
The Carrier Frequency of Two SMN1 Genes in Parents of Symptomatic Children with SMA and the Significance of SMN1 Exon 8 in Carriers

 and
and
Abstract
:1. Introduction
2. Aims
- To investigate the genotype of SMA carriers using current carrier screening techniques. We aim to determine the proportion of silent carriers (2+0 and 1+1D) in an Australian state-based population and compare this to international data;
- To assess the relationship between parental SMN1 exon 8 copy number and proband genotype.
3. Methods
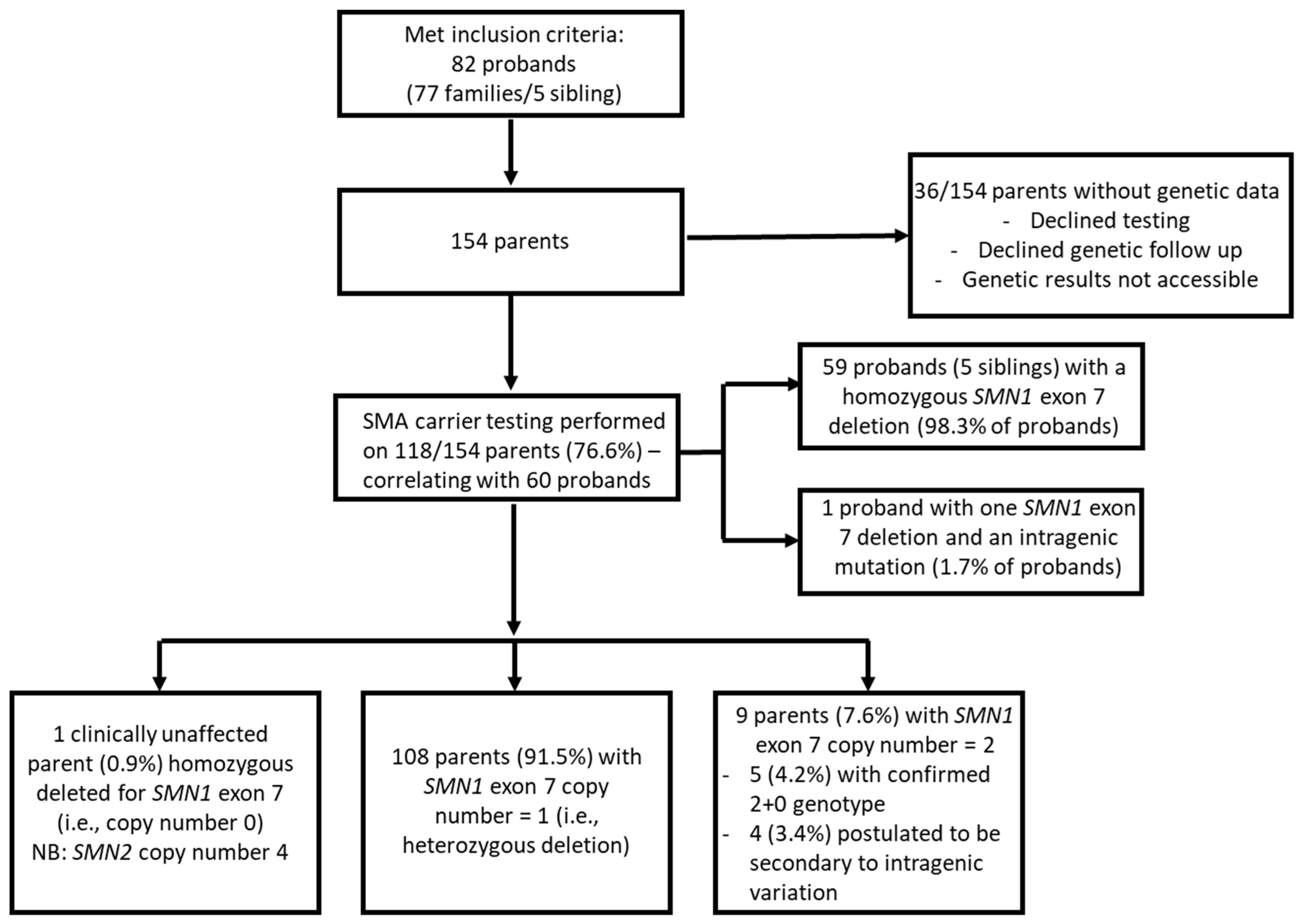
3.1. Study Design and Participants
3.2. Literature Review
3.3. Statistical Analysis
4. Results
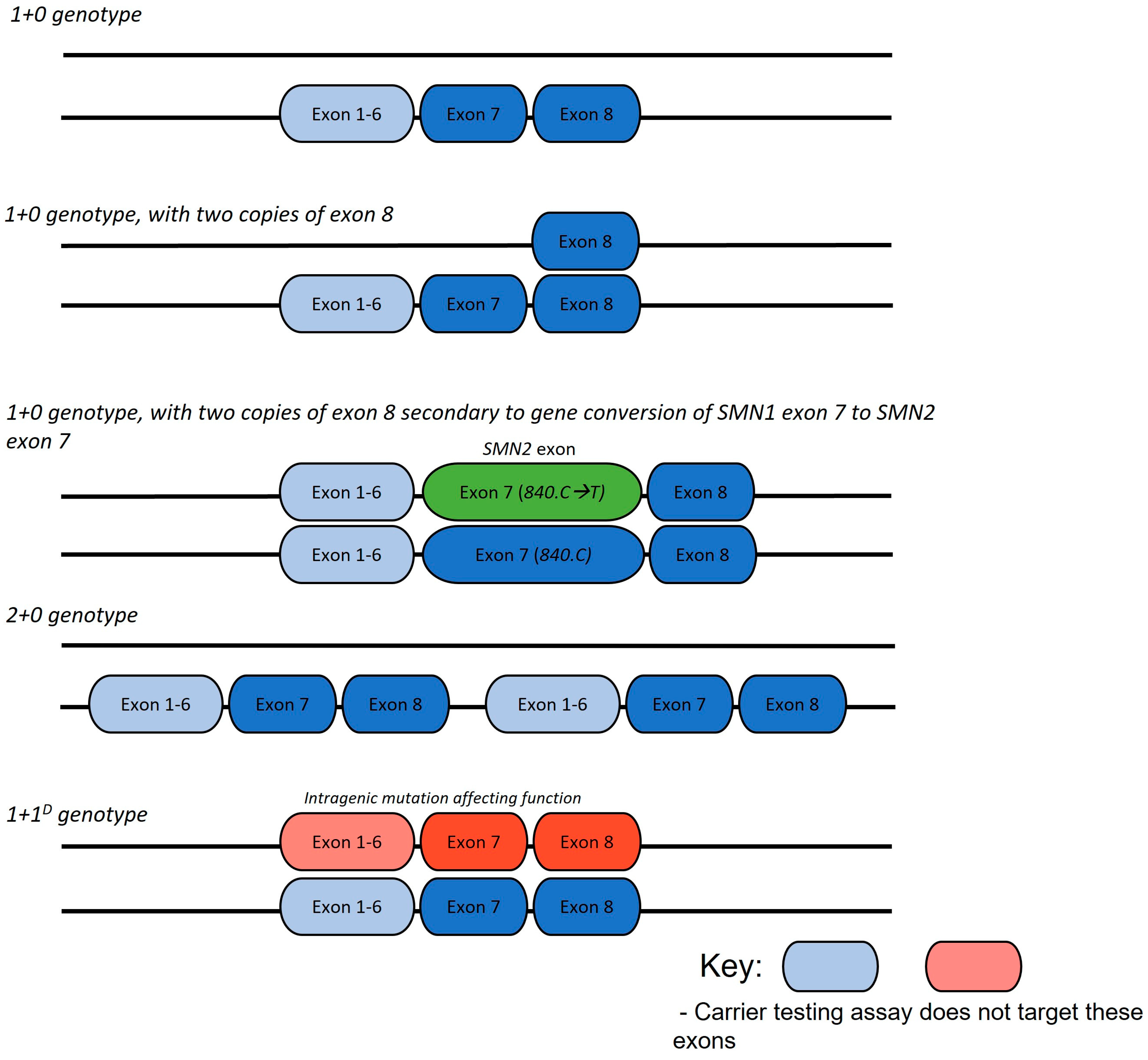
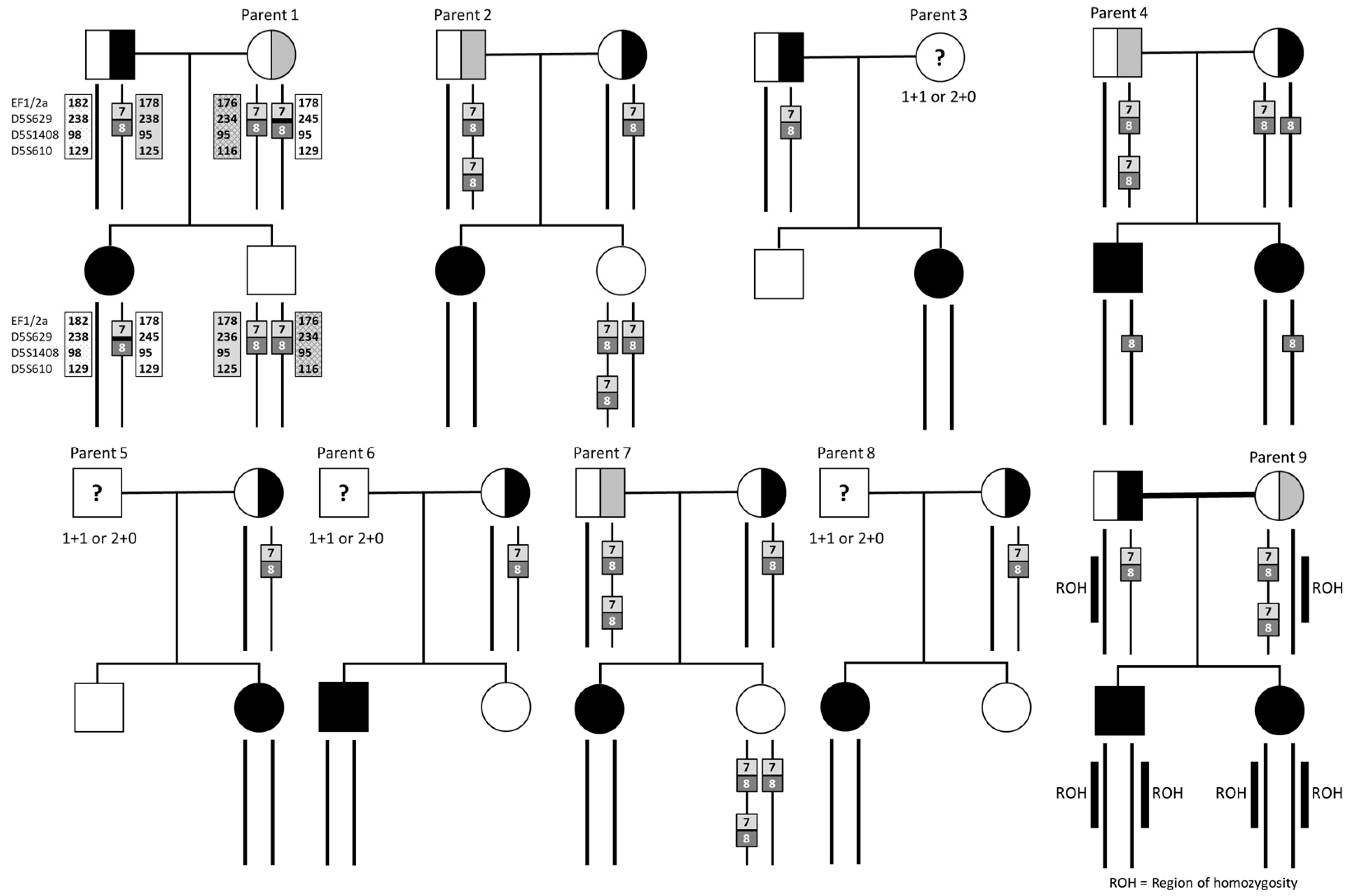
4.1. Parents with Two Copies of SMN1 Exon 7
4.2. Results in the Context of Worldwide Published Literature
4.3. Analysis of Parents with a Heterozygous Deletion of SMN1 Exon 7 and Two Copies of Exon 8
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- D’Amico, A.; Mercuri, E.; Tiziano, F.D.; Bertini, E. Spinal muscular atrophy. Orphanet J. Rare Dis. 2011, 6, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carey, K.A.; Farrar, M.A.; Kasparian, N.A.; Street, D.J.; De Abreu Lourenco, R. Family, healthcare professional, and societal preferences for the treatment of infantile spinal muscular atrophy: A discrete choice experiment. Dev. Med. Child Neurol. 2022, 64, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Farrar, M.A.; Kiernan, M.C. The Genetics of Spinal Muscular Atrophy: Progress and Challenges. Neurotherapeutics 2015, 12, 290–302. [Google Scholar] [CrossRef] [Green Version]
- Burghes, A.H.; Beattie, C.E. Spinal muscular atrophy: Why do low levels of survival motor neuron protein make motor neurons sick? Nat. Rev. Neurosci. 2009, 10, 597–609. [Google Scholar] [CrossRef] [Green Version]
- Rouzier, C.; Chaussenot, A.; Paquis-Flucklinger, V. Molecular diagnosis and genetic counseling for spinal muscular atrophy (SMA). Arch. Pediatr. 2020, 27, 7s9–7s14. [Google Scholar] [CrossRef]
