
Genetic Differentiation and Widespread Mitochondrial Heteroplasmy among Geographic Populations of the Gourmet Mushroom Thelephora ganbajun from Yunnan, China


 ,
,
Abstract
:1. Introduction
2. Materials and Methods
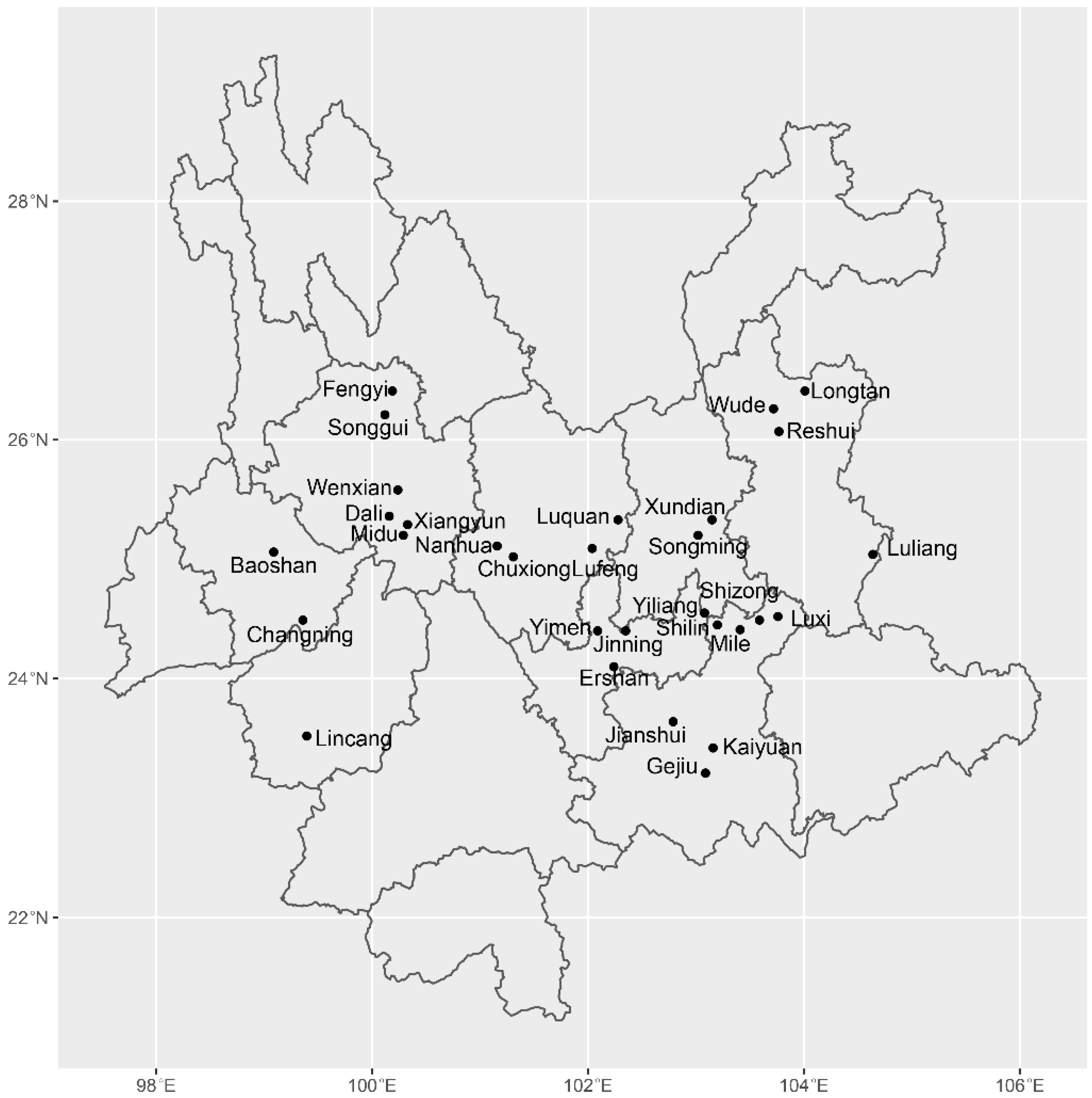
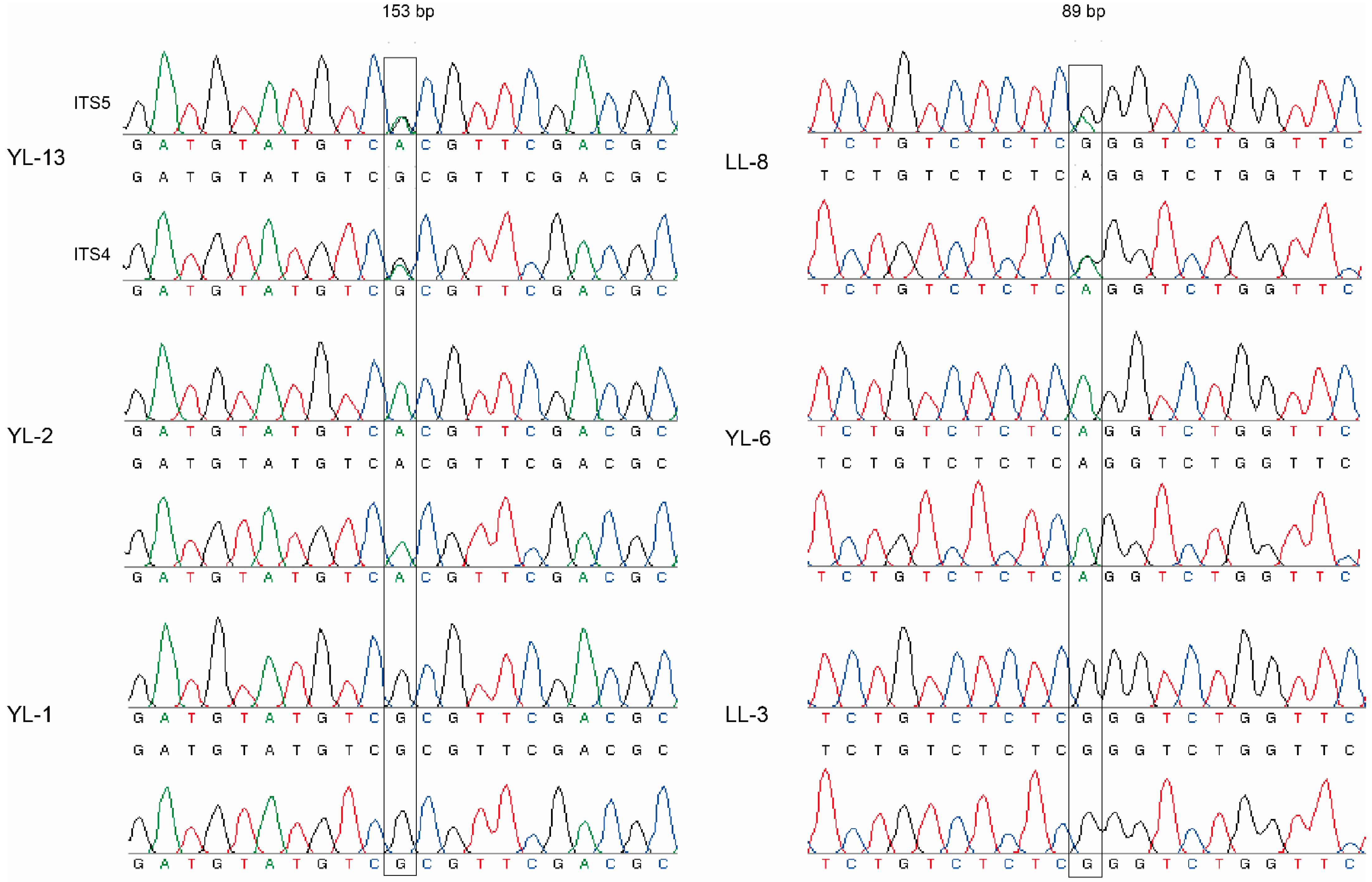
2.1. Sampling, DNA Extraction, PCR Amplification, and Sequencing
2.2. Data Analysis
2.2.1. Haplotype Inference of ITS Sequences
2.2.2. ITS Genetic Diversity and Population Structure
2.2.3. Phylogenetic Analyses
3. Results
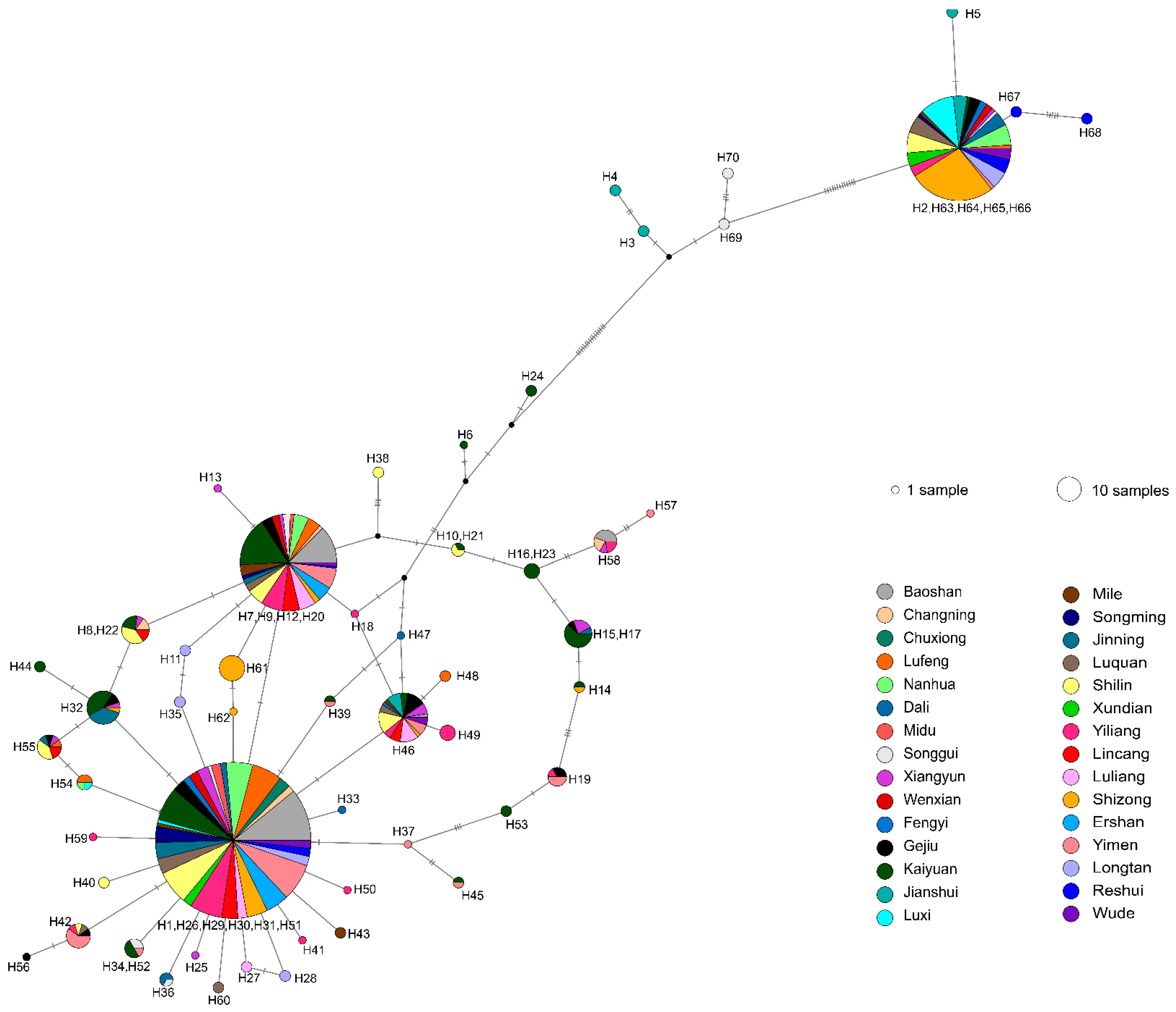
3.1. ITS Haplotypes’ Distribution and Diversity
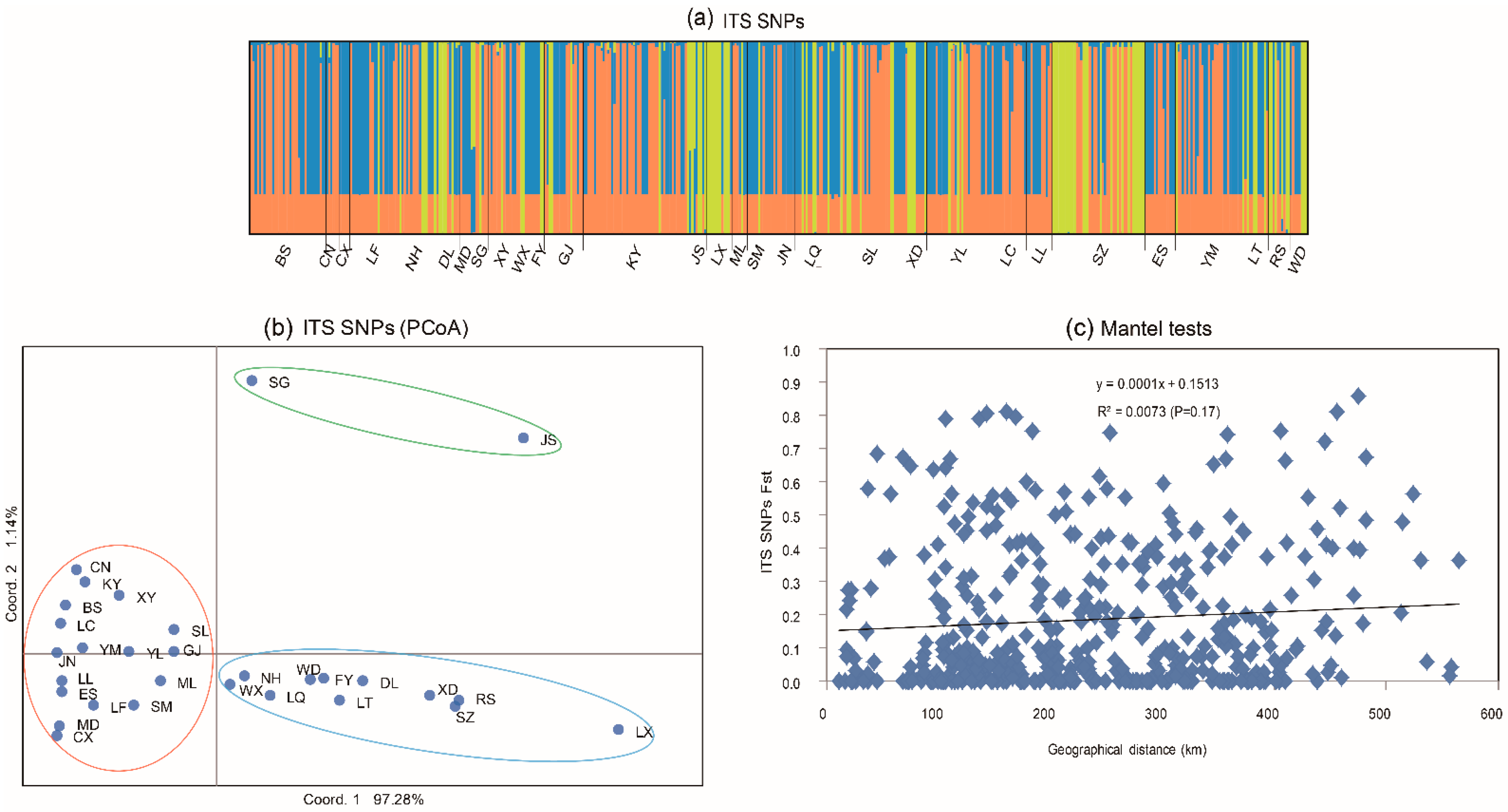
3.2. Population Structure Based on ITS Sequences
3.3. Genetic Differentiation of ITS
3.4. Intraspecific Clustering of ITS Sequences
3.5. Frequencies of α and β Type of Introns in cox1 Gene
4. Discussion
4.1. Distinct Intraspecific Differentiation Based on ITS Sequences
4.2. Genetic Variation and Differentiation Revealed by ITS Sequences
