Probiotic Lactobacillus Paracasei Expressing a Nucleic Acid-Hydrolyzing Minibody (3D8 Scfv) Enhances Probiotic Activities in Mice Intestine as Revealed by Metagenomic Analyses

Abstract
:1. Introduction
2. Methods
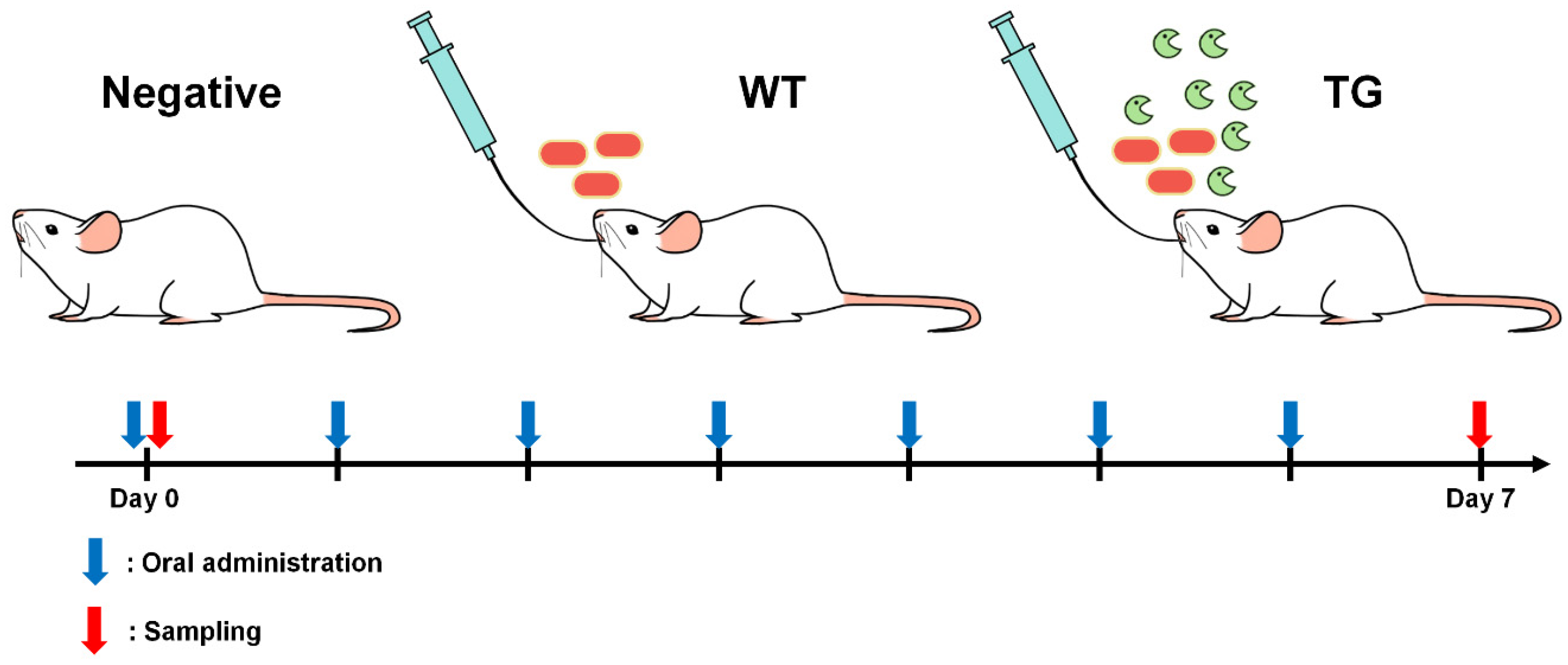
2.1. Animals and Bacteria
2.2. Construction of a Vector Expressing Recombinant Protein and Transformation
2.3. Oral Administration of Lactobacillus to BALB/c mice
2.4. Sample Preparation and DNA Extraction
2.5. Preparation of Libraries for 16S Ribosomal RNA Sequencing and Paired-End Sequencing Using the MiSeq System
2.6. Preprocessing of Raw Data, Clustering, and Taxonomic Assignment
2.7. Statistical Analyses
3. Results
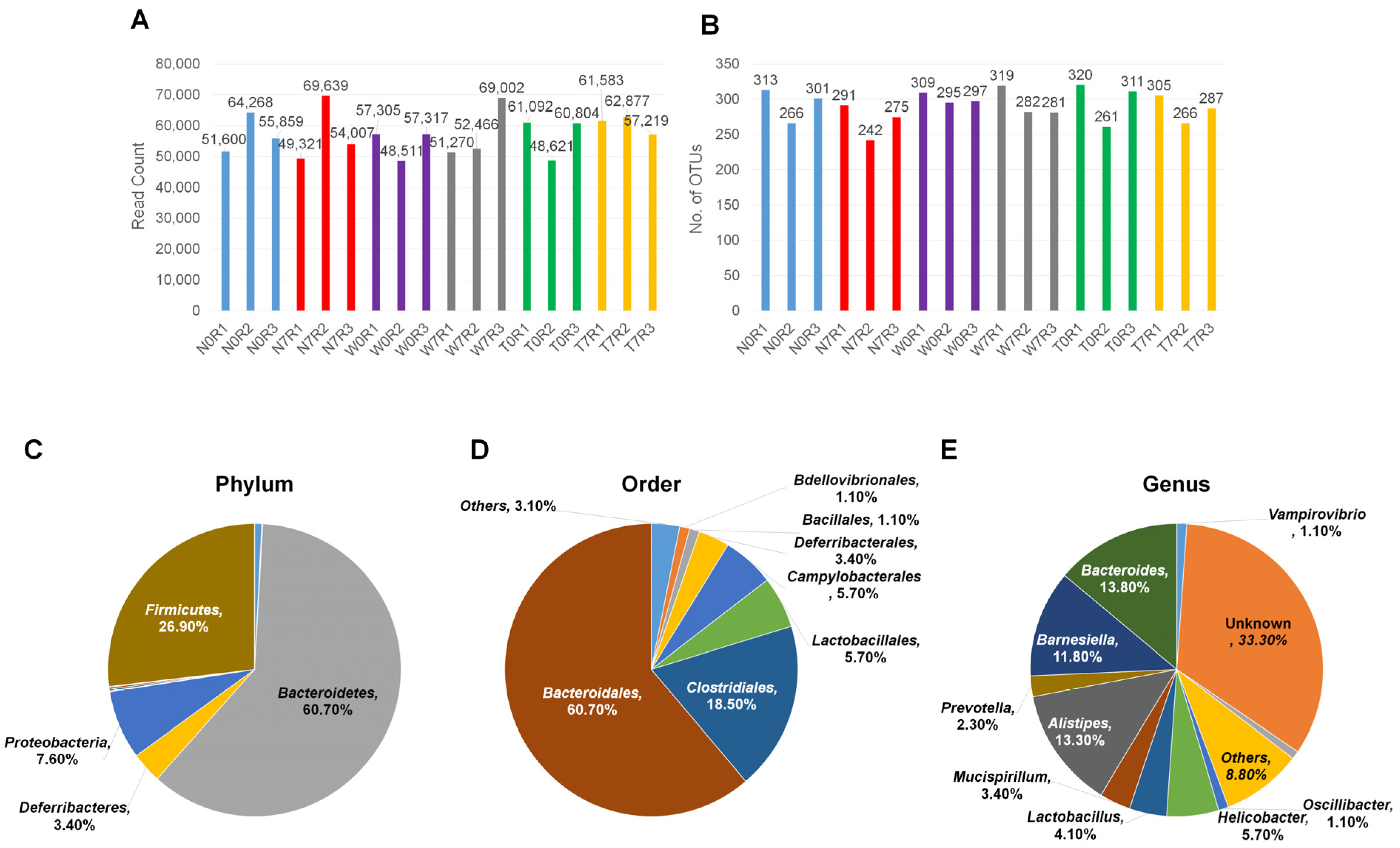
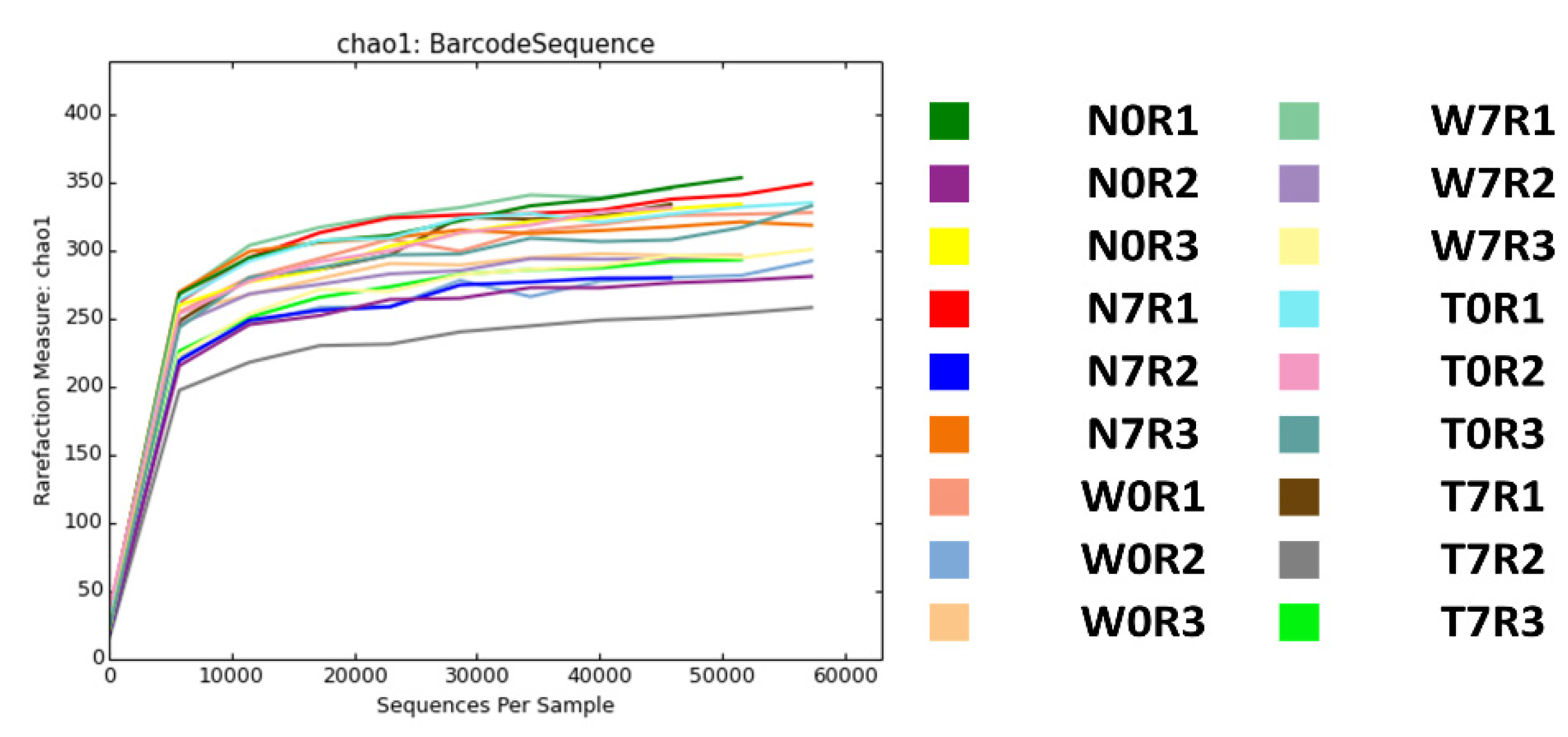
3.1. Sample Preparation and High-Throughput 16S ribosomal RNA Metagenomic Analyses Using MiSeq System
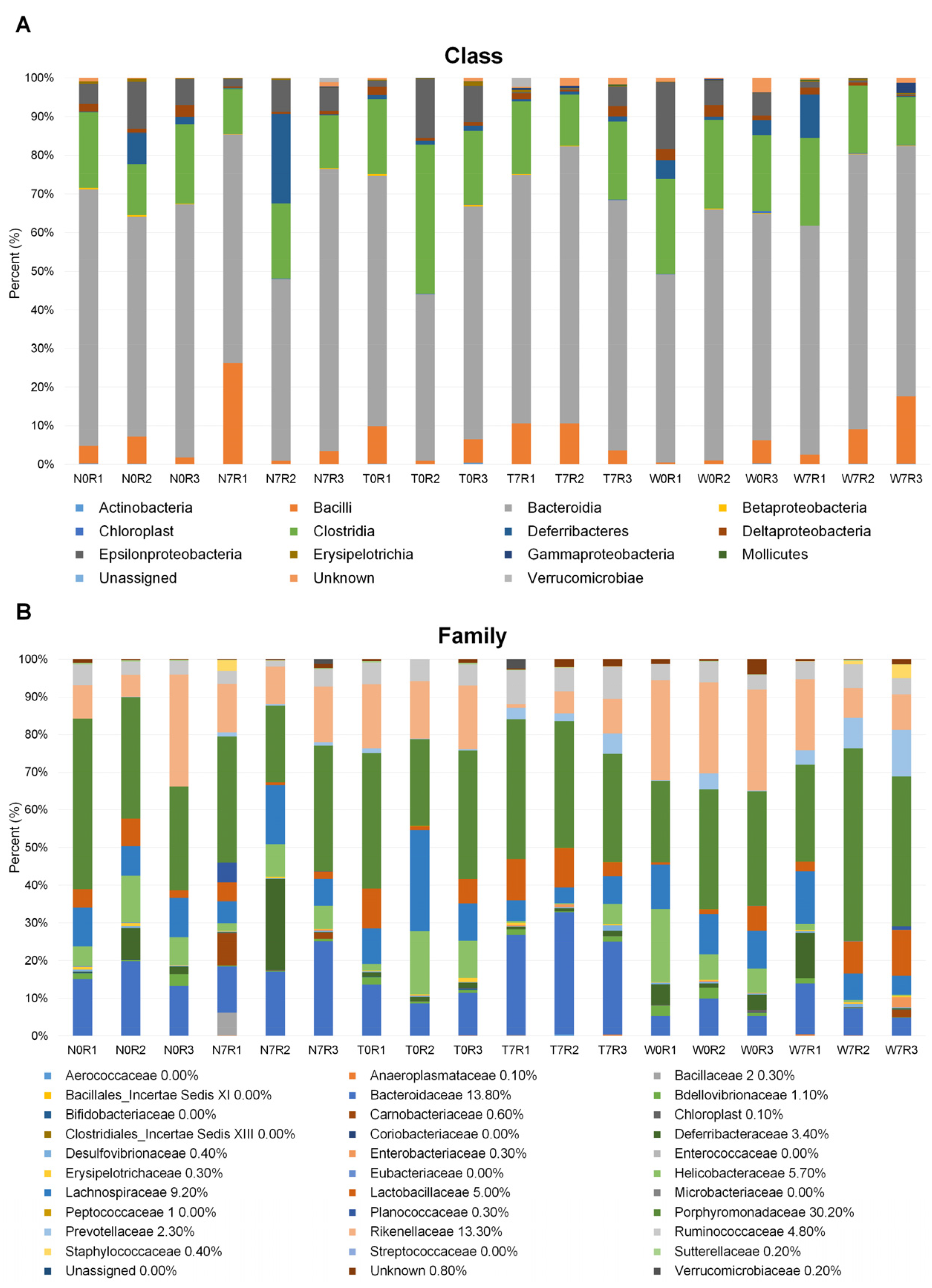
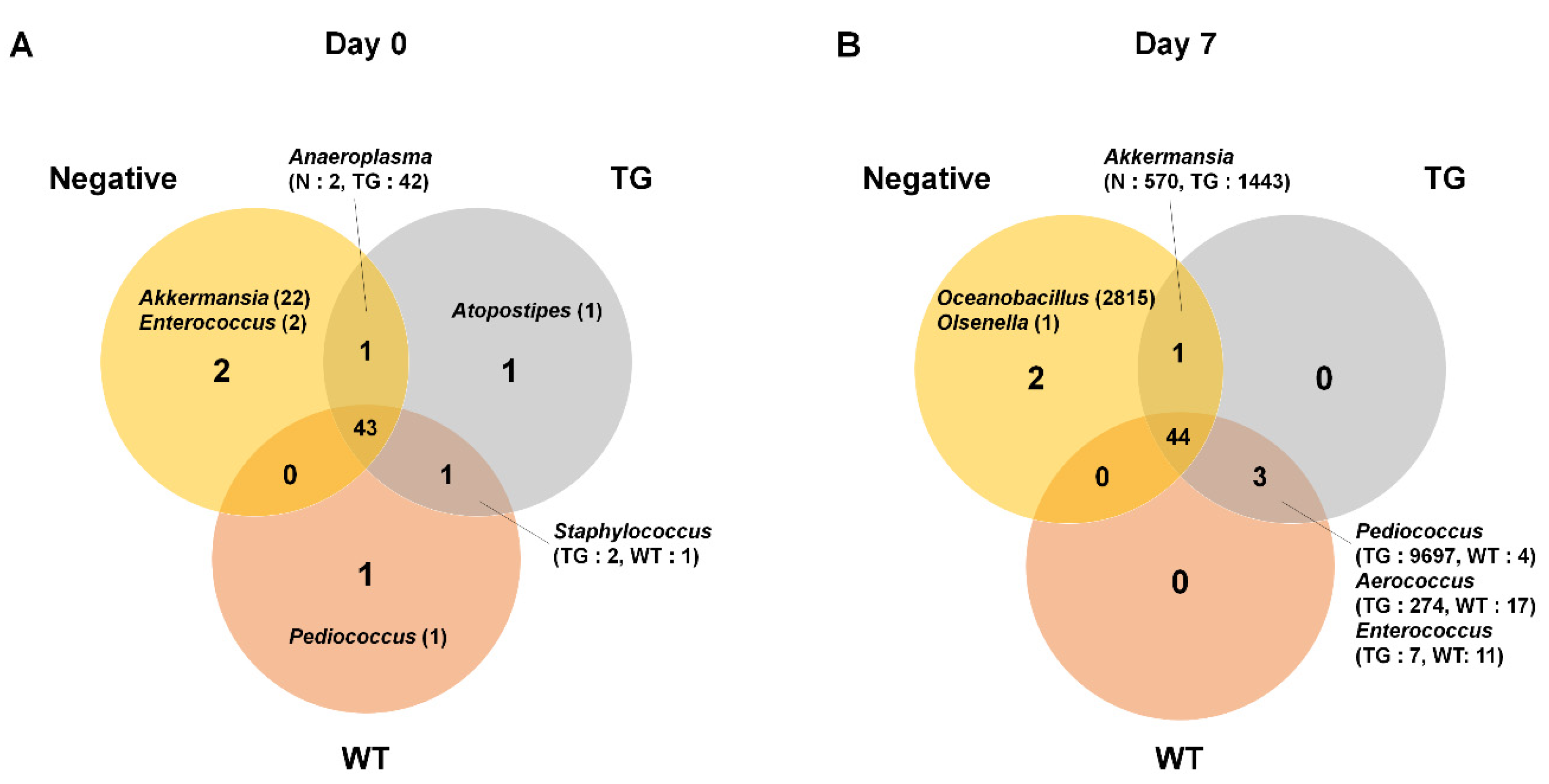
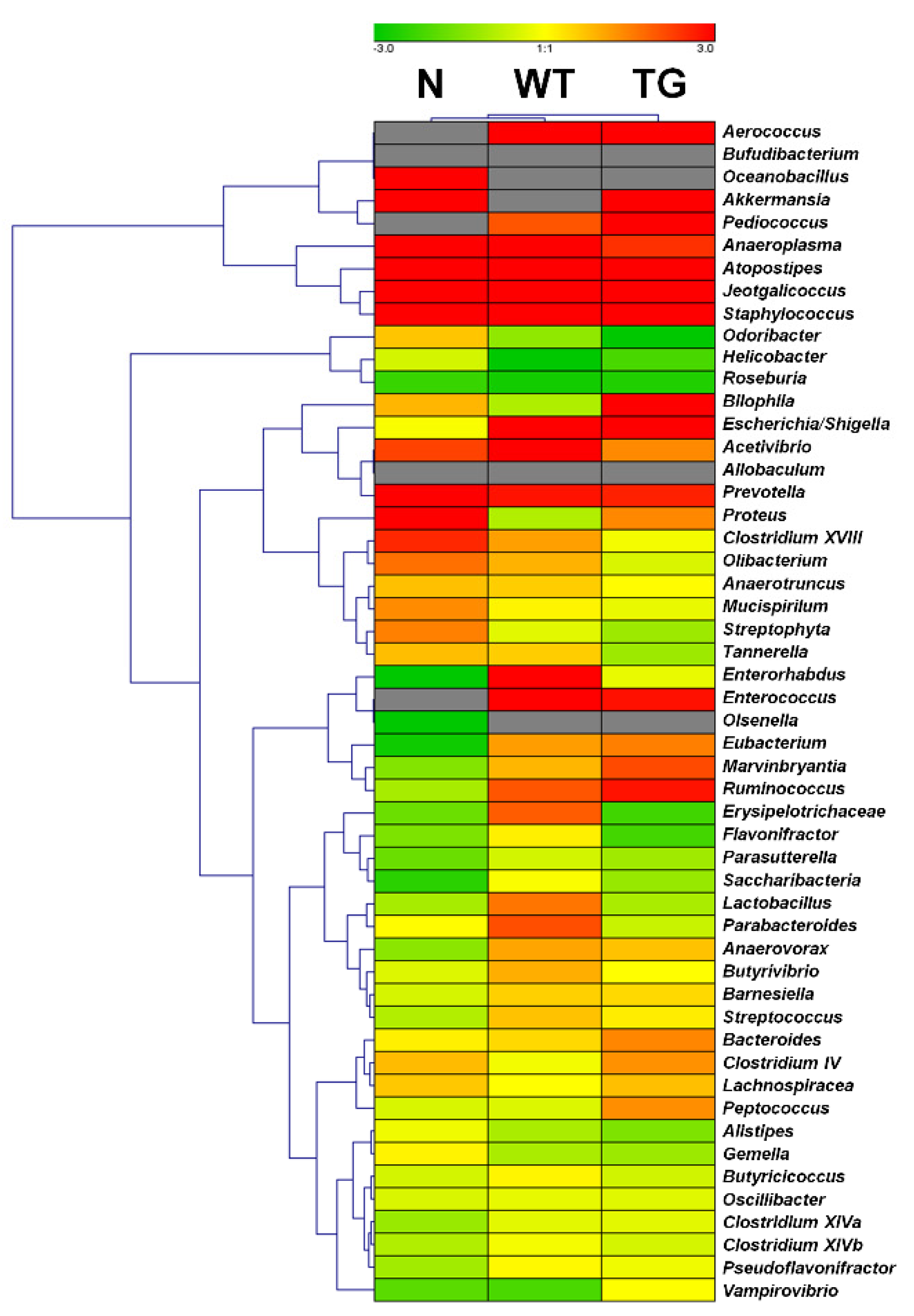
3.2. Microbial Composition in Mice Intestine
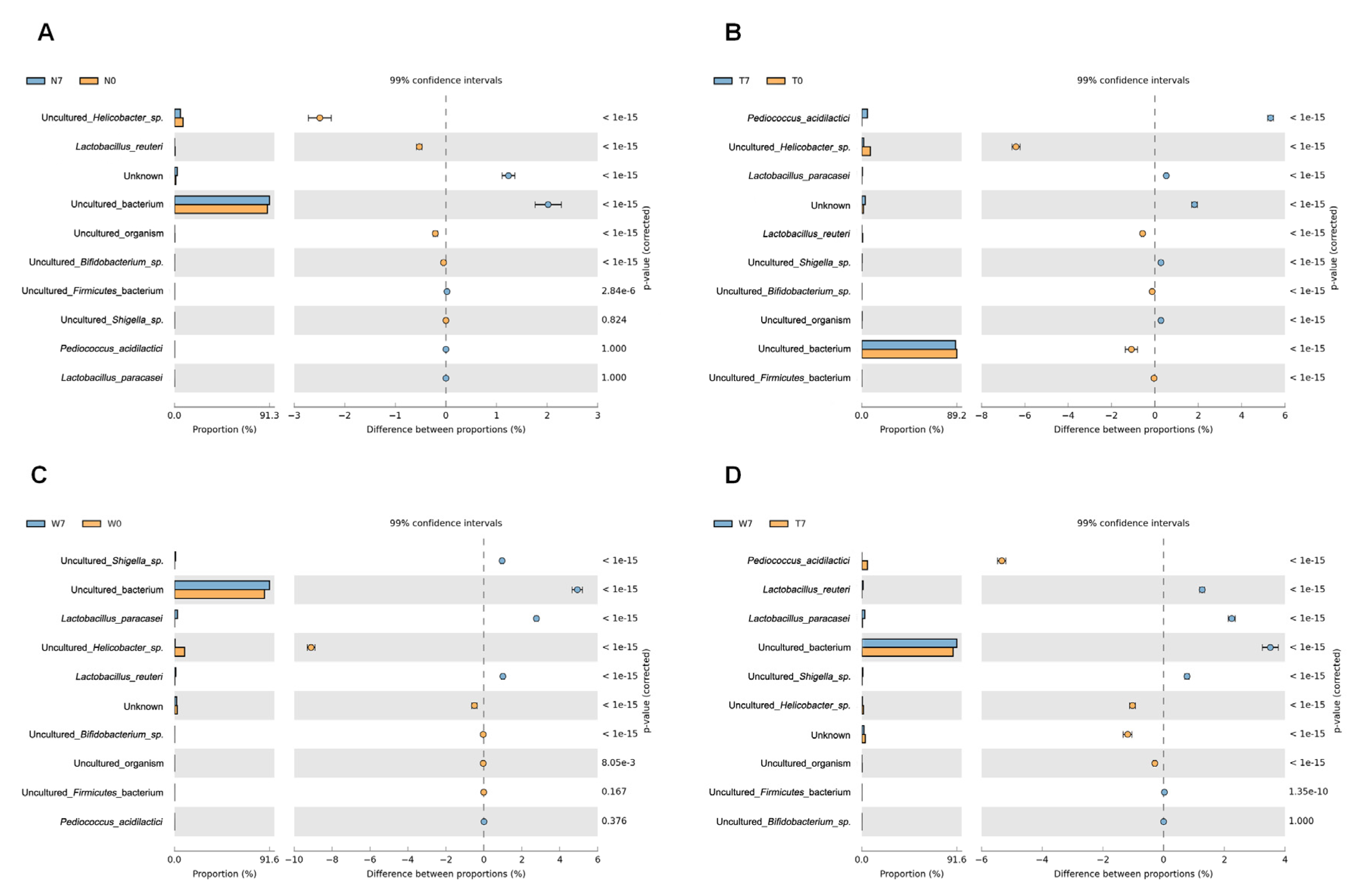
3.3. Comparison of Bacterial Compositions Among the Three Different Conditions
4. Discussion
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Data Availability
References
- Hill, C.; Guarner, F.; Reid, G.; Gibson, G.R.; Merenstein, D.J.; Pot, B.; Morelli, L.; Canani, R.B.; Flint, H.J.; Salminen, S. Expert consensus document: The international scientific association for probiotics and prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Parvez, S.; Malik, K.; Ah Kang, S.; Kim, H.Y. Probiotics and their fermented food products are beneficial for health. J. Appl. Microbiol. 2006, 100, 1171–1185. [Google Scholar] [CrossRef] [PubMed]
- Bartkiene, E.; Zavistanaviciute, P.; Lele, V.; Ruzauskas, M.; Bartkevics, V.; Bernatoniene, J.; Gallo, P.; Tenore, G.C.; Santini, A. Lactobacillus plantarum LUHS135 and paracasei LUHS244 as functional starter cultures for the food fermentation industry: Characterisation, mycotoxin-reducing properties, optimisation of biomass growth and sustainable encapsulation by using dairy by-products. LWT Food Sci. Technol. 2018, 93, 649–658. [Google Scholar]
