Novel Insights into the Genetic Landscape of Nonalcoholic Fatty Liver Disease

 ,
,  ,
,
Abstract
:1. Nonalcoholic Fatty Liver Disease
2. The Overall Role on Inherited Factors
3. PNPLA3 and HSD17B13: The Role of Lipid Droplets and Retinol Metabolism
4. Impact of Genetic Variation in Nuclear-Encoded Mitochondrial Protein
5. Impact of Genetic Variation Influencing Inflammation and Fibrogenesis
6. Other Common Genetic Determinants of NAFLD
7. Rare Genetic Determinants of NAFLD
8. Therapeutic Perspectives
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Estes, C.; Anstee, Q.M.; Arias-Loste, M.T.; Bantel, H.; Bellentani, S.; Caballeria, J.; Colombo, M.; Craxi, A.; Crespo, J.; Day, C.P.; et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016–2030. J. Hepatol. 2018, 69, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, G.; Brizi, M.; Morselli-Labate, A.M.; Bianchi, G.; Bugianesi, E.; McCullough, A.J.; Forlani, G.; Melchionda, N. Association of nonalcoholic fatty liver disease with insulin resistance. Am. J. Med. 1999, 107, 450–455. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Yang, L.; McCall, S.; Huang, J.; Yu, X.X.; Pandey, S.K.; Bhanot, S.; Monia, B.P.; Li, Y.-X.; Diehl, A.M. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 2007, 45, 1366–1374. [Google Scholar] [CrossRef] [PubMed]
- Mashek, D.G.; Khan, S.A.; Sathyanarayan, A.; Ploeger, J.M.; Franklin, M.P. Hepatic lipid droplet biology: Getting to the root of fatty liver. Hepatology 2015, 62, 964–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.-C.; Torbenson, M.S.; Yeh, M.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef]
- Eslam, M.; Valenti, L.; Romeo, S. Genetics and epigenetics of NAFLD and NASH: Clinical impact. J. Hepatol. 2018, 68, 268–279. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Donati, B.; Fares, R.; Lombardi, R.; Mancina, R.M.; Romeo, S.; Valenti, L. PNPLA3 I148M polymorphism and progressive liver disease. World J. Gastroenterol. 2013, 19, 6969–6978. [Google Scholar] [CrossRef]
- Valenti, L.V.C.; Baselli, G.A. Genetics of nonalcoholic fatty liver disease: A 2018 update. Curr. Pharm. Des. 2018, 24, 4566–4573. [Google Scholar] [CrossRef]
- Makkonen, J.; Pietiläinen, K.H.; Rissanen, A.; Kaprio, J.; Yki-Järvinen, H. Genetic factors contribute to variation in serum alanine aminotransferase activity independent of obesity and alcohol: A study in monozygotic and dizygotic twins. J. Hepatol. 2009, 50, 1035–1042. [Google Scholar] [CrossRef]
- Loomba, R.; Schork, N.; Chen, C.-H.; Bettencourt, R.; Bhatt, A.; Ang, B.; Nguyen, P.; Hernandez, C.; Richards, L.; Salotti, J.; et al. Heritability of hepatic fibrosis and steatosis based on a prospective twin study. Gastroenterology 2015, 149, 1784–1793. [Google Scholar] [CrossRef]
- Guerrero, R.; Vega, G.L.; Grundy, S.M.; Browning, J.D. Ethnic differences in hepatic steatosis: An insulin resistance paradox? Hepatology 2009, 49, 791–801. [Google Scholar] [CrossRef]
- Caussy, C.; Soni, M.; Cui, J.; Bettencourt, R.; Schork, N.; Chen, C.-H.; Al Ikhwan, M.; Bassirian, S.; Cepin, S.; Gonzalez, M.P.; et al. Nonalcoholic fatty liver disease with cirrhosis increases familial risk for advanced fibrosis. J. Clin. Investig. 2017, 127, 2697–2704. [Google Scholar] [CrossRef] [Green Version]
