Different Clinical Presentations and Management in Complete Androgen Insensitivity Syndrome (CAIS)



Abstract
:1. Introduction
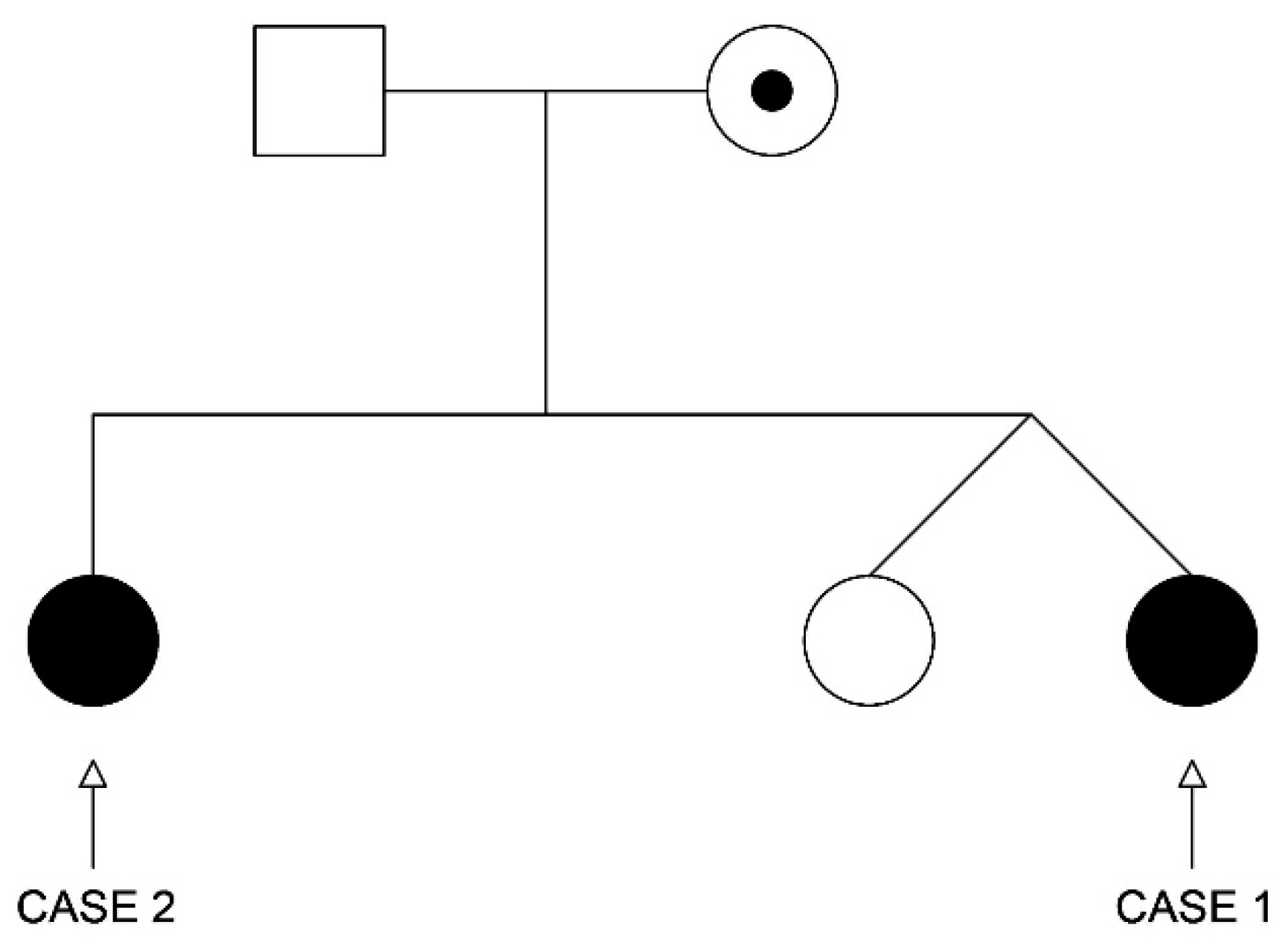
2. Clinical Presentation
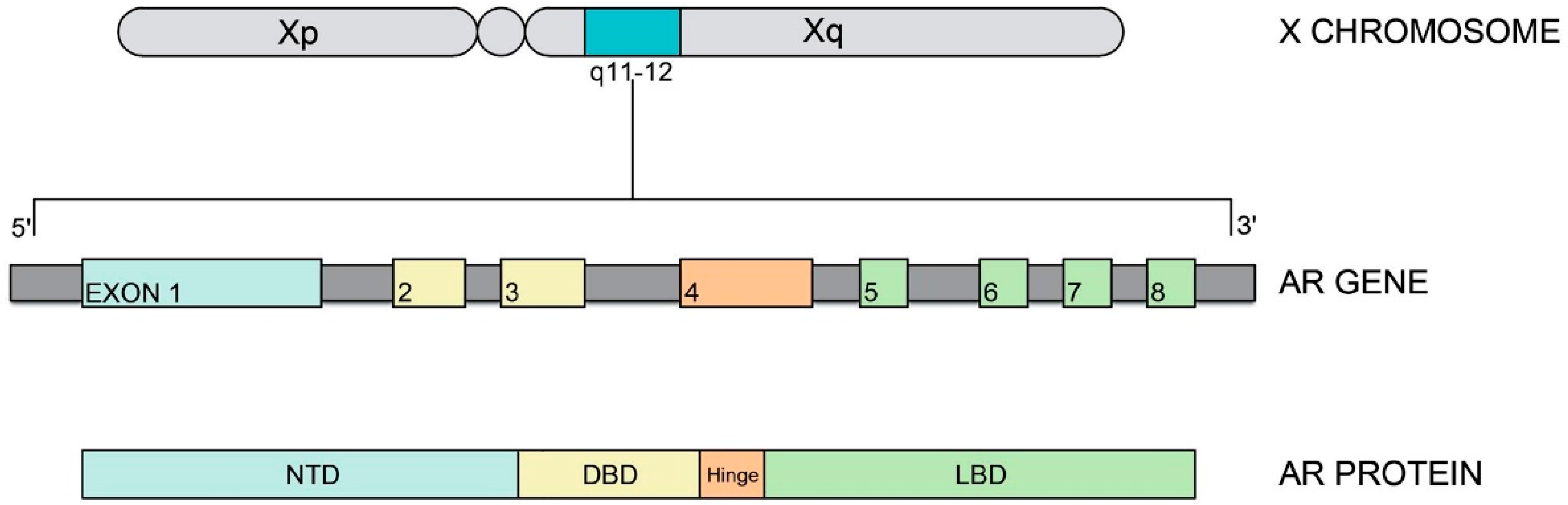
3. Androgen Receptor (AR) Gene and Protein
4. Time of Gonadectomy and Risk of Malignancy
5. Follow-Up of Retained Testes
6. Hormonal Replacement Therapy (HRT)
7. Bone Mineral Density and Body Composition
8. Differential Diagnosis in Clinical Practice
9. Challenges in Diagnosis and Management
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lubahn, D.B.; Joseph, D.R.; Sullivan, P.M.; Willard, H.F.; French, F.S.; Wilson, E.M. Cloning of human androgen receptor complementary DNA and localization to the X. chromosome. Science 1988, 240, 327–330. [Google Scholar] [CrossRef]
- Radpour, R.; Falah, M.; Aslani, A.; Zhong, X.Y.; Saleki, A. Identification of a Critical Novel Mutation in the Exon 1 of Androgen Receptor Gene in 2 Brothers with Complete Androgen Insensitivity Syndrome. J. Androl. 2009, 30, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Hughes, I.A.; Davies, J.D.; Bunch, T.I.; Pasterski, V.; Mastroyannopoulou, K.; Macdougall, J. Androgen insensitivity syndrome. Lancet 2012, 380, 1419–1428. [Google Scholar] [CrossRef] [Green Version]
- Papadimitriou, D.T.; Linglart, A.; Morel, Y.; Chaussain, J.-L. Puberty in Subjects with Complete Androgen Insensitivity Syndrome. Horm. Res. Paediatr. 2006, 65, 126–131. [Google Scholar] [CrossRef]
- Bruce Gottlieb, M.A.T. Androgen Insensitivity Syndrome; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; NCBI Books; University of Washington: Seattle, WA, USA, 2017. [Google Scholar]
- Morris, J.M. The syndrome of testicular feminization in male pseudohermaphrodites. Am. J. Obstet. Gynecol. 1953, 65, 1192–1211. [Google Scholar] [CrossRef]
- Oakes, M.B.; Eyvazzadeh, A.D.; Quint, E.; Smith, Y.R. Complete Androgen Insensitivity Syndrome-A Review. J. Pediatr. Adolesc. Gynecol. 2008, 21, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Hughes, I.A.; Deeb, A. Androgen resistance. Best Pract. Res. Clin. Endocrinol. Metab. 2006, 20, 577–598. [Google Scholar] [CrossRef] [PubMed]
- Gulía, C.; Baldassarra, S.; Zangari, A.; Briganti, V.; Gigli, S.; Gaffi, M.; Signore, F.; Vallone, C.; Nucciotti, R.; Costantini, F.M.; et al. Androgen insensitivity syndrome. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 3873–3887. [Google Scholar] [CrossRef] [PubMed]
- Petroli, R.J.; Hiort, O.; Struve, D.; Gesing, J.K.; Soardi, F.C.; Spínola-Castro, A.M.; Melo, K.; Prado Arnhold, I.J.; Maciel-Guerra, A.T.; Guerra-Junior, G.; et al. Functional impact of novel androgen receptor mutations on the clinical manifestation of androgen insensitivity syndrome. Sex. Dev. 2018, 11, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Boehmer, A.L.M.; Brüggenwirth, H.; van Assendelft, C.; Otten, B.J.; Verleun-Mooijman, M.C.; Niermeijer, M.F.; Brunner, H.G.; Rouwé, C.W.; Waelkens, J.J.; Oostdijk, W.; et al. Genotype Versus Phenotype in Families with Androgen Insensitivity Syndrome. J. Clin. Endocrinol. Metab. 2001, 86, 4151–4160. [Google Scholar] [CrossRef]
- Gottlieb, B.; Beitel, L.K.; Nadarajah, A.; Paliouras, M.; Trifiro, M. The androgen receptor gene mutations database: 2012 update. Hum. Mutat. 2012, 33, 887–894. [Google Scholar] [CrossRef] [Green Version]
- Mendonca, B.B.; Domenice, S.; Arnhold, I.J.P.; Costa, E.M.F. 46, XY disorders of sex development (DSD). Clin. Endocrinol. 2009, 70, 173–187. [Google Scholar] [CrossRef]
- Lanciotti, L.; Cofini, M.; Leonardi, A.; Penta, L.; Esposito, S. Up-To-Date Review About Minipuberty and Overview on Hypothalamic-Pituitary-Gonadal Axis Activation in Fetal and Neonatal Life. Front. Endocrinol. 2018, 9, 410. [Google Scholar] [CrossRef]
- Batista, R.L.; Costa, E.M.F.; Rodrigues, A.D.; Gomes, N.L.; Faria, J.A., Jr.; Nishi, M.Y.; Arnhold, I.J.P.; Domenice, S.; Mendonca, B.B. Androgen insensitivity syndrome: A review. Arch. Endocrinol. Metab. 2018, 62, 227–235. [Google Scholar] [CrossRef]
- Dey, R.; Biswas, S.C.; Chattopadhvav, N.; Gupta, D.; RoyBiswas, R.; Mukhopadhyay, A. The XY Female (Androgen Insensitivity Syndrome)—Runs in the Family. J. Obstet. Gynecol. India 2012, 62, 332–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottlieb, B.; Beitel, L.K.T.M. Androgen Insensitivity Syndrome. 2011; pp. 432–442. Available online: http://www.ncbi.nlm.nih.gov/books/NBK1429/ (accessed on 13 March 2019).
- Zachmann, M.; Prader, A.; Sobel, E.H.; Crigler, J.F., Jr.; Ritzén, E.M.; Atarés, M.; Ferrandez, A. Pubertal growth in patients with androgen insensitivity: Indirect evidence for the importance of estrogens in pubertal growth of girls. J. Pediatr. 1986, 108, 694–697. [Google Scholar] [CrossRef]
- Ogata, T.; Matsuo, N. Comparison of adult height between patients with XX and XY gonadal dysgenesis: Support for a Y specific growth gene(s). J. Med. Genet. 1992, 29, 539–541. [Google Scholar] [CrossRef] [PubMed]
- Ritzén, E.M. Pubertal growth in genetic disorders of sex hormone action and secretion. Acta Paediatr. Suppl. 1992, 383, 22–25. [Google Scholar] [PubMed]
- Danilovic, D.L.S.; Correa, P.H.S.; Costa, E.M.F.; Melo, K.F.S.; Mendonca, B.B.; Arnhold, I.J.P. Height and bone mineral density in androgen insensitivity syndrome with mutations in the androgen receptor gene. Osteoporos. Int. 2007, 18, 369–374. [Google Scholar] [CrossRef]
- Melo, K.F.S.; Mendonca, B.B.; Billerbeck, A.E.C.; Costa, E.M.; Inácio, M.; Silva, F.A.; Leal, A.M.; Latronico, A.C.; Arnhold, I.J. Clinical, hormonal, behavioral, and genetic characteristics of androgen insensitivity syndrome in a Brazilian cohort: Five novel mutations in the androgen receptor gene. J. Clin. Endocrinol. Metab. 2003, 88, 3241–3250. [Google Scholar] [CrossRef]