- Calucho, M.; Bernal, S.; Alías, L.; March, F.; Venceslá, A.; Rodríguez-Álvarez, F.J.; Aller, E.; Fernández, R.M.; Borrego, S.; Millán, J.M.; et al. Correlation between SMA type and SMN2 copy number revisited: An analysis of 625 unrelated Spanish patients and a compilation of 2834 reported cases. Neuromuscul. Disord. 2018, 28, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Sonic Genetics. Reproductive Carrier Screening Panel (CF, SMA and Fragile X) Macquarie Park; NSW: Sonic Healthcare, Austin, TX, USA, 2021; Available online: https://www.sonicgenetics.com.au/our-tests/all-tests/reproductive-carrier-screening-panel-cf-sma-and-fragile-x/ (accessed on 23 May 2023).
- Prior, T.W. Carrier screening for spinal muscular atrophy. Genet. Med. 2008, 10, 840–842. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.; Zhang, C.; Teng, Y.; Zeng, S.; Chen, S.; Liang, D.; Li, Z.; Wu, L. Detection of Spinal Muscular Atrophy Using a Duplexed Real-Time PCR Approach with Locked Nucleic Acid-Modified Primers. Ann. Lab. Med. 2021, 41, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Campbell, L.; Daniels, R.J.; Dubowitz, V.; Davies, K.E. Maternal mosaicism for a second mutational event in a type I spinal muscular atrophy family. Am. J. Hum. Genet. 1998, 63, 37–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eggermann, T.; Zerres, K.; Anhuf, D.; Kotzot, D.; Fauth, C.; Rudnik-Schöneborn, S. Somatic mosaicism for a heterozygous deletion of the survival motor neuron (SMN1) gene. Eur. J. Hum. Genet. 2005, 13, 309–313. [Google Scholar] [CrossRef] [Green Version]
- Verhaart, I.E.C.; Robertson, A.; Leary, R.; McMacken, G.; König, K.; Kirschner, J.; Jones, C.C.; Cook, S.F.; Lochmüller, H. A multi-source approach to determine SMA incidence and research ready population. J. Neurol. 2017, 264, 1465–1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendrickson, B.C.; Donohoe, C.; Akmaev, V.R.; Sugarman, E.A.; Labrousse, P.; Boguslavskiy, L.; Flynn, K.; Rohlfs, E.M.; Walker, A.; Allitto, B.; et al. Differences in SMN1 allele frequencies among ethnic groups within North America. J. Med. Genet. 2009, 46, 641–644. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.; Calabro, V.; Chong, B.; Gardiner, N.; Cowie, S.; du Sart, D. Population screening and cascade testing for carriers of SMA. Eur. J. Hum. Genet. 2007, 15, 759–766. [Google Scholar] [CrossRef] [Green Version]
