
Confirmation of a Phenotypic Entity for TSPEAR Variants in Egyptian Ectodermal Dysplasia Patients and Role of Ethnicity

 , ,
, ,
Abstract
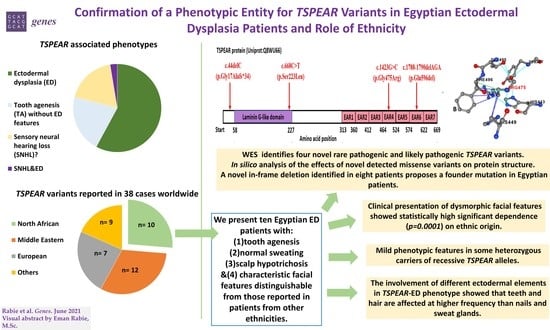
:
1. Introduction
2. Materials and Methods
2.1. Ethical Aspects
2.2. Patient Phenotyping and Inclusion Criteria
2.3. Molecular Analyses
2.3.1. DNA Extraction and Whole Exome Sequencing (WES)
2.3.2. Variant Annotation and Filtration
2.3.3. Variant Segregation
2.3.4. In Silico Prediction of Protein Structural Alterations Caused by TSPEAR Variants
2.4. Comparison of Phenotypic Variabilities among TSPEAR Cohorts in Relation to Ethnicities
3. Results
3.1. Clinical Features
3.2. Molecular Data
3.2.1. TSPEAR Variants’ Identification, Segregation and MAF
3.2.2. In Silico Predicted Protein Structural Alterations of Missense TSPEAR Variants
3.3. Comparison of Clinical Phenotypes and Dysmorphic Facial Features of TSPEAR Cohorts of Different Ethnicities
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wright, J.T.; Fete, M.; Schneider, H.; Zinser, M.; Koster, M.I.; Clarke, A.J.; Hadj-Rabia, S.; Tadini, G.; Pagnan, N.; Visinoni, A.F.; et al. Ectodermal dysplasias: Classification and organization by phenotype, genotype and molecular pathway. Am. J. Med. Genet. Part A 2019, 179, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Freire-Maia, N. Ectodermal dysplasias. Hum. Hered. 1971, 131, 309–312. [Google Scholar] [CrossRef] [PubMed]
- Freire-Maia, N. Ectodermal dysplasias revisited. Acta Genet. Med. Gemellol. 1977, 26, 121–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freire-Maia, N.; Lisboa-Costa, T.; Pagnan, N.A. Ectodermal dysplasias: How many? Am. J. Med. Genet. 2001, 104, 84. [Google Scholar] [CrossRef]
- Pinheiro, M.; Freire-Maia, N. Ectodermal dysplasias: A clinical classification and a causal review. Am. J. Med. Genet. 1994, 53, 153–162. [Google Scholar] [CrossRef]
- Nguyen-Nielsen, M.; Skovbo, S.; Svaneby, D.; Pedersen, L.; Fryzek, J. The prevalence of X-linked hypohidrotic ectodermal dysplasia (XLHED) in Denmark, 1995–2010. Eur. J. Med. Genet. 2013, 56, 236–242. [Google Scholar] [CrossRef]
- Asai, Y.; Irvine, A.D. Ectodermal dysplasias. In Harper’s Textbook of Pediatric Dermatology; Wiley Online Books: Hoboken, NJ, USA, 2011; pp. 127.1–127.104. ISBN 9781444345384. [Google Scholar]
- Pagnan, N.A.B.; Visinoni, Á.F. Update on ectodermal dysplasias clinical classification. Am. J. Med. Genet. Part A 2014, 164, 2415–2423. [Google Scholar] [CrossRef]
- Visinoni, A.F.; Lisboa-Costa, T.; Pagnan, N.A.B.; Chautard-Freire-Maia, E.A. Ectodermal dysplasias: Clinical and molecular review. Am. J. Med. Genet. Part A 2009, 149, 1980–2002. [Google Scholar] [CrossRef]
- Patthey, C.; Gunhaga, L. Signaling pathways regulating ectodermal cell fate choices. Exp. Cell Res. 2014, 321, 11–16. [Google Scholar] [CrossRef] [Green Version]
- Mikkola, M.L. Molecular aspects of hypohidrotic ectodermal dysplasia. Am. J. Med. Genet. Part A 2009, 149A, 2031–2036. [Google Scholar] [CrossRef]
- Mikkola, M.L.; Thesleff, I. Ectodysplasin signaling in development. Cytokine Growth Factor Rev. 2003, 14, 211–224. [Google Scholar] [CrossRef]
