
Transcriptomic Analysis of Long Non-Coding RNA during Candida albicans Infection


1
Center for Biotechnology, Federal University of Rio Grande do Sul, Porto Alegre 91501-970, Brazil
2
School of Health and Life Sciences, Pontifical Catholic University of Rio Grande do Sul, Porto Alegre 90619-900, Brazil
3
National Institute of Science and Technology—Forensic Science, Porto Alegre 91501-970, Brazil
4
Institute of Informatics, Federal University of Rio Grande do Sul, Porto Alegre 91501-970, Brazil
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Genes 2023, 14(2), 251; https://doi.org/10.3390/genes14020251
Submission received: 1 November 2022
/
Revised: 7 January 2023
/
Accepted: 16 January 2023
/
Published: 18 January 2023
(This article belongs to the Section RNA)
Abstract
:Candida albicans is one of the most commonly found species in fungal infections. Due to its clinical importance, molecular aspects of the host immune defense against the fungus are of interest to biomedical sciences. Long non-coding RNAs (lncRNAs) have been investigated in different pathologies and gained widespread attention regarding their role as gene regulators. However, the biological processes in which most lncRNAs perform their function are still unclear. This study investigates the association between lncRNAs with host response to C. albicans using a public RNA-Seq dataset from lung samples of female C57BL/6J wild-type Mus musculus with induced C. albicans infection. The animals were exposed to the fungus for 24 h before sample collection. We selected lncRNAs and protein-coding genes related to the host immune response by combining the results from different computational approaches used for gene selection: differential expression gene analysis, co-expression genes network analysis, and machine learning-based gene selection. Using a guilt by association strategy, we inferred connections between 41 lncRNAs and 25 biological processes. Our results indicated that nine up-regulated lncRNAs were associated with biological processes derived from the response to wounding: 1200007C13Rik, 4833418N02Rik, Gm12840, Gm15832, Gm20186, Gm38037, Gm45774, Gm4610, Mir22hg, and Mirt1. Additionally, 29 lncRNAs were related to genes involved in immune response, while 22 lncRNAs were associated with processes related to reactive species production. These results support the participation of lncRNAs during C. albicans infection, and may contribute to new studies investigating lncRNA functions in the immune response.
1. Introduction
Species of Candida are widely found in the human microbiota. Although mostly harmless for healthy individuals, these fungi can cause severe infection in immunocompromised individuals [1]. Candidemia (bloodstream candidiasis) and invasive candidiasis are life-threatening and recurrent in hospital environments, affecting patients undergoing immunosuppressive therapy, hemodialysis, recovering from organ transplantation, and even debilitated low-birth-weight infants [2,3]. Although different Candida species are clinically relevant, such as Candida glabrata, C. parapsilosis, C. tropicalis [4], and C. auris [5], C. albicans remains the most common pathogen responsible for candidemia [6].
Long non-coding RNA (lncRNA) are molecules with a length longer than 200 nucleotides, subdivided according to their genomic localization or transcription process: antisense, bidirectional, intergenic, intronic, or sense-overlapping lncRNA [7]. These transcripts are essential to several cellular processes related to gene regulation, such as enhancement or interference during transcription, as well as post-transcriptional modifications [8]. A single lncRNA can also target different genes and molecules and therefore participate in more than one form of regulation.
The extent of lncRNA regulatory contribution to host defense against pathogens, or eventually aggravating the infection, remains a research topic [9]. Some reports indicate specific lncRNAs as potential transcription regulators during infections promoted by pathogens such as Mycobacterium tuberculosis [10] and SARS-CoV-2 [11]. Only a few studies have examined lncRNA’s association with fungal infections. Human lncRNAs RP11-588G21.2 and RP11-394I13.1 have been identified and proposed as potential biomarkers for C. albicans infection [12]. Hou et al. [13] recently reported the lncRNA lnc-CCL3L3-1:1 as potentially related to the human inflammatory response to -glucan, while analyzing gene expression of CD14+ monocytes stimulated by C. albicans cell wall polysaccharide.
According to Ensembl BioMart data [14], 42% of the lncRNAs in Homo sapiens (GRCh38.p13) are annotated as novel transcripts, indicating these molecules are yet to be characterized. At the same time, the number reaches 64% for the model organism Mus musculus (GRCm39) when also including novel transcripts from predicted genes. Even among annotated lncRNA, less than 3% have known functions [15].
To understand how lncRNAs can participate in host immune response during C. albicans infection, we propose new associations between these molecules and biological processes in M. musculus by using a public RNA-seq dataset and multiple bioinformatic approaches. Our results suggest the association of 41 lncRNAs not previously described in C. albicans infection. The present study shows that these lncRNAs are associated with multiple biological processes related to inflammation, response to wounding, and host defense.
2. Materials and Methods
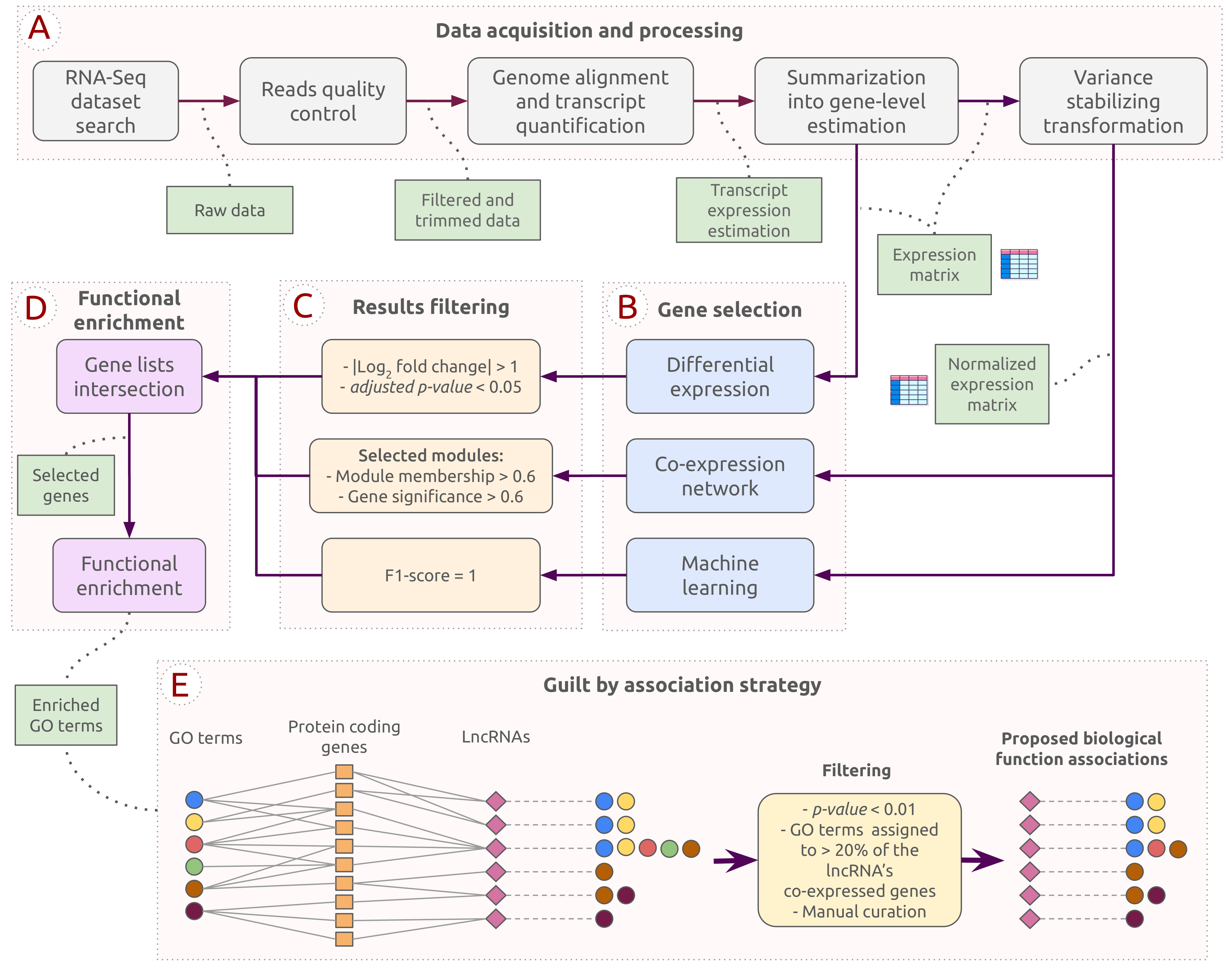
The pipeline employed in this work was designed to explore potential functional roles of lncRNAs during C. albicans infection. Figure 1 provides the steps for all analyses performed and the chosen thresholds. The pipeline was divided into stages consisting of data acquisition and processing, the three methods for gene selection, results filtering and intersection, functional enrichment analysis, and a guilt by association strategy. This section will subsequently detail the tools and parameters determined for each step. The source code and hyperparameters used in the experiments and results can be accessed on GitHub: https://github.com/sbcblab/candida-infection-pipeline, (accessed on 3 May 2021).
