
Seven Additional Patients with SOX17 Related Pulmonary Arterial Hypertension and Review of the Literature
 , , , , , , , , , , and
, , , , , , , , , , and
Abstract
:1. Introduction
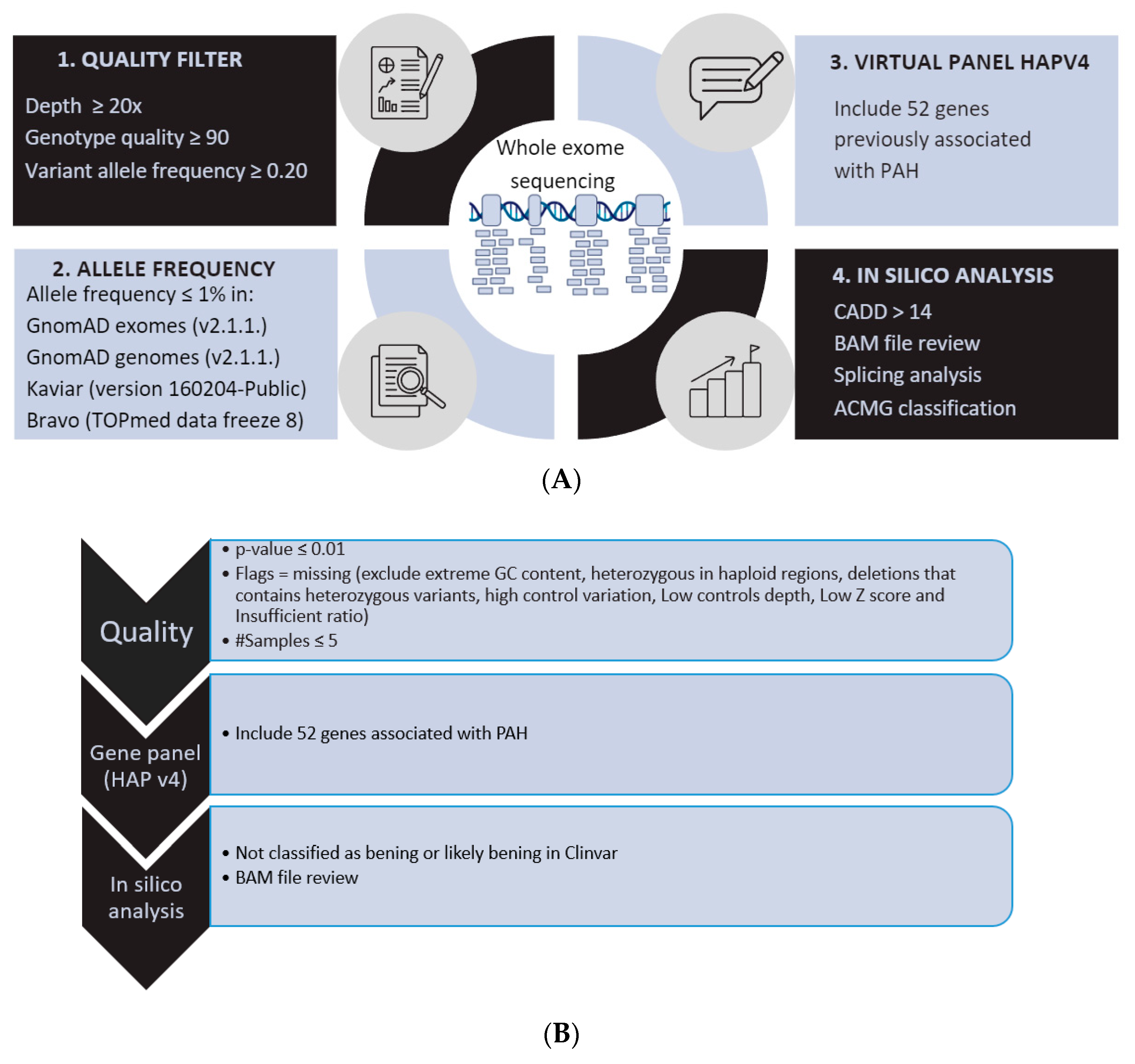
2. Materials and Methods
2.1. Patients
2.2. Genetic Analysis
3. Results
3.1. Clinical Description of New PAH Patients with Variants in SOX17
3.1.1. Patient 1
3.1.2. Patient 2
3.1.3. Patient 3
3.1.4. Patient 4
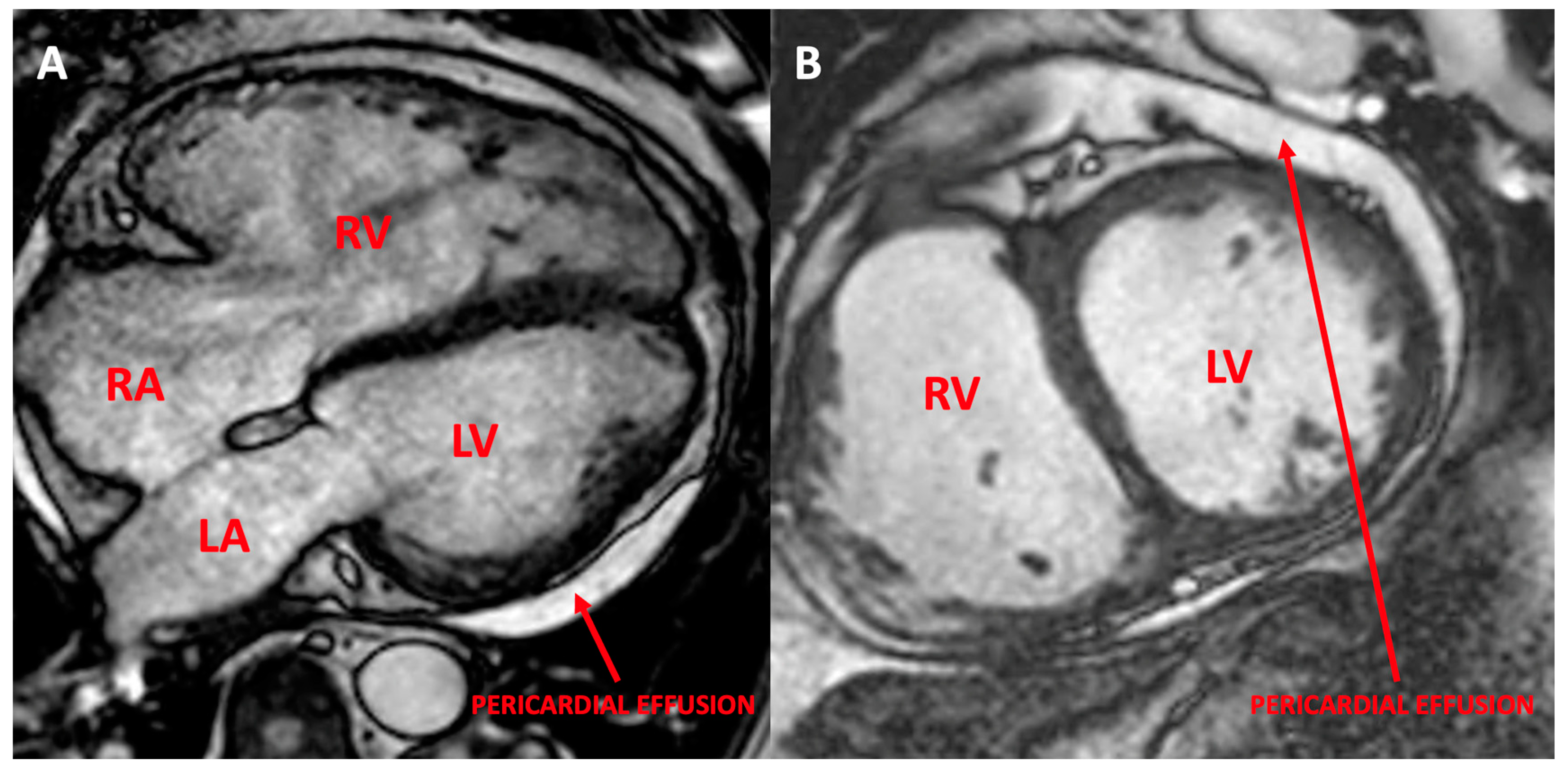
3.1.5. Patient 5
3.1.6. Patient 6
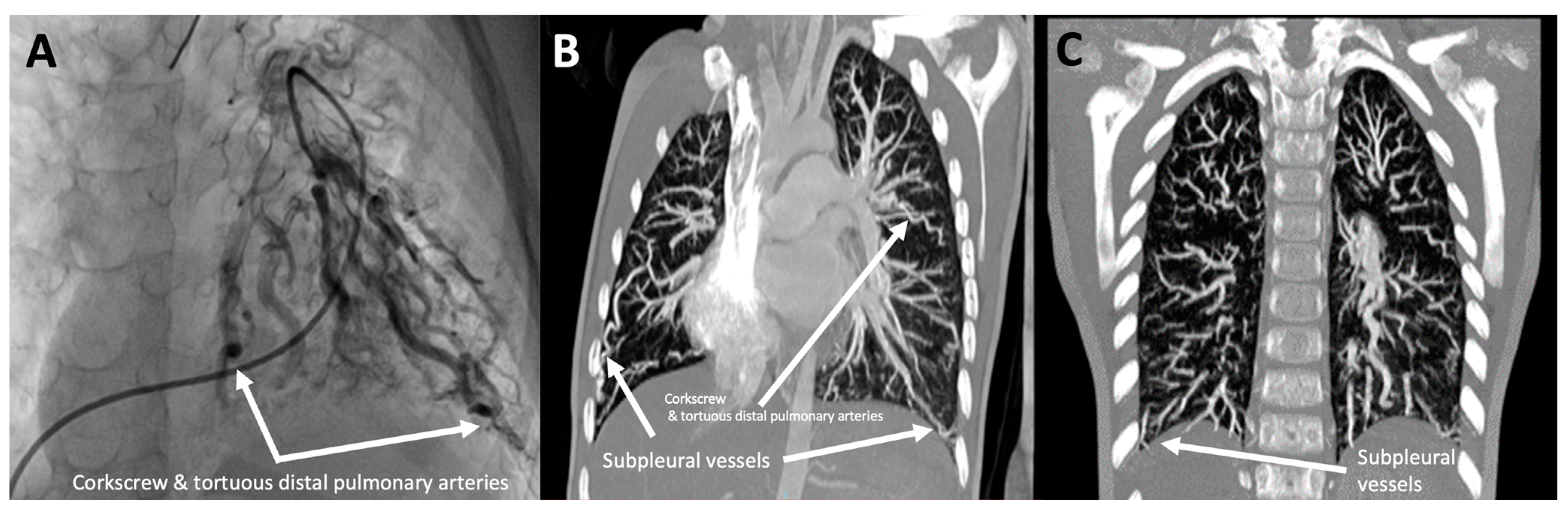
3.1.7. Patient 7
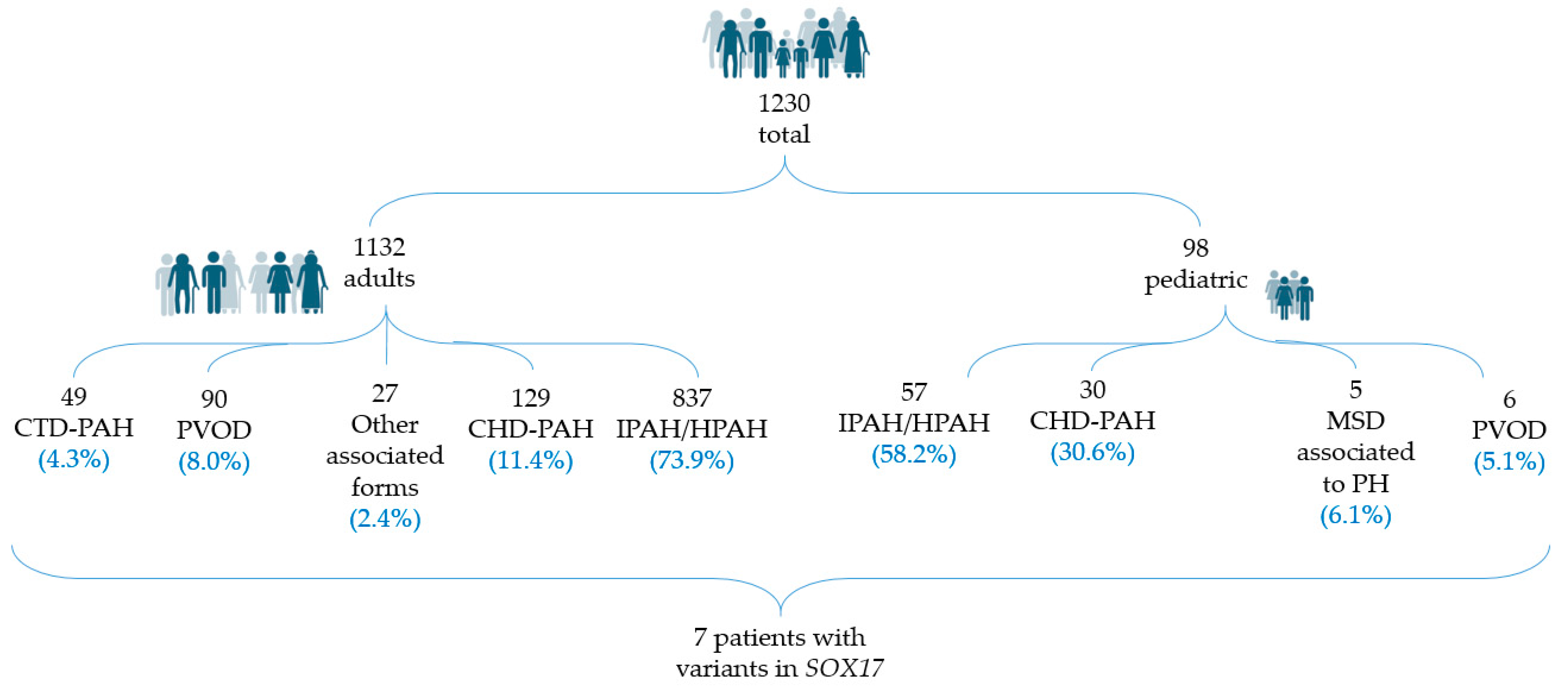
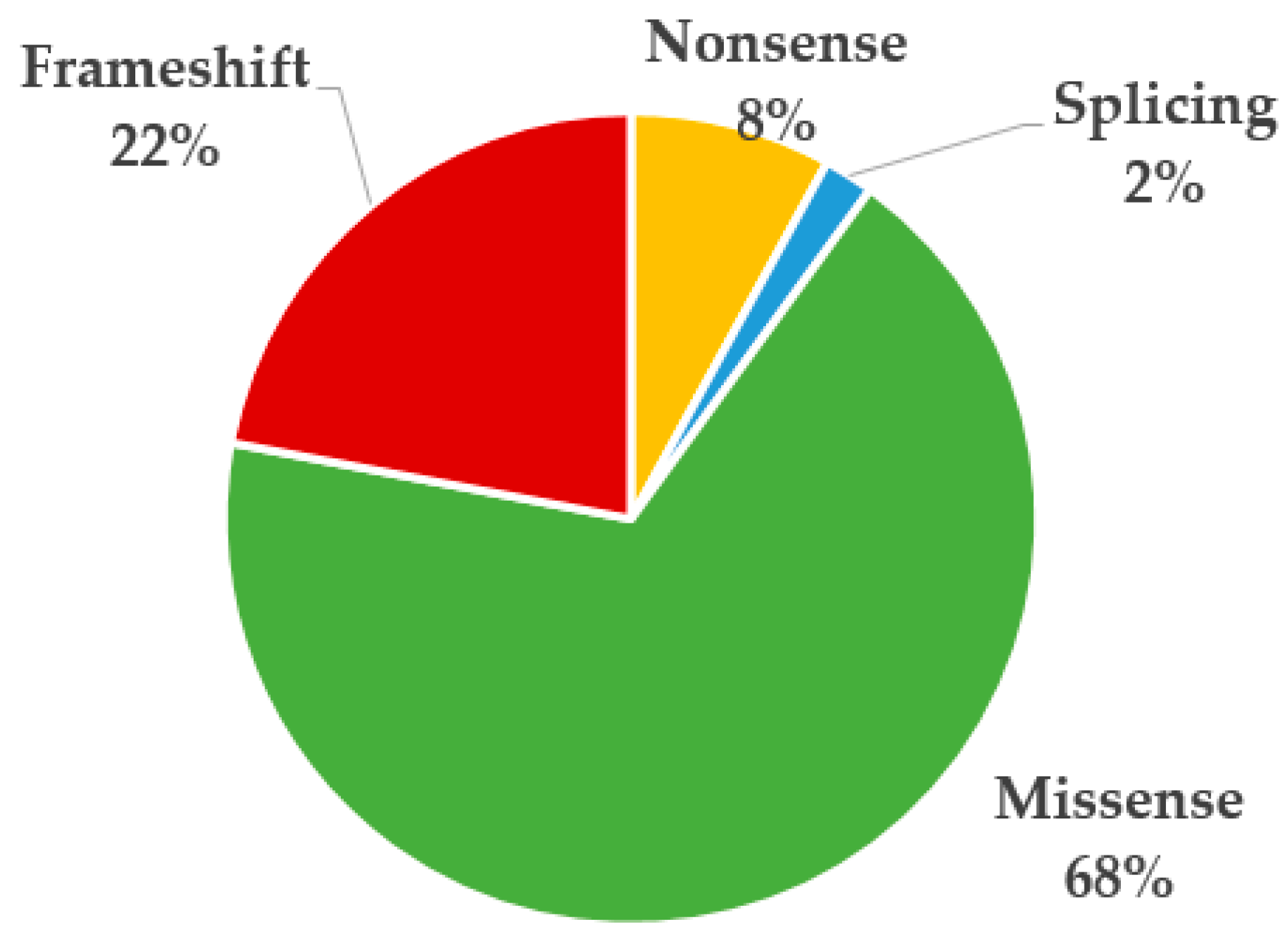
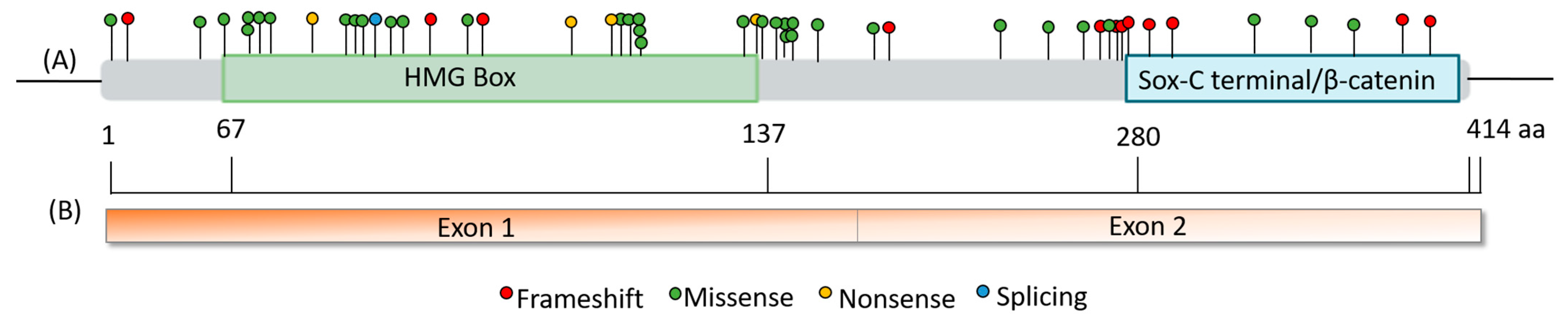
3.2. Genetic and Clinical Review of the Total Number of PAH Patients with Variants in SOX17
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hemnes, A.R.; Humbert, M. Pathobiology of pulmonary arterial hypertension: Understanding the roads less travelled. Eur. Respir. Rev. 2017, 26, 170093. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Kovacs, G.; Hoeper, M.M.; Badagliacca, R.; Berger, R.M.; Brida, M.; Carlsen, J.; Coats, A.J.; Escribano-Subias, P.; Ferrari, P. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: Developed by the task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS). Endorsed by the International Society for Heart and Lung Transplantation (ISHLT) and the European Reference Network on rare respiratory diseases (ERN-LUNG). Eur. Heart J. 2022, 43, 3618–3731. [Google Scholar] [PubMed]