- Farrar, M.A.; Johnston, H.M.; Grattan-Smith, P.; Turner, A.; Kiernan, M.C. Spinal muscular atrophy: Molecular mechanisms. Curr. Mol. Med. 2009, 9, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Sheng-Yuan, Z.; Xiong, F.; Chen, Y.-J.; Yan, T.-Z.; Zeng, J.; Li, L.; Zhang, Y.-N.; Chen, W.-Q.; Bao, X.-H.; Zhang, C.; et al. Molecular characterization of SMN copy number derived from carrier screening and from core families with SMA in a Chinese population. Eur. J. Hum. Genet. EJHG 2010, 18, 978–984. [Google Scholar] [CrossRef] [PubMed]
- Ar Rochmah, M.A.; Awano, H.; Awaya, T.; Harahap, N.I.F.; Morisada, N.; Bouike, Y.; Saito, T.; Kubo, Y.; Saito, K.; Lai, P.S.; et al. Spinal muscular atrophy carriers with two SMN1 copies. Brain Dev. 2017, 39, 851–860. [Google Scholar] [CrossRef]
- Mailman, M.; Hemingway, T.; Darsey, R.; Glasure, C.; Huang, Y.; Chadwick, R.; Heinz, J.; Papp, A.; Snyder, P.; Sedra, M.; et al. Hybrids monosomal for human chromosome 5 reveal the presence of a spinal muscular atrophy (SMA) carrier with two SMN1 copies on one chromosome. Hum. Genet. 2001, 108, 109–115. [Google Scholar] [CrossRef]
- Alías, L.; Barceló, M.; Bernal, S.; Martínez-Hernández, R.; Also-Rallo, E.; Vázquez, C.; Santana, A.; Millán, J.; Baiget, M.; Tizzano, E. Improving detection and genetic counseling in carriers of spinal muscular atrophy with two copies of the SMN1 gene. Clin. Genet. 2014, 85, 470–475. [Google Scholar] [CrossRef]
- Yoon, S.; Lee, C.H.; Lee, K.A. Determination of SMN1 and SMN2 copy numbers in a Korean population using multiplex ligation-dependent probe amplification. Korean J. Lab. Med. 2010, 30, 93–96. [Google Scholar] [CrossRef] [Green Version]
- Sangaré, M.; Hendrickson, B.; Sango, H.A.; Chen, K.; Ba, J.N.; Amara, A.; Dutra, A.; Schindler, A.B.; Guindo, A.; Traoré, M.; et al. Genetics of low spinal muscular atrophy carrier frequency in sub-Saharan Africa. Ann. Neurol. 2014, 75, 525–532. [Google Scholar] [CrossRef] [Green Version]
- Farrar, M.A.; Kiernan, M.C. Spinal muscular atrophy-the dawning of a new era. Nat. Rev. Neurology 2020, 16, 593–594. [Google Scholar] [CrossRef] [PubMed]