2.1. Samples and Data Preprocessing
The dataset used in this study is available at NCBI Gene Expression Omnibus (GEO) under the identifier GSE119853 [16]. It consists of 19 lung samples from female C57BL/6J wild-type M. musculus—11 samples from control animals and 8 samples from animals exposed to C. albicans for 24 h previous to sample collection.
For preprocessing the raw data, tools FastQC (v. 0.11.09, https://www.bioinformatics.babraham.ac.uk/projects/fastqc, accessed on 3 May 2021) and Trimmomatic (v. 0.39) [17] were applied to perform quality control and trimming or removal of low-quality sequence reads. Adapter sequences indicated by FastQC were also removed.
2.2. Gene Expression Matrix and Analysis
After preprocessing, raw reads were mapped against the M. musculus reference genome (GRCm39) with the STAR (v. 2.7.9A) [18] aligner through RSEM (v. 1.17) [19]. RSEM was used to quantify the transcripts previously found. By using the tximport package (v. 1.24.0) [20] implemented in R, RSEM results were summarized in a gene-level matrix. The summarized matrix (m genes x n samples) was imported to the DESeq2 package (v. 1.36.0) [21] for gene expression analysis. Genes with and p-value < 0.05 were considered differentially expressed genes (DEGs). The Benjamini–Hochberg method was used to adjust the p-value.
A normalized version of the gene expression matrix was obtained using the varianceStabilizingTransformation function (VST) from the DESeq2 package. The resulting normalized matrix was further used as input to construct the co-expression networks and perform the machine learning analysis.
2.3. Co-Expression Network Construction and Analysis
The WGCNA package (v. 1.71) [22] was employed to construct and analyze a weighted gene co-expression network. The normalized matrix was utilized as input, and the goodSamplesGenes function was applied to remove genes with low variance or those infrequent amongst all samples. The following settings were chosen for the generation of the network: signed network type; (soft-threshold parameter chosen based on the scale-free topology criterion, > 0.8); ; ; .
A Pearson correlation between trait information and module eigengene (ME) was performed to investigate whether the network modules were significantly associated with the clinical condition. ME is considered the first principal component of the module and summarizes the module’s overall expression. For further analysis, we selected the module with the strongest correlation coefficient related to the disease and control condition.
To identify the most significant genes related to the clinical conditions, we considered the measures of module membership (MM), which represents the correlation between gene expression and ME, and the gene significance (GS), which describes the correlation between gene expression and the external trait. The mentioned measures were used to filter the most relevant genes for each module using cut-offs of and .
2.4. Support Vector Classification
A machine learning approach was employed to select the genes most likely related to each condition. With the normalized matrix serving as input, each gene was individually evaluated by a support vector classification (SVC) algorithm available at SciKit Learn (v. 1.1.2, https://scikit-learn.org, accessed on 30 July 2021) [23]. Due to the reduced sample size, the leave-one-out method was used as a cross-validation strategy to divide the normalized gene expression values into train and test subsets. Using the f1-score to evaluate classification success, we selected those genes whose prediction reached f1-score = 1.0. This rigorous threshold was established to reinforce the selection of only genes for which the expression value range did not overlap between healthy and infected samples, i.e., genes distinctly expressed under all the samples from both conditions.
2.5. Functional Enrichment Analysis
The selected genes from both co-expression modules were submitted to functional enrichment analysis using the topGO package (v. 2.48.0) [24] to investigate the biological processes associated with the data. Only Gene Ontology (GO) terms with an p-value < 0.05 were retrieved. The method used to correct the p-value was the false discovery rate (FDR). The topGO results were manually investigated for further discussion.
Moreover, we explore the functional enrichment results associated with the lncRNAs. To perform that, we created a subnetwork considering only the lncRNAs and their protein-coding genes’ first-degree neighbors. These lncRNA–coding gene interactions must show a moderate or strong correlation coefficient (≥0.4). After, functional enrichment was performed on the subnetwork. The GO terms assigned to the co-expressed protein-coding genes were summarized for each lncRNA considering only the resulting GO terms with p-value < 0.01 and enriched for at least of protein-coding genes interacting with each lncRNA. These results were manually inspected and selected for the guilt by association strategy.
2.6. Gene Biotype Classification
The biomaRt package (v. 2.52.0) [25] provided biological classification information about the different transcripts identified. Afterward, we performed analyses with CPC (v. 2.0 beta) [26] and RNAsamba (web version, https://rnasamba.lge.ibi.unicamp.br, accessed on 6 June 2022) [27] to validate the genes classified as lncRNAs. We used the genes’ complementary DNA (cDNA) sequences as input in both tools. Predictions for each sequence were summarized to establish a consensus for each gene, considering every isoform and both tools. Manual curation was performed using NCBI Gene database (https://www.ncbi.nlm.nih.gov/gene, accessed on 9 June 2022) considering “Mus musculus” and “Gene type” information for those with “RefSeq Status = Validated”. lncRNAs with missing information were disregarded.
3. Results
The selected dataset was processed and analyzed to identify lncRNAs expressed during C. albicans infection. After the data acquisition and the processing steps described in Section 2.1 and Section 2.2 (see also Figure 1A), we obtained an expression matrix of 27,677 genes. The expression matrix was used as input for the three gene selection approaches (differential expression analysis, co-expression network analysis, and machine learning-based gene selection; Figure 1B, C). The resulting genes from each approach were intersected for further investigations (Figure 1D). A potential biological function was attributed to each lncRNA by considering their co-expressed first-degree neighbors using a guilt by association strategy. In the end, 64 lncRNAs were selected and investigated to infer which biological processes they could be associated with (Figure 1E).
3.1. Co-Expression Networks
We constructed a co-expression network to uncover the main biological pathways related to the host response during C. albicans infection (Figure 1B). Highly interconnected genes were clustered into eight modules represented by different colors (Figure 2A). The co-expression network approach allows the inference of gene-gene relationships by identifying modules strongly related to the health condition (infection or control). The results showed that multiple modules are related to each condition, whereas turquoise (, p-value = ) and blue (, p-value = ) modules presented the highest correlation coefficient values associated with infection and control, respectively. To further explore these results and confirm the significance of turquoise and blue modules, eigengene expression across individual samples was evaluated, while heatmaps show the gene expression of the selected modules (Figure 1D and Figure 2C). Based on these results, we consider the blue and turquoise modules for further analysis.
In these modules, MM and GS measures allowed the identification of genes strongly related to the infection and control conditions (GS and MM > 0.6) (Figure 2E,F), resulting in a total of 10,938 genes selected from the turquoise module (1876 lncRNAs) and 8896 genes from the blue module (1207 lncRNAs).
3.2. Differentially Expressed Genes
The gene expression profiles of infection samples were compared to control samples, resulting in 5576 DEGs (3509 up- and 2463 down-regulated) identified between the conditions (Figure 1C). Protein-coding genes were the most abundant biotype identified in the DEGs (57.4%), with 1710 genes up-regulated and 1719 down-regulated in Candida-infected samples, as exhibited by the volcano plot in Figure 3A. The lncRNA group represents 20.8% of all DEGs, where 800 were up-regulated and 441 down-regulated in the infected animals (Figure 3B). In the volcano plot figures, all DEGs are colored according to their assigned co-expression module. Up-regulated protein-coding and lncRNA DEGs were only found in the turquoise module, while all down-regulated protein-coding and lncRNA DEGs belong to the blue module. The remaining biotypes identified correspond to pseudogenes, genes to be experimentally confirmed, immunoglobulin variable chains, and T-cell receptors.