- Girerd, B.; Lau, E.; Montani, D.; Humbert, M. Genetics of pulmonary hypertension in the clinic. Curr. Opin. Pulm. Med. 2017, 23, 386–391. [Google Scholar] [CrossRef]
- Castaño, J.A.T.; Hernández-Gonzalez, I.; Gallego, N.; Pérez-Olivares, C.; Ochoa Parra, N.; Arias, P.; Granda, E.; Acebo, G.G.; Lago-Docampo, M.; Palomino-Doza, J. Customized massive parallel sequencing panel for diagnosis of pulmonary arterial hypertension. Genes 2020, 11, 1158. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.M.; Badesch, D.B.; Ghofrani, H.A.; Gibbs, J.S.R.; Gomberg-Maitland, M.; McLaughlin, V.V.; Preston, I.R.; Souza, R.; Waxman, A.B.; Grünig, E. Phase 3 trial of sotatercept for treatment of pulmonary arterial hypertension. N. Engl. J. Med. 2023, 388, 1478–1490. [Google Scholar] [CrossRef]
- Gräf, S.; Haimel, M.; Bleda, M.; Hadinnapola, C.; Southgate, L.; Li, W.; Hodgson, J.; Liu, B.; Salmon, R.M.; Southwood, M. Identification of rare sequence variation underlying heritable pulmonary arterial hypertension. Nat. Commun. 2018, 9, 1416. [Google Scholar] [CrossRef]
- Welch, C.L.; Aldred, M.A.; Balachandar, S.; Dooijes, D.; Eichstaedt, C.A.; Graf, S.; Houweling, A.C.; Machado, R.D.; Pandya, D.; Prapa, M. Defining the Clinical Validity of Genes Reported to Cause Pulmonary Arterial Hypertension. medRxiv 2023, 25, 100925. [Google Scholar] [CrossRef]