- Doehnert, U.; Bertelloni, S.; Werner, R.; Dati, E.; Hiort, O. Characteristic features of reproductive hormone profiles in late adolescent and adult females with complete androgen insensitivity syndrome. Sex. Dev. 2015, 9, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Lahlou, N.; Bouvattier, C.; Linglart, A.; Rodrigue, D.; Teinturier, C. The role of gonadal peptides in clinical investigation. Ann. Biol. Clin. 2009, 67, 283–292. [Google Scholar] [CrossRef]
- Mainwaring, W.I. The mechanism of action of androgens. Monogr. Endocrinol. 1977, 10, 1–178. [Google Scholar] [PubMed]
- Behre, H.M.; Nieschlag, S. Testosterone: Action, Deficiency and Substitution; Springer: New York, NY, USA, 1998. [Google Scholar]
- Yamada, G.; Suzuki, K.; Haraguchi, R.; Miyagawa, S.; Satoh, Y.; Kamimura, M.; Nakagata, N.; Kataoka, H.; Kuroiwa, A.; Chen, Y. Molecular genetic cascades for external genitalia formation: An emerging organogenesis program. Dev. Dyn. 2006, 235, 1738–1752. [Google Scholar] [CrossRef] [Green Version]
- Verhoeven, G.; Swinnen, J.V. Indirect mechanisms and cascades of androgen action. Mol. Cell. Endocrinol. 1999, 151, 205–212. [Google Scholar] [CrossRef]
- Chamberlain, N.L.; Whitacre, D.C.; Miesfeld, R.L. Delineation of two distinct type 1 activation functions in the androgen receptor amino-terminal domain. J. Biol. Chem. 1996, 271, 26772–26778. [Google Scholar] [CrossRef]
- Sack, J.S.; Kish, K.F.; Wang, C.; Attar, R.M.; Kiefer, S.E.; An, Y.; Wu, G.Y.; Scheffler, J.E.; Salvati, M.E.; Krystek, S.R., Jr.; et al. Crystallographic structures of the ligand-binding domains of the androgen receptor and its T877A mutant complexed with the natural agonist dihydrotestosterone. Proc. Natl. Acad. Sci. USA 2001, 98, 4904–4909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matias, P.M.; Donner, P.; Coelho, R.; Thomaz, M.; Peixoto, C.; Macedo, S.; Otto, N.; Joschko, S.; Scholz, P.; Wegg, A.; et al. Structural Evidence for Ligand Specificity in the Binding Domain of the Human Androgen Receptor. J. Biol. Chem. 2000, 275, 26164–26171. [Google Scholar] [CrossRef] [Green Version]
- Tsai, M.; O’Malley, B.W. Molecular Mechanisms of Action of Steroid/Thyroid Receptor Superfamily Members. Annu. Rev. Biochem. 1994, 63, 451–486. [Google Scholar] [CrossRef]
- Mangelsdorf, D.J.; Thummel, C.; Beato, M.; Herrlich, P.; Schütz, G.; Umesono, K.; Blumberg, B.; Kastner, P.; Mark, M.; Chambon, P.; et al. The nuclear receptor superfamily: The second decade. Cell 1995, 83, 835–839. [Google Scholar] [CrossRef] [Green Version]
- Evans, R.M. The steroid and thyroid hormone receptor superfamily. Science 1988, 240, 889–895. [Google Scholar] [CrossRef]
- Jenster, G.; van der Korput, H.A.G.M.; van Vroonhoven, C.; van der Kwast, T.H.; Trapman, J.; Brinkmann, A.O. Domains of the Human Androgen Receptor Involved in Steroid Binding, Transcriptional Activation, and Subcellular Localization. Mol. Endocrinol. 1991, 5, 1396–1404. [Google Scholar] [CrossRef] [Green Version]
- Pereira de Jésus-Tran, K.; Côté, P.-L.; Cantin, L.; Blanchet, J.; Labrie, F.; Breton, R. Comparison of crystal structures of human androgen receptor ligand-binding domain complexed with various agonists reveals molecular determinants responsible for binding affinity. Protein Sci. 2006, 15, 987–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myung, J.-K.; Banuelos, C.A.; Fernandez, J.G.; Mawji, N.R.; Wang, J.; Tien, A.H.; Yang, Y.C.; Tavakoli, I.; Haile, S.; Watt, K.; et al. An androgen receptor N-terminal domain antagonist for treating prostate cancer. J. Clin. Investig. 2013, 123, 2948–2960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazemi-Esfarjani, P.; Trifiro, M.A.; Pinsky, L. Evidence for a repressive function of the long polyglutamine tract in the human androgen receptor: Possible pathogenetic relevance for the (CAG)n-expanded neuronopathies. Hum. Mol. Genet. 1995, 4, 523–527. [Google Scholar] [CrossRef] [PubMed]
- McPhaul, M.J.; Marcelli, M.; Tilley, W.D.; Griffin, J.E.; Isidro-Gutierrez, R.F.; Wilson, J.D. Molecular basis of androgen resistance in a family with a qualitative abnormality of the androgen receptor and responsive to high-dose androgen therapy. J. Clin. Investig. 1991, 87, 1413–1421. [Google Scholar] [CrossRef]
- Choong, C.S.; Quigley, C.A.; French, F.S.; Wilson, E.M. A novel missense mutation in the amino-terminal domain of the human androgen receptor gene in a family with partial androgen insensitivity syndrome causes reduced efficiency of protein translation. J. Clin. Investig. 1996, 98, 1423–1431. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, P.W.; Lin, D.L.; Nakao, R.; Chang, C. The linkage of Kennedy’s neuron disease to ARA24, the first identified androgen receptor polyglutamine region-associated coactivator. J. Biol. Chem. 1999, 274, 20229–20234. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; Marcelli, M.; McPhaul, M.J. Transcriptional activation and transient expression of the human androgen receptor. J. Steroid Biochem. Mol. Biol. 1996, 59, 9–20. [Google Scholar] [CrossRef]
- Sasaki, M.; Kaneuchi, M.; Sakuragi, N.; Fujimoto, S.; Carroll, P.R.; Dahiya, R. The polyglycine and polyglutamine repeats in the androgen receptor gene in Japanese and Caucasian populations. Biochem. Biophys. Res. Commun. 2003, 312, 1244–1247. [Google Scholar] [CrossRef]
- Beato, M.; Chávez, S.; Truss, M. Transcriptional regulation by steroid hormones. Steroids 1996, 61, 240–251. [Google Scholar] [CrossRef]