- D’Silva, A.M.; Kariyawasam, D.S.T.; Best, S.; Wiley, V.; Farrar, M.A.; Group, N.S.N.S. Integrating newborn screening for spinal muscular atrophy into health care systems: An Australian pilot programme. Dev. Med. Child Neurol. 2022, 64, 625–632. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, W.K.; Hamilton, D.; Kuhle, S. SMA carrier testing: A meta-analysis of differences in test performance by ethnic group. Prenat. Diagn. 2014, 34, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Wirth, B.; Herz, M.; Wetter, A.; Moskau, S.; Hahnen, E.; Rudnik-Schöneborn, S.; Wienker, T.; Zerres, K. Quantitative analysis of survival motor neuron copies: Identification of subtle SMN1 mutations in patients with spinal muscular atrophy, genotype-phenotype correlation, and implications for genetic counseling. Am. J. Hum. Genet. 1999, 64, 1340–1356. [Google Scholar] [CrossRef] [Green Version]
- Wirth, B.; Schmidt, T.; Hahnen, E.; Rudnik-Schöneborn, S.; Krawczak, M.; Müller-Myhsok, B.; Schönling, J.; Zerres, K. De novo rearrangements found in 2% of index patients with spinal muscular atrophy: Mutational mechanisms, parental origin, mutation rate, and implications for genetic counseling. Am. J. Hum. Genet. 1997, 61, 1102–1111. [Google Scholar] [CrossRef] [Green Version]
- Archibald, A.D.; McClaren, B.J.; Caruana, J.; Tutty, E.; King, E.A.; Halliday, J.L.; Best, S.; Kanga-Parabia, A.; Bennetts, B.H.; Cliffe, C.C.; et al. The Australian Reproductive Genetic Carrier Screening Project (Mackenzie’s Mission): Design and Implementation. J. Pers. Medicine 2022, 12, 1781. [Google Scholar] [CrossRef]
- Li, S.; Han, X.; Xu, Y.; Chang, C.; Gao, L.; Li, J.; Lu, Y.; Mao, A.; Wang, Y. Comprehensive Analysis of Spinal Muscular Atrophy: SMN1 Copy Number, Intragenic Mutation, and 2 + 0 Carrier Analysis by Third-Generation Sequencing. J. Mol. Diagn. 2022, 24, 1009–1020. [Google Scholar] [CrossRef]
- Schofield, D.; Lee, E.; Parmar, J.; Kelly, S.; Hobbs, M.; Laing, N.; Mumford, J.; Shrestha, R. Economic evaluation of population-based, expanded reproductive carrier screening for genetic diseases in Australia. Genet. Med. Off. J. Am. Coll. Med. Genetics 2023, 25, 100813. [Google Scholar] [CrossRef]
- Luo, M.; Liu, L.; Peter, I.; Zhu, J.; Scott, S.A.; Zhao, G.; Eversley, C.; Kornreic, R.; Desnick, R.J.; Edelmann, L. An Ashkenazi Jewish SMN1 haplotype specific to duplication alleles improves pan-ethnic carrier screening for spinal muscular atrophy. Genet. Med. Off. J. Am. Coll. Med. Genetics 2014, 16, 149–156. [Google Scholar] [CrossRef] [Green Version]