3.3. SCV-Based Gene Selection
Selecting genes with a consistent expression value range among all samples was essential to increase the chances of including only genes associated with each health condition. Every gene was individually classified with the sci-kit learn SVC algorithm according to its VST normalized expression matrix (Figure 1B,C). For the turquoise module, the prediction of sample condition reached f1-score = 1 for 1438 genes, meaning that those genes could perfectly separate infected and control samples during the tests. As for the blue module, 1095 genes classified the health condition with maximum accuracy.
3.4. Candidate Genes Prioritization
To narrow down the candidate genes of interest, we cross-referenced the results from all approaches previously applied: DEGs analysis, module identification followed by gene selection using and as the cut-off, and machine learning-based gene selection (Figure 1C,D). The intersected gene lists resulted in 820 genes from the turquoise module, including 65 lncRNAs. Figure 4A presents the number of selected genes through each approach and its intersections considering the turquoise module. The same intersection was performed for the blue module, where 793 genes were selected, including 60 lncRNAs (Figure 4B).
Afterward, the 125 selected genes classified as lncRNAs were validated to confirm their assigned gene type, resulting in the exclusion of 12 lncRNAs. Fifty-eight lncRNA from the turquoise module and fifty-five from the blue module remained.
The confirmed lncRNAs were used as seeds to filter the blue and turquoise modules and create different subnetworks. These subnetworks included only the lncRNAs’ first-degree neighbors, showing a moderate or strong correlation coefficient () [28]. When the subnetworks were created, not all lncRNAs showed first-degree interactions following the cut-off criterion. Consequentially, 51 lncRNAs from the turquoise network remained, forming a subnetwork with 657 nodes and 4479 edges. The blue subnetwork included 111 nodes (14 representing lncRNAs) and 129 edges. Only the genes included in those networks were considered for the following analyses.
3.5. Functional Enrichment Analysis
Functional enrichment analysis was performed to investigate the biological processes related to the previously selected genes from both networks and how they might be associated with C. albicans infection. We highlighted enriched GO terms, such as response to molecules of fungal origin, inflammatory response, and response to wounding, among others. The GO terms are represented in Figure 5, which includes information on the regulation associated with each gene set. Genes not presented in any GO term were not used to infer lncRNA-associated roles.
In total, 471 protein-coding genes from the turquoise module were identified in the functional enrichment. Those genes formed the final subnetwork alongside the remaining 51 lncRNAs and 3568 edges, as shown in Figure 6A. For the blue module, one lncRNA lost all its connections and was excluded, leaving the subnetwork with 13 lncRNA genes, in addition to the 50 protein-coding genes and 66 edges (Figure 6B).
4. Discussion
Unraveling the molecular mechanisms related to lncRNAs has been challenging, primarily due to the heterogeneous nature of this molecule and its relatively low levels of gene expression [29]. Furthermore, lncRNA sequences are less conserved across species than protein-coding genes [29], and structural domains that determine their functionality must be better understood.
Here, we take several steps to ensure data quality and combine stringent filtering criteria using various integrative approaches to achieve our results. None of the 64 selected lncRNAs (51 up- and 13 down-regulated) have been previously associated with C. albicans infection. However, 18 lncRNAs were mentioned in studies related to different health conditions (Table 1), including infections, lung diseases, and injury.
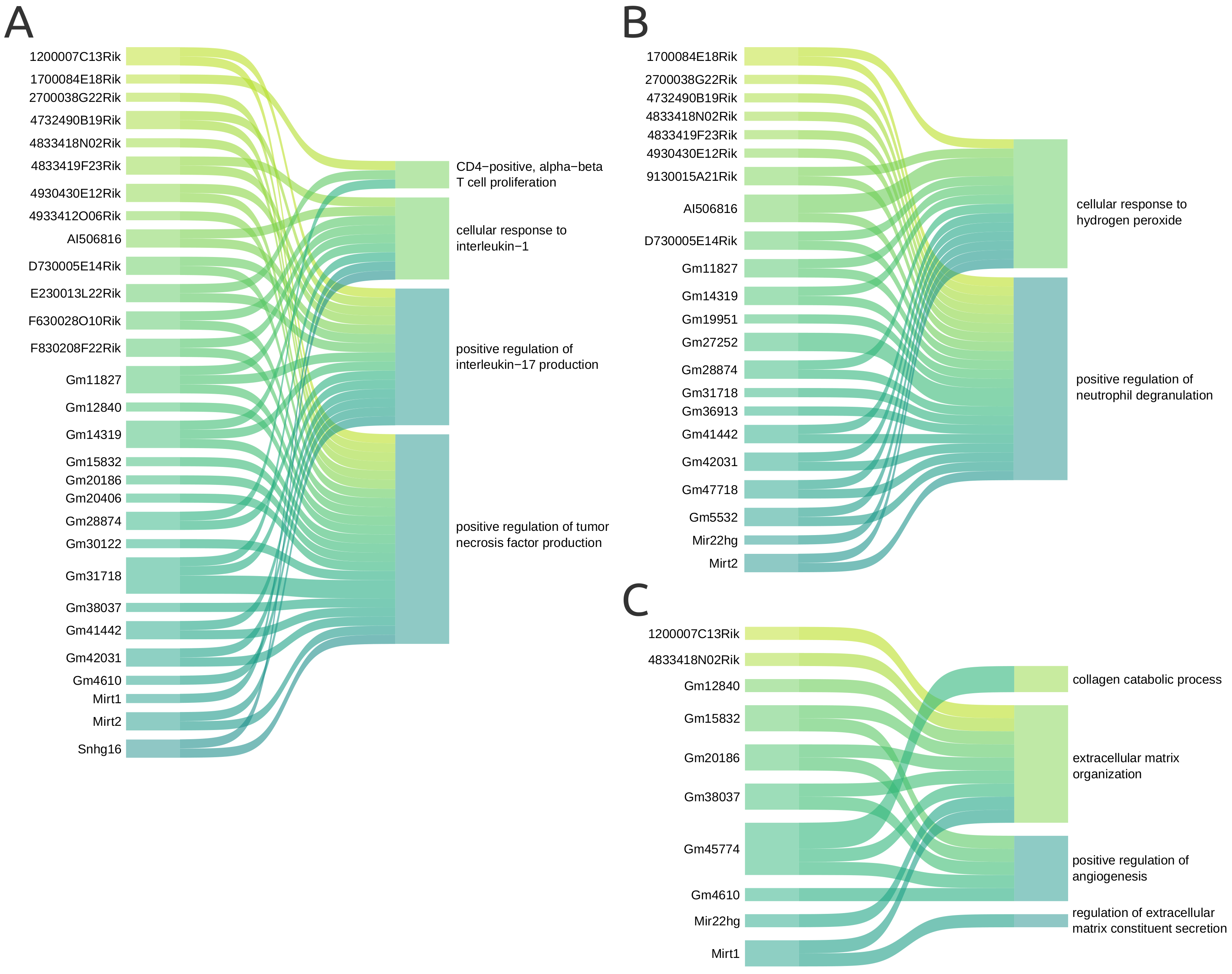
For the guilt by association approach, we selected representative GO terms corresponding to the protein-coding genes interacting with each lncRNA in the co-expression network. This approach assumes that genes that share some interaction or expression pattern are more likely to participate in similar processes or even the same functions [30], and has been previously used to explore lncRNAs without annotated functions [31]. It also enables the inference of unknown biological processes for genes when their co-expressed pairs already have described functions. The most relevant associations are shown in Figure 7 and will be discussed below. Supplementary Figure S1 contains all the proposed associations between the 41 lncRNAs and 25 GO terms.
4.1. Inflammatory and Immune Response Might Be Promoted by lncRNA
According to the results, F630028O10Rik interacted with protein-coding genes enriched in inflammatory cytokine-derived GO terms, such as positive regulation of tumor necrosis factor (TNF) production and cellular response to interleukin-1 (IL-1). Both TNF and IL-1 are pro-inflammatory cytokines produced and secreted by cells such as macrophages and mediated through the NF-B pathway. As presented in Figure 7A, we propose that some lncRNAs may be involved in both biological processes, namely Snhg16, F830208F22Rik, Gm14319, Gm11827, and Gm31718.