4.3. Distribution of Heteroplasmic Mitochondrial cox1 Gene
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sha, T.; Xu, J.P.; Palanichamy, M.G.; Zhang, H.B.; Li, T.; Zhao, Z.W.; Zhang, Y.P. Genetic diversity of the endemic gourmet mushroom Thelephora ganbajun from southwestern China. Microbiol.-SGM 2008, 154, 3460–3468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brundrett, M. Diversity and classification of mycorrhizal associations. Biol. Rev. 2004, 79, 473–495. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Mo, M.Z.; Yang, L.; Mi, F.; Cao, Y.; Liu, C.L.; Tang, X.Z.; Wang, P.F.; Xu, J.P. Exploring the Species Diversity of Edible Mushrooms in Yunnan, Southwestern China, by DNA Barcoding. J. Fungi 2021, 7, 310. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.P.; Wang, P.F. Mitochondrial inheritance in basidiomycete fungi. Fungal Biol. Rev. 2015, 29, 209–219. [Google Scholar] [CrossRef]
- Barr, C.M.; Neiman, M.; Taylor, D.R. Inheritance and recombination of mitochondrial genomes in plants, fungi and animals. New Phytol. 2005, 168, 39–50. [Google Scholar] [CrossRef]
- Cheng, J.; Luo, Q.; Ren, Y.H.; Luo, Z.; Liao, W.L.; Wang, X.; Li, Q. Panorama of intron dynamics and gene rearrangements in the phylum Basidiomycota as revealed by the complete mitochondrial genome of Turbinellus floccosus. Appl. Microbiol. Biot. 2021, 105, 2017–2032. [Google Scholar] [CrossRef]
- Huang, W.L.; Feng, H.Y.; Tu, W.Y.; Xiong, C.; Jin, X.; Li, P.; Wang, X.; Li, Q. Comparative Mitogenomic Analysis Reveals Dynamics of Intron Within and Between Tricholoma Species and Phylogeny of Basidiomycota. Front. Genet. 2021, 12, 534871. [Google Scholar] [CrossRef]
- Li, Q.; Yang, L.; Xiang, D.; Wan, Y.; Wu, Q.; Huang, W.; Zhao, G. The complete mitochondrial genomes of two model ectomycorrhizal fungi (Laccaria): Features, intron dynamics and phylogenetic implications. Int. J. Biol. Macromol. 2020, 145, 974–984. [Google Scholar] [CrossRef]