- Alías, L.; Bernal, S.; Calucho, M.; Martínez, E.; March, F.; Gallano, P.; Fuentes-Prior, P.; Abuli, A.; Serra-Juhe, C.; Tizzano, E.F. Utility of two SMN1 variants to improve spinal muscular atrophy carrier diagnosis and genetic counselling. Eur. J. Hum. Genet. EJHG 2018, 26, 1554–1557. [Google Scholar] [CrossRef]
- Vidal-Folch, N.; Gavrilov, D.; Raymond, K.; Rinaldo, P.; Tortorelli, S.; Matern, D.; Oglesbee, D. Multiplex Droplet Digital PCR Method Applicable to Newborn Screening, Carrier Status, and Assessment of Spinal Muscular Atrophy. Clin. Chem. 2018, 64, 1753–1761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azad, A.K.; Huang, C.K.; Jin, H.; Zou, H.; Yanakakis, L.; Du, J.; Fiddler, M.; Naeem Goldstein, Y. Enhanced Carrier Screening for Spinal Muscular Atrophy: Detection of Silent (SMN1: 2 + 0) Carriers Utilizing a Novel TaqMan Genotyping Method. Lab. Med. 2020, 51, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Ge, X.; Meng, L.; Scull, J.; Li, J.; Tian, X.; Zhang, T.; Jin, W.; Cheng, H.; Wang, X.; et al. The next generation of population-based spinal muscular atrophy carrier screening: Comprehensive pan-ethnic SMN1 copy-number and sequence variant analysis by massively parallel sequencing. Anesthesia Analg. 2017, 19, 936–944. [Google Scholar] [CrossRef] [PubMed]
- Ceylan, A.C.; Erdem, H.B.; Şahin, İ.; Agarwal, M. SMN1 gene copy number analysis for spinal muscular atrophy (SMA) in a Turkish cohort by CODE-SEQ technology, an integrated solution for detection of SMN1 and SMN2 copy numbers and the “2+0” genotype. Neurol. Sci. 2020, 41, 2575–2584. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Sanchis-Juan, A.; French, C.E.; Connell, A.J.; Delon, I.; Kingsbury, Z.; Chawla, A.; Halpern, A.L.; Taft, R.J.; Bentley, D.R.; et al. Spinal muscular atrophy diagnosis and carrier screening from genome sequencing data. Anesthesia Analg. 2020, 22, 945–953. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Genetic Heritage | Total Number of Parents with Genetic Data | Parents with Two SMN1 Copies | 2+0 Genotype | Point Mutation | Not Determined |
|---|---|---|---|---|---|
| Caucasian | 76 (64.4%) | 7/76 (9.2%) | 2/76 (2.6%) | 1/76 (1.3%) | 4/76 (5.3%) |
| Hispanic | 1 (0.8%) | 0 | - | - | - |