- Lamartine, J. Towards a new classification of ectodermal dysplasias. Clin. Exp. Dermatol. Clin. Dermatol. 2003, 28, 351–355. [Google Scholar] [CrossRef]
- Priolo, M.; Laganà, C. Ectodermal dysplasias: A new clinical-genetic classification. J. Med. Genet. 2001, 38, 579–585. [Google Scholar] [CrossRef]
- Priolo, M.; Silengo, M.; Lerone, M.; Ravazzolo, R. Ectodermal dysplasias: Not only ‘skin’deep. Clin. Genet. 2000, 58, 415–430. [Google Scholar] [CrossRef]
- Itin, P.H.; Fistarol, S.K. Ectodermal dysplasias. Am. J. Med. Genet. Part C Semin. Med. Genet. 2004, 131C, 45–51. [Google Scholar] [CrossRef]
- Itin, P.H. Etiology and pathogenesis of ectodermal dysplasias. Am. J. Med. Genet. Part A 2014, 164, 2472–2477. [Google Scholar] [CrossRef]
- Salinas, C.F.; Irvine, A.D.; Itin, P.H.; Di Giovanna, J.J.; Schneider, H.; Clarke, A.J.; McGovern, L.S.; Fete, M. Second international conference on a classification of ectodermal dysplasias: Development of a multiaxis model. Am. J. Med. Genet. Part A 2014, 164, 2482–2489. [Google Scholar] [CrossRef]
- Mortier, K.; Wackens, G. Ectodermal dysplasia, anhidrotic. Orphanet Encycl. 2004, 3, 1–6. [Google Scholar]
- Dellavia, C.; Catti, F.; Sforza, C.; Tommasi, D.G.; Ferrario, V.F. Craniofacial growth in ectodermal dysplasia: An 8 year longitudinal evaluation of Italian subjects. Angle Orthod. 2010, 80, 733–739. [Google Scholar] [CrossRef] [Green Version]
- Cluzeau, C.; Hadj-Rabia, S.; Jambou, M.; Mansour, S.; Guigue, P.; Masmoudi, S.; Bal, E.; Chassaing, N.; Vincent, M.-C.; Viot, G.; et al. Only four genes (EDA1, EDAR, EDARADD, and WNT10A) account for 90% of hypohidrotic/anhidrotic ectodermal dysplasia cases. Hum. Mutat. 2011, 32, 70–72. [Google Scholar] [CrossRef] [Green Version]
- Bohring, A.; Stamm, T.; Spaich, C.; Haase, C.; Spree, K.; Hehr, U.; Hoffmann, M.; Ledig, S.; Sel, S.; Wieacker, P. WNT10A mutations are a frequent cause of a broad spectrum of ectodermal dysplasias with sex-biased manifestation pattern in heterozygotes. Am. J. Hum. Genet. 2009, 85, 97–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adaimy, L.; Chouery, E.; Mégarbané, H.; Mroueh, S.; Delague, V.; Nicolas, E.; Belguith, H.; de Mazancourt, P.; Mégarbané, A. Mutation in WNT10A is associated with an autosomal recessive ectodermal dysplasia: The odonto-onycho-dermal dysplasia. Am. J. Hum. Genet. 2007, 81, 821–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowles, B.; Ferrer, A.; Nishimura, C.J.; Pinto e Vairo, F.; Rey, T.; Leheup, B.; Sullivan, J.; Schoch, K.; Stong, N.; Agolini, E. TSPEAR variants are primarily associated with ectodermal dysplasia and tooth agenesis but not hearing loss: A novel cohort study. Am. J. Med. Genet. Part A 2021, 185, 2417–2433. [Google Scholar] [CrossRef] [PubMed]
- Delmaghani, S.; Aghaie, A.; Michalski, N.; Bonnet, C.; Weil, D.; Petit, C. Defect in the gene encoding the EAR/EPTP domain-containing protein TSPEAR causes DFNB98 profound deafness. Hum. Mol. Genet. 2012, 21, 3835–3844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheel, H.; Tomiuk, S.; Hofmann, K. A common protein interaction domain links two recently identified epilepsy genes. Hum. Mol. Genet. 2002, 11, 1757–1762. [Google Scholar] [CrossRef] [Green Version]