- Zhu, N.; Welch, C.L.; Wang, J.; Allen, P.M.; Gonzaga-Jauregui, C.; Ma, L.; King, A.K.; Krishnan, U.; Rosenzweig, E.B.; Ivy, D.D. Rare variants in SOX17 are associated with pulmonary arterial hypertension with congenital heart disease. Genome Med. 2018, 10, 56. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Kamachi, Y.; Kondoh, H. Sox proteins: Regulators of cell fate specification and differentiation. Development 2013, 140, 4129–4144. [Google Scholar] [CrossRef]
- Sarkar, A.; Hochedlinger, K. The sox family of transcription factors: Versatile regulators of stem and progenitor cell fate. Cell Stem Cell 2013, 12, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Pfister, S.; Jones, V.J.; Power, M.; Truisi, G.L.; Khoo, P.; Steiner, K.A.; Kanai-Azuma, M.; Kanai, Y.; Tam, P.P.; Loebel, D.A. Sox17-dependent gene expression and early heart and gut development in Sox17-deficient mouse embryos. Int. J. Dev. Biol. 2011, 55, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Francois, M.; Koopman, P.; Beltrame, M. SoxF genes: Key players in the development of the cardio-vascular system. Int. J. Biochem. Cell Biol. 2010, 42, 445–448. [Google Scholar] [CrossRef] [PubMed]
- Lange, A.W.; Haitchi, H.M.; LeCras, T.D.; Sridharan, A.; Xu, Y.; Wert, S.E.; James, J.; Udell, N.; Thurner, P.J.; Whitsett, J.A. Sox17 is required for normal pulmonary vascular morphogenesis. Dev. Biol. 2014, 387, 109–120. [Google Scholar] [CrossRef]
- Sakamoto, Y.; Hara, K.; Kanai-Azuma, M.; Matsui, T.; Miura, Y.; Tsunekawa, N.; Kurohmaru, M.; Saijoh, Y.; Koopman, P.; Kanai, Y. Redundant roles of Sox17 and Sox18 in early cardiovascular development of mouse embryos. Biochem. Biophys. Res. Commun. 2007, 360, 539–544. [Google Scholar] [CrossRef]
