tsai, M.J.; O’Malley, B.W. Identification of two factors required for transcription of the ovalbumin gene. Mol. Cell. Biol. 1986, 6, 4259–4267. [Google Scholar] [PubMed]
- Achatz, G.; Holzl, B.; Speckmayer, R.; Hauser, C.; Sandhofer, F.; Paulweber, B. Functional domains of the human orphan receptor ARP-1/COUP-TFII involved in active repression and transrepression. Mol. Cell. Biol. 1997, 17, 4914–4932. [Google Scholar] [PubMed]
- Leng, X.; Cooney, A.J.; Tsai, S.Y.; Tsai, M.J. Molecular mechanisms of COUP-TF-mediated transcriptional repression: Evidence for transrepression and active repression. Mol. Cell. Biol. 1996, 16, 2332–2340. [Google Scholar] [PubMed]
- Zhang, P.; Bennoun, M.; Gogard, C.; Bossard, P.; Leclerc, I.; Kahn, A.; Vasseur-Cognet, M. Expression of COUP-TFII in metabolic tissues during development. Mech. Dev. 2002, 119, 109–114. [Google Scholar] [CrossRef]
- Wang, L.H.; Ing, N.H.; Tsai, S.Y.; O’Malley, B.W.; Tsai, M.J. The COUP-TFs compose a family of functionally related transcription factors. Gene Expr. 1991, 1, 207–216. [Google Scholar] [PubMed]
- Ladias, J.A.; Karathanasis, S.K. Regulation of the apolipoprotein AI gene by ARP-1, a novel member of the steroid receptor superfamily. Science 1991, 251, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Krishnan, V.; Pereira, F.A.; Tsai, S.Y.; Tsai, M.J. Chicken ovalbumin upstream promoter-transcription factors and their regulation. J. Steroid Biochem. Mol. Biol. 1996, 56, 81–85. [Google Scholar] [CrossRef]
- Pereira, F.A.; Qiu, Y.; Zhou, G.; Tsai, M.J.; Tsai, S.Y. The orphan nuclear receptor COUP-TFII is required for angiogenesis and heart development. Genes Dev. 1999, 13, 1037–1049. [Google Scholar] [CrossRef] [PubMed]
- Takamoto, N.; Kurihara, I.; Lee, K.; Demayo, F.J.; Tsai, M.J.; Tsai, S.Y. Haploinsufficiency of chicken ovalbumin upstream promoter transcription factor II in female reproduction. Mol. Endocrinol. 2005, 19, 2299–2308. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, S.L.; van Horssen, A.M.; Chang, C.; Burbach, J.P. Expression of nuclear hormone receptors in the rat supraoptic nucleus. Endocrinology 1995, 136, 2276–2283. [Google Scholar]
- Sabra-Makke, L.; Tourrel-Cuzin, C.; Denis, R.G.; Moldes, M.; Pegorier, J.P.; Luquet, S.; Vasseur-Cognet, M.; Bossard, P. The nutritional induction of COUP-TFII gene expression in ventromedial hypothalamic neurons is mediated by the melanocortin pathway. PLoS ONE 2010, 5, e13464. [Google Scholar] [CrossRef] [PubMed]
- Bardoux, P.; Zhang, P.; Flamez, D.; Perilhou, A.; Lavin, T.A.; Tanti, J.F.; Hellemans, K.; Gomas, E.; Godard, C.; Andreelli, F.; et al. Essential role of chicken ovalbumin upstream promoter-transcription factor II in insulin secretion and insulin sensitivity revealed by conditional gene knockout. Diabetes 2005, 54, 1357–1363. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.K.; Vatamaniuk, M.Z.; Lee, C.S.; Flaschen, R.C.; Fulmer, J.T.; Matschinsky, F.M.; Duncan, S.A.; Kaestner, K.H. The MODY1 gene HNF4α regulates selected genes involved in insulin secretion. J. Clin. Investig. 2005, 115, 1006–1015. [Google Scholar] [CrossRef] [PubMed]
- Pearson, E.R.; Boj, S.F.; Steele, A.M.; Barrett, T.; Stals, K.; Shield, J.P.; Ellard, S.; Ferrer, J.; Hattersley, A.T. Macrosomia and hyperinsulinaemic hypoglycaemia in patients with heterozygous mutations in the HNF4α gene. PLoS Med. 2007, 4, e118. [Google Scholar] [CrossRef] [PubMed]