| East Asian 1 | 6 (5.1%) | 1/6 (16.7%) | 1/6 (16.7%) | - | - |
| South Asian 2 | 14 (11.9%) | 1/14 (7.1%) | 1/14 (7.1%) | - | - |
| West Asian 3 | 14 (11.9%) | 0 | - | - | - |
| African | 5 (4.2%) | 0 | - | - | - |
| Polynesian | 2 (1.7%) | 0 | - | - | - |
| Author | Population | Proband Genetics | Number Analysed | Result | Genotype |
|---|---|---|---|---|---|
| Sheng-Yuan et al. (2010) [16] | Chinese | 8% compound heterozygote 92% homozygous deletion | 44 parents | 4/44 (9.1%) had two SMN1 exon 7 copies | 2 (4.5%) intragenic mutation 2 (4.5%) 2+0 genotype |
| Ar Rochmah et al. (2017) [17] | Japanese | 3% compound heterozygote 97% homozygous deletion | 65 parents | 3/65 (4.6%) had two SMN1 exon 7 copies | 1 (1.5%) intragenic mutation 1 (1.5%) 2+0 genotype 1 (1.5%) not further studied |
| Total Combined | 6.4% (7/109) had 2 SMN1 exon 7 copies | ||||
| This Study | Australian (NSW) | 1.7% compound heterozygote 98.3% homozygous deletion | 118 parents | 9/118 (7.6%) had 2 SMN1 exon 7 copies | 1 (0.9%) intragenic mutation 4 (3.4%) probable 2+0 genotype 4 (3.4%) not further studied |
| Total Combined | 16/227 (7.1%) had 2 SMN1 exon 7 copies | ||||
| Author | Population | Number Analysed | Result | Genotype |
|---|---|---|---|---|
| Mailman et al. (2001) [18] | North American | 100 parents | 4/100 (4.0%) had 2 SMN1 exon 7 copies | 1 (1%) 2+0 genotype 1 (1%) de novo mutation 2 (2%) not further studied |
| Smith et al. (2007) [14] | Australian (Victoria) | 117 parents | 7/117 (6.0%) had 2 SMN1 exon 7 copies | 2 (1.7%) 2+0 genotype 2 (1.7%) de novo mutation 3 (2.6%) undetermined |
| Sheng-Yuan et al. (2010) [16] | Chinese | 40 parents | 2/40 (5.0%) had 2 SMN1 exon 7 copies | 2 (5.0%) 2+0 genotype |
| Alias et al. (2014) [19] | Spanish | 488 parents | 21/488 (4.3%) had 2 SMN1 exon 7 copies | 15 (3.1%) 2+0 genotype 5 (1.0%) de novo mutation 1 (0.2%) undetermined |
| Ar Rochmah et al. (2017) [17] | Japanese | 63 parents | 2/63 (3.2%) had 2 SMN1 exon 7 copies | 1 (1.6%) 2+0 genotype 1 (1.6%) not further studied |
| Statistics | Median: 4.5%, Mean: 4.5%, SD: 1.05%, 95% CI: 3.63–5.46% | |||
| Total Combined | 36/808 (4.5%) had 2 SMN1 exon 7 copies | |||