- Long, M.T.; Gurary, E.B.; Massaro, J.M.; Ma, J.; Hoffmann, U.; Chung, R.T.; Benjamin, E.J.; Loomba, R. Parental non-alcoholic fatty liver disease increases risk of non-alcoholic fatty liver disease in offspring. Liver Int. 2018, 39, 740–747. [Google Scholar] [CrossRef]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [Green Version]
- Pirola, C.J.; Garaycoechea, M.; Flichman, D.; Arrese, M.; San Martino, J.; Gazzi, C.; Castaño, G.O.; Sookoian, S. Splice variant rs72613567 prevents worst histologic outcomes in patients with nonalcoholic fatty liver disease. J. Lipid Res. 2019, 60, 176–185. [Google Scholar] [CrossRef] [Green Version]
- Eslam, M.; Hashem, A.M.; Leung, R.; Romero-Gomez, M.; Berg, T.; Dore, G.J.; Chan, H.L.; Irving, W.L.; Sheridan, D.; Abate, M.L.; et al. International hepatitis C genetics consortium (IHCGC). interferon-λ rs12979860 genotype and liver fibrosis in viral and non-viral chronic liver disease. Nat. Commun. 2015, 6, 6422. [Google Scholar] [CrossRef]
- Luukkonen, P.K.; Zhou, Y.; Nidhina Haridas, P.A.; Dwivedi, O.P.; Hyötyläinen, T.; Ali, A.; Juuti, A.; Leivonen, M.; Tukiainen, T.; Ahonen, L.; et al. Impaired hepatic lipid synthesis from polyunsaturated fatty acids in TM6SF2 E167K variant carriers with NAFLD. J. Hepatol. 2017, 67, 128–136. [Google Scholar] [CrossRef] [Green Version]
- Mancina, R.M.; Dongiovanni, P.; Petta, S.; Pingitore, P.; Meroni, M.; Rametta, R.; Borén, J.; Montalcini, T.; Pujia, A.; Wiklund, O.; et al. The MBOAT7-TMC4 variant rs641738 increases risk of nonalcoholic fatty liver disease in individuals of european descent. Gastroenterology 2016, 150, 1219–1230. [Google Scholar] [CrossRef]
- Luukkonen, P.K.; Zhou, Y.; Hyötyläinen, T.; Leivonen, M.; Arola, J.; Orho-Melander, M.; Orešič, M.; Yki-Järvinen, H. The MBOAT7 variant rs641738 alters hepatic phosphatidylinositols and increases severity of non-alcoholic fatty liver disease in humans. J. Hepatol. 2016, 65, 1263–1265. [Google Scholar] [CrossRef] [Green Version]
- Amarapurkar, D.N.; Hashimoto, E.; Lesmana, L.A.; Sollano, J.D.; Chen, P.-J.; Goh, K.-L. Asia-pacific working party on NAFLD. How common is non-alcoholic fatty liver disease in the Asia-Pacific region and are there local differences? J. Gastroenterol. Hepatol. 2007, 22, 788–793. [Google Scholar] [CrossRef]
- Di Filippo, M.; Moulin, P.; Roy, P.; Samson-Bouma, M.E.; Frachon, S.C.; Chebel-Dumont, S.; Peretti, N.; Dumortier, J.; Zoulim, F.; Fontanges, T.; et al. Homozygous MTTP and APOB mutations may lead to hepatic steatosis and fibrosis despite metabolic differences in congenital hypocholesterolemia. J. Hepatol. 2014, 61, 891–902. [Google Scholar] [CrossRef]
- Sookoian, S.; Pirola, C.J. Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology 2011, 53, 1883–1894. [Google Scholar] [CrossRef]
- Valenti, L.; Al-Serri, A.; Daly, A.; Galmozzi, E.; Rametta, R.; Dongiovanni, P.; Nobili, V.; Mozzi, E.; Roviaro, G.; Vanni, E.; et al. 127 homozygosity for the PNPLA3/adiponutrin I148M polymorphism influences liver fibrosis in patients with nonalcoholic fatty liver disease. J. Hepatol. 2010, 52, S57. [Google Scholar] [CrossRef]