- Perilhou, A.; Tourrel-Cuzin, C.; Kharroubi, I.; Henique, C.; Fauveau, V.; Kitamura, T.; Magnan, C.; Postic, C.; Prip-Buus, C.; Vasseur-Cognet, M. The transcription factor COUP-TFII is negatively regulated by insulin and glucose via Foxo1- and ChREBP-controlled pathways. Mol. Cell. Biol. 2008, 28, 6568–6579. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Xie, X.; Qin, J.; Jeha, G.S.; Saha, P.K.; Yan, J.; Haueter, C.M.; Chan, L.; Tsai, S.Y.; Tsai, M.J. The nuclear orphan receptor COUP-TFII plays an essential role in adipogenesis, glucose homeostasis, and energy metabolism. Cell Metab. 2009, 9, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Yu, S.; Hsu, C.H.; Eguchi, J.; Rosen, E.D. The orphan nuclear receptor chicken ovalbumin upstream promoter-transcription factor II is a critical regulator of adipogenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 2421–2426. [Google Scholar] [CrossRef] [PubMed]
- Myers, S.A.; Wang, S.C.; Muscat, G.E. The chicken ovalbumin upstream promoter-transcription factors modulate genes and pathways involved in skeletal muscle cell metabolism. J. Biol. Chem. 2006, 281, 24149–24160. [Google Scholar] [CrossRef] [PubMed]
- Crowther, L.M.; Wang, S.C.; Eriksson, N.A.; Myers, S.A.; Murray, L.A.; Muscat, G.E. Chicken ovalbumin upstream promoter-transcription factor II regulates nuclear receptor, myogenic, and metabolic gene expression in skeletal muscle cells. Physiol. Genomics 2011, 43, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Okamura, M.; Kudo, H.; Wakabayashi, K.; Tanaka, T.; Nonaka, A.; Uchida, A.; Tsutsumi, S.; Sakakibara, I.; Naito, M.; Osborne, T.F.; et al. COUP-TFII acts downstream of Wnt/β-catenin signal to silence PPARγ gene expression and repress adipogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 5819–5824. [Google Scholar] [CrossRef] [PubMed]
- Benoit, G.; Cooney, A.; Giguere, V.; Ingraham, H.; Lazar, M.; Muscat, G.; Perlmann, T.; Renaud, J.P.; Schwabe, J.; Sladek, F.; et al. International union of pharmacology. LXVI. Orphan nuclear receptors. Pharmacol. Rev. 2006, 58, 798–836. [Google Scholar] [CrossRef] [PubMed]
- Perilhou, A.; Tourrel-Cuzin, C.; Zhang, P.; Kharroubi, I.; Wang, H.; Fauveau, V.; Scott, D.K.; Wollheim, C.B.; Vasseur-Cognet, M. The MODY1 gene for hepatocyte nuclear factor 4α and a feedback loop control COUP-TFII expression in pancreatic β cells. Mol. Cell. Biol. 2008, 28, 4588–4597. [Google Scholar] [CrossRef] [PubMed]
- Park, J.I.; Tsai, S.Y.; Tsai, M.J. Molecular mechanism of chicken ovalbumin upstream promoter-transcription factor (COUP-TF) actions. Keio J. Med. 2003, 52, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Malik, S.; Karathanasis, S. Transcriptional activation by the orphan nuclear receptor ARP-1. Nucleic Acids Res. 1995, 23, 1536–1543. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Tsai, S.Y.; Tsai, M.J. COUP-TF an orphan member of the steroid/thyroid hormone receptor superfamily. Trends Endocrinol. Metab. 1994, 5, 234–239. [Google Scholar] [CrossRef]
- Marcus, S.L.; Winrow, C.J.; Capone, J.P.; Rachubinski, R.A. A p56lck ligand serves as a coactivator of an orphan nuclear hormone receptor. J. Biol. Chem. 1996, 271, 27197–27200. [Google Scholar] [CrossRef] [PubMed]
- Jonk, L.J.; de Jonge, M.E.; Pals, C.E.; Wissink, S.; Vervaart, J.M.; Schoorlemmer, J.; Kruijer, W. Cloning and expression during development of three murine members of the COUP family of nuclear orphan receptors. Mech. Dev. 1994, 47, 81–97. [Google Scholar] [CrossRef]
- Tsai, S.Y.; Tsai, M.J. Chick ovalbumin upstream promoter-transcription factors (COUP-TFs): Coming of age. Endocr. Rev. 1997, 18, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Litchfield, L.M.; Riggs, K.A.; Hockenberry, A.M.; Oliver, L.D.; Barnhart, K.G.; Cai, J.; Pierce, W.M., Jr.; Ivanova, M.M.; Bates, P.J.; Appana, S.N.; et al. Identification and characterization of nucleolin as a COUP-TFII coactivator of retinoic acid receptor β transcription in breast cancer cells. PLoS ONE 2012, 7, e38278. [Google Scholar] [CrossRef] [PubMed]