- Peled, A.; Sarig, O.; Samuelov, L.; Bertolini, M.; Ziv, L.; Weissglas-Volkov, D.; Eskin-Schwartz, M.; Adase, C.A.; Malchin, N.; Bochner, R. Mutations in TSPEAR, encoding a regulator of notch signaling, affect tooth and hair follicle morphogenesis. PLoS Genet. 2016, 12, e1006369. [Google Scholar] [CrossRef]
- Du, R.; Dinckan, N.; Song, X.; Coban-Akdemir, Z.; Jhangiani, S.N.; Guven, Y.; Aktoren, O.; Kayserili, H.; Petty, L.E.; Muzny, D.M. Identification of likely pathogenic and known variants in TSPEAR, LAMB3, BCOR, and WNT10A in four Turkish families with tooth agenesis. Hum. Genet. 2018, 137, 689–703. [Google Scholar] [CrossRef]
- Alshegifi, H.A.; Alamoudi, A.M.; Alrougi, A.; Alshaikh, H.; Alamri, A.; Shawli, A.M. Ectodermal Dysplasia: A Case Report. Cureus 2022, 14, e21184. [Google Scholar] [CrossRef]
- Zhou, M.; Zhang, H.; Camhi, H.; Seymen, F.; Koruyucu, M.; Kasimoglu, Y.; Kim, J.-W.; Kim-Berman, H.; Yuson, N.M.R.; Benke, P.J. Analyses of oligodontia phenotypes and genetic etiologies. Int. J. Oral Sci. 2021, 13, 32. [Google Scholar] [CrossRef]
- De La Dure-Molla, M.; Fournier, B.P.; Manzanares, M.C.; Acevedo, A.C.; Hennekam, R.C.; Friedlander, L.; Boy-Lefèvre, M.; Kerner, S.; Toupenay, S.; Garrec, P. Elements of morphology: Standard terminology for the teeth and classifying genetic dental disorders. Am. J. Med. Genet. Part A 2019, 179, 1913–1981. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Lei, R.; Ding, S.-W.; Zhu, S. Skewer: A fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinform. 2014, 15, 182. [Google Scholar] [CrossRef]
- Reid, J.G.; Carroll, A.; Veeraraghavan, N.; Dahdouli, M.; Sundquist, A.; English, A.; Bainbridge, M.; White, S.; Salerno, W.; Buhay, C. Launching genomics into the cloud: Deployment of Mercury, a next generation sequence analysis pipeline. BMC Bioinform. 2014, 15, 30. [Google Scholar] [CrossRef] [Green Version]
- Wohler, E.; Martin, R.; Griffith, S.; Rodrigues, E.d.S.; Antonescu, C.; Posey, J.E.; Coban-Akdemir, Z.; Jhangiani, S.N.; Doheny, K.F.; Lupski, J.R. PhenoDB, GeneMatcher and VariantMatcher, tools for analysis and sharing of sequence data. Orphanet J. Rare Dis. 2021, 16, 365. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Sherry, S.T.; Ward, M.-H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [Green Version]
- Auton, A.; Abecasis, G.R.; Altshuler, D.M.; Durbin, R.M.; Abecasis, G.R.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Donnelly, P.; Eichler, E.E.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Sim, N.-L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef] [Green Version]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Cooper, G.M.; Stone, E.A.; Asimenos, G.; Green, E.D.; Batzoglou, S.; Sidow, A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005, 15, 901–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coban-Akdemir, Z.; White, J.J.; Song, X.; Jhangiani, S.N.; Fatih, J.M.; Gambin, T.; Bayram, Y.; Chinn, I.K.; Karaca, E.; Punetha, J. Identifying genes whose mutant transcripts cause dominant disease traits by potential gain-of-function alleles. Am. J. Hum. Genet. 2018, 103, 171–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, H.A.; El-Kamah, G.Y.; Rabie, E.; Mostafa, M.I.; Abouzaid, M.R.; Hassib, N.F.; Mehrez, M.I.; Abdel-Kader, M.A.; Mohsen, Y.H.; Zada, S.K. Gene Mutations of the Three Ectodysplasin Pathway Key Players (EDA, EDAR, and EDARADD) Account for More than 60% of Egyptian Ectodermal Dysplasia: A Report of Seven Novel Mutations. Genes 2021, 12, 1389. [Google Scholar] [CrossRef]