| This study | Australian (NSW) | 116 parents | 8/116 (6.9%) had 2 SMN1 exon 7 copies | 3 (2.6%) probable 2+0 genotype 5 (4.3%) not further studied |
| Total Combined | 44/924 (4.8%) had 2 SMN1 exon 7 copies | |||
| Probands with Two or Less SMN2 Copies | Probands with Three or More SMN2 Copies | Total | |
|---|---|---|---|
| Carrier with a 1:1 copy number ratio of SMN1 exon 7 to exon 8 | 15 | 21 | 36 |
| Carrier with heterozygous deletion of SMN1 exon 7 and 2 copies exon 8 | 0 | 6 | 6 |
| Total | 15 | 27 | 42 |
| Author | Methods | Result |
|---|---|---|
| Luo et al. (2014) [30] | Microsatellite analysis identified haplotype blocks and next-generation sequencing identified specific SNPs associated with the 2+0 genotype in those with Ashkenazi Jewish heritage. | Identified g.27134T>G in intron 7 and g.27706_27707delAT in exon 8 as polymorphisms associated with 2+0 carrier status. This also had predictive value in those of an African American, Asian, Hispanic, and Caucasian background. |
| Alias et al. (2018) [31] | Examined g.27134T>G and g.27706_27707delAT in a Spanish population | It was found that the presence of the SNPs increased or reduced the residual carrier risk in this population. |
| PCR-Based Technologies | ||
| Vidal-Folch et al. (2018) [32] | Used digital droplet PCR to simultaneously screen for SMN1 and SMN2 copy numbers as well as the g.27134T>G SNP. | Allowed for accurate screening of carriers without requiring standard curves. Interassay imprecision was <7.1% CV and interassay imprecision was <6.0% CV. Testing was 100% specific and sensitive in SMA. |
| Azad et al. (2020) [33] | Custom SNP-specific assay using TaqMan genotyping technology to determine CNV of SMN1, SMN2 and presence of g.27134T>G SNP | Results are 100% concordant with standard PCR in 21 pilot samples. Good reproducibility with a 1–4% CV for all genotypes. |
| Next-Generation Sequencing Technologies | ||
| Feng et al. (2017) [34] | Paralogous gene copy-number analysis by ratio and sum to detect SMN1, SMN2 copy numbers, and g.27134T>G SNP on short-read next-generation sequence | Results are 100% sensitive and 99.6% specific to detect 1 copy of SMN1, and 100% concordant detection of g.27134T>G SNP compared to RFLP assay. Carrier detection rates increased according to ethnic heritage; African American (70.5% to 90.3%), Ashkenazi Jewish (90.5% to 92.8%), Asian (93.3% to 93.6%), Caucasian (94.8% to 95%), and Hispanic (90% to 92.6%) |