Furthermore, 17 lncRNAs were connected to positive regulation of interleukin-17 (IL-17) production (Figure 7A). IL-17 is a pro-inflammatory cytokine secreted by T helper 17 cells, which derive from the T helper (also known as CD4-positive). The GO terms CD4-positive and alpha-beta T cell proliferation were also enriched during the functional analysis, and the lncRNAs 1700084E18Rik, E230013L22Rik, and Gm28874 are connected to it through the protein-coding genes they interact with. This indicates those lncRNAs may participate in both proliferation and differentiation of CD4-positive cells. In addition, lncRNAs cell-specific expression patterns have been observed during proliferation in human T helper cell cultures [32].
The lncRNA Mirt2 was associated with positive regulation of IL-17 and TNF production, as shown in Figure 7A. In this context, Mirt2 regulates excessive inflammatory response in mice macrophage after lipopolysaccharide stimulus [33] (Table 1). Meanwhile, the lncRNA E230013L22Rik was linked to the GO term inflammatory response to antigenic stimulus. E230013L22Rik was previously reported as a potential regulator in the development of sepsis in mice [34]. Zou et al. inferred the lncRNA association with pro- and anti-inflammatory cytokines, besides inflammasome activation (Table 1).
GO terms related to immune response activation were also enriched in our analysis. Candidalysin can activate immune response by inducing the MAPK signaling pathway via MEK1/2 and ERK1/2 in endothelial cells [35]. Likewise, it can activate the immune system by inducing chemokines that mediate neutrophil and monocyte chemotaxis to the wound site [35,36]. At last, C. albicans cell wall components such as -1,3-glucans, -1,6-glucans, and mannoproteins can be identified by pattern recognition receptors (PRR) and trigger the immune response [37].
4.2. lncRNAs Related to Reactive Species Production during Host Defense
Neutrophils and macrophages are the first recruited cells to neutralize C. albicans by producing cytotoxic molecules, such as reactive oxygen species (ROS) and nitric oxide (NO) [38]. Reactive species generation is effective in fighting against microbial infections, being produced when the pathogen is recognized and during the response process [39]. Macrophages can create a toxic environment for the engulfed pathogen with an excessive amount of copper ion, generating ROS [40]. The 2500002B13Rik lncRNA, now denominated iNOS Transcriptional Regulatory Intergenic LncRNA Locus (Nostrill), was recently characterized [41]. Mathy et al. discovered that Nostrill overexpression was associated with up-regulation of the inducible nitric oxide synthase (iNOS) gene and NO production upon challenging microglia cells with bacterial LPS (Table 1).
Similarly, neutrophils also utilize reactive species by using both O2-dependent and O2-independent mechanisms to attack invading microorganisms [39]. Unlike macrophages, they take action by externalizing granules containing cytotoxic substances, performing a process known as degranulation [42]. Positive regulation of neutrophil degranulation was the second most frequent GO term associated with the lncRNA from our network (Figure 7B). Neutrophils have a critical role in host defense against fungal infections, killing the pathogen by different strategies, such as phagocytosis, oxidative bursts, and forming extracellular traps. All the mentioned strategies rely on reactive species [43,44]. Phagocytosis stimulates the generation of ROS such as hydrogen peroxide, observed in Figure 7B. Several lncRNAs also interact with genes enriched in the cellular response to the hydrogen peroxide () process (Figure 7B). is essential to immune response, as it promotes neutrophil recruitment and chemotaxis to the wounding site [45]. At low levels, supports normal physiological processes by inducing signaling pathways [46]. However, under certain stimuli, such as growth factors or chemokines, production is increased, leading to reversible oxidation of specific proteins, which alters their activity, localization, and interactions [46]. Consequently, these modified proteins contribute to orchestrating different processes, such as cell proliferation, differentiation, migration, and angiogenesis [46]. However, prolonged exposure to high ROS levels causes non-specific oxidation, impairing macromolecule function and leading to tissue damage [46].
4.3. Response to Wounding Healing Is Potentially Mediated by lncRNA
The functional enrichment analysis revealed critical biological processes associated with the host’s response to infection. From the sixty-four lncRNAs, only two were directly found in the GO enrichment gene sets, both up-regulated and assigned to the “response to wounding” GO term: Mir22hg (ENSMUSG00000085148) and AI506816 (ENSMUSG00000105987). Response to wounding is expected, given that infection by C. albicans requires invasion of epithelial and endothelial tissues [47]. After adhering to the epithelial cells, C. albicans hyphae secrete candidalysin, a toxin known to damage epithelial cells and cause cellular stress to initiate the infection [48,49]. Eventually, tissue damage can be aggravated due to an exaggerated inflammatory response to control the infection. Additionally, during pathogen infection, rapid generation of reactive species is crucial to the host defense, but prolonged exposure is detrimental to the surrounding tissue [49]. The wound healing process includes angiogenesis, extracellular matrix (ECM) organization, and inflammation processes also enriched among the lncRNAs.
The Sankey plot in Figure 7C represents the associations between GO terms involved in processes related to wounding response and the lncRNAs. Nine lncRNAs interact with protein-coding genes involved in ECM organization: 1200007C13Rik, 4833418N02Rik, Gm12840, Gm15832, Gm20186, Gm38037, Gm45774, Mir22hg, and Mirt1. Four of the mentioned lncRNAs also interact with genes associated with positive regulation of angiogenesis, namely Gm15832, Gm4610, Gm38037, and Gm45774, with the addition of Gm4610. Meanwhile, Gm45774 interacts with genes involved in the collagen catabolic process and Gm4610 with a gene linked to regulating extracellular matrix constituent secretion. Mir22hg was already directly associated with response to wounding within the functional enrichment results, which supports the result obtained with the designed strategy.
The angiogenic process is essential to effective wound healing as it promotes vascularization for the new tissue being assembled. A study associated angiogenesis with C. albicans and candidalysin secretion due to increased levels of fibroblast growth factor (FGF-2) in infected animals [50]. Moreover, the authors observed a greater mortality rate in animals treated with FGF-2 and hypothesized that this process might improve the fungus pathogenicity. Yet, fibroblasts are crucial to wounding response, as these molecules synthesize a new ECM [36]. In addition, the negative regulation of the apoptotic process was enriched in our analysis. While this biological process can be linked to the endothelial cell proliferation that occurs during wound healing, as previously reported [51,52], it has been observed that apoptosis inhibition could also be a host solution to keep more viable macrophages, increasing its defense [52].
F630028O10Rik was associated with reduced angiogenesis in lung cancer [53]. This lncRNA was also associated with increased spine chord injury severity due to the inflammatory response [54]. Xu et al. observed that the gene acted in TLR4-induced pyroptosis and served as a competing endogenous RNA; its overexpression favored the up-regulation of Col1a1 (collagen, type I, alpha 1), a gene involved in inflammation. Interestingly, up-regulation of F630028O10Rik was also observed in our analysis, while the TLR4 gene was not considered a DEG due to its expression values (). As a result of our functional enrichment analysis, no pyroptosis-derived GO term was assigned. Additionally, Col1a1 was slightly down-regulated (, p-value = ) in the C. albicans infected mice. Although both datasets describe different health conditions, the divergent transcript patterns mentioned indicate that F630028O10Rik may also regulate other genes, as lncRNAs can interact with different targets [55].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Information regarding the lncRNA reported in the literature, as well as the module, gene significance (GS), module membership (MM), and () from all selected lncRNA. Myocardial infarction: MI; lipopolysaccharide, LPS.
Table 1.