- Valeriano, V.D.V.; Balolong, M.P.; Kang, D.K. Probiotic roles of Lactobacillus spp. in swine: Insights from gut microbiota. J. Appl. Microbiol. 2016, 122, 554–567. [Google Scholar] [CrossRef] [PubMed]
- Lähteinen, T.; Malinen, E.; Koort, J.M.; Mertaniemi-Hannus, U.; Hankimo, T.; Karikoski, N.; Pakkanen, S.; Laine, H.; Sillanpää, H.; Söderholm, H. Probiotic properties of Lactobacillus isolates originating from porcine intestine and feces. Anaerobe 2010, 16, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Claesson, M.J.; Van Sinderen, D.; O’Toole, P.W. The genus Lactobacillus—A genomic basis for understanding its diversity. FEMS Microbiol. Lett. 2007, 269, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-R.; Kim, J.-S.; Lee, S.-H.; Lee, W.-R.; Sohn, J.-N.; Chung, Y.-C.; Shim, H.-K.; Lee, S.-C.; Kwon, M.-H.; Kim, Y.-S. Heavy and light chain variable single domains of an anti-DNA binding antibody hydrolyze both double-and single-stranded DNAs without sequence specificity. J. Biol. Chem. 2006, 281, 15287–15295. [Google Scholar] [CrossRef] [PubMed]
- Hoang, P.M.; Cho, S.; Kim, K.E.; Byun, S.J.; Lee, T.-K.; Lee, S. Development of Lactobacillus paracasei harboring nucleic acid-hydrolyzing 3D8 scFv as a preventive probiotic against murine norovirus infection. Appl. Microbiol. Biotechnol. 2015, 99, 2793–2803. [Google Scholar] [CrossRef] [PubMed]
- Jun, H.-R.; Pham, C.D.; Lim, S.-I.; Lee, S.-C.; Kim, Y.-S.; Park, S.; Kwon, M.-H. An RNA-hydrolyzing recombinant antibody exhibits an antiviral activity against classical swine fever virus. Biochem. Biophys. Res. Commun. 2010, 395, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Cho, S.; Hoang, P.M.; Kim, D.; Lee, Y.; Kil, E.-J.; Byun, S.-J.; Lee, T.-K.; Kim, D.