- Den Dunnen, J.T.; Dalgleish, R.; Maglott, D.R.; Hart, R.K.; Greenblatt, M.S.; McGowan-Jordan, J.; Roux, A.; Smith, T.; Antonarakis, S.E.; Taschner, P.E.M. HGVS recommendations for the description of sequence variants: 2016 update. Hum. Mutat. 2016, 37, 564–569. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [CrossRef]
- Chen, Y.; Lu, H.; Zhang, N.; Zhu, Z.; Wang, S.; Li, M. PremPS: Predicting the impact of missense mutations on protein stability. PLoS Comput. Biol. 2020, 16, e1008543. [Google Scholar] [CrossRef]
- Ittisoponpisan, S.; Islam, S.A.; Khanna, T.; Alhuzimi, E.; David, A.; Sternberg, M.J.E. Can predicted protein 3D structures provide reliable insights into whether missense variants are disease associated? J. Mol. Biol. 2019, 431, 2197–2212. [Google Scholar] [CrossRef]
- Song, J.-S.; Bae, M.; Kim, J.-W. Novel TSPEAR mutations in non-syndromic oligodontia. Oral Dis. 2020, 26, 847–849. [Google Scholar] [CrossRef]
- Sloan-Heggen, C.M.; Bierer, A.O.; Shearer, A.E.; Kolbe, D.L.; Nishimura, C.J.; Frees, K.L.; Ephraim, S.S.; Shibata, S.B.; Booth, K.T.; Campbell, C.A. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum. Genet. 2016, 135, 441–450. [Google Scholar] [CrossRef] [Green Version]
- ClinGen Hearing Loss Expert Panel: TSPEAR—Nonsyndromic Genetic Hearing Loss. Available online: https://search.clinicalgenome.org/kb/gene-validity/CGGV:assertion_f0928b06-d3bb-41fe-8222-d7f0e6c0a25a-2017-09-26T040000.000Z (accessed on 10 January 2022).
- Smith, R.; University of Iowa, Iowa City, Iowa, US. Personal Communication, 2022.
- Rehm, H.L.; Berg, J.S.; Brooks, L.D.; Bustamante, C.D.; Evans, J.P.; Landrum, M.J.; Ledbetter, D.H.; Maglott, D.R.; Martin, C.L.; Nussbaum, R.L. ClinGen—the clinical genome resource. N. Engl. J. Med. 2015, 372, 2235–2242. [Google Scholar] [CrossRef] [Green Version]
- Savey, L.; Resche-Rigon, M.; Wechsler, B.; Comarmond, C.; Piette, J.C.; Cacoub, P.; Saadoun, D. Ethnicity and association with disease manifestations and mortality in Behçet’s disease. Orphanet J. Rare Dis. 2014, 9, 42. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.Y.; Levy, A.N.; Trivedi, H.D.; Chan, F.K.L.; Ng, S.C.; Ananthakrishnan, A.N. Ethnicity influences phenotype and outcomes in inflammatory bowel disease: A systematic review and meta-analysis of population-based studies. Clin. Gastroenterol. Hepatol. 2018, 16, 190–197. [Google Scholar] [CrossRef] [Green Version]
- Bondarets, N.; McDonald, F. Analysis of the vertical facial form in patients with severe hypodontia. Am. J. Phys. Anthropol. 2000, 111, 177–184. [Google Scholar] [CrossRef]
- Al-Abdallah, M.; AlHadidi, A.; Hammad, M.; Al-Ahmad, H. Prevalence and distribution of dental anomalies: A comparison between maxillary and mandibular tooth agenesis. Am. J. Orthod. Dentofac. Orthop. 2015, 148, 793–798. [Google Scholar] [CrossRef]