- Clarke, R.L.; Yzaguirre, A.D.; Yashiro-Ohtani, Y.; Bondue, A.; Blanpain, C.; Pear, W.S.; Speck, N.A.; Keller, G. The expression of Sox17 identifies and regulates haemogenic endothelium. Nat. Cell Biol. 2013, 15, 502–510. [Google Scholar] [CrossRef]
- Yang, H.; Lee, S.; Lee, S.; Kim, K.; Yang, Y.; Kim, J.H.; Adams, R.H.; Wells, J.M.; Morrison, S.J.; Koh, G.Y. Sox17 promotes tumor angiogenesis and destabilizes tumor vessels in mice. J. Clin. Investig. 2012, 123, 418–431. [Google Scholar] [CrossRef]
- Montani, D.; Lechartier, B.; Girerd, B.; Eyries, M.; Ghigna, M.; Savale, L.; Jaïs, X.; Seferian, A.; Jevnikar, M.; Boucly, A. An emerging phenotype of pulmonary arterial hypertension patients carrying SOX17 variants. Eur. Respir. J. 2022, 60, 2200656. [Google Scholar] [CrossRef]
- Saba, R.; Kitajima, K.; Rainbow, L.; Engert, S.; Uemura, M.; Ishida, H.; Kokkinopoulos, I.; Shintani, Y.; Miyagawa, S.; Kanai, Y. Sox17 expression in endocardium precursor cells regulates heart development in mice. Sci. Rep. 2019, 9, 11953. [Google Scholar] [CrossRef]
- Liu, D.; Liu, Q.; Guan, L.; Jiang, X.; Zhou, D.; Beghetti, M.; Qu, J.; Jing, Z. BMPR2 mutation is a potential predisposing genetic risk factor for congenital heart disease associated pulmonary vascular disease. Int. J. Cardiol. 2016, 211, 132–136. [Google Scholar] [CrossRef]
- Roberts, K.E.; McElroy, J.J.; Wong, W.; Yen, E.; Widlitz, A.; Barst, R.J.; Knowles, J.A.; Morse, J.H. BMPR2 mutations in pulmonary arterial hypertension with congenital heart disease. Eur. Respir. J. 2004, 24, 371–374. [Google Scholar] [CrossRef] [PubMed]