-H.; Kim, S. Therapeutic strategy for the prevention of pseudorabies virus infection in C57BL/6 mice by 3D8 scFv with intrinsic nuclease activity. Mol. Cells 2015, 38, 773. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Yu, J.; Cho, S.; Byun, S.-J.; Kim, D.H.; Lee, T.-K.; Kwon, M.-H.; Lee, S. A nucleic-acid hydrolyzing single chain antibody confers resistance to DNA virus infection in HeLa cells and C57BL/6 mice. PLoS Pathog. 2014, 10, e1004208. [Google Scholar] [CrossRef] [PubMed]
- Scholz, M.B.; Lo, C.-C.; Chain, P.S. Next generation sequencing and bioinformatic bottlenecks: The current state of metagenomic data analysis. Curr. Opin. Biotechnol. 2012, 23, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.N.; Pace, N.R. Gastrointestinal microbiology enters the metagenomics era. Curr. Opin. Gastroenterol. 2008, 24, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Feng, Q.; Liang, S.; Sonne, S.B.; Xia, Z.; Qiu, X.; Li, X.; Long, H.; Zhang, J.; Zhang, D. A catalog of the mouse gut metagenome. Nat. Biotechnol. 2015, 33, 1103–1108. [Google Scholar] [CrossRef] [PubMed]
- Droege, M.; Hill, B. The genome sequencer flx™ system—Longer reads, more applications, straight forward bioinformatics and more complete data sets. J. Biotechnol. 2008, 136, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Oulas, A.; Pavloudi, C.; Polymenakou, P.; Pavlopoulos, G.A.; Papanikolaou, N.; Kotoulas, G.; Arvanitidis, C.; Iliopoulos, I. Metagenomics: Tools and insights for analyzing next-generation sequencing data derived from biodiversity studies. Bioinform. Biol. Insights 2015, 9, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oozeer, R.; Furet, J.; Goupil-Feuillerat, N.; Anba, J.; Mengaud, J.; Corthier, G. Differential activities of four Lactobacillus casei promoters during bacterial transit through the gastrointestinal tracts of human-microbiota-associated mice. Appl. Environ. Microbiol. 2005, 71, 1356–1363. [Google Scholar] [CrossRef] [PubMed]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Fu, L.; Niu, B.; Wu, S.; Wooley, J. Ultrafast clustering algorithms for metagenomic sequence analysis. Brief Bioinform. 