- Ikonen, T.; Palvimo, J.J.; Jänne, O.A. Interaction between the amino- and carboxyl-terminal regions of the rat androgen receptor modulates transcriptional activity and is influenced by nuclear receptor coactivators. J. Biol. Chem. 1997, 272, 29821–29828. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Kemppainen, J.A.; Voegel, J.J.; Gronemeyer, H.; Wilson, E.M. Activation function 2 in the human androgen receptor ligand binding domain mediates interdomain communication with the NH(2)-terminal domain. J. Biol. Chem. 1999, 274, 37219–37225. [Google Scholar] [CrossRef]
- Moilanen, A.M.; Karvonen, U.; Poukka, H.; Yan, W.; Toppari, J.; Jänne, O.A.; Palvimo, J.J. A testis-specific androgen receptor coregulator that belongs to a novel family of nuclear proteins. J. Biol. Chem. 1999, 274, 3700–3704. [Google Scholar] [CrossRef] [PubMed]
- Helsen, C.; Kerkhofs, S.; Clinckemalie, L.; Spans, L.; Laurent, M.; Boonen, S.; Vanderschueren, D.; Claessens, F. Structural basis for nuclear hormone receptor DNA binding. Mol. Cell. Endocrinol. 2012, 348, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Luisi, B.F.; Xu, W.X.; Otwinowski, Z.; Freedman, L.P.; Yamamoto, K.R.; Sigler, P.B. Crystallographic analysis of the interaction of the glucocorticoid receptor with DNA. Nature 1991, 352, 497–505. [Google Scholar] [CrossRef]
- Freedmann, L.P. Anatomy of the Steroid Receptor Zinc Finger Region. Endocr. Rev. 1992, 13, 129–145. [Google Scholar] [CrossRef] [PubMed]
- Jenster, G.; Trapman, J.; Brinkmann, A.O. Nuclear import of the human androgen receptor. Biochem. J. 1993, 293 Pt 3, 761–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cutress, M.L.; Whitaker, H.C.; Mills, I.G.; Stewart, M.; Neal, D.E. Structural basis for the nuclear import of the human androgen receptor. J. Cell Sci. 2008, 121, 957–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.X.; Kemppainen, J.A.; Wilson, E.M. Identification of three proline-directed phosphorylation sites in the human androgen receptor. Mol. Endocrinol. 1995, 9, 605–615. [Google Scholar] [CrossRef]
- Chang, C.-Y.; McDonnell, D.P. Evaluation of Ligand-Dependent Changes in AR Structure Using Peptide Probes. Mol. Endocrinol. 2002, 16, 647–660. [Google Scholar] [CrossRef] [Green Version]
- Brown, T.R. Human androgen insensitivity syndrome. J. Androl. 1995, 16, 299–303. [Google Scholar] [PubMed]
- Infante, J.B.; Alvelos, M.I.; Bastos, M.; Carrilho, F.; Lemos, M.C. Complete androgen insensitivity syndrome caused by a novel splice donor site mutation and activation of a cryptic splice donor site in the androgen receptor gene. J. Steroid Biochem. Mol. Biol. 2016, 155 Pt A, 63–66. [Google Scholar] [CrossRef]
- Werner, R.; Zhan, J.; Gesing, J.; Struve, D.; Hiort, O. In-vitro Characterization of Androgen Receptor Mutations Associated with Complete Androgen Insensitivity Syndrome Reveals Distinct Functional Deficits. Sex. Dev. 2008, 2, 73–83. [Google Scholar] [CrossRef]
- Hellwinkel, O.J.; Holterhus, P.M.; Struve, D.; Marschke, C.; Homburg, N.; Hiort, O. A unique exonic splicing mutation in the human androgen receptor gene indicates a physiologic relevance of regular androgen receptor transcript variants. J. Clin. Endocrinol. Metab. 2001, 86, 2569–2575. [Google Scholar] [CrossRef]
- Gelmann, E.P. Androgen receptor mutations in prostate cancer. Cancer Treat. Res. 1996, 87, 285–302. [Google Scholar] [PubMed]
- Gast, A.; Neuschmid-Kaspar, F.; Klocker, H.; Cato, A.C. A single amino acid exchange abolishes dimerization of the androgen receptor and causes Reifenstein syndrome. Mol. Cell. Endocrinol. 1995, 111, 93–98. [Google Scholar] [CrossRef]
- Lobaccaro, J.M.; Poujol, N.; Chiche, L.; Lumbroso, S.; Brown, T.R.; Sultan, C. Molecular modeling and in vitro investigations of the human androgen receptor DNA-binding domain: Application for the study of two mutations. Mol. Cell. Endocrinol. 1996, 116, 137–147. [Google Scholar] [CrossRef]
- Brüggenwirth, H.T.; Boehmer, A.L.; Lobaccaro, J.M.; Chiche, L.; Sultan, C.; Trapman, J.; Brinkmann, A.O. Substitution of Ala564 in the first zinc cluster of the deoxyribonucleic acid (DNA)-binding domain of the androgen receptor by Asp, Asn, or Leu exerts differential effects on DNA binding. Endocrinology 1998, 139, 103–110. [Google Scholar] [CrossRef]
- Greep, R.O. Reproductive endocrinology: Concepts and perspectives, an overview. Recent Prog. Horm. Res. 1978, 34, 1–23. [Google Scholar]
- Parkin, D.M.; Whelan, S.L.; Ferlay, J.; Teppo, L.; Thomas, D.B. Cancer Incidence in Five Continents; IARC Scientific Publications: Lyon, France, 2002; Volume 8. [Google Scholar]
- Deans, R.; Creighton, S.M.; Liao, L.M.; Conway, G.S. Timing of gonadectomy in adult women with complete androgen insensitivity syndrome (CAIS): Patient preferences and clinical evidence. Clin. Endocrinol. 2012, 76, 894–898. [Google Scholar] [CrossRef] [PubMed]
- Hurt, W.G.; Bodurtha, J.N.; McCall, J.B.; Ali, M.M. Seminoma in pubertal patient with androgen insensitivity syndrome. Am. J. Obstet. Gynecol. 1989, 161, 530–531. [Google Scholar] [CrossRef]
- Moch, H.; Cubilla, A.L.; Humphrey, P.A.; Reuter, V.E.; Ulbright, T.M. The 2016 WHO Classification of Tumours of the Urinary System and Male Genital Organs—Part A: Renal, Penile, and Testicular Tumours. Eur. Urol. 2016, 70, 93–105. [Google Scholar] [CrossRef]
- Cools, M.; Wolffenbuttel, K.P.; Hersmus, R.; Mendonca, B.B.; Kaprová, J.; Drop, S.L.S.; Stoop, H.; Gillis, A.J.M.; Oosterhuis, J.W.; Costa, E.M.F.; et al. Malignant testicular germ cell tumors in postpubertal individuals with androgen insensitivity: Prevalence, pathology and relevance of single nucleotide polymorphism-based susceptibility profiling. Hum. Reprod. 2017, 32, 2561–2573. [Google Scholar] [CrossRef] [PubMed]