- Choi, S.J.; Lee, J.W.; Song, J.H. Dental anomaly patterns associated with tooth agenesis. Acta Odontol. Scand. 2017, 75, 161–165. [Google Scholar] [CrossRef]
- Lloyd-Lewis, B.; Mourikis, P.; Fre, S. Notch signalling: Sensor and instructor of the microenvironment to coordinate cell fate and organ morphogenesis. Curr. Opin. Cell Biol. 2019, 61, 16–23. [Google Scholar] [CrossRef]
- Artavanis-Tsakonas, S.; Rand, M.D.; Lake, R.J. Notch signaling: Cell fate control and signal integration in development. Science 1999, 284, 770–776. [Google Scholar] [CrossRef] [Green Version]
- Borggrefe, T.; Liefke, R. Fine-tuning of the intracellular canonical Notch signaling pathway. Cell Cycle 2012, 11, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Felszeghy, S.; Suomalainen, M.; Thesleff, I. Notch signalling is required for the survival of epithelial stem cells in the continuously growing mouse incisor. Differentiation 2010, 80, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.-Y.; Kao, C.-H.; Lin, K.M.-C.; Kaartinen, V.; Yang, L.-T. Notch Signaling Regulates Late-Stage Epidermal Differentiation and Maintains Postnatal Hair Cycle Homeostasis. PLoS ONE 2011, 6, e15842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanpain, C.; Lowry, W.E.; Pasolli, H.A.; Fuchs, E. Canonical notch signaling functions as a commitment switch in the epidermal lineage. Genes Dev. 2006, 20, 3022–3035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.; Lin, M.-H.; Tian, X.; Cheng, H.-T.; Gridley, T.; Shen, J.; Kopan, R. γ-Secretase functions through Notch signaling to maintain skin appendages but is not required for their patterning or initial morphogenesis. Dev. Cell 2004, 7, 731–743. [Google Scholar] [CrossRef] [Green Version]
- Aubin-Houzelstein, G. Notch signaling and the developing hair follicle. In Notch Signaling in Embryology and Cancer. Advances in Experimental Medicine and Biology; Reichrath, J., Reichrath, S., Eds.; Springer US: New York, NY, USA, 2012; pp. 142–160. [Google Scholar] [CrossRef]
- Wang, Y.; Yao, B.; Duan, X.; Li, J.; Song, W.; Jirigala, E.; Li, Z.; Yuan, X.; Kong, Y.; Zhang, Y. Notch1 down-regulation in lineage-restricted niches of mouse eccrine sweat glands [Preprint]. Res. Sq. 2021. [Google Scholar] [CrossRef]
- Dang, H.; Lin, A.L.; Zhang, B.; Zhang, H.; Katz, M.S.; Yeh, C. Role for Notch signaling in salivary acinar cell growth and differentiation. Dev. Dyn. 2009, 238, 724–731. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.-H.; Kopan, R. Long-range, nonautonomous effects of activated Notch1 on tissue homeostasis in the nail. Dev. Biol. 2003, 263, 343–359. [Google Scholar] [CrossRef] [Green Version]
- Staub, E.; Pérez-Tur, J.; Siebert, R.; Nobile, C.; Moschonas, N.K.; Deloukas, P.; Hinzmann, B. The novel EPTP repeat defines a superfamily of proteins implicated in epileptic disorders. Trends Biochem. Sci. 2002, 27, 441–444. [Google Scholar] [CrossRef]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Chapman, M.; Evans, K.; Azevedo, L.; Hayden, M.; Heywood, S.; Millar, D.S.; Phillips, A.D.; et al. The Human Gene Mutation Database (HGMD®): Optimizing its use in a clinical diagnostic or research setting. Hum. Genet. 2020, 139, 1197–1207. [Google Scholar] [CrossRef]