2012, 13, 656–668. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Faul, F.; Erdfelder, E.; Buchner, A.; Lang, A.-G. Statistical power analyses using G*Power 3.1: Tests for correlation and regression analyses. Behav Res Methods 2009, 41, 1149–1160. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Beiko, R.G. Identifying biologically relevant differences between metagenomic communities. Bioinformatics 2010, 26, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.; Godovannyi, A.; Ma, C.; Zhang, Y.; Ahmadi-Vand, Z.; Dai, C.; Gorzelak, M.A.; Chan, Y.; Chan, J.M.; Lochner, A. Prolonged antibiotic treatment induces a diabetogenic intestinal microbiome that accelerates diabetes in nod mice. ISME J. 2015. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Zhang, X.-X.; Wu, B.; Xian, Q. Metagenomic insights into tetracycline effects on microbial community and antibiotic resistance of mouse gut. Ecotoxicology 2015, 24, 2125–2132. [Google Scholar] [CrossRef] [PubMed]
- Nobel, Y.R.; Cox, L.M.; Kirigin, F.F.; Bokulich, N.A.; Yamanishi, S.; Teitler, I.; Chung, J.; Sohn, J.; Barber, C.M.; Goldfarb, D.S. Metabolic and metagenomic outcomes from early-life pulsed antibiotic treatment. Nat. Commun. 2015, 6, 7486. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Mazcorro, J.F.; Ivanov, I.; Mills, D.A.; Noratto, G. Influence of whole-wheat consumption on fecal microbial community structure of obese diabetic mice. PeerJ 2016, 4, e1702. [Google Scholar] [CrossRef] [PubMed]
- Everard, A.; Lazarevic, V.; Gaïa, N.; Johansson, M.; Ståhlman, M.; Backhed, F.; Delzenne, N.M.; Schrenzel, J.; François, P.; Cani, P.D. Microbiome of prebiotic-treated mice reveals novel targets involved in host response during obesity. ISME J. 2014, 8, 2116–2130. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Kim, J.; Park, S.