- Kravarusic, D.; Seguier-Lipszyc, E.; Feigin, E.; Nimri, R.; Nagelberg, N.; Freud, E. Androgen insensitivity syndrome: Risk of malignancy and timing of surgery in a paediatric and adolescent population. Afr. J. Paediatr. Surg. 2011, 8, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Gómez García, I.; Romero Molina, M.; López-García Moreno, A.; Buendía González, E.; Rubio Hidalgo, E.; Bolufer, E.; Sampietro Crespo, A.; Gómez Rodríguez, A. Sertoli cell tumor, a rare testicular tumor, our experience and review of the literature. Arch. Esp. Urol. 2010, 63, 392–395. [Google Scholar] [CrossRef] [PubMed]
- Kaprova-Pleskacova, J.; Stoop, H.; Brüggenwirth, H.; Cools, M.; Wolffenbuttel, K.P.; Drop, S.L.; Snajderova, M.; Lebl, J.; Oosterhuis, J.W.; Looijenga, L.H. Complete androgen insensitivity syndrome: Factors influencing gonadal histology including germ cell pathology. Mod. Pathol. 2014, 27, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Cools, M.; Drop, S.L.S.; Wolffenbuttel, K.P.; Oosterhuis, J.W.; Looijenga, L.H.J. Germ Cell Tumors in the Intersex Gonad: Old Paths, New Directions, Moving Frontiers. Endocr. Rev. 2006, 27, 468–484. [Google Scholar] [CrossRef] [Green Version]
- Cools, M.; van Aerde, K.; Kersemaekers, A.-M.; Boter, M.; Drop, S.L.; Wolffenbuttel, K.P.; Steyerberg, E.W.; Oosterhuis, J.W.; Looijenga, L.H. Morphological and immunohistochemical differences between gonadal maturation delay and early germ cell neoplasia in patients with undervirilization syndromes. J. Clin. Endocrinol. Metab. 2005, 90, 5295–5303. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.X.; Shi, H.Y.; Cai, Z.J.; Liu, A.; Zhang, D.; Huang, H.F.; Jin, H.M. Increased risk of gonadal malignancy and prophylactic gonadectomy: A study of 102 phenotypic female patients with y chromosome or Y-derived sequences. Hum. Reprod. 2014, 29, 1413–1419. [Google Scholar] [CrossRef] [PubMed]
- Döhnert, U.; Wünsch, L.; Hiort, O. Gonadectomy in Complete Androgen Insensitivity Syndrome: Why and When? Sex. Dev. 2017, 11, 171–174. [Google Scholar] [CrossRef] [Green Version]
- Herman, M.; Wernicke, G.A.; Yan, W.; Nori, D.; Parashar, B. Pure seminoma in the setting of androgen insensitivity syndrome treated with surgical resection and para-aortic radiation: A case report and review of literature. J. Cancer Res. Ther. 2010, 6, 318–320. [Google Scholar] [CrossRef]
- Dieckmann, K.P.; Skakkebaek, N.E. Carcinoma in situ of the testis: Review of biological and clinical features. Int. J. Cancer 1999, 83, 815–822. [Google Scholar] [CrossRef]
- Cheikhelard, A.; Morel, Y.; Thibaud, E.; Lortat-Jacob, S.; Jaubert, F.; Polak, M.; Nihoul-Fekete, C. Long-Term Followup and Comparison Between Genotype and Phenotype in 29 Cases of Complete Androgen Insensitivity Syndrome. J. Urol. 2008, 180, 1496–1501. [Google Scholar] [CrossRef]
- Audi, L.; Fernández-Cancio, M.; Carrascosa, A.; Andaluz, P.; Torán, N.; Piró, C.; Vilaró, E.; Vicens-Calvet, E.; Gussinyé, M.; Albisu, M.A.; et al. Novel (60%) and recurrent (40%) androgen receptor gene mutations in a series of 59 patients with a 46,XY disorder of sex development. J. Clin. Endocrinol. Metab. 2010, 95, 1876–1888. [Google Scholar] [CrossRef]
- Cools, M.; Looijenga, L. Update on the Pathophysiology and Risk Factors for the Development of Malignant Testicular Germ Cell Tumors in Complete Androgen Insensitivity Syndrome. Sex. Dev. 2017, 11, 175–181. [Google Scholar] [CrossRef] [Green Version]
- Tack, L.J.W.; Maris, E.; Looijenga, L.H.J.; Hannema, S.E.; Audi, L.; Köhler, B.; Holterhus, P.M.; Riedl, S.; Wisniewski, A.; Flück, C.E.; et al. Management of Gonads in Adults with Androgen Insensitivity: An International Survey. Horm. Res. Paediatr. 2018, 90, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Hashmi, A.; Hanif, F.; Hanif, S.M.; Abdullah, F.E.; Shamim, M.S. Complete Androgen Insensitivity Syndrome. J. Coll. Phys. Surg. Pak. 2008, 18, 442–444. [Google Scholar]
- Mendoza, N.; Motos, M.A. Androgen insensitivity syndrome. Gynecol. Endocrinol. 2013, 29, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Mouriquand, P.D.E.; Gorduza, D.B.; Gay, C.-L.; Meyer-Bahlburg, H.F.; Baker, L.; Baskin, L.S.; Bouvattier, C.; Braga, L.H.; Caldamone, A.C.; Duranteau, L.; et al. Surgery in disorders of sex development (DSD) with a gender issue: If (why), when, and how? J. Pediatr. Urol. 2016, 12, 139–149. [Google Scholar] [CrossRef]
- Lee, P.A.; Nordenström, A.; Houk, C.P.; Ahmed, S.F.; Auchus, R.; Baratz, A.; Baratz Dalke, K.; Liao, L.M.; Lin-Su, K.; Looijenga, L.H.; et al. Global Disorders of Sex Development Update since 2006: Perceptions, Approach and Care. Horm. Res. Paediatr. 2016, 85, 158–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannema, S.; Scott, I.; Rajpert-De Meyts, E.; Skakkebæk, N.; Coleman, N.; Hughes, I. Testicular development in the complete androgen insensitivity syndrome. J. Pathol. 2006, 208, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Kratz, C.P.; Greene, M.H.; Bratslavsky, G.; Shi, J. A stratified genetic risk assessment for testicular cancer. Int. J. Androl. 2011, 34, e98–e102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, C.C.; Kanetsky, P.A.; Wang, Z.; Hildebrandt, M.A.; Koster, R.; Skotheim, R.I.; Kratz, C.P.; Turnbull, C.; Cortessis, V.K.; Bakken, A.C.; et al. Meta-analysis identifies four new loci associated with testicular germ cell tumor. Nat. Genet. 2013, 45, 680–685. [Google Scholar] [CrossRef] [Green Version]