- Doi, T.; Sugimoto, K.; Puri, P. Prenatal retinoic acid up-regulates pulmonary gene expression of COUP-TFII, FOG2, and GATA4 in pulmonary hypoplasia. J. Pediatr. Surg. 2009, 44, 1933–1937. [Google Scholar] [CrossRef] [PubMed]
- Fjose, A.; Weber, U.; Mlodzik, M. A novel vertebrate svp-related nuclear receptor is expressed as a step gradient in developing rhombomeres and is affected by retinoic acid. Mech. Dev. 1995, 52, 233–246. [Google Scholar] [CrossRef]
- Malpel, S.; Mendelsohn, C.; Cardoso, W.V. Regulation of retinoic acid signaling during lung morphogenesis. Development 2000, 127, 3057–3067. [Google Scholar] [PubMed]
- Prahalad, P.; Dakshanamurthy, S.; Ressom, H.; Byers, S.W. Retinoic acid mediates regulation of network formation by COUP-TFII and VE-cadherin expression by TGFβ receptor kinase in breast cancer cells. PLoS ONE 2010, 5, e10023. [Google Scholar] [CrossRef] [PubMed]
- Neels, J.G.; Grimaldi, P.A. Physiological functions of peroxisome proliferator-activated receptor beta. Physiol. Rev. 2014, 94, 795–858. [Google Scholar] [CrossRef] [PubMed]
- Sher, T.; Yi, H.F.; McBride, O.W.; Gonzalez, F.J. cDNA cloning, chromosomal mapping, and functional characterization of the human peroxisome proliferator activated receptor. Biochemistry 1993, 32, 5598–5604. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, T.; Brkanac, Z.; Dupont, B.R.; Xing, G.Q.; Leach, R.J.; Detera-Wadleigh, S.D. Assignment of the human nuclear hormone receptor, NUC1 (PPARD), to chromosome 6p21.1-p21.2. Genomics 1996, 35, 637–638. [Google Scholar] [CrossRef] [PubMed]
- Greene, M.E.; Blumberg, B.; McBride, O.W.; Yi, H.F.; Kronquist, K.; Kwan, K.; Hsieh, L.; Greene, G.; Nimer, S.D. Isolation of the human peroxisome proliferator activated receptor γ cDNA: Expression in hematopoietic cells and chromosomal mapping. Gene Expr. 1995, 4, 281–299. [Google Scholar] [PubMed]
- Berger, J.; Moller, D.E. The mechanisms of action of PPARs. Annu. Rev. Med. 2002, 53, 409–435. [Google Scholar] [CrossRef] [PubMed]
- Issemann, I.; Prince, R.A.; Tugwood, J.D.; Green, S. The peroxisome proliferator-activated receptor: Retinoid X receptor heterodimer is activated by fatty acids and fibrate hypolipidaemic drugs. J. Mol. Endocrinol. 1993, 11, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Qi, C.; Korenberg, J.R.; Chen, X.N.; Noya, D.; Rao, M.S.; Reddy, J.K. Structural organization of mouse peroxisome proliferator-activated receptor γ (mPPAR γ) gene: Alternative promoter use and different splicing yield two mPPAR γ isoforms. Proc. Natl. Acad. Sci. USA 1995, 92, 7921–7925. [Google Scholar] [CrossRef] [PubMed]
- Tontonoz, P.; Hu, E.; Graves, R.A.; Budavari, A.I.; Spiegelman, B.M. mPPAR γ 2: Tissue-specific regulator of an adipocyte enhancer. Genes Dev. 1994, 8, 1224–1234. [Google Scholar] [CrossRef] [PubMed]
- Escher, P.; Braissant, O.; Basu-Modak, S.; Michalik, L.; Wahli, W.; Desvergne, B. Rat PPARs: Quantitative analysis in adult rat tissues and regulation in fasting and refeeding. Endocrinology 2001, 142, 4195–4202. [Google Scholar] [CrossRef] [PubMed]
- Holst, D.; Luquet, S.; Nogueira, V.; Kristiansen, K.; Leverve, X.; Grimaldi, P.A. Nutritional regulation and role of peroxisome proliferator-activated receptor δ in fatty acid catabolism in skeletal muscle. Biochim. Biophys. Acta 2003, 1633, 43–50. [Google Scholar] [CrossRef]