- Valenti, L.; Alisi, A.; Galmozzi, E.; Bartuli, A.; Del Menico, B.; Alterio, A.; Dongiovanni, P.; Fargion, S.; Nobili, V. I148M patatin-like phospholipase domain-containing 3 gene variant and severity of pediatric nonalcoholic fatty liver disease. Hepatology 2010, 52, 1274–1280. [Google Scholar] [CrossRef]
- Stender, S.; Kozlitina, J.; Nordestgaard, B.G.; Tybjærg-Hansen, A.; Hobbs, H.H.; Cohen, J.C. Adiposity amplifies the genetic risk of fatty liver disease conferred by multiple loci. Nat. Genet. 2017, 49, 842–847. [Google Scholar] [CrossRef]
- Basuray, S.; Smagris, E.; Cohen, J.C.; Hobbs, H.H. The PNPLA3 variant associated with fatty liver disease (I148M) accumulates on lipid droplets by evading ubiquitylation. Hepatology 2017, 66, 1111–1124. [Google Scholar] [CrossRef] [Green Version]
- Horton, J.D.; Bashmakov, Y.; Shimomura, I.; Shimano, H. Regulation of sterol regulatory element binding proteins in livers of fasted and refed mice. Proc. Natl. Acad. Sci. USA 1998, 95, 5987–5992. [Google Scholar] [CrossRef] [Green Version]
- Bruschi, F.V.; Tardelli, M.; Claudel, T.; Trauner, M. PNPLA3 expression and its impact on the liver: Current perspectives. Hepatic Med. Évid. Res. 2017, 9, 55–66. [Google Scholar] [CrossRef]
- Huang, Y.; He, S.; Li, J.Z.; Seo, Y.-K.; Osborne, T.F.; Cohen, J.C.; Hobbs, H.H. A feed-forward loop amplifies nutritional regulation of PNPLA3. Proc. Natl. Acad. Sci. USA 2010, 107, 7892–7897. [Google Scholar] [CrossRef] [Green Version]
- Dubuquoy, C.; Robichon, C.; Lasnier, F.; Langlois, C.; Dugail, I.; Foufelle, F.; Girard, J.; Burnol, A.-F.; Postic, C.; Moldes, M. Distinct regulation of adiponutrin/PNPLA3 gene expression by the transcription factors ChREBP and SREBP1c in mouse and human hepatocytes. J. Hepatol. 2011, 55, 145–153. [Google Scholar] [CrossRef]
- Mitsche, M.A.; Hobbs, H.H.; Cohen, J.C. Patatin-like phospholipase domain-containing protein 3 promotes transfers of essential fatty acids from triglycerides to phospholipids in hepatic lipid droplets. J. Biol. Chem. 2018, 293, 6958–6968. [Google Scholar] [CrossRef]
- Donati, B.; Motta, B.M.; Pingitore, P.; Meroni, M.; Pietrelli, A.; Alisi, A.; Petta, S.; Xing, C.; Dongiovanni, P.; del Menico, B.; et al. The rs2294918 E434K variant modulates patatin-like phospholipase domain-containing 3 expression and liver damage. Hepatology 2016, 63, 787–798. [Google Scholar] [CrossRef]
- Basuray, S.; Wang, Y.; Smagris, E.; Cohen, J.C.; Hobbs, H.H. Accumulation of PNPLA3 on lipid droplets is the basis of associated hepatic steatosis. Proc. Natl. Acad. Sci. USA 2019, 116, 9521–9526. [Google Scholar] [CrossRef] [Green Version]
- Mondul, A.; Mancina, R.M.; Merlo, A.; Dongiovanni, P.; Rametta, R.; Montalcini, T.; Valenti, L.; Albanes, D.; Romeo, S. PNPLA3 I148M variant influences circulating retinol in NPLA3 I148M variant influences circulating retinol in adults with nonalcoholic fatty liver disease or Obesity. J. Nutr. 2015, 145, 1687–1691. [Google Scholar] [CrossRef]
- Abul-Husn, N.S.; Cheng, X.; Li, A.H.; Xin, Y.; Schurmann, C.; Stevis, P.; Liu, Y.; Kozlitina, J.; Stender, S.; Wood, G.C.; et al. A protein-truncating HSD17B13 variant and protection from chronic liver disease. N. Engl. J. Med. 2018, 378, 1096–1106. [Google Scholar] [CrossRef]