- Van der Hout, A.H.; Oudesluijs, G.G.; Venema, A.; Verheij, J.B.G.M.; Mol, B.G.J.; Rump, P.; Brunner, H.G.; Vos, Y.J.; van Essen, A.J. Mutation screening of the Ectodysplasin-A receptor gene EDAR in hypohidrotic ectodermal dysplasia. Eur. J. Hum. Genet. 2008, 16, 673–679. [Google Scholar] [CrossRef] [Green Version]
- Bodle, E.E.; Zhu, W.; Velez-Bartolomei, F.; Tesi-Rocha, A.; Liu, P.; Bernstein, J.A. Combined Genome Sequencing and RNA Analysis Reveals and Characterizes a Deep Intronic Variant in IGHMBP2 in a Patient with Spinal Muscular Atrophy with Respiratory Distress Type 1. Pediatr. Neurol. 2021, 114, 16–20. [Google Scholar] [CrossRef]
- Larrue, R.; Chamley, P.; Bardyn, T.; Lionet, A.; Gnemmi, V.; Cauffiez, C.; Glowacki, F.; Pottier, N.; Broly, F. Diagnostic utility of whole-genome sequencing for nephronophthisis. NPJ Genom. Med. 2020, 5, 38. [Google Scholar] [CrossRef]
- Di Scipio, M.; Tavares, E.; Deshmukh, S.; Audo, I.; Green-Sanderson, K.; Zubak, Y.; Zine-Eddine, F.; Pearson, A.; Vig, A.; Tang, C.Y.; et al. Phenotype Driven Analysis of Whole Genome Sequencing Identifies Deep Intronic Variants that Cause Retinal Dystrophies by Aberrant Exonization. Investig. Ophthalmol. Vis. Sci. 2020, 61, 36. [Google Scholar] [CrossRef]
- Temtamy, S.; Aglan, M. Consanguinity and genetic disorders in Egypt. Middle East J. Med. Genet. 2012, 1, 12–17. [Google Scholar] [CrossRef]
- Shawky, R.M.; Elsayed, N.S.; Ibrahim, D.S.; Seifeldin, N.S. Profile of genetic disorders prevalent in northeast region of Cairo, Egypt. Egypt. J. Med. Hum. Genet. 2012, 13, 45–62. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Exon Number | Forward (F) and Reverse (R) Primers Sequences (5′ to 3′) | Annealing Temperature (°C) |
|---|---|---|
| Exon 1 | F-ACCTCTGTCCCCGCCTTAG | 61 |
| R-CCATCTCCACAGGGTGCTAC | ||
| Exon 5 | F-AAGCTCAGTGGTCGCCTCC | 62 |
| R-ACACGAGAGGGGCTGAGAG | ||
| Exon 9 | F-TGGGAATAGCACCTGTGATG | 59.5 |
| R-AGAGCAGCACTAGGTTTGGC | ||
| Exon11 | F-CCCCGGCTCCTCCTCTATAA | 61.5 |
| R-CCTCGGCAGCTCATTACCT |
| Fa | P | Consanguinity | Sex | Age | Sweating | Hair | Skin | Nails | Teeth | Conical Shaped Teeth | Other Oral Features | Others | Genotype /Variant | Affected Ectodermal Organs |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | + | M | 16 y | N | High anterior hairline | Dry skin, severe palmoplanter hyperkeratosis, and keratosis pilaris. | D | O | + | Thick lips, everted lips, macroglossia, median grooved tongue, broad uvula, enamel hypocalcification, retained deciduous teeth, and delayed eruption. | Skeletal abnormalities: talipes equinovarus, pes cavus, low inserted third toes, clinodactyly (toes), arachnodactyly (hands), and no ejaculation. Mother has hypodontia. | Homozygous/ c.44delC p.(Gly17Alafs*34) | Hair, teeth and nails |
| 2 | 2 | + | M | 10 y | N | N | Dry palms | D | O | + | Thick lower lip, everted lower lip, macrostomia, short broad philtrum, and very narrow V shaped palate. | Both parents have hypodontia | Heterozygous/ c.668C>T p.(Ser223Leu) | Teeth and nails |