- Petty, R.E.; Southwood, T.R.; Manners, P.; Baum, J.; Glass, D.N.; Goldenberg, J.; He, X.; Maldonado-Cocco, J.; Orozco-Alcala, J.; Prieur, A. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: Second revision, Edmonton, 2001. J. Rheumatol. 2004, 31, 390–392. [Google Scholar]
- Weiss, J.E.; Lee, T.; Rabinovich, C.E.; Wallace, C.A.; Oliveira, S.K.; Onel, K.; Anton, J.; Kimura, Y. Life-threatening pulmonary hypertension (PH) and alveolar proteinosis (AP) in systemic JIA (sJIA). In Arthritis and Rheumatism; Wiley-Liss Div John Wiley & Sons Inc.: Hoboken, NJ, USA, 2008. [Google Scholar]
- Kimura, Y.; Weiss, J.E.; Haroldson, K.L.; Lee, T.; Punaro, M.; Oliveira, S.; Rabinovich, E.; Riebschleger, M.; Antón, J.; Blier, P.R. Pulmonary hypertension and other potentially fatal pulmonary complications in systemic juvenile idiopathic arthritis. Arthritis Care Res. 2013, 65, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Pascual, V.; Allantaz, F.; Arce, E.; Punaro, M.; Banchereau, J. Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J. Exp. Med. 2005, 201, 1479–1486. [Google Scholar] [CrossRef] [PubMed]
- Irigoyen, P.I.; Olson, J.; Hom, C.; Ilowite, N.T. Treatment of systemic onset juvenile rheumatoid arthritis with anakinra. In Arthritis and Rheumatism; Wiley-Liss Div John Wiley & Sons Inc.: Hoboken, NJ, USA, 2004. [Google Scholar]