-J. High-throughput 16s rRNA gene sequencing reveals alterations of mouse intestinal microbiota after radiotherapy. Anaerobe 2015, 33, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Yap, I.K.; Kho, M.T.; Lim, S.H.E.; Ismail, N.H.; Yam, W.K.; Chong, C.W. Acclimatisation-induced stress influenced host metabolic and gut microbial composition change. Mol. BioSyst. 2015, 11, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Liu, S.; Wang, Z.; Zhang, X.-X.; Li, M.; Wu, B. Metagenomic profiles and antibiotic resistance genes in gut microbiota of mice exposed to arsenic and iron. Chemosphere 2014, 112, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Faith, J.J.; Ahern, P.P.; Ridaura, V.K.; Cheng, J.; Gordon, J.I. Identifying gut microbe–host phenotype relationships using combinatorial communities in gnotobiotic mice. Sci. Transl. Med. 2014, 6, 220ra211. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Linnenbrink, M.; Künzel, S.; Fernandes, R.; Nadeau, M.-J.; Rosenstiel, P.; Baines, J.F. Dietary history contributes to enterotype-like clustering and functional metagenomic content in the intestinal microbiome of wild mice. Proc. Natl. Acad. Sci. USA 2014, 111, E2703–E2710. [Google Scholar] [CrossRef] [PubMed]
- Wexler, H.M. Bacteroides: The good, the bad, and the nitty-gritty. Clin. Microbiol. Rev. 2007, 20, 593–621. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wu, J.; Li, J.V.; Zhou, N.-Y.; Tang, H.; Wang, Y. Gut microbiota composition modifies fecal metabolic profiles in mice. J. Proteome Res. 2013, 12, 2987–2999. [Google Scholar] [CrossRef] [PubMed]
- Zanotti, I.; Turroni, F.; Piemontese, A.; Mancabelli, L.; Milani, C.; Viappiani, A.; Prevedini, G.; Sanchez, B.; Margolles, A.; Elviri, L. Evidence for cholesterol-lowering activity by Bifidobacterium bifidum PRL2010 through gut microbiota modulation. Appl. Microbiol. Biotechnol. 2015, 99, 6813–6829. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Arioli, S.; Wang, A.; Villa, C.R.; Jahani, R.; Song, Y.S.; Mora, D.; Guglielmetti, S.; Comelli, E.M. Impact of Bifidobacterium bifidum MIMBB75 on mouse intestinal microorganisms. FEMS Microbiol. Ecol. 2013, 85, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Park, D.