| 3 | 3 | - | F | 9 y 6 m | N | Sparse scalp hair *, and high anterior hairline | N | N | O | + | Thick lips, everted lips, deep labiomental sulcus, and dimpled chin. | Compound heterozygous/ c.[1423G>C]; [1788-1790delAGA] p.([Gly475Arg];[Glu596del]) | Hair and teeth | |
| 3 | 4 | - | M | 6 y | N | Sparse scalp hair *, and high anterior hairline | N | N | O | + | Thick lower lip, and everted lips. | Hair and teeth | ||
| 4 | 5 | + | F | 8 y | Hyperhidrosis of palms and soles | Sparse scalp hair *, high anterior hairline and sparse eyebrows | Hyperkeratosis | N | O | + | Thick lips, prominent philtrum, lower pseudolabial cleft, highly attached upper labial frenum, long uvula, and wide overjet. | Homozygous/ c.1788-1790delAGAp.(Glu596del) | Hair, teeth and sweat glands | |
| 5 | 6 | + | M | 3 y 8 m | N | Sparse scalp hair, high anterior hairline, and sparse eyebrows. | N | N | O | + | Everted lower lip, and bow shape upper lip. | Delayed motor and mental milestones | Homozygous/ c.1788-1790delAGA p.(Glu596del) | Hair and teeth |
| 6 | 7 | + | F | 25 y | N | Sparse scalp hair *, high anterior hairline, and sparse eyebrows. | N | N | O | N/A (completely edentulous) | Thick lips, everted lower lip, short philtrum, mandibular prognathism, fissured tongue, and decreased salivary flow rate. | Homozygous/ c.1788-1790delAGA p.(Glu596del) | Hair and teeth | |
| 6 | 8 | + | F | 21 y | N | Sparse scalp hair *, high anterior hairline, and absent eyebrows. | N | D | O | N/A (missing all anterior teeth) | Short philtrum, and thick lower lip. | Familial Mediterranean fever, and bilateral syndactyly between 2nd and 3rd toes. | Homozygous/ c.1788-1790delAGA p.(Glu596del) | Hair, teeth and nails |
| 7 | 9 | + | F | 3 y 6 m | N | Sparse scalp hair, high anterior hairline, and sparse eyebrows. | Dry skin | D | O | + | Everted lower lip, deep labiomental sulcus, asymmetry of the lower lip, and bifid tip of the tongue. | Homozygous/ c.1788-1790delAGA p.(Glu596del) | Hair, teeth and nails | |
| 8 | 10 | + | F | 12 y 8 m | N | Sparse scalp hair * and high anterior hairline. | N | N | O | + | Thick lips, everted lips, narrow philtrum, narrow mandibular arch, lower pseudolabial cleft, retained deciduous teeth, and delayed eruption. | Homozygous/ c.1788-1790delAGA p.(Glu596del) | Hair and teeth |
| TSPEAR Variant Name NM_144991.3 NP_659428.2 | Minor Allele Frequencies (MAF) | Polyphen-2 | MutationTaster2 | PROVEAN | SIFT | Mutation Assessor | CADD | GERP | ClinVar Clinical Significance | ACMG Classification/Evidence |
|---|---|---|---|---|---|---|---|---|---|---|
| c.44delC p.(Gly17Alafs*34 | N/A | N/A | Disease-causing (Probability = 1) | N/A | N/A | N/A | N/A | 1.91 | N/A | Pathogenic (PVS1, PM2, PP3) |
| c.668C>T p.(Ser223Leu) | gnomAD:0.0007259 1000G:0.0008 dbSNP:0.000982 (rs149481227) | Possibly damaging (0.521) | Disease-causing (Probability = 0.99, score = 145) | Deleterious (−2.962) | Deleterious (0.02) | Medium (0.828) | 25.9 | 5.11 | Conflicting interpretations of pathogenicity | Likely pathogenic (PM1, PM2, PP3, PP4) |
| c.1788-1790delAGA p.(Glu596del) | gnomAD:0.0001204 1000G: N/A dbSNP: 0.00026 (rs782084367) | N/A | Disease-causing (Probability = 0.99) | N/A | N/A | N/A | N/A | 3.44 | N/A | Pathogenic (PS4, PM1, PM2, PM3, PP1) |