- Price, L.C.; Wort, S.J.; Perros, F.; Dorfmüller, P.; Huertas, A.; Montani, D.; Cohen-Kaminsky, S.; Humbert, M. Inflammation in pulmonary arterial hypertension. Chest 2012, 141, 210–221. [Google Scholar] [CrossRef]
- Wang, T.; Wang, S.; Xu, Y.; Zhao, C.; Qiao, X.; Yang, C.; Liu, X.; Yang, Y. SOX17 loss-of-function mutation underlying familial pulmonary arterial hypertension. Int. Heart J. 2021, 62, 566–574. [Google Scholar] [CrossRef]
- Miyagawa, K.; Shi, M.; Chen, P.; Hennigs, J.K.; Zhao, Z.; Wang, M.; Li, C.G.; Saito, T.; Taylor, S.; Sa, S. Smooth muscle contact drives endothelial regeneration by BMPR2-notch1–mediated metabolic and epigenetic changes. Circ. Res. 2019, 124, 211–224. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 BMP/TGFβ |  Channels |  Transcription Factors |  Other |
|---|---|---|---|
| ACVRL1 BMPR2 ENG GDF2 SMAD9 CAV1 BMP10 BMPR1A BMPR1B SMAD1 SMAD4 | ATP13A3 KCNK3 ABCC8 AQP1 | EIF2AK4 SOX17 TBX4 KLF2 | KDR TET2 GGCX FBLN2 KLK1 PDGFD NOTCH3 |
| Patient ID | HTP1301 Patient 1 | HTP973 Patient 2 | HTP1089 Patient 3 | HTP964 Patient 4 | HTP1010 Patient 5 | HTP379 Patient 6 | SPO Patient 7 |
|---|---|---|---|---|---|---|---|
| Sex | Male | Male | Male | Female | Female | Male | Male |
| PAH diagnosis | IPAH | IPAH | APAH-CHD | APAH-CTD | APAH-CHD | IPAH | IPAH |
| Age at diagnosis (years) | 0.9 (11 months) | 6 | 0.2 (3 months) | 34 | 2 | 54 | 11 |
| NYHA | IV | III | II-III | IV | III | II | III |
| Family history | No | No | No | No | No | No | No |
| Genetic information | |||||||