- Wang, Y.X.; Lee, C.H.; Tiep, S.; Yu, R.T.; Ham, J.; Kang, H.; Evans, R.M. Peroxisome-proliferator-activated receptor δ activates fat metabolism to prevent obesity. Cell 2003, 113, 159–170. [Google Scholar] [CrossRef]
- Luquet, S.; Lopez-Soriano, J.; Holst, D.; Fredenrich, A.; Melki, J.; Rassoulzadegan, M.; Grimaldi, P.A. Peroxisome proliferator-activated receptor δ controls muscle development and oxidative capability. FASEB J. 2003, 17, 2299–2301. [Google Scholar] [PubMed]
- Wang, Y.X.; Zhang, C.L.; Yu, R.T.; Cho, H.K.; Nelson, M.C.; Bayuga-Ocampo, C.R.; Ham, J.; Kang, H.; Evans, R.M. Regulation of muscle fiber type and running endurance by PPARδ. PLoS Biol. 2004, 2, e294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, T.; Yamamoto, J.; Iwasaki, S.; Asaba, H.; Hamura, H.; Ikeda, Y.; Watanabe, M.; Magoori, K.; Ioka, R.X.; Tachibana, K.; et al. Activation of peroxisome proliferator-activated receptor δ induces fatty acid β-oxidation in skeletal muscle and attenuates metabolic syndrome. Proc. Natl. Acad. Sci. USA 2003, 100, 15924–15929. [Google Scholar] [CrossRef] [PubMed]
- IJpenberg, A.; Jeannin, E.; Wahli, W.; Desvergne, B. Polarity and specific sequence requirements of peroxisome proliferator-activated receptor (PPAR)/retinoid X receptor heterodimer binding to DNA. A functional analysis of the malic enzyme gene PPAR response element. J. Biol. Chem. 1997, 272, 20108–20117. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.N.; Hsu, M.H.; Griffin, H.J.; Johnson, E.F. Novel sequence determinants in peroxisome proliferator signaling. J. Biol. Chem. 1995, 270, 16114–16121. [Google Scholar] [CrossRef] [PubMed]
- Kliewer, S.A.; Umesono, K.; Noonan, D.J.; Heyman, R.A.; Evans, R.M. Convergence of 9-cis retinoic acid and peroxisome proliferator signalling pathways through heterodimer formation of their receptors. Nature 1992, 358, 771–774. [Google Scholar] [CrossRef] [PubMed]
- Takata, Y.; Liu, J.; Yin, F.; Collins, A.R.; Lyon, C.J.; Lee, C.H.; Atkins, A.R.; Downes, M.; Barish, G.D.; Evans, R.M.; et al. PPARδ-mediated antiinflammatory mechanisms inhibit angiotensin II-accelerated atherosclerosis. Proc. Natl. Acad. Sci. USA 2008, 105, 4277–4282. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.Y.; Wang, X.; Pixley, F.J.; Yu, J.J.; Dent, A.L.; Broxmeyer, H.E.; Stanley, E.R.; Ye, B.H. BCL-6 negatively regulates macrophage proliferation by suppressing autocrine IL-6 production. Blood 2005, 105, 1777–1784. [Google Scholar] [CrossRef] [PubMed]
- Ali, F.Y.; Hall, M.G.; Desvergne, B.; Warner, T.D.; Mitchell, J.A. PPARβ/δ agonists modulate platelet function via a mechanism involving PPAR receptors and specific association/repression of PKCα—Brief report. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1871–1873. [Google Scholar] [CrossRef] [PubMed]
- Rochette-Egly, C. Retinoic acid signaling and mouse embryonic stem cell differentiation: Cross talk between genomic and non-genomic effects of RA. Biochim. Biophys. Acta 2015, 1851, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Shaw, N.; Elholm, M.; Noy, N. Retinoic acid is a high affinity selective ligand for the peroxisome proliferator-activated receptor β/δ. J. Biol. Chem. 2003, 278, 41589–41592. [Google Scholar] [CrossRef] [PubMed]
- Thulasiraman, P.; McAndrews, D.J.; Mohiudddin, I.Q. Curcumin restores sensitivity to retinoic acid in triple negative breast cancer cells. BMC Cancer 2014, 14, 724. [Google Scholar] [CrossRef] [PubMed]