Information regarding the lncRNA reported in the literature, as well as the module, gene significance (GS), module membership (MM), and () from all selected lncRNA. Myocardial infarction: MI; lipopolysaccharide, LPS.
| Gene Symbol | Literature | Expression Data | |||||
|---|---|---|---|---|---|---|---|
| Biological Process | Condition | Organism | Module | GS | MM | ||
| 1200007C13Rik | Turquoise | −0.97 | 0.93 | 5.59 | |||
| 1700084E18Rik | Turquoise | −0.92 | 0.80 | 1.87 | |||
| 2500002B13Rik | Increases expression of the inducible nitric oxide synthase (iNOS) gene and nitric oxide production [41] | LPS stimulus | M. musculus | Turquoise | −0.91 | 0.72 | 1.84 |
| 2700038G22Rik | Turquoise | −0.88 | 0.81 | 1.27 | |||
| 4732490B19Rik | Turquoise | −0.94 | 0.87 | 4.00 | |||
| 4833418N02Rik | Turquoise | −0.93 | 0.90 | 1.52 | |||
| 4833419F23Rik | Turquoise | −0.93 | 0.81 | 2.31 | |||
| 4833438C02Rik | Turquoise | −0.92 | 0.83 | 1.17 | |||
| 4930430E12Rik | Turquoise | −0.91 | 0.84 | 3.43 | |||
| 4930551O13Rik | Turquoise | −0.85 | 0.81 | 2.40 | |||
| 4933412O06Rik | Turquoise | −0.95 | 0.92 | 4.09 | |||
| 9130015A21Rik | Turquoise | −0.96 | 0.81 | 3.40 | |||
| A530013C23Rik | Associated with gut microbiota-dysbiosis [56] | Gut microbiota transplant | M. musculus, H. sapiens | Turquoise | −0.84 | 0.92 | 1.89 |
| AI506816 | Up-regulated during mice second pregnancy in comparison to first pregnancy [57] | Multiparity | M. musculus | Turquoise | −0.96 | 0.85 | 1.32 |
| D730005E14Rik | Turquoise | −0.97 | 0.87 | 2.84 | |||
| E230013L22Rik | Up-regulated during sepsis [34] | Septic acute lung injury | Turquoise | −0.96 | 0.97 | 3.09 | |
| F630028O10Rik | Promotes angiogenesis inhibition [53] | Lung cancer | M. musculus | Turquoise | −0.95 | 0.92 | 2.16 |
| Enhanced microglial pyroptosis by activating the PI3K/AKT pathway | Spinal cord injury | M. musculus, H. sapiens | |||||
| F830208F22Rik | Turquoise | −0.90 | 0.94 | 3.14 | |||
| Gm11827 | Turquoise | −0.97 | 0.83 | 3.20 | |||
| Gm12840 | Turquoise | −0.95 | 0.92 | 2.56 | |||
| Gm14319 | Turquoise | −0.91 | 0.81 | 2.14 | |||
| Gm15832 | Up-regulated in infarct border zone samples [58] | MI | M. musculus | Turquoise | −0.96 | 0.93 | 2.76 |
| Gm16685 (NAIL) | Proinflammatory response regulation [59] | Ulcerative colitis | M. musculus | Turquoise | −0.92 | 0.97 | 3.96 |
| Gm19951 | Turquoise | −0.93 | 0.77 | 1.74 | |||
| Gm20186 | Turquoise | −0.94 | 0.97 | 2.84 | |||
| Gm20406 | Turquoise | −0.95 | 0.93 | 5.30 | |||
| Gm27252 | Turquoise | −0.97 | 0.86 | 2.62 | |||
| Gm28874 | Turquoise | −0.93 | 0.81 | 1.82 | |||
| Gm29292 | Turquoise | −0.81 | 0.88 | 2.42 | |||
| Gm30122 | Turquoise | −0.95 | 0.89 | 1.48 | |||
| Gm31683 | Turquoise | −0.88 | 0.91 | 2.40 | |||
| Gm31718 | Turquoise | −0.96 | 0.85 | 3.14 | |||
| Gm36913 | Turquoise | −0.94 | 0.80 | 4.77 | |||
| Gm38037 | Turquoise | −0.91 | 0.91 | 1.93 | |||
| Gm39321 | Turquoise | −0.90 | 0.83 | 2.26 | |||
| Gm40578 | Turquoise | −0.87 | 0.93 | 3.71 | |||
| Gm41442 | Potential regulator during sepsis [34] | Sepsis | M. musculus | Turquoise | −0.95 | 0.97 | 4.03 |
| Gm42031 | Turquoise | −0.98 | 0.88 | 3.65 | |||
| Gm45606 | Turquoise | −0.90 | 0.85 | 1.12 | |||
| Gm45774 | Turquoise | −0.96 | 0.96 | 5.29 | |||
| Gm4610 | Turquoise | −0.96 | 0.88 | 2.80 | |||
| Gm47709 | Turquoise | −0.90 | 0.94 | 4.82 | |||
| Gm47718 | Turquoise | −0.95 | 0.78 | 3.28 | |||
| Gm5532 | Turquoise | −0.89 | 0.76 | 1.87 | |||
| Il1bos | Turquoise | −0.93 | 0.95 | 2.95 | |||
| Mir22hg | Response to cycloheximide and cisplatin stress [60,61] | Chemical stress | H. sapiens | Turquoise | −0.98 | 0.93 | 1.76 |
| Mirt1 | Association with genes involved in left ventricular remodeling [62] | MI | M. musculus | Turquoise | −0.96 | 0.89 | 2.52 |
| NF-B activation [63] | MI | M. musculus | |||||
| Mirt2 | Association with genes involved in left ventricular remodeling [62] | MI | M. musculus | Turquoise | −0.96 | 0.89 | 3.60 |
| Reduced inflammatory response [33] | LPS stimulus | M. musculus | |||||
| Reduced apoptosis [64] | Accute MI | M. musculus | |||||
| Platr7 | Turquoise | −0.87 | 0.86 | 3.03 | |||
| Snhg16 | Enhanced the LPS-induced inflammatory pathway [65] | LPS stimulus | M. musculus | Turquoise | −0.89 | 0.96 | 1.03 |
| Snhg5 | Decreased target mRNA degradation by STAU1 [66] | Colorectal cancer | M. musculus | Turquoise | −0.88 | 0.93 | 1.12 |
| 2610027K06Rik | Blue | 0.96 | 0.97 | −2.01 | |||
| 4632428C04Rik | Blue | 0.95 | 0.96 | −2.08 | |||
| 9630028I04Rik | Blue | 0.96 | 0.98 | −3.23 | |||
| A230057D06Rik | Blue | 0.95 | 0.90 | −2.43 | |||
| AI504432 | Blue | 0.97 | 0.94 | −1.73 | |||
| AI838599 | Prediction: regulation of the TNF-NF-B signaling pathway [67] | Diabetic neuropathy | M. musculus | Blue | 0.97 | 0.93 | −2.30 |
| C430049B03Rik | Demarking between embryonic and extra-embryonic mesoderm [68] | Cardiogenesis | M. musculus | Blue | 0.96 | 0.92 | −2.26 |
| D630024D03Rik | Blue | 0.96 | 0.94 | −2.83 | |||
| E030013I19Rik | Blue | 0.93 | 0.88 | −2.05 | |||
| Gm12474 | Unknown. Possibly has a role in regulating the gsdmd gene [69] | Diabetic periodontitis | M. musculus | Blue | 0.94 | 0.94 | −2.59 |
| Gm45332 | Blue | 0.94 | 0.96 | −1.78 | |||
| Gm48662 | Blue | 0.98 | 0.96 | −4.40 | |||
| Sfta3-ps | Blue | 0.95 | 0.99 | −1.46 | |||
5. Conclusions and Perspectives
The understanding of C. albicans–host interactions is crucial to investigate new forms of diagnostic and therapeutic methodologies. Molecules such as lncRNAs are still little studied in this context, which results in limited reports asserting their importance during the immune response to fungal infections. A recent review discussed how lncRNAs reportedly relevant for host defense against other pathogens could also participate in the immune response against C. albicans, as well as be used in host-derived therapies [70]. The authors proposed that lncRNAs could be interesting targets for therapy focusing on different defense mechanisms, such as tissue barrier and inflammation. Mirt2 was proposed as a lncRNA potentially related to the induction of cytokines, an essential process during the immune response to C. albicans.
In this study, we infer biological functions in which lncRNAs may participate during host immune defense against C. albicans infection. For that, we used a guilt by association strategy. Many of the selected lncRNAs are not described in the literature, and none have been reported in fungal infection.
Our presented results suggest that 46 lncRNAs might participate in processes related to 25 selected GO terms (Supplementary Figure S1) and should be further investigated. Among the nine lncRNAs associated with wounding healing processes, Gm45774 was linked to three of the four GO terms selected, as shown in Figure 7C. Three out of the twenty-nine associated lncRNAs could be highlighted in the inflammation processes: Gm31718, Gm11827, and Gm14319. While Gm31718 was linked to all the selected GO terms, Gm11827 and Gm14319 were associated with the GO terms regarding cytokines: cellular response to interleukin-1, positive regulation of interleukin-17 production, and positive regulation of tumor necrosis factor production (Figure 7A). Some GO terms were associated with several lncRNAs, highlighting positive regulation of tumor necrosis factor production, positive regulation of neutrophil degranulation, and immunoglobulin-mediated immune response (Supplementary Figure S1). These findings might provide insight into the role of lncRNA during C. albicans infection. While the selected lncRNAs are up-regulated during the host’s defense against C. albicans, we can not determine whether they are expressed due to the presence of the fungi or as part of the general immune response. More data analysis, including different infected organs samples and periods throughout the infection, would be necessary to evaluate lncRNA specificity.