| Genomic position | 8:55370907 | 8:55370916 | 8:55370942 | 8:55371799 | 8:55371798 | 8:55371798 | 8:54459532 |
| * cDNA position (NM_022454.4) | c.209G>T | c.218A>C | c.244G>T | c.489G>C | c.499_520del | c.499_520del | c.788dup |
| Protein position | p.Arg70Leu | p.Asn73Thr | p.Glu82Ter | p.Gln163His | p.Leu167TrpfsTer213 | p.Leu167TrpfsTer213 | p.Glu264Glyfs101 |
| Hemodynamic parameters at diagnosis | |||||||
| MPAP (mmHg) | No RHC | 52 | 52 | 53 | No RHC *** | 56 | 68 |
| PAWP (mmHg) | No RHC | 5 | 12 | 14 | No RHC | 12 | 9 |
| PVR (Wood units) | No RHC | 16.0 | 16.5 | 15.6 | No RHC | 6.96 | 17.6 |
| 6MWT (meters) | No 6MWT | 530 | 545 | 393 | No 6MWT (developmental delay) | 580 | 450 |
| Associated conditions | Choledochal cyst PFO Small PDA | PFO Positive ANA | Left isomerism, inferior vena cava draining through hemiazygos vein to coronary sinus and right atrium ASD | Juvenile RA T-cell large granular lymphocyte leukemia Hypothyroidism | Big ASD and PDA in Eisenmenger Autism | Severe LVSD and AT at the age of 67 | PFO Hypercoagulability OSA |
| Treatment and follow-up | |||||||
| § Baseline risk (according to the 2022 ESC/ERS Guidelines) | - | - | - | Intermediate risk | - | Intermediate risk | - |
| § Outcomes and current risk according to the 4-strata 2022 ESC/ERS risk score at follow-up | VA-ECMO Lung transplantation | Death | Alive | High risk. Dead at 35 years despite triple sequential therapy including systemic prostacyclins | Lung transplant at age 17 years | Intermediate–low risk under monotherapy with PDE5i | Alive |