- Ma, Y.; Belyaeva, O.; Brown, P.M.; Fujita, K.; Valles, K.; Karki, S.; S de Boer, Y.; Koh, C.J.; Chen, Y.; Du, X.; et al. HSD17B13 is a hepatic retinol dehydrogenase associated with histological features of non-alcoholic fatty liver disease. Hepatology 2019, 69, 1504–1519. [Google Scholar] [CrossRef]
- Pirazzi, C.; Valenti, L.; Motta, B.M.; Pingitore, P.; Hedfalk, K.; Mancina, R.M.; Burza, M.A.; Indiveri, C.; Ferro, Y.; Montalcini, T.; et al. PNPLA3 has retinyl-palmitate lipase activity in human hepatic stellate cells. Hum. Mol. Genet. 2014, 23, 4077–4085. [Google Scholar] [CrossRef] [Green Version]
- Masarone, M.; Rosato, V.; Dallio, M.; Gravina, A.G.; Aglitti, A.; Loguercio, C.; Federico, A.; Persico, M. Role of oxidative stress in pathophysiology of nonalcoholic fatty liver disease. Oxid. Med. Cell. Longev. 2018, 2018, 9547613. [Google Scholar] [CrossRef]
- Stickel, F.; Hampe, J. Dissecting the evolutionary genetics of iron overload in non-alcoholic fatty liver disease. J. Hepatol. 2010, 53, 793–794. [Google Scholar] [CrossRef]
- Valenti, L.; Fracanzani, A.L.; Bugianesi, E.; Dongiovanni, P.; Galmozzi, E.; Vanni, E.; Canavesi, E.; Lattuada, E.; Roviaro, G.; Marchesini, G.; et al. HFE genotype, parenchymal iron accumulation, and liver fibrosis in patients with nonalcoholic fatty liver disease. Gastroenterology 2010, 138, 905–912. [Google Scholar] [CrossRef]
- Al-Serri, A.; Anstee, Q.M.; Valenti, L.; Nobili, V.; Leathart, J.B.; Dongiovanni, P.; Patch, J.; Fracanzani, A.L.; Fargion, S.; Day, C.P.; et al. The SOD2 C47T polymorphism influences NAFLD fibrosis severity: Evidence from case-control and intra-familial allele association studies. J. Hepatol. 2012, 56, 448–454. [Google Scholar] [CrossRef]
- Namikawa, C.; Shu-Ping, Z.; Vyselaar, J.R.; Nozaki, Y.; Nemoto, Y.; Ono, M.; Akisawa, N.; Saibara, T.; Hiroi, M.; Enzan, H.; et al. Polymorphisms of microsomal triglyceride transfer protein gene and manganese superoxide dismutase gene in non-alcoholic steatohepatitis. J. Hepatol. 2004, 40, 781–786. [Google Scholar] [CrossRef]
- Matsubayashi, S.-S.; Matsumine, H.; Kobayashi, T.; Hattori, Y.-N.; Shimizu, Y.; Mizuno, Y. Structural dimorphism in the mitochondrial targeting sequence in the human manganese superoxide dismutase gene: A predictive evidence for conformational change to influence mitochondrial transport and a study of allelic association in Parkinson’s disease. Biochem. Biophys. Res. Commun. 1996, 229, 361. [Google Scholar] [CrossRef]
- Sutton, A.; Imbert, A.; Igoudjil, A.; Descatoire, V.; Cazanave, S.; Pessayre, D.; Degoul, F. The manganese superoxide dismutase Ala16Val dimorphism modulates both mitochondrial import and mRNA stability. Pharmacogenet. Genomics 2005, 15, 311–319. [Google Scholar] [CrossRef]
- Boland, M.L.; Oldham, S.; Boland, B.B.; Will, S.; Lapointe, J.-M.; Guionaud, S.; Rhodes, C.J.; Trevaskis, J.L. Nonalcoholic steatohepatitis severity is defined by a failure in compensatory antioxidant capacity in the setting of mitochondrial dysfunction. World J. Gastroenterol. 2018, 24, 1748–1765. [Google Scholar] [CrossRef]
- Otabe, S.; Clement, K.; Dina, C.; Pelloux, V.; Guy-Grand, B.; Froguel, P.; Vasseur, F. A genetic variation in the 5’ flanking region of the UCP3 gene is associated with body mass index in humans in interaction with physical activity. Diabetologia 2000, 43, 245–249. [Google Scholar] [CrossRef]