Mortality related to C. albicans infection is a challenge worldwide, with significant incidence and recurrence rates in vulnerable patients. Growing evidence suggests that lncRNAs play a critical role in the immune response during infections. Exploring these molecules further could give us strategic options for therapeutic treatments for fungal infections. The lncRNAs uncovered in this work can be used as a starting point to pave the way for future research to illustrate the role of these molecules in defense mechanisms. Furthermore, designing a more extensive investigation to help unravel the different modes of action of lncRNAs and understanding the miRNA–lncRNA–mRNA axis may help to solve the role of lncRNAs in the immune response and as promising molecules for future targeted therapy.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/genes14020251/s1, Figure S1: Sankey plot representing each selected lncRNA (right side) and the GO terms (left side) enriched by genes they interacted with in the coexpression network.
Author Contributions
Conceptualization, data curation, analysis, results interpretation, writing, G.F.G.; conceptualization, methodology design, results interpretation, writing, J.d.F.P.; funding acquisition, project administration, supervision, methodology design, writing, M.D. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants from the Fundação de Amparo à Pesquisa do Estado do Rio Grande do Sul—FAPERGS (19/2551-0001906-8 and 17/2551-0000520-1), Conselho Nacional de Desenvolvimento Científico e Tecnológico—CNPq (314082/2021-2, 440279/2022-4 and 465450/2014-8), and the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—CAPES/STICAMSUD (88881.522073/2020-01) and DAAD/CAPES PROBRAL 88881.198766/2018-01). This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Brasil (CAPES)—Finance Code 001.
Data Availability Statement
The source code used in the experiments can be accessed on GitHub: https://github.com/sbcblab/candida-infection-pipeline, accessed on 3 May 2021.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Richardson, J.P.; Moyes, D.L. Adaptive immune responses to Candida albicans infection. Virulence 2015, 6, 327–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamin, D.K.; Stoll, B.J.; Gantz, M.G.; Walsh, M.C.; Sánchez, P.J.; Das, A.; Shankaran, S.; Higgins, R.D.; Auten, K.J.; Miller, N.A.; et al. Neonatal candidiasis: Epidemiology, risk factors, and clinical judgment. Pediatrics 2010, 126, e865–e873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doi, A.M.; Pignatari, A.C.C.; Edmond, M.B.; Marra, A.R.; Camargo, L.F.A.; Siqueira, R.A.; da Mota, V.P.; Colombo, A.L. Epidemiology and microbiologic characterization of nosocomial candidemia from a Brazilian national surveillance program. PLoS ONE 2016, 11, e0146909. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Sae-Tia, S.; Fries, B.C. Candidiasis and mechanisms of antifungal resistance. Antibiotics 2020, 9, 312. [Google Scholar] [CrossRef] [PubMed]
- Calvo, B.; Melo, A.S.; Perozo-Mena, A.; Hernandez, M.; Francisco, E.C.; Hagen, F.; Meis, J.F.; Colombo, A.L. First report of Candida auris in America: Clinical and microbiological aspects of 18 episodes of candidemia. J. Infect. 2016, 73, 369–374. [Google Scholar] [CrossRef]
- Nash, A.K.; Auchtung, T.A.; Wong, M.C.; Smith, D.P.; Gesell, J.R.; Ross, M.C.; Stewart, C.J.; Metcalf, G.A.; Muzny, D.M.; Gibbs, R.A.; et al. The gut mycobiome of the Human Microbiome Project healthy cohort. Microbiome 2017, 5, 153. [Google Scholar] [CrossRef]
- Statello, L.; Guo, C.J.; Chen, L.L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef]
- Fernandes, J.C.; Acuña, S.M.; Aoki, J.I.; Floeter-Winter, L.M.; Muxel, S.M. Long non-coding RNAs in the regulation of gene expression: Physiology and disease. Non-Coding RNA 2019, 5, 17. [Google Scholar] [CrossRef] [Green Version]
- Agliano, F.; Rathinam, V.A.; Medvedev, A.E.; Vanaja, S.K.; Vella, A.T. Long noncoding RNAs in host–pathogen interactions. Trends Immunol. 2019, 40, 492–510. [Google Scholar] [CrossRef]
- Fu, Y.; Xu, X.; Xue, J.; Duan, W.; Yi, Z. Deregulated lncRNAs in B cells from patients with active tuberculosis. PLoS ONE 2017, 12, e0170712. [Google Scholar] [CrossRef]
- Saha, C.; Laha, S.; Chatterjee, R.; Bhattacharyya, N.P. Co-Regulation of Protein Coding Genes by Transcription Factor and Long Non-Coding RNA in SARS-CoV-2 Infected Cells: An In Silico Analysis. Non-Coding RNA 2021, 7, 74. [Google Scholar] [CrossRef]
- Riege, K.; Hölzer, M.; Klassert, T.E.; Barth, E.; Bräuer, J.; Collatz, M.; Hufsky, F.; Mostajo, N.; Stock, M.; Vogel, B.; et al. Massive effect on LncRNAs in human monocytes during fungal and bacterial infections and in response to vitamins A and D. Sci. Rep. 2017, 7, 40598. [Google Scholar] [CrossRef] [Green Version]
- Hou, J.s.; He, Y.y.; Du, L.l.; Duan, Z.m.; Chen, X.; Li, M. Gene expression profile and long noncoding RNA analysis in Candida albicans insoluble β-glucan-stimulated CD14+ monocytes and THP-1 cells. Microb. Pathog. 2021, 157, 104963. [Google Scholar] [CrossRef]
- Kinsella, R.J.; Kähäri, A.; Haider, S.; Zamora, J.; Proctor, G.; Spudich, G.; Almeida-King, J.; Staines, D.; Derwent, P.; Kerhornou, A.; et al. Ensembl BioMarts: A hub for data retrieval across taxonomic space. Database 2011, 2011, bar030. [Google Scholar] [CrossRef]
- Robinson, E.K.; Covarrubias, S.; Carpenter, S. The how and why of lncRNA function: An innate immune perspective. Biochim. Biophys. Acta 2020, 1863, 194419. [Google Scholar] [CrossRef]
- Singhania, A.; Graham, C.M.; Gabryšová, L.; Moreira-Teixeira, L.; Stavropoulos, E.; Pitt, J.M.; Chakravarty, P.; Warnatsch, A.; Branchett, W.J.; Conejero, L.; et al. Transcriptional profiling unveils type I and II interferon networks in blood and tissues across diseases. Nat. Commun. 2019, 10, 2887. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Research 2015, 4, 1521. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Alexa, A.; Rahnenfuhrer, J. topGO: Enrichment Analysis for Gene Ontology. R Package Version 2.48.0. 2022, Volume 2. Available online: https://bioconductor.org/packages/topGO/ (accessed on 3 May 2021).