-Y.; Ahn, Y.-T.; Park, S.-H.; Huh, C.-S.; Yoo, S.-R.; Yu, R.; Sung, M.-K.; McGregor, R.A.; Choi, M.-S. Supplementation of Lactobacillus curvatus KY1032 in diet-induced obese mice is associated with gut microbial changes and reduction in obesity. PLoS ONE 2013, 8, 59470. [Google Scholar]
- Bull-Otterson, L.; Feng, W.; Kirpich, I.; Wang, Y.; Qin, X.; Liu, Y.; Gobejishvili, L.; Joshi-Barve, S.; Ayvaz, T.; Petrosino, J. Metagenomic analyses of alcohol induced pathogenic alterations in the intestinal microbiome and the effect of Lactobacillus rhamnosus GG treatment. PloS ONE 2013, 8, e53028. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Sung, C.Y.J.; Lee, N.; Ni, Y.; Pihlajamäki, J.; Panagiotou, G.; El-Nezami, H. Probiotics modulated gut microbiota suppresses hepatocellular carcinoma growth in mice. Proc. Natl. Acad. Sci. USA 2016, 113, E1306–E1315. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-L.; Lai, Y.-W.; Chen, C.-S.; Chu, T.-W.; Lin, W.; Yen, C.-C.; Lin, M.-F.; Tu, M.-Y.; Chen, C.-M. Probiotic Lactobacillus casei expressing human lactoferrin elevates antibacterial activity in the gastrointestinal tract. Biometals 2010, 23, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Haakensen, M.; Dobson, C.M.; Hill, J.E.; Ziola, B. Reclassification of Pediococcus dextrinicus (coster and white 1964) back 1978 (approved lists 1980) as Lactobacillus dextrinicus comb. Nov., and emended description of the genus Lactobacillus. Int. J. Syst. Evolut. Microbiol. 2009, 59, 615–621. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition | Day | Replicate | Abbreviation | Total Bases (bp) | Read Count |
|---|---|---|---|---|---|
| N | 0 | 1 | N0R1 | 117,395,034 | 259,801 |
| 2 | N0R2 | 129,075,064 | 285,466 | ||
| 3 | N0R3 | 128,072,181 | 283,889 | ||
| 7 | 1 | N7R1 | 102,570,095 | 224,927 | |
| 2 | N7R2 | 147,077,671 | 325,948 | ||