- Schrauwen, P.; Xia, J.; Walder, K.; Snitker, S.; Ravussin, E. A novel polymorphism in the proximal UCP3 promoter region: Effect on skeletal muscle UCP3 mRNA expression and obesity in male non-diabetic Pima Indians. Int. J. Obes. 1999, 23, 1242–1245. [Google Scholar] [CrossRef]
- Meirhaeghe, A.; Amouyel, P.; Helbecque, N.; Cottel, D.; Otabe, S.; Froguel, P.; Vasseur, F. An uncoupling protein 3 gene polymorphism associated with a lower risk of developing Type II diabetes and with atherogenic lipid profile in a French cohort. Diabetologia 2000, 43, 1424–1428. [Google Scholar] [CrossRef] [Green Version]
- Aller, R.; De Luis, D.A.; Izaola, O.; Sagrado, M.G.; Conde, R.; Alvarez, T.; Pacheco, D.; Velasco, M.C. Role of -55CT polymorphism of UCP3 gene on non alcoholic fatty liver disease and insulin resistance in patients with obesity. Nutr. Hosp. 2010, 25, 572–576. [Google Scholar]
- Fares, R.; Petta, S.; Lombardi, R.; Grimaudo, S.; Dongiovanni, P.; Pipitone, R.; Rametta, R.; Fracanzani, A.L.; Mozzi, E.; Craxì, A.; et al. The UCP2 -866 G>A promoter region polymorphism is associated with nonalcoholic steatohepatitis. Liver Int. 2015, 35, 1574–1580. [Google Scholar] [CrossRef]
- Emdin, C.A.; Haas, M.; Khera, A.V.; Aragam, K.; Chaffin, M.; Jiang, L.; Wei, W.; Feng, Q.; Karjalainen, J.; Havulinna, A.; et al. A missense variant in mitochondrial amidoxime reducing component 1 gene and protection against liver disease. BioRxiv 2019. [Google Scholar] [CrossRef]
- Sparacino-Watkins, C.E.; Tejero, J.; Sun, B.; Gauthier, M.C.; Thomas, J.; Ragireddy, V.; Merchant, B.A.; Wang, J.; Azarov, I.; Basu, P.; et al. Nitrite reductase and nitric-oxide synthase activity of the mitochondrial molybdopterin enzymes mARC1 and mARC2. J. Biol. Chem. 2014, 289, 10345–10358. [Google Scholar] [CrossRef]
- Schneider, J.; Girreser, U.; Havemeyer, A.; Bittner, F.; Clement, B. Detoxification of trimethylamine N-oxide by the mitochondrial amidoxime reducing component mARC. Chem. Res. Toxicol. 2018, 31, 447–453. [Google Scholar] [CrossRef]
- Petta, S.; Grimaudo, S.; Cammà, C.; Cabibi, D.; Di Marco, V.; Licata, G.; Pipitone, R.; Craxì, A. 1315 IL28B and PNPLA3 polymorphisms affect histological liver damage in patients with non-alcoholic fatty liver disease. J. Hepatol. 2012, 56, S518. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Meroni, M.; Baselli, G.A.; Bassani, G.A.; Rametta, R.; Pietrelli, A.; Maggioni, M.; Facciotti, F.; Trunzo, V.; Badiali, S.; et al. Insulin resistance promotes lysyl oxidase like 2 induction and fibrosis accumulation in non-alcoholic fatty liver disease. Clin. Sci. 2017, 131, 1301–1315. [Google Scholar] [CrossRef]
- Eriksson, J.W.; Lundkvist, P.; Jansson, P.-A.; Johansson, L.; Kvarnström, M.; Moris, L.; Miliotis, T.; Forsberg, G.-B.; Risérus, U.; Lind, L.; et al. Effects of dapagliflozin and n-3 carboxylic acids on non-alcoholic fatty liver disease in people with type 2 diabetes: A double-blind randomised placebo-controlled study. Diabetologia 2018, 61, 1923–1934. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Valenti, L.; Rametta, R.; Daly, A.K.; Nobili, V.; Mozzi, E.; Leathart, J.B.S.; Pietrobattista, A.; Burt, A.D.; Maggioni, M.; et al. Genetic variants regulating insulin receptor signalling are associated with the severity of liver damage in patients with non-alcoholic fatty liver disease. Gut 2010, 59, 267–273. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.S.; Jackson, A.U.; Li, Y.K.; Stringham, H.M.; Kuusisto, J.; Kangas, A.J.; Soininen, P.; Ala-Korpela, M.; Burant, C.F.; Salomaa, V.; et al. Novel association of TM6SF2 rs58542926 genotype with increased serum tyrosine levels and decreased apoB-100 particles in Finns. J. Lipid Res. 2017, 58, 1471–1481. [Google Scholar] [CrossRef] [Green Version]
- Kozlitina, J.; Smagris, E.; Stender, S.; Nordestgaard, B.G.; Zhou, H.H.; Tybjærg-Hansen, A.; Vogt, T.F.; Hobbs, H.H.; Cohen, J.C. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2014, 46, 352–356. [Google Scholar] [CrossRef] [Green Version]
- Dongiovanni, P.; Petta, S.; Maglio, C.; Fracanzani, A.L.; Pipitone, R.; Mozzi, E.; Motta, B.M.; Kaminska, D.; Rametta, R.; Grimaudo, S.; et al. Transmembrane 6 superfamily member 2 gene variant disentangles nonalcoholic steatohepatitis from cardiovascular disease. Hepatology 2015, 61, 506–514. [Google Scholar] [CrossRef]
- Liu, Y.-L.; Reeves, H.L.; Burt, A.D.; Tiniakos, D.; McPherson, S.; Leathart, J.B.S.; Allison, M.E.D.; Alexander, G.J.; Piguet, A.-C.; Anty, R.; et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat. Commun. 2014, 5, 4309. [Google Scholar] [CrossRef] [Green Version]
- Donati, B.; Dongiovanni, P.; Romeo, S.; Meroni, M.; McCain, M.; Miele, L.; Petta, S.; Maier, S.; Rosso, C.; De Luca, L.; et al. MBOAT7 rs641738 variant and hepatocellular carcinoma in non-cirrhotic individuals. Sci. Rep. 2017, 7, 4492. [Google Scholar] [CrossRef]
- Williams, C.D.; Stengel, J.; Asike, M.I.; Torres, D.M.; Shaw, J.; Contreras, M.; Landt, C.L.; Harrison, S.A. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: A prospective study. Gastroenterology 2011, 140, 124–131. [Google Scholar] [CrossRef]
- Wong, V.W.-S.; Chan, H.L.-Y.; Hui, A.Y.; Chan, K.-F.; Liew, C.-T.; Chan, F.K.; Sung, J.J.-Y. Clinical and histological features of non-alcoholic fatty liver disease in Hong Kong Chinese. Aliment. Pharmacol. Ther. 2004, 20, 45–49. [Google Scholar] [CrossRef]
- Rozario, R.; Ramakrishna, B. Histopathological study of chronic hepatitis B and C: A comparison of two scoring systems. J. Hepatol. 2003, 38, 223–229. [Google Scholar] [CrossRef]
- Altlparmak, E.; Köklü, S.; Yallnklllc, M.; Yüksel, O.; Cicek, B.; Kayacetin, E.; Sahin, T. Viral and host causes of fatty liver in chronic hepatitis B. World J. Gastroenterol. 2005, 11, 3056–3059. [Google Scholar] [CrossRef]
- Gordon, A.; McLean, C.A.; Pedersen, J.S.; Bailey, M.J.; Roberts, S.K. Hepatic steatosis in chronic hepatitis B and C: Predictors, distribution and effect on fibrosis. J. Hepatol. 2005, 43, 38–44. [Google Scholar] [CrossRef]
- Thomopoulos, K.C.; Arvaniti, V.; Tsamantas, A.C.; Dimitropoulou, D.; Gogos, C.A.; Siagris, D.; Theocharis, G.J.; Labropoulou-Karatza, C. Prevalence of liver steatosis in patients with chronic hepatitis B: A study of associated factors and of relationship with fibrosis. Eur. J. Gastroenterol. Hepatol. 2006, 18, 233–237. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Meroni, M.; Mancina, R.M.; Baselli, G.; Rametta, R.; Pelusi, S.; Mannisto, V.; Fracanzani, A.L.; Badiali, S.; Miele, L.; et al. Protein phosphatase 1 regulatory subunit 3B gene variation protects against hepatic fat accumulation and fibrosis in individuals at high risk of nonalcoholic fatty liver disease. Hepatol. Commun. 2018, 2, 666–675. [Google Scholar] [CrossRef] [Green Version]