- Stoop, H.; Honecker, F.; van de Geijn, G.; Gillis, A.J.; Cools, M.C.; de Boer, M.; Bokemeyer, C.; Wolffenbuttel, K.P.; Drop, S.L.; de Krijger, R.R.; et al. Stem cell factor as a novel diagnostic marker for early malignant germ cells. J. Pathol. 2008, 216, 43–54. [Google Scholar] [CrossRef]
- Page, D.C. Hypothesis: A Y-chromosomal gene causes gonadoblastoma in dysgenetic gonads. Development 1987, 10, 151–155. [Google Scholar]
- Lau, Y.F. Gonadoblastoma, testicular and prostate cancers, and the TSPY gene. Am. J. Hum. Genet. 1999, 64, 921–927. [Google Scholar] [CrossRef]
- Lau, Y.-F.C.; Lau, H.W.; Kömüves, L.G. Expression pattern of a gonadoblastoma candidate gene suggests a role of the Y chromosome in prostate cancer. Cytogenet. Genome Res. 2003, 101, 250–260. [Google Scholar] [CrossRef]
- Lau, Y.F.; Zhang, J. Expression analysis of thirty one Y chromosome genes in human prostate cancer. Mol. Carcinog. 2000, 27, 308–321. [Google Scholar] [CrossRef]
- Kersemaekers, A.-M.F.; Honecker, F.; Stoop, H.; Cools, M.; Molier, M.; Wolffenbuttel, K.; Bokemeyer, C.; Li, Y.; Lau, Y.F.; Oosterhuis, J.W.; et al. Identification of germ cells at risk for neoplastic transformation in gonadoblastoma. Hum. Pathol. 2005, 36, 512–521. [Google Scholar] [CrossRef]
- Rajpert-De Meyts, E. Developmental model for the pathogenesis of testicular carcinoma in situ: Genetic and environmental aspects. Hum. Reprod. Update 2006, 12, 303–323. [Google Scholar] [CrossRef] [PubMed]
- Van der Zwan, Y.G.; Biermann, K.; Wolffenbuttel, K.P.; Cools, M.; Looijenga, L.H.J. Gonadal maldevelopment as risk factor for germ cell cancer: Towards a clinical decision model. Eur. Urol. 2015, 67, 692–701. [Google Scholar] [CrossRef] [PubMed]
- Rey, R.A. Mini-puberty and true puberty: Differences in testicular function. Ann. Endocrinol. 2014, 75, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Cools, M.; Wolffenbuttel, K.P.; Drop, S.L.S.; Oosterhuis, J.W.; Looijenga, L.H.J. Gonadal Development and Tumor Formation at the Crossroads of Male and Female Sex Determination. Sex. Dev. 2011, 5, 167–180. [Google Scholar] [CrossRef] [PubMed]
- O’Shaughnessy, P.J. Hormonal control of germ cell development and spermatogenesis. Semin. Cell Dev. Biol. 2014, 29, 55–65. [Google Scholar] [CrossRef]
- Hannema, S.E.; Scott, I.S.; Hodapp, J.; Martin, H.; Coleman, N.; Schwabe, J.W.; Hughes, I.A. Residual activity of mutant androgen receptors explains wolffian duct development in the complete androgen insensitivity syndrome. J. Clin. Endocrinol. Metab. 2004, 89, 5815–5822. [Google Scholar] [CrossRef]
- Schmoll, H.J.; Souchon, R.; Krege, S.; Albers, P.; Beyer, J.; Kollmannsberger, C.; Fossa, S.D.; Skakkebaek, N.E.; de Wit, R.; Fizazi, K.; et al. European consensus on diagnosis and treatment of germ cell cancer: A report of the European Germ Cell Cancer Consensus Group (EGCCCG). Ann. Oncol. 2004, 15, 1377–1399. [Google Scholar] [CrossRef]
- Patel, V.; Casey, R.K.; Gomez-Lobo, V. Timing of Gonadectomy in Patients with Complete Androgen Insensitivity Syndrome-Current Recommendations and Future Directions. J. Pediatr. Adolesc. Gynecol. 2016, 29, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Voorhoeve, P.M.; le Sage, C.; Schrier, M.; Gillis, A.J.; Stoop, H.; Nagel, R.; Liu, Y.P.; van Duijse, J.; Drost, J.; Griekspoor, A.; et al. A Genetic Screen Implicates miRNA-372 and miRNA-373 As Oncogenes in Testicular Germ Cell Tumors. Cell 2006, 124, 1169–1181. [Google Scholar] [CrossRef] [Green Version]
- Looijenga, L.H.J.; Gillis, A.J.M.; Stoop, H.; Hersmus, R.; Oosterhuis, J.W. Relevance of microRNAs in normal and malignant development, including human testicular germ cell tumours. Int. J. Androl. 2007, 30, 304–315. [Google Scholar] [CrossRef] [PubMed]
- Palmer, R.D.; Murray, M.J.; Saini, H.K.; van Dongen, S.; Abreu-Goodger, C.; Muralidhar, B.; Pett, M.R.; Thornton, C.M.; Nicholson, J.C.; Enright, A.J.; et al. Malignant Germ Cell Tumors Display Common MicroRNA Profiles Resulting in Global Changes in Expression of Messenger RNA Targets. Cancer Res. 2010, 70, 2911–2923. [Google Scholar] [CrossRef] [PubMed]
- Murray, M.J.; Coleman, N. A new generation of biomarkers for malignant germ cell tumours. Nat. Rev. Urol. 2012, 9, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Novotny, G.W.; Belling, K.C.; Bramsen, J.B.; Nielsen, J.E.; Bork-Jensen, J.; Almstrup, K.; Sonne, S.B.; Kjems, J.; Rajpert-De Meyts, E.; Leffers, H. MicroRNA expression profiling of carcinoma in situ cells of the testis. Endocr. Relat. Cancer 2012, 19, 365–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rijlaarsdam, M.A.; van Agthoven, T.; Gillis, A.J.M.; Patel, S.; Hayashibara, K.; Lee, K.Y.; Looijenga, L.H. Identification of known and novel germ cell cancer-specific (embryonic) miRs in serum by high-throughput profiling. Andrology 2015, 3, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Van Agthoven, T.; Looijenga, L.H.J. Accurate primary germ cell cancer diagnosis using serum based microRNA detection (ampTSmiR test). Oncotarget 2017, 8, 58037–58049. [Google Scholar] [CrossRef]
- Kim, W.; Rosen, M.A.; Langer, J.E.; Banner, M.P.; Siegelman, E.S.; Ramchandani, P. US MR imaging correlation in pathologic conditions of the scrotum. Radiographics 2007, 27, 1239–1253. [Google Scholar] [CrossRef] [PubMed]
- Elzinga-Tinke, J.E.; Sirre, M.E.; Looijenga, L.H.J.; van Casteren, N.; Wildhagen, M.F.; Dohle, G.R. The predictive value of testicular ultrasound abnormalities for carcinoma in situ of the testis in men at risk for testicular cancer. Int. J. Androl. 2010, 33, 597–603. [Google Scholar] [CrossRef]
- Heinemann, V.; Frey, U.; Linke, J.; Dieckmann, K.-P. Testicular microlithiasis—One case and four points to note. Scand. J. Urol. Nephrol. 2003, 37, 515–518. [Google Scholar] [CrossRef] [PubMed]