| c.1423G>C p.(Gly475Arg) | gnomAD:0.00003183 1000G: N/A dbSNP: 0.00005 (rs782056388) | Probably damaging (0.994) | Disease-causing (Probability = 0.99, score = 56) | Deleterious (−6.024) | Deleterious (0.01) | Medium (0.828) | 24.5 | 5.18 | Uncertain significance | Likely pathogenic (PM1, PM2, PM3, PP1, PP3) |
| Reported Ethnicity | North African | Middle Eastern | European | Others * | Total | p-Value |
|---|---|---|---|---|---|---|
| Number of patients | 10/38 (26.3%) | 12/38 (31.6%) | 9/38 (23.7%) | 7/38 (18.4%) | 38/38 (100%) | - |
| TSPEAR-associated phenotypes ** | ||||||
| ED | 10/10 (100%) | 4/12 (33.3%) | 6/9 (66.7%) | 2/7 (28.6%) | 22/38 (57.9%) | 0.0046 |
| TA without other ectodermal features | 0/10 (0%) | 3/12 (25%) | 3/9 (33.3%) | 2/7 (28.6%) | 8/38 (21.1%) | 0.2799 |
| SNHL | 0/10 (0%) | 5/12 (41.7%) | 0/9 (0%) | 2/7 (28.6%) | 7/38 (18.4%) | 0.0282 |
| SNHL & ED | 0/10 (0%) | 0/12 (0%) | 0/9 (0%) | 1/7 (14.3%) | 1/38 (2.6) | 0.2080 |
| Dysmorphic facial features | ||||||
| Dysmorphic facial features | 10/10 (100%) | 5/12 (41.7%) | 0/9 (0%) | 2/7 (28.6%) | 17/38 (44.7%) | 0.0001 |
| Detailed dysmorphic facial features | Ten ED patients of Egyptian origin featured:
| Five reported cases: 1- Three ED cases of Palestinian origin featured:
2- One ED case of Saudi origin reported typical ED facial features including
3- One isolated TA case of Turkish origin had microcephaly, and reported faces were:
| N/A | Two reported cases: 1- One ED case of Ashkenazi Jewish ancestry showed:
2- The other case is African American who had TA, and featured:
| ||
| Ectodermal elements involvement in ED phenotype *** | ||||||
| Teeth | 10/10 (100%) | 4/4 (100%) | 3/6 (50%) | 2/3 (66.7%) | 19/23 (82.6%) | 0.1737 |
| Hair | 9/10 (90%) | 4/4 (100%) | 3/6 (50%) | 2/3 (66.7%) | 18/23 (78.3%) | 0.0477 |
| Sweat glands | 0/10 (0%) | 4/4 (100%) | 4/6 (66.7%) | 1/3 (33.3%) | 9/23 (39.1%) | 0.0022 |
| Nails | 4/10 (40%) | 0/4 (0%) | 4/6 (66.7%) | 2/3 (66.7%) | 10/23 (43.5%) | 0.1649 |
| References | Current study | [24,25,27,28,29] | [24] | [24,27,55,56] | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rabie, E.A.; Sayed, I.S.M.; Amr, K.; Ahmed, H.A.; Mostafa, M.I.; Hassib, N.F.; El-Sayed, H.; Zada, S.K.; El-Kamah, G. Confirmation of a Phenotypic Entity for TSPEAR Variants in Egyptian Ectodermal Dysplasia Patients and Role of Ethnicity. Genes 2022, 13, 1056. https://doi.org/10.3390/genes13061056
Rabie EA, Sayed ISM, Amr K, Ahmed HA, Mostafa MI, Hassib NF, El-Sayed H, Zada SK, El-Kamah G. Confirmation of a Phenotypic Entity for TSPEAR Variants in Egyptian Ectodermal Dysplasia Patients and Role of Ethnicity. Genes. 2022; 13(6):1056. https://doi.org/10.3390/genes13061056
Chicago/Turabian StyleRabie, Eman A., Inas S. M. Sayed, Khalda Amr, Hoda A. Ahmed, Mostafa I. Mostafa, Nehal F. Hassib, Heba El-Sayed, Suher K. Zada, and Ghada El-Kamah. 2022. "Confirmation of a Phenotypic Entity for TSPEAR Variants in Egyptian Ectodermal Dysplasia Patients and Role of Ethnicity" Genes 13, no. 6: 1056. https://doi.org/10.3390/genes13061056