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Y.J.; Yang, D.C.; Kong, L.; Hou, M.; Meng, Y.Q.; Wei, L.; Gao, G. CPC2: A fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 2017, 45, W12–W16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camargo, A.P.; Sourkov, V.; Pereira, G.A.G.; Carazzolle, M.F. RNAsamba: Neural network-based assessment of the protein-coding potential of RNA sequences. NAR Genom. Bioinform. 2020, 2, lqz024. [Google Scholar] [CrossRef] [Green Version]
- Evans, J.D. Straightforward Statistics for the Behavioral Sciences; Thomson Brooks/Cole Publishing Co.: Minocqua, WI, USA, 1996. [Google Scholar]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [Green Version]
- Gillis, J.; Pavlidis, P. The impact of multifunctional genes on “guilt by association” analysis. PLoS ONE 2011, 6, e17258. [Google Scholar] [CrossRef] [Green Version]
- Pauli, A.; Valen, E.; Lin, M.F.; Garber, M.; Vastenhouw, N.L.; Levin, J.Z.; Fan, L.; Sandelin, A.; Rinn, J.L.; Regev, A.; et al. Systematic identification of long noncoding RNAs expressed during zebrafish embryogenesis. Genome Res. 2012, 22, 577–591. [Google Scholar] [CrossRef] [Green Version]
- Hu, G.; Tang, Q.; Sharma, S.; Yu, F.; Escobar, T.M.; Muljo, S.A.; Zhu, J.; Zhao, K. Expression and regulation of intergenic long noncoding RNAs during T cell development and differentiation. Nat. Immunol. 2013, 14, 1190–1198. [Google Scholar] [CrossRef]
- Du, M.; Yuan, L.; Tan, X.; Huang, D.; Wang, X.; Zheng, Z.; Mao, X.; Li, X.; Yang, L.; Huang, K.; et al. The LPS-inducible lncRNA Mirt2 is a negative regulator of inflammation. Nat. Commun. 2017, 8, 2049. [Google Scholar] [CrossRef] [Green Version]
- Zou, L.; Yu, Q.; Zhang, L.; Yuan, X.; Fang, F.; Xu, F. Identification of inflammation related lncRNAs and Gm33647 as a potential regulator in septic acute lung injury. Life Sci. 2021, 282, 119814. [Google Scholar] [CrossRef]
- Swidergall, M.; Khalaji, M.; Solis, N.V.; Moyes, D.L.; Drummond, R.A.; Hube, B.; Lionakis, M.S.; Murdoch, C.; Filler, S.G.; Naglik, J.R. Candidalysin is required for neutrophil recruitment and virulence during systemic Candida albicans infection. J. Infect. Dis. 2019, 220, 1477–1488. [Google Scholar] [CrossRef] [Green Version]
- Gardner, A.; Borthwick, L.A.; Fisher, A.J. Lung epithelial wound healing in health and disease. Expert Rev. Respir. Med. 2010, 4, 647–660. [Google Scholar] [CrossRef]
- Vendele, I.; Willment, J.A.; Silva, L.M.; Palma, A.S.; Chai, W.; Liu, Y.; Feizi, T.; Spyrou, M.; Stappers, M.H.; Brown, G.D.; et al. Mannan detecting C-type lectin receptor probes recognise immune epitopes with diverse chemical, spatial and phylogenetic heterogeneity in fungal cell walls. PLoS Pathog. 2020, 16, e1007927. [Google Scholar] [CrossRef] [Green Version]
- Navarathna, D.H.; Lionakis, M.S.; Roberts, D.D. Endothelial nitric oxide synthase limits host immunity to control disseminated Candida albicans infections in mice. PLoS ONE 2019, 14, e0223919. [Google Scholar] [CrossRef] [Green Version]
- Andrés, C.M.C.; Pérez de la Lastra, J.M.; Juan, C.A.; Plou, F.J.; Pérez-Lebeña, E. The Role of Reactive Species on Innate Immunity. Vaccines 2022, 10, 1735. [Google Scholar] [CrossRef]
- Culbertson, E.M.; Khan, A.A.; Muchenditsi, A.; Lutsenko, S.; Sullivan, D.J.; Petris, M.J.; Cormack, B.P.; Culotta, V.C. Changes in mammalian copper homeostasis during microbial infection. Metallomics 2020, 12, 416–426. [Google Scholar] [CrossRef]
- Mathy, N.W.; Burleigh, O.; Kochvar, A.; Whiteford, E.R.; Behrens, M.; Marta, P.; Tian, C.; Gong, A.Y.; Drescher, K.M.; Steyger, P.S.; et al. A novel long intergenic non-coding RNA, Nostrill, regulates iNOS gene transcription and neurotoxicity in microglia. J. Neuroinflammat. 2021, 18, 16. [Google Scholar] [CrossRef]
- Lacy, P. Mechanisms of degranulation in neutrophils. Allergy Asthma Clin. Immunol. 2006, 2, 1–11. [Google Scholar] [CrossRef]
- Zhong, H.; Lu, R.Y.; Wang, Y. Neutrophil extracellular traps in fungal infections: A seesaw battle in hosts. Front. Immunol. 2022, 13, 977493. [Google Scholar] [CrossRef] [PubMed]
- Desai, J.V.; Lionakis, M.S. The role of neutrophils in host defense against invasive fungal infections. Curr. Clin. Microbiol. Rep. 2018, 5, 181–189. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, S.; Rosowski, E.E.; Huttenlocher, A. Neutrophil migration in infection and wound repair: Going forward in reverse. Nat. Rev. Immunol. 2016, 16, 378–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canton, M.; Sánchez-Rodríguez, R.; Spera, I.; Venegas, F.C.; Favia, M.; Viola, A.; Castegna, A. Reactive oxygen species in macrophages: Sources and targets. Front. Immunol. 2021, 12, 734229. [Google Scholar] [CrossRef]
- Maza, P.K.; Bonfim-Melo, A.; Padovan, A.C.; Mortara, R.A.; Orikaza, C.M.; Ramos, L.M.D.; Moura, T.R.; Soriani, F.M.; Almeida, R.S.; Suzuki, E.; et al. Candida albicans: The ability to invade epithelial cells and survive under oxidative stress is unlinked to hyphal length. Front. Microbiol. 2017, 8, 1235. [Google Scholar] [CrossRef] [Green Version]
- Moyes, D.L.; Wilson, D.; Richardson, J.P.; Mogavero, S.; Tang, S.X.; Wernecke, J.; Höfs, S.; Gratacap, R.L.; Robbins, J.; Runglall, M.; et al. Candidalysin is a fungal peptide toxin critical for mucosal infection. Nature 2016, 532, 64–68. [Google Scholar] [CrossRef] [Green Version]
- Blagojevic, M.; Camilli, G.; Maxson, M.; Hube, B.; Moyes, D.L.; Richardson, J.P.; Naglik, J.R. Candidalysin triggers epithelial cellular stresses that induce necrotic death. Cell. Microbiol. 2021, 23, e13371. [Google Scholar] [CrossRef]
- Vellanki, S.; Huh, E.Y.; Saville, S.P.; Lee, S.C. Candida albicans morphology-dependent host FGF-2 response as a potential therapeutic target. J. Fungi 2019, 5, 22. [Google Scholar] [CrossRef] [Green Version]
- Barker, K.S.; Park, H.; Phan, Q.T.; Xu, L.; Homayouni, R.; Rogers, P.D.; Filler, S.G. Transcriptome profile of the vascular endothelial cell response to Candida albicans. J. Infect. Dis. 2008, 198, 193–202. [Google Scholar] [CrossRef] [Green Version]
- Chin, V.K.; Lee, T.Y.; Rusliza, B.; Chong, P.P. Dissecting Candida albicans infection from the perspective of C. albicans virulence and omics approaches on host–pathogen interaction: A review. Int. J. Mol. Sci. 2016, 17, 1643. [Google Scholar] [CrossRef]
- Qin, L.; Zhong, M.; Adah, D.; Qin, L.; Chen, X.; Ma, C.; Fu, Q.; Zhu, X.; Li, Z.; Wang, N.; et al. A novel tumour suppressor lncRNA F630028O10Rik inhibits lung cancer angiogenesis by regulating miR-223-3p. J. Cell. Mol. Med. 2020, 24, 3549–3559. [Google Scholar] [CrossRef]
- Xu, S.; Wang, J.; Jiang, J.; Song, J.; Zhu, W.; Zhang, F.; Shao, M.; Xu, H.; Ma, X.; Lyu, F. TLR4 promotes microglial pyroptosis via lncRNA-F630028O10Rik by activating PI3K/AKT pathway after spinal cord injury. Cell Death Dis. 2020, 11, 693. [Google Scholar] [CrossRef]
- Rinn, J.L.; Chang, H.Y. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Wang, H.; Chen, X.; Zhang, Y.; Li, W.; Rao, X.; Liu, Y.; Zhao, L.; Pu, J.; Gui, S.; et al. Integrative Analysis of Long Non-coding RNAs, Messenger RNAs, and MicroRNAs Indicates the Neurodevelopmental Dysfunction in the Hippocampus of Gut Microbiota-Dysbiosis Mice. Front. Mol. Neurosci. 2021, 14, 745437. [Google Scholar] [CrossRef]
- Liu, J.L.; Zuo, R.J.; Peng, Y.; Fu, Y.S. The impact of multiparity on uterine gene expression and decidualization in mice. Reprod. Sci. 2016, 23, 687–694. [Google Scholar] [CrossRef]
- Liu, B.; Cheng, Y.; Tian, J.; Zhang, L.; Cui, X. Upregulated lncRNA Pvt1 may be important for cardiac remodeling at the infarct border zone. Mol. Med. Rep. 2020, 22, 2605–2616. [Google Scholar] [CrossRef]
- Akıncılar, S.C.; Wu, L.; Ng, Q.F.; Chua, J.Y.H.; Unal, B.; Noda, T.; Chor, W.H.J.; Ikawa, M.; Tergaonkar, V. NAIL: An evolutionarily conserved lncRNA essential for licensing coordinated activation of p38 and NFκB in colitis. Gut 2021, 70, 1857–1871. [Google Scholar] [CrossRef]