| Last known PAH therapies | Epoprostenol | ** Triple sequential therapy | ** Triple sequential therapy | ** Triple sequential therapy | ** Triple sequential therapy | * Tadalafil | Quadruple therapy: Tadalafil, macicentan, Sc treprostinil + sotatercept |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gallego-Zazo, N.; Miranda-Alcaraz, L.; Cruz-Utrilla, A.; del Cerro Marín, M.J.; Álvarez-Fuente, M.; del Mar Rodríguez Vázquez del Rey, M.; Guillén Rodríguez, I.; Becerra-Munoz, V.M.; Moya-Bonora, A.; Ochoa Parra, N.; et al. Seven Additional Patients with SOX17 Related Pulmonary Arterial Hypertension and Review of the Literature. Genes 2023, 14, 1965. https://doi.org/10.3390/genes14101965
Gallego-Zazo N, Miranda-Alcaraz L, Cruz-Utrilla A, del Cerro Marín MJ, Álvarez-Fuente M, del Mar Rodríguez Vázquez del Rey M, Guillén Rodríguez I, Becerra-Munoz VM, Moya-Bonora A, Ochoa Parra N, et al. Seven Additional Patients with SOX17 Related Pulmonary Arterial Hypertension and Review of the Literature. Genes. 2023; 14(10):1965. https://doi.org/10.3390/genes14101965
Chicago/Turabian StyleGallego-Zazo, Natalia, Lucía Miranda-Alcaraz, Alejandro Cruz-Utrilla, María Jesús del Cerro Marín, María Álvarez-Fuente, María del Mar Rodríguez Vázquez del Rey, Inmaculada Guillén Rodríguez, Victor Manuel Becerra-Munoz, Amparo Moya-Bonora, Nuria Ochoa Parra, and et al. 2023. "Seven Additional Patients with SOX17 Related Pulmonary Arterial Hypertension and Review of the Literature" Genes 14, no. 10: 1965. https://doi.org/10.3390/genes14101965