- Stender, S.; Smagris, E.; Lauridsen, B.K.; Kofoed, K.F.; Nordestgaard, B.G.; Tybjaerg-Hansen, A.; Pennacchio, L.A.; Dickel, D.E.; Cohen, J.C.; Hobbs, H.H. Relationship between genetic variation at PPP1R3B and levels of liver glycogen and triglyceride. Hepatology 2018, 67, 2182–2195. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Stender, S.; Pietrelli, A.; Mancina, R.M.; Cespiati, A.; Petta, S.; Pelusi, S.; Pingitore, P.; Badiali, S.; Maggioni, M.; et al. Causal relationship of hepatic fat with liver damage and insulin resistance in nonalcoholic fatty liver. J. Intern. Med. 2018, 283, 356–370. [Google Scholar] [CrossRef]
- Cefalu’, A.B.; Pirruccello, J.P.; Noto, D.; Gabriel, S.; Valenti, V.; Gupta, N.; Spina, R.; Tarugi, P.; Kathiresan, S.; Averna, M.R. A novel APOB mutation identified by exome sequencing cosegregates with steatosis, liver cancer and hypocholesterolemia. Arter. Thromb. Vasc. Biol. 2013, 33, 2021–2025. [Google Scholar] [CrossRef]
- Zhou, Y.; Mägi, R.; Milani, L.; Lauschke, V.M. Global genetic diversity of human apolipoproteins and effects on cardiovascular disease risk. J. Lipid Res. 2018, 59, 1987–2000. [Google Scholar] [CrossRef] [Green Version]
- Pelusi, S.; Baselli, G.; Pietrelli, A.; Dongiovanni, P.; Donati, B.; McCain, M.V.; Meroni, M.; Fracanzani, A.L.; Romagnoli, R.; Petta, S.; et al. Rare pathogenic variants predispose to hepatocellular carcinoma in nonalcoholic fatty liver disease. Sci. Rep. 2019, 9, 3682. [Google Scholar] [CrossRef]
- Nass, K.J.; van den Berg, E.H.; Faber, K.N.; Schreuder, T.C.M.A.; Blokzijl, H.; Dullaart, R.P.F. High prevalence of apolipoprotein B dyslipoproteinemias in non-alcoholic fatty liver disease: The lifelines cohort study. Metabolism 2017, 72, 37–46. [Google Scholar] [CrossRef]
- Donati, B.; Pietrelli, A.; Pingitore, P.; Dongiovanni, P.; Caddeo, A.; Walker, L.; Baselli, G.; Pelusi, S.; Rosso, C.; Vanni, E.; et al. Telomerase reverse transcriptase germline mutations and hepatocellular carcinoma in patients with nonalcoholic fatty liver disease. Cancer Med. 2017, 6, 1930–1940. [Google Scholar] [CrossRef]
- Calado, R.T.; Regal, J.A.; Kleiner, D.E.; Schrump, D.S.; Peterson, N.R.; Pons, V.; Chanock, S.J.; Lansdorp, P.M.; Young, N.S. A spectrum of severe familial liver disorders associate with telomerase mutations. PLoS ONE 2009, 4, e7926. [Google Scholar] [CrossRef]
- Pericleous, M.; Kelly, C.; Wang, T.; Livingstone, C.; Ala, A. Wolman’s disease and cholesteryl ester storage disorder: The phenotypic spectrum of lysosomal acid lipase deficiency. Lancet Gastroenterol. Hepatol. 2017, 2, 670–679. [Google Scholar] [CrossRef]
- Ma, J.; Hennein, R.; Liu, C.; Long, M.T.; Hoffmann, U.; Jacques, P.F.; Lichtenstein, A.H.; Hu, F.B.; Levy, D. Improved diet quality associates with reduction in liver fat—particularly in individuals with high genetic risk scores for nonalcoholic fatty liver disease. Gastroenterology 2018, 155, 107–117. [Google Scholar] [CrossRef]