| Ceylan et al. (2020) [35] | Used CODE-SEQ technology; an NGS assay of 18 pairs of coded oligonucleotides coupled with a unique probe to target SMA-related loci and reference regions | Results show 100% correlation with the MLPA results in all 80 samples tested for exon 7 SMN1 CN. Unable to test the accuracy in the detection of the g.27134T>G SNP as none were present in the sample. |
| Chen et al. (2020) [36] | Analysed read depth and used eight reference genome differences to identify copy number of SMN1 and SMN2 as well as g.27134T>G SNP. | Accuracy of 99.8% and 99.7% for SMN1 and SMN2 CN compared to qPCR and MLPA, precision of 100% for both SMA and 1+0 carrier status. Carrier detection rates increased with SNP by 21.3% in those with African heritage and 2% at most for all other heritages. |
| Li et al. (2022) [28] | Utilised long-range PCR and third-generation sequencing of full-length and downstream regions of SMN1 and SMN2. | Improved detection rates of SMA carriers in a Chinese population from 91% to 98%, including three SMN1 intragenic mutations and an in-frame mutation to SMN2. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davidson, J.E.; Russell, J.S.; Martinez, N.N.; Mowat, D.R.; Jones, K.J.; Kirk, E.P.; Kariyawasam, D.; Farrar, M.; D’Silva, A. The Carrier Frequency of Two SMN1 Genes in Parents of Symptomatic Children with SMA and the Significance of SMN1 Exon 8 in Carriers. Genes 2023, 14, 1403. https://doi.org/10.3390/genes14071403
Davidson JE, Russell JS, Martinez NN, Mowat DR, Jones KJ, Kirk EP, Kariyawasam D, Farrar M, D’Silva A. The Carrier Frequency of Two SMN1 Genes in Parents of Symptomatic Children with SMA and the Significance of SMN1 Exon 8 in Carriers. Genes. 2023; 14(7):1403. https://doi.org/10.3390/genes14071403
Chicago/Turabian StyleDavidson, Joanne E, Jacqueline S Russell, Noelia Nunez Martinez, David R Mowat, Kristi J Jones, Edwin P Kirk, Didu Kariyawasam, Michelle Farrar, and Arlene D’Silva. 2023. "The Carrier Frequency of Two SMN1 Genes in Parents of Symptomatic Children with SMA and the Significance of SMN1 Exon 8 in Carriers" Genes 14, no. 7: 1403. https://doi.org/10.3390/genes14071403