| 3 | N7R3 | 113,530,929 | 250,331 | ||
| WT | 0 | 1 | W0R1 | 129,201,892 | 288,118 |
| 2 | W0R2 | 125,601,235 | 278,505 | ||
| 3 | W0R3 | 140,795,712 | 311,553 | ||
| 7 | 1 | W7R1 | 121,644,286 | 269,049 | |
| 2 | W7R2 | 123,340,544 | 271,652 | ||
| 3 | W7R3 | 148,592,201 | 326,110 | ||
| TG | 0 | 1 | T0R1 | 138,288,303 | 305,165 |
| 2 | T0R2 | 107,080,820 | 237,986 | ||
| 3 | T0R3 | 135,852,937 | 300,959 | ||
| 7 | 1 | T7R1 | 139,324,032 | 307,834 | |
| 2 | T7R2 | 131,123,910 | 287,958 | ||
| 3 | T7R3 | 123,911,242 | 274,262 |
| Sample | OTUs | Chao1 | Shannon | Simpson | Goods Coverage |
|---|---|---|---|---|---|
| N0R1 | 313 | 352.667 | 5.8142597 | 0.9593455 | 0.999321705 |
| N0R2 | 266 | 282.235 | 5.0649192 | 0.9344073 | 0.999626564 |
| N0R3 | 301 | 332.316 | 5.5026827 | 0.9550186 | 0.999373422 |
| N7R1 | 291 | 338.3 | 5.6458194 | 0.9655083 | 0.999107885 |
| N7R2 | 242 | 264.235 | 4.4974754 | 0.9035892 | 0.999597926 |
| N7R3 | 275 | 295.313 | 5.4140089 | 0.9521503 | 0.999518581 |
| W0R1 | 309 | 335.4 | 5.4151359 | 0.9425895 | 0.999424134 |
| W0R2 | 295 | 334 | 5.8569545 | 0.9670265 | 0.999175445 |
| W0R3 | 297 | 332.769 | 5.6102921 | 0.955642 | 0.999459148 |
| W7R1 | 319 | 338.833 | 5.6387481 | 0.9569166 | 0.99931734 |
| W7R2 | 282 | 295.043 | 5.610285 | 0.9572774 | 0.999523501 |
| W7R3 | 281 | 307.105 | 5.6519495 | 0.9641348 | 0.999536245 |
| T0R1 | 320 | 349 | 5.8469024 | 0.963316 | 0.999508937 |
| T0R2 | 261 | 282.368 | 4.9104504 | 0.9228563 | 0.99940355 |
| T0R3 | 311 | 320 | 5.810696 | 0.9640873 | 0.999703967 |
| T7R1 | 305 | 330.588 | 5.631011 | 0.9554437 | 0.999512853 |
| T7R2 | 266 | 295.077 | 5.140159 | 0.9231 | 0.999554686 |
| T7R3 | 287 | 297.5 | 5.7108717 | 0.9602213 | 0.999632989 |
| All * | N0 | N7 | W0 | W7 | T0 | T7 | |
|---|---|---|---|---|---|---|---|
| Lactobacillus paracasei | 5743 (0.6%) | 0 | 0 | 0 | 4790 (2.8%) | 0 | 953 (0.5%) |
| Lactobacillus reuteri | 6120 (0.6%) | 1068 (0.6%) | 163 (0.1%) | 733 (0.4%) | 2518 (1.5%) | 1295 (0.8%) | 343 (0.2%) |