- Nakhal, R.S.; Hall-Craggs, M.; Freeman, A.; Kirkham, A.; Conway, G.S.; Arora, R.; Woodhouse, C.R.; Wood, D.N.; Creighton, S.M. Evaluation of Retained Testes in Adolescent Girls and Women with Complete Androgen Insensitivity Syndrome. Radiology 2013, 268, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Wünsch, L.; Holterhus, P.M.; Wessel, L.; Hiort, O. Patients with disorders of sex development (DSD) at risk of gonadal tumour development: Management based on laparoscopic biopsy and molecular diagnosis. BJU Int. 2012, 110, E958–E965. [Google Scholar] [CrossRef] [PubMed]
- Warne, G.L.; Grover, S.; Zajac, J.D. Hormonal therapies for individuals with intersex conditions: Protocol for use. Treat. Endocrinol. 2005, 4, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Bertelloni, S.; Dati, E.; Baroncelli, G.I. Disorders of sex development: Hormonal management in adolescence. Gynecol. Endocrinol. 2008, 24, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Hiort, O.; Reinecke, S.; Thyen, U.; Jürgensen, M.; Holterhus, P.M.; Schön, D.; Richter-Appelt, H. Puberty in disorders of somatosexual differentiation. J. Pediatr. Endocrinol. Metab. 2003, 16 (Suppl. 2), 297–306. [Google Scholar] [PubMed]
- Arnhold, I.J.P.; Melo, K.; Costa, E.M.F.; Danilovic, D.; Inacio, M.; Domenice, S.; Mendonca, B.B. 46,XY Disorders of Sex Development (46,XY DSD) due to Androgen Receptor Defects: Androgen Insensitivity Syndrome. In Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2011. [Google Scholar]
- Bertelloni, S.; Dati, E.; Baroncelli, G.I.; Hiort, O. Hormonal management of complete androgen insensitivity syndrome from adolescence onward. Horm. Res. Paediatr. 2011, 76, 428–433. [Google Scholar] [CrossRef]
- Drobac, S.; Rubin, K.; Rogol, A.D.; Rosenfield, R.L. A workshop on pubertal hormone replacement options in the United States. J. Pediatr. Endocrinol. Metab. 2006, 19, 55–64. [Google Scholar] [CrossRef]
- Kopper, N.W.; Gudeman, J.; Thompson, D.J. Transdermal hormone therapy in postmenopausal women: A review of metabolic effects and drug delivery technologies. Drug Des. Dev. Ther. 2009, 2, 193–202. [Google Scholar] [CrossRef]
- Schenck-Gustafsson, K.; Brincat, M.; Erel, C.T.; Gambacciani, M.; Lambrinoudaki, I.; Moen, M.H.; Tremollieres, F.; Vujovic, S.; Rozenberg, S.; Rees, M. EMAS position statement: Managing the menopause in the context of coronary heart disease. Maturitas 2011, 68, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, W.; Marshall, L.; Werner, R.; Kulle, A.; Holterhus, P.M.; Rall, K.; Köhler, B.; Richter-Unruh, A.; Hartmann, M.F.; Wudy, S.A.; et al. Oestrogen versus androgen in hormone-replacement therapy for complete androgen insensitivity syndrome: A multicentre, randomised, double-dummy, double-blind crossover trial. Lancet Diabetes Endocrinol. 2018, 8587, 1–10. [Google Scholar] [CrossRef]
- Fliegner, M.; Krupp, K.; Brunner, F.; Rall, K.; Brucker, S.Y.; Briken, P.; Richter-Appelt, H. Sexual Life and Sexual Wellness in Individuals with Complete Androgen Insensitivity Syndrome (CAIS) and Mayer-Rokitansky-Küster-Hauser Syndrome (MRKHS). J. Sex. Med. 2014, 11, 729–742. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.F.; Cheng, A.; Hughes, I.A. Assessment of the gonadotrophin-gonadal axis in androgen insensitivity syndrome. Arch. Dis. Child. 1999, 80, 324–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, I.; Werner, R.; Bunch, T.; Hiort, O. Androgen Insensitivity Syndrome. Semin. Reprod. Med. 2012, 30, 432–442. [Google Scholar] [CrossRef]
- Seeman, E. Sexual Dimorphism in Skeletal Size, Density, and Strength. J. Clin. Endocrinol. Metab. 2001, 86, 4576–4584. [Google Scholar] [CrossRef] [PubMed]
- Baroncelli, G.I. Osteoporosis in Men Chapter: The Effects of Sex Steroids on Bone Growth, 2nd ed.; Orwoll, E., Bilezikian, J., Vanderschueren, D., Eds.; Elsevier Inc.: New York, NY, USA, 2009. [Google Scholar]
- Vanderschueren, D.; Vandenput, L.; Boonen, S.; Lindberg, M.K.; Bouillon, R.; Ohlsson, C. Androgens and Bone. Endocr. Rev. 2004, 25, 389–425. [Google Scholar] [CrossRef] [Green Version]
- Rochira, V.; Carani, C. Aromatase deficiency in men: A clinical perspective. Nat. Rev. Endocrinol. 2009, 5, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Laurent, M.; Sinnesael, M.; Antonio, L.; Dubois, V.; Gielen, E.; Classens, F.; Vanderschueren, D. Androgens and estrogens in skeletal sexual dimorphism. Asian J. Androl. 2014, 16, 213. [Google Scholar] [CrossRef] [PubMed]
- Callewaert, F.; Boonen, S.; Vanderschueren, D. Sex steroids and the male skeleton: A tale of two hormones. Trends Endocrinol. Metab. 2010, 21, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Bertelloni, S.; Baroncelli, G.I.; Mora, S. Bone Health in Disorders of Sex Differentiation. Sex. Dev. 2010, 4, 270–284. [Google Scholar] [CrossRef] [PubMed]
- Bertelloni, S.; Meriggiola, M.C.; Dati, E.; Balsamo, A.; Baroncelli, G.I. Bone Mineral Density in Women Living with Complete Androgen Insensitivity Syndrome and Intact Testes or Removed Gonads. Sex. Dev. 2017, 11, 182–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertelloni, S.; Baroncelli, G.I.; Federico, G.; Cappa, M.; Lala, R.; Saggese, G. Altered Bone Mineral Density in Patients with Complete Androgen Insensitivity Syndrome. Horm. Res. 1998, 50, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Soule, S.G.; Conway, G.; Prelevic, G.M.; Prentice, M.; Ginsburg, J.; Jacobs, H.S. Osteopenia as a feature of the androgen insensitivity syndrome. Clin. Endocrinol. 1995, 43, 671–675. [Google Scholar] [CrossRef]