- Budhu, A.; Gillilan, R.; Noy, N. Localization of the RAR interaction domain of cellular retinoic acid binding protein-II. J. Mol. Biol. 2001, 305, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Budhu, A.S.; Noy, N. Direct channeling of retinoic acid between cellular retinoic acid-binding protein II and retinoic acid receptor sensitizes mammary carcinoma cells to retinoic acid-induced growth arrest. Mol. Cell. Biol. 2002, 22, 2632–2641. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Levi, L.; Siegel, R.; Noy, N. Retinoic acid induces neurogenesis by activating both retinoic acid receptors (RARs) and peroxisome proliferator-activated receptor β/δ (PPARβ/δ). J. Biol. Chem. 2012, 287, 42195–42205. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.D.; Ostapchuk, P.; Hearing, P. Functional interaction of nuclear factors EF-C, HNF4, and RXRα with hepatitis B virus enhancer I. J. Virol. 1993, 67, 3940–3950. [Google Scholar] [PubMed]
- Power, S.C.; Cereghini, S. Positive regulation of the vHNF1 promoter by the orphan receptors COUP-TF1/Ear3 and COUP-TFII/Arp1. Mol. Cell. Biol. 1996, 16, 778–791. [Google Scholar] [PubMed]
- Hertz, R.; Bishara-Shieban, J.; Bar-Tana, J. Mode of action of peroxisome proliferators as hypolipidemic drugs. Suppression of apolipoprotein C-III. J. Biol. Chem. 1995, 270, 13470–13475. [Google Scholar] [CrossRef] [PubMed]
- Hertz, R.; Seckbach, M.; Zakin, M.M.; Bar-Tana, J. Transcriptional suppression of the transferrin gene by hypolipidemic peroxisome proliferators. J. Biol. Chem. 1996, 271, 218–224. [Google Scholar] [PubMed]
- Pineda Torra, I.; Jamshidi, Y.; Flavell, D.M.; Fruchart, J.C.; Staels, B. Characterization of the human PPARα promoter: Identification of a functional nuclear receptor response element. Mol. Endocrinol. 2002, 16, 1013–1028. [Google Scholar] [CrossRef] [PubMed]
- Mogilenko, D.A.; Dizhe, E.B.; Shavva, V.S.; Lapikov, I.A.; Orlov, S.V.; Perevozchikov, A.P. Role of the nuclear receptors HNF4 α, PPAR α, and LXRs in the TNF α-mediated inhibition of human apolipoprotein A–I gene expression in HepG2 cells. Biochemistry 2009, 48, 11950–11960. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.C.; Puigserver, P.; Chen, G.; Donovan, J.; Wu, Z.; Rhee, J.; Adelmant, G.; Stafford, J.; Kahn, C.R.; Granner, D.K.; et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature 2001, 413, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, R.; Li, Y.; Chen, W.; Zhao, S.; Chen, G. Vitamin A status affects obesity development and hepatic expression of key genes for fuel metabolism in Zucker fatty rats. Biochem. Cell Biol. 2012, 90, 548–557. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, T.; Takenaka, M.; Yamada, K.; Matsuda, T.; Hashimoto, M.; Tanaka, T. Characterization of the 5′ flanking region of rat glucokinase gene. Biochem. Biophys. Res. Commun. 1989, 164, 1247–1252. [Google Scholar] [CrossRef]
- Chen, G.; Zhang, Y.; Lu, D.; Li, N.Q.; Ross, A.C. Retinoids synergize with insulin to induce hepatic Gck expression. Biochem. J. 2009, 419, 645–653. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, R.; Wang, Y.; Li, R.; Chen, G. Transcriptional Factors Mediating Retinoic Acid Signals in the Control of Energy Metabolism. Int. J. Mol. Sci. 2015, 16, 14210-14244. https://doi.org/10.3390/ijms160614210
Zhang R, Wang Y, Li R, Chen G. Transcriptional Factors Mediating Retinoic Acid Signals in the Control of Energy Metabolism. International Journal of Molecular Sciences. 2015; 16(6):14210-14244. https://doi.org/10.3390/ijms160614210
Chicago/Turabian StyleZhang, Rui, Yueqiao Wang, Rui Li, and Guoxun Chen. 2015. "Transcriptional Factors Mediating Retinoic Acid Signals in the Control of Energy Metabolism" International Journal of Molecular Sciences 16, no. 6: 14210-14244. https://doi.org/10.3390/ijms160614210