- Tani, H.; Torimura, M. Identification of short-lived long non-coding RNAs as surrogate indicators for chemical stress response. Biochem. Biophys. Res. Commun. 2013, 439, 547–551. [Google Scholar] [CrossRef]
- Tani, H.; Onuma, Y.; Ito, Y.; Torimura, M. Long non-coding RNAs as surrogate indicators for chemical stress responses in human-induced pluripotent stem cells. PLoS ONE 2014, 9, e106282. [Google Scholar] [CrossRef]
- Zangrando, J.; Zhang, L.; Vausort, M.; Maskali, F.; Marie, P.Y.; Wagner, D.R.; Devaux, Y. Identification of candidate long non-coding RNAs in response to myocardial infarction. BMC Genom. 2014, 15, 460. [Google Scholar] [CrossRef]
- Li, X.; Zhou, J.; Huang, K. Inhibition of the lncRNA Mirt1 attenuates acute myocardial infarction by suppressing NF-κB activation. Cell. Physiol. Biochem. 2017, 42, 1153–1164. [Google Scholar] [CrossRef]
- Zhu, F.; Li, Q.; Li, J.; Li, B.; Li, D. Long noncoding Mirt2 reduces apoptosis to alleviate myocardial infarction through regulation of the miR-764/PDK1 axis. Lab. Investig. 2021, 101, 165–176. [Google Scholar] [CrossRef]
- Wang, W.; Lou, C.; Gao, J.; Zhang, X.; Du, Y. LncRNA SNHG16 reverses the effects of miR-15a/16 on LPS-induced inflammatory pathway. Biomed. Pharmacother. 2018, 106, 1661–1667. [Google Scholar] [CrossRef] [PubMed]
- Damas, N.D.; Marcatti, M.; Côme, C.; Christensen, L.L.; Nielsen, M.M.; Baumgartner, R.; Gylling, H.M.; Maglieri, G.; Rundsten, C.F.; Seemann, S.E.; et al. SNHG5 promotes colorectal cancer cell survival by counteracting STAU1-mediated mRNA destabilization. Nat. Commun. 2016, 7, 13875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Zhou, Y.; Zhou, F.; Yu, X.; Liu, J.; Liu, Y.; Zhu, Y.; Wang, W.; Chen, N. Altered expression of long noncoding and messenger RNAs in diabetic nephropathy following treatment with rosiglitazone. Biomed. Res. Int. 2020, 2020, 1360843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, M.R.; Duan, Q.; Nagle, A.; Kathiriya, I.S.; Huang, Y.; Rao, K.; Haldar, S.M.; Bruneau, B.G. Minimal in vivo requirements for developmentally regulated cardiac long intergenic non-coding RNAs. Development 2019, 146, dev185314. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, Q.; Nie, L.; Zhang, P.; Zhao, P.; Yuan, Q.; Ji, N.; Ding, Y.; Wang, Q. Metformin ameliorates the NLPP3 inflammasome mediated pyroptosis by inhibiting the expression of NEK7 in diabetic periodontitis. Arch. Oral Biol. 2020, 116, 104763. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, H.; Chen, N.; Yang, J.; Zhou, H. LncRNA: A Potential Target for Host-Directed Therapy of Candida Infection. Pharmaceutics 2022, 14, 621. [Google Scholar] [CrossRef]
Figure 1.
Pipeline representing the analyses performed in this study, including the following steps: (A) Data acquisition and processing; (B) Gene selection; (C) Results filtering; (D) Intersection and functional enrichment analysis; and (E) Guilt by association strategy.
Figure 1.
Pipeline representing the analyses performed in this study, including the following steps: (A) Data acquisition and processing; (B) Gene selection; (C) Results filtering; (D) Intersection and functional enrichment analysis; and (E) Guilt by association strategy.

Figure 2.
(A) Cluster dendrogram representation of the co-expression network modules. (B) Correlation between the eigengene modules and the health condition trait. (C) Heatmap presenting the sample gene expression for the turquoise module. The barplot shows the module eigengene expression value associated with each sample. (D) Heatmap presenting the sample gene expression for the blue module. The barplot shows the module eigengene expression value associated with each sample. (E) Correlation between gene significance (GS) and MM for genes in the turquoise module. (F) Correlation between gene significance (GS) and MM for genes in the blue module.
Figure 2.
(A) Cluster dendrogram representation of the co-expression network modules. (B) Correlation between the eigengene modules and the health condition trait. (C) Heatmap presenting the sample gene expression for the turquoise module. The barplot shows the module eigengene expression value associated with each sample. (D) Heatmap presenting the sample gene expression for the blue module. The barplot shows the module eigengene expression value associated with each sample. (E) Correlation between gene significance (GS) and MM for genes in the turquoise module. (F) Correlation between gene significance (GS) and MM for genes in the blue module.

Figure 3.
Volcano plots showing DEGs results, with adjusted p-value in the y-axis and in the x-axis. Genes are divided according to their biotype: (A) protein-coding or (B) long non-coding RNA. Blue and turquoise colors represent DEGs assigned to blue (control condition) or turquoise (infection condition) modules.
Figure 3.
Volcano plots showing DEGs results, with adjusted p-value in the y-axis and in the x-axis. Genes are divided according to their biotype: (A) protein-coding or (B) long non-coding RNA. Blue and turquoise colors represent DEGs assigned to blue (control condition) or turquoise (infection condition) modules.

Figure 4.
Venn diagram illustrating the number of genes selected by different approaches: DEGs, and , ML, and filtering for lncRNA biotype. (A) All genes from the turquoise module. (B) All genes from the blue module.
Figure 4.
Venn diagram illustrating the number of genes selected by different approaches: DEGs, and , ML, and filtering for lncRNA biotype. (A) All genes from the turquoise module. (B) All genes from the blue module.

Figure 5.
The table on the right side shows the top 15 GO terms enriched for the selected genes from both modules analyzed. On the left, the outer circle’s scatter plots represent the GO terms identified and the regulation of its associated genes, based on the values. The z-scores showed in the inner circle were calculated for each BP as z---.
Figure 5.
The table on the right side shows the top 15 GO terms enriched for the selected genes from both modules analyzed. On the left, the outer circle’s scatter plots represent the GO terms identified and the regulation of its associated genes, based on the values. The z-scores showed in the inner circle were calculated for each BP as z---.

Figure 6.
Subnetworks representing the selected protein-coding genes (in blue) and lncRNAs (in red). (A) Turquoise module. (B) Blue module.
Figure 6.
Subnetworks representing the selected protein-coding genes (in blue) and lncRNAs (in red). (A) Turquoise module. (B) Blue module.

Figure 7.
Sankey plot representing the GO terms inferred to the selected lncRNAs. The GO terms were grouped according to its biological function. (A) GO terms associated with the inflammatory response. (B) GO terms associated with reactive oxygen species.(C) GO terms involved in response to wounding.
Figure 7.
Sankey plot representing the GO terms inferred to the selected lncRNAs. The GO terms were grouped according to its biological function. (A) GO terms associated with the inflammatory response. (B) GO terms associated with reactive oxygen species.(C) GO terms involved in response to wounding.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Gonçalves, G.F.; de Faria Poloni, J.; Dorn, M. Transcriptomic Analysis of Long Non-Coding RNA during Candida albicans Infection. Genes 2023, 14, 251. https://doi.org/10.3390/genes14020251
AMA Style
Gonçalves GF, de Faria Poloni J, Dorn M. Transcriptomic Analysis of Long Non-Coding RNA during Candida albicans Infection. Genes. 2023; 14(2):251. https://doi.org/10.3390/genes14020251
Chicago/Turabian StyleGonçalves, Gabriela Flores, Joice de Faria Poloni, and Márcio Dorn. 2023. "Transcriptomic Analysis of Long Non-Coding RNA during Candida albicans Infection" Genes 14, no. 2: 251. https://doi.org/10.3390/genes14020251
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.