- Vered, I.; Kaiserman, I.; Sela, B.-A.; Sack, J. Cross Genotype Sex Hormone Treatment in Two Cases of Hypogonadal Osteoporosis. J. Clin. Endocrinol. Metab. 1997, 82, 576–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcus, R.; Leary, D.; Schneider, D.L.; Shane, E.; Favus, M.; Quigley, C.A. The contribution of testosterone to skeletal development and maintenance: Lessons from the androgen insensitivity syndrome. J. Clin. Endocrinol. Metab. 2000, 85, 1032–1037. [Google Scholar] [CrossRef] [PubMed]
- Tian, Q.; Dai, Z.; Yu, W.; Tian, J.-P.; Lang, J. Study of bone mineral density in complete androgen insensitivity syndrome patients. Zhonghua Fu Chan Ke Za Zhi 2005, 40, 799–802. [Google Scholar] [PubMed]
- Sobel, V.; Schwartz, B.; Zhu, Y.-S.; Cordero, J.J.; Imperato-McGinley, J. Bone Mineral Density in the Complete Androgen Insensitivity and 5α-Reductase-2 Deficiency Syndromes. J. Clin. Endocrinol. Metab. 2006, 91, 3017–3023. [Google Scholar] [CrossRef] [Green Version]
- Han, T.S.; Goswami, D.; Trikudanathan, S.; Creighton, S.M.; Conway, G.S. Comparison of bone mineral density and body proportions between women with complete androgen insensitivity syndrome and women with gonadal dysgenesis. Eur. J. Endocrinol. 2008, 159, 179–185. [Google Scholar] [CrossRef] [Green Version]
- Taes, Y.; Lapauw, B.; Vandewalle, S.; Zmierczak, H.; Goemaere, S.; Vanderschueren, D.; Kaufman, J.M.; T’Sjoen, G. Estrogen-specific action on bone geometry and volumetric bone density: Longitudinal observations in an adult with complete androgen insensitivity. Bone 2009, 45, 392–397. [Google Scholar] [CrossRef]
- Sinnesael, M.; Claessens, F.; Laurent, M.; Dubois, V.; Boonen, S.; Deboel, L.; Vanderschueren, D. Androgen receptor (AR) in osteocytes is important for the maintenance of male skeletal integrity: Evidence from targeted AR disruption in mouse osteocytes. J. Bone Miner. Res. 2012, 27, 2535–2543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birnbaum, W.; Bertelloni, S. Sex Hormone Replacement in Disorders of Sex Development. Endocr. Dev. 2014, 27, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Nabhan, Z.M.; DiMeglio, L.A.; Qi, R.; Perkins, S.M.; Eugster, E.A. Conjugated Oral versus Transdermal Estrogen Replacement in Girls with Turner Syndrome: A Pilot Comparative Study. J. Clin. Endocrinol. Metab. 2009, 94, 2009–2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, T.F.J.; Wat, W.Z.M.; Creighton, S.M.; Conway, G.S. Bone mineral density in complete androgen insensitivity syndrome and the timing of gonadectomy. Clin. Endocrinol. 2017, 87, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Yanase, T.; Nomura, M.; Okabe, T.; Goto, K.; Sato, T.; Kawano, H.; Kato, S.; Nawata, H. Androgen receptor null male mice develop late-onset obesity caused by decreased energy expenditure and lipolytic activity but show normal insulin sensitivity with high adiponectin secretion. Diabetes 2005, 54, 1000–1008. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.-Y.; Xu, Q.; Yeh, S.; Wang, R.-S.; Sparks, J.D.; Chang, C. Insulin and leptin resistance with hyperleptinemia in mice lacking androgen receptor. Diabetes 2005, 54, 1717–1725. [Google Scholar] [CrossRef] [PubMed]
- Yanase, T.; Fan, W.; Kyoya, K.; Min, L.; Takayanagi, R.; Kato, S.; Nawata, H. Androgens and metabolic syndrome: Lessons from androgen receptor knock out (ARKO) mice. J. Steroid Biochem. Mol. Biol. 2008, 109, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Yu, I.-C.; Lin, H.-Y.; Liu, N.-C.; Wang, R.S.; Sparks, J.D.; Yeh, S.; Chang, C. Hyperleptinemia without Obesity in Male Mice Lacking Androgen Receptor in Adipose Tissue. Endocrinology 2008, 149, 2361–2368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dati, E.; Baroncelli, G.I.; Mora, S.; Russo, G.; Baldinotti, F.; Parrini, D.; Erba, P.; Simi, P.; Bertelloni, S. Body Composition and Metabolic Profile in Women with Complete Androgen Insensitivity Syndrome. Sex. Dev. 2009, 3, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, J.; Sun, L.; Kumar, T.R.; Blair, H.C.; Zaidi, M. Follicle-stimulating hormone stimulates TNF production from immune cells to enhance osteoblast and osteoclast formation. Proc. Natl. Acad. Sci. USA 2006, 103, 14925–14930. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.-L.; Blair, H.; Cao, J.; Yuen, T.; Latif, R.; Guo, L.; Tourkova, I.L.; Li, J.; Davies, T.F.; Sun, L.; et al. Blocking antibody to the-subunit of FSH prevents bone loss by inhibiting bone resorption and stimulating bone synthesis. Proc. Natl. Acad. Sci. USA 2012, 109, 14574–14579. [Google Scholar] [CrossRef]
- Sun, L.; Peng, Y.; Sharrow, A.C.; Iqbal, J.; Zhang, Z.; Papachristou, D.J.; Zaidi, S.; Zhu, L.L.; Yaroslavskiy, B.B.; Zhou, H.; et al. FSH directly regulates bone mass. Cell 2006, 125, 247–260. [Google Scholar] [CrossRef]
- Ferlin, A.; De Toni, L.; Sandri, M.; Foresta, C. Relaxin and insulin-like peptide 3 in the musculoskeletal system: From bench to bedside. Br. J. Pharmacol. 2017, 174, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Tanner, J.M.; Whitehouse, R.H. Clinical longitudinal standards for height, weight, height velocity, weight velocity, and stages of puberty. Arch. Dis. Child. 1976, 51, 170–179. [Google Scholar] [CrossRef]
- Yue, L.; Wu, P.; Xia, Z.; Fan, C.; Xia, Q. A novel deletion mutation in AR gene causes complete androgen insensitivity syndrome in a Chinese family. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2010, 27, 631–633. [Google Scholar] [CrossRef]
- Batch, J.A.; Williams, D.M.; Davies, H.R.; Brown, B.D.; Evans, B.A.; Hughes, I.A.; Patterson, M.N. Androgen receptor gene mutations identified by SSCP in fourteen subjects with androgen insensitivity syndrome. Hum. Mol. Genet. 1992, 1, 497–503. [Google Scholar] [CrossRef]