- Valenti, L.; Romeo, S. Destined to develop NAFLD? The predictors of fatty liver from birth to adulthood. J. Hepatol. 2016, 65, 668–670. [Google Scholar] [CrossRef] [Green Version]
- Musunuru, K.; Kathiresan, S. Genetics of common, complex coronary artery disease. Cell 2019, 177, 132–145. [Google Scholar] [CrossRef]
- Guzman, C.B.; Duvvuru, S.; Akkari, A.; Bhatnagar, P.; Battioui, C.; Foster, W.; Zhang, X.M.; Shankar, S.S.; Deeg, M.A.; Chalasani, N.; et al. Coding variants in PNPLA3 and TM6SF2 are risk factors for hepatic steatosis and elevated serum alanine aminotransferases caused by a glucagon receptor antagonist. Hepatol. Commun. 2018, 2, 561–570. [Google Scholar] [CrossRef]
- Pillai, S.; Duvvuru, S.; Bhatnagar, P.; Foster, W.; Farmen, M.; Shankar, S.; Harris, C.; Bastyr, E., 3rd; Hoogwerf, B.; Haupt, A. The PNPLA3 I148M variant is associated with transaminase elevations in type 2 diabetes patients treated with basal insulin peglispro. Pharmacogenomics J. 2018, 18, 487–493. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Petta, S.; Männistö, V.; Mancina, R.M.; Pipitone, R.M.; Kärjä, V.; Maggioni, M.; Käkelä, P.; Wiklund, O.; Mozzi, E.; et al. Statin use and non-alcoholic steatohepatitis in at risk individuals. J. Hepatol. 2015, 63, 705–712. [Google Scholar] [CrossRef]
- Lindén, D.; Ahnmark, A.; Pingitore, P.; Ciociola, E.; Ahlstedt, I.; Andréasson, A.-C.; Sasidharan, K.; Madeyski-Bengtson, K.; Zurek, M.; Mancina, R.M.; et al. Pnpla3 silencing with antisense oligonucleotides ameliorates nonalcoholic steatohepatitis and fibrosis in Pnpla3 I148M knock-in mice. Mol. Metab. 2019, 22, 49–61. [Google Scholar] [CrossRef]


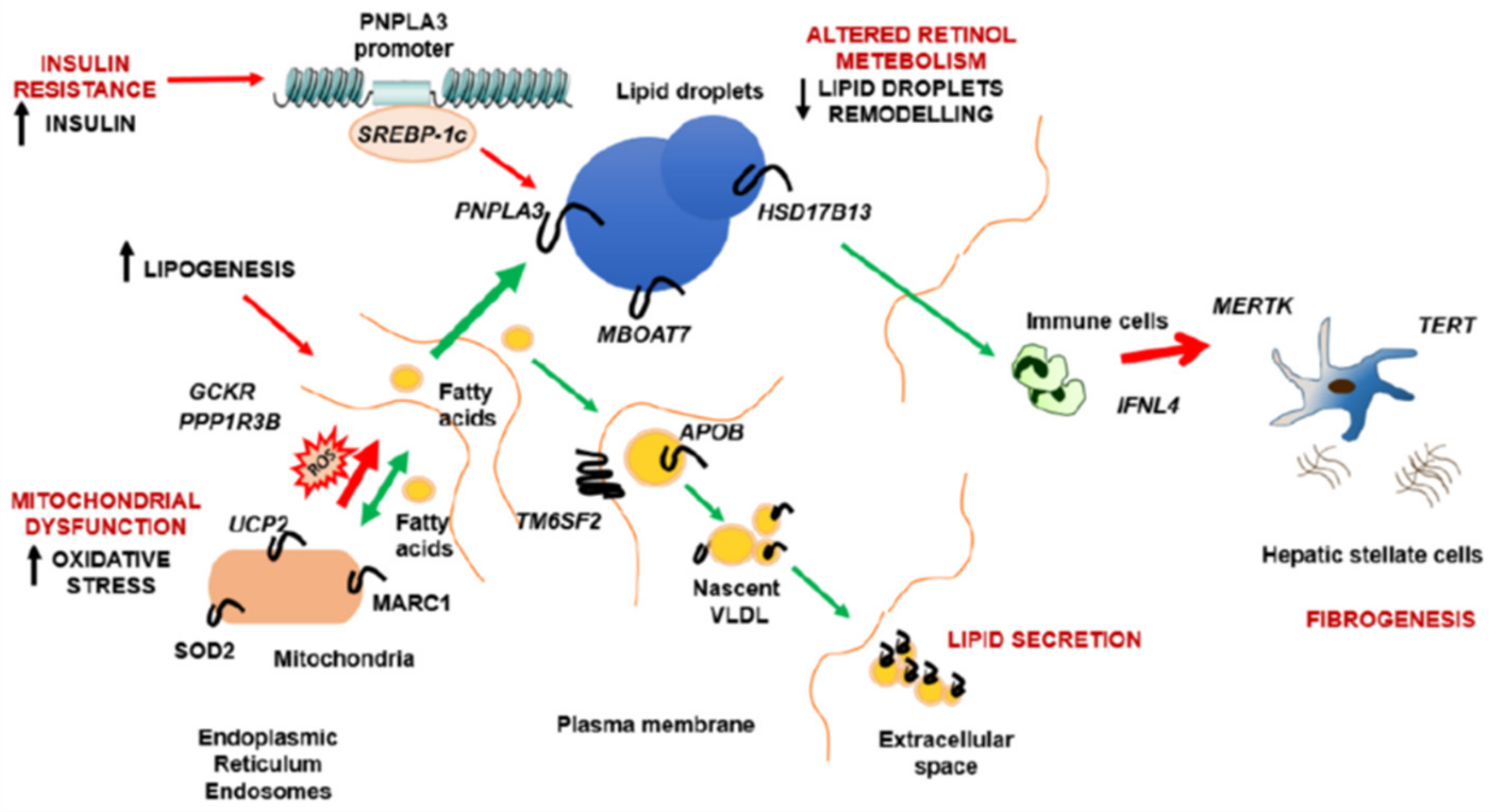{kind=link}
| Gene | Variant | Effect Size | Direction | Steatosis | NASH | Fibrosis | HCC | Mortality in NAFLD | Minor Allele Frequency in Italy |
|---|---|---|---|---|---|---|---|---|---|
| PNPLA3 | I148M | +++ | ←↑ | + | + | + | + | + | 0.27 |
| TM6SF2 | E167K | +++ | ←↑ | + | + | + | + | 0.06 | |
| GCKR | P446L | + | ←↑ | + | 0.30 | ||||
| MBOAT7 | rs641738 | + | ←↑ | + | + | + | 0.44 | ||
| HSD17B13 | rs72613567 | ++ | ↓ | + | + | + | 0.22 | ||
| IL28B (IFNL3/4) | rs12979860 | + | ↓ | + | 0.36 | ||||
| MERTK | rs4374383 | + | ↓ | + | 0.36 | ||||
| APOB | several | +++ | ←↑ | + | + | + | <0.01 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taliento, A.E.; Dallio, M.; Federico, A.; Prati, D.; Valenti, L. Novel Insights into the Genetic Landscape of Nonalcoholic Fatty Liver Disease. Int. J. Environ. Res. Public Health 2019, 16, 2755. https://doi.org/10.3390/ijerph16152755
Taliento AE, Dallio M, Federico A, Prati D, Valenti L. Novel Insights into the Genetic Landscape of Nonalcoholic Fatty Liver Disease. International Journal of Environmental Research and Public Health. 2019; 16(15):2755. https://doi.org/10.3390/ijerph16152755
Chicago/Turabian StyleTaliento, Alice Emma, Marcello Dallio, Alessandro Federico, Daniele Prati, and Luca Valenti. 2019. "Novel Insights into the Genetic Landscape of Nonalcoholic Fatty Liver Disease" International Journal of Environmental Research and Public Health 16, no. 15: 2755. https://doi.org/10.3390/ijerph16152755