| Pediococcus acidilactici | 9702 (0.9%) | 0 | 0 | 1 | 4 | 0 | 9697 (5.3%) |
| Uncultured bacterium | 922,984 (89.4%) | 153,395 (89.3%) | 158,000 (91.3%) | 141,368 (86.7%) | 158,205 (91.6%) | 152,017 (89.2%) | 159,999 (88.1%) |
| Uncultured Bifidobacterium sp. | 360 | 82 | 0 | 53 | 0 | 225 (0.1%) | 0 |
| Uncultured Firmicutes bacterium | 275 | 14 | 52 | 65 | 53 | 84 | 7 |
| Uncultured Helicobacter sp. | 58,145 (5.6%) | 14,060 (8.2%) | 9849 (5.7%) | 16,001 (9.8%) | 1213 (0.7) | 13,879 (8.1%) | 3143 (1.7%) |
| Uncultured organism | 2469 (0.2%) | 594 (0.3%) | 236 (0.1%) | 307 (0.2%) | 260 (0.2%) | 274 (0.2%) | 798 (0.4%) |
| Uncultured Shigella sp. | 2669 (0.3%) | 41 | 39 | 168 (0.1%) | 1843 (1.1%) | 40 | 538 (0.3%) |
| Unknown | 24,294 (2.4%) | 2473 (1.4%) | 4628 (2.7%) | 4437 (2.7%) | 3852 (2.2%) | 2703 (1.6%) | 6201 (3.4%) |
| Total | 1,032,761 | 171,727 | 172,967 | 163,133 | 172,738 | 170,517 | 181,679 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cho, S.; Kim, D.; Lee, Y.; Kil, E.-J.; Cho, M.-J.; Byun, S.-J.; Cho, W.K.; Lee, S. Probiotic Lactobacillus Paracasei Expressing a Nucleic Acid-Hydrolyzing Minibody (3D8 Scfv) Enhances Probiotic Activities in Mice Intestine as Revealed by Metagenomic Analyses. Genes 2018, 9, 276. https://doi.org/10.3390/genes9060276
Cho S, Kim D, Lee Y, Kil E-J, Cho M-J, Byun S-J, Cho WK, Lee S. Probiotic Lactobacillus Paracasei Expressing a Nucleic Acid-Hydrolyzing Minibody (3D8 Scfv) Enhances Probiotic Activities in Mice Intestine as Revealed by Metagenomic Analyses. Genes. 2018; 9(6):276. https://doi.org/10.3390/genes9060276
Chicago/Turabian StyleCho, Seungchan, Dongjun Kim, Yongjun Lee, Eui-Joon Kil, Mun-Ju Cho, Sung-June Byun, Won Kyong Cho, and Sukchan Lee. 2018. "Probiotic Lactobacillus Paracasei Expressing a Nucleic Acid-Hydrolyzing Minibody (3D8 Scfv) Enhances Probiotic Activities in Mice Intestine as Revealed by Metagenomic Analyses" Genes 9, no. 6: 276. https://doi.org/10.3390/genes9060276