- Ledig, S.; Jakubiczka, S.; Neulen, J.; Aulepp, U.; Burck-Lehmann, U.; Mohnike, K.; Thiele, H.; Zierler, H.; Brewer, C.; Wieacker, P. Novel and Recurrent Mutations in Patients with Androgen Insensitivity Syndromes. Horm. Res. Paediatr. 2005, 63, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Lobaccaro, J.M.; Lumbroso, S.; Poujol, N.; Georget, V.; Brinkmann, A.O.; Malpuech, G.; Sultan, C. Complete androgen insensitivity syndrome due to a new frameshift deletion in exon 4 of the androgen receptor gene: Functional analysis of the mutant receptor. Mol. Cell. Endocrinol. 1995, 111, 21–28. [Google Scholar] [CrossRef]
- Galani, A.; Sofocleous, C.; Karahaliou, F.; Papathanasiou, A.; Kitsiou-Tzeli, S.; Kalpini-Mavrou, A. Sex-reversed phenotype in association with two novel mutations c.2494delA and c.T3004C in the ligand-binding domain of the androgen receptor gene. Fertil. Steril. 2008, 90, 2008.e1–2008.e4. [Google Scholar] [CrossRef]
- Avila, D.M.; Wilson, C.M.; Nandi, N.; Griffin, J.E.; McPhaul, M.J. Immunoreactive AR and Genetic Alterations in Subjects with Androgen Resistance and Undetectable AR Levels in Genital Skin Fibroblast Ligand-Binding Assays. J. Clin. Endocrinol. Metab. 2002, 87, 182–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacLean, H.E.; Chu, S.; Warne, G.L.; Zajac, J.D. Related individuals with different androgen receptor gene deletions. J. Clin. Investig. 1993, 91, 1123–1128. [Google Scholar] [CrossRef] [PubMed]
- Baldazzi, L.; Baroncini, C.; Pirazzoli, P.; Balsamo, A.; Capelli, M.; Marchetti, G.; Bernardi, F.; Cacciari, E. Two mutations causing complete androgen insensitivity: A frame-shift in the steroid binding domain and a Cys→Phe substitution in the second zinc finger of the androgen receptor. Hum. Mol. Genet. 1994, 3, 1169–1170. [Google Scholar] [CrossRef] [PubMed]
- Vilchis, F.; Ramos, L.; Kofman-Alfaro, S.; Zenteno, J.C.; Méndez, J.P.; Chávez, B. Extreme androgen resistance in a kindred with a novel insertion/deletion mutation in exon 5 of the androgen receptor gene. J. Hum. Genet. 2003, 48, 346–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, H.W.; Kim, S.C.; Kim, H.L. Frame-shift mutation in hormone binding domain of human androgen receptor gene causes complete androgen insensitivity. Mol. Cells 1998, 8, 741–745. [Google Scholar] [PubMed]
- Soriano Guillén, L.; Muñoz Calvo, M.T.; Martinez Pérez, J.; Pozo Román, J.; Martín Sobrino, M.A.; González Medeiro, I.; Argente Oliver, J. Deletion of thymine at position 2298 in exon 5 of the androgenic receptor gene causing complete androgen insensitivity syndrome. An. Esp. Pediatr. 2002, 56, 347–352. [Google Scholar] [CrossRef]
- T’Sjoen, G.; De Cuypere, G.; Monstrey, S.; Hoebeke, P.; Freedman, F.K.; Appari, M.; Holterhus, P.M.; Van Borsel, J.; Cools, M. Male Gender Identity in Complete Androgen Insensitivity Syndrome. Arch. Sex. Behav. 2011, 40, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Jeske, Y.W.A.; McGown, I.N.; Cowley, D.M.; Oley, C.; Thomsett, M.J.; Choong, C.S.; Cotterill, A.M. Androgen receptor genotyping in a large Australasian cohort with androgen insensitivity syndrome; identification of four novel mutations. J. Pediatr. Endocrinol. Metab. 2007, 20, 893–908. [Google Scholar] [CrossRef] [PubMed]
- Köhler, B.; Lumbroso, S.; Leger, J.; Audran, F.; Grau, E.S.; Kurtz, F.; Pinto, G.; Salerno, M.; Semitcheva, T.; Czernichow, P.; et al. Androgen insensitivity syndrome: Somatic mosaicism of the androgen receptor in seven families and consequences for sex assignment and genetic counseling. J. Clin. Endocrinol. Metab. 2005, 90, 106–111. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Hormone | At first Evaluation | Reference Values |
|---|---|---|
| LH (mUI/mL) | 23.82 | 2.3–3.5 |
| FSH (mUI/mL) | 130 | 2.4–5.2 |
| Testosterone (ng/mL) | 0.16 | 0.10–0.75 |
| Oestradiol (pg/mL) | <20 | 21–85 |
| Prolactin (ng/mL) | 26.9 | 3.3–26.7 |
| DHEA-S (ug/dL) | 203.6 | 15–260 |
| 4-Androstenedione (ng/mL) | 0.7 | 0.24–0.38 |
| TSH (mUI/mL) | 1.76 | 0.34–5.6 |
| FT4 (ng/dL) | 0.77 | 0.54–1.24 |
| Hormone | At Diagnosis | After Surgery | Reference Values |
|---|---|---|---|
| LH (mUI/Ml) | 25.81 | 34.57 | 5.3–10.5 |
| FSH (mUI/mL) | 3.8 | 105.20 | 5.8–8.6 |
| Testosterone (ng/mL) | 4.9 | 0.10–0.75 | |
| Oestradiol (pg/mL) | 23 | 48 | 21–85 |
| Prolactin (ng/mL) | 15.5 | 3–24 | |
| 17-Hydroxyprogesterone (ng/mL) | 1.5 | 0.16–2.31 | |
| DHEA-S (µg/dL) | 261.8 | 35–535 | |
| 4-Androstenedione (ng/mL) | 4 | 0.3–3.5 | |
| β-HCG (mUI/mL) | 1.92 | 0–5 | |
| α-Fetoprotein (ng/mL) | 1.4 | 0.6–8.1 | |
| TSH (mUI/mL) | 1.540 | 0.34–5.6 | |
| FT4 (ng/dL) | 0.72 | 0.54–1.24 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lanciotti, L.; Cofini, M.; Leonardi, A.; Bertozzi, M.; Penta, L.; Esposito, S. Different Clinical Presentations and Management in Complete Androgen Insensitivity Syndrome (CAIS). Int. J. Environ. Res. Public Health 2019, 16, 1268. https://doi.org/10.3390/ijerph16071268
Lanciotti L, Cofini M, Leonardi A, Bertozzi M, Penta L, Esposito S. Different Clinical Presentations and Management in Complete Androgen Insensitivity Syndrome (CAIS). International Journal of Environmental Research and Public Health. 2019; 16(7):1268. https://doi.org/10.3390/ijerph16071268
Chicago/Turabian StyleLanciotti, Lucia, Marta Cofini, Alberto Leonardi, Mirko Bertozzi, Laura Penta, and Susanna Esposito. 2019. "Different Clinical Presentations and Management in Complete Androgen Insensitivity Syndrome (CAIS)" International Journal of Environmental Research and Public Health 16, no. 7: 1268. https://doi.org/10.3390/ijerph16071268