- Li, Q.; Wu, P.; Li, L.J.; Feng, H.Y.; Tu, W.Y.; Bao, Z.J.; Xiong, C.; Gui, M.Y.; Huang, W.L. The first eleven mitochondrial genomes from the ectomycorrhizal fungal genus (Boletus) reveal intron loss and gene rearrangement. Int. J. Biol. Macromol. 2021, 172, 560–572. [Google Scholar] [CrossRef]
- Xu, J.P.; Li, H. Current perspectives on mitochondrial inheritance in fungi. Cell Health Cytoskel. 2015, 7, 143–154. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Wang, Q.F.; Chen, C.; Jin, X.; Chen, Z.Q.; Xiong, C.; Li, P.; Zhao, J.; Huang, W.L. Characterization and comparative mitogenomic analysis of six newly sequenced mitochondrial genomes from ectomycorrhizal fungi (Russula) and phylogenetic analysis of the Agaricomycetes. Int. J. Biol. Macromol. 2018, 119, 792–802. [Google Scholar] [CrossRef] [PubMed]
- Beaudet, D.; Nadimi, M.; Iffis, B.; Hijri, M. Rapid Mitochondrial Genome Evolution through Invasion of Mobile Elements in Two Closely Related Species of Arbuscular Mycorrhizal Fungi. PLoS ONE 2013, 8, e60768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wang, S.J.; Li, H.X.; Liu, C.L.; Mi, F.; Wang, R.R.; Mo, M.Z.; Xu, J.P. Evidence for Persistent Heteroplasmy and Ancient Recombination in the Mitochondrial Genomes of the Edible Yellow Chanterelles From Southwestern China and Europe. Front. Microbiol. 2021, 12, 699598. [Google Scholar] [CrossRef] [PubMed]
- Birky, C.W. The Inheritance of Genes in Mitochondria and Chloroplasts: Laws, Mechanisms, and Models. Annu. Rev. Genet. 2001, 35, 125–148. [Google Scholar] [CrossRef] [PubMed]
- Billiard, S.; López-Villavicencio, M.; Devier, B.; Hood, M.E.; Fairhead, C.; Giraud, T. Having sex, yes, but with whom? Inferences from fungi on the evolution of anisogamy and mating types. Biol. Rev. 2011, 86, 421–442. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.; Vissing, J. Paternal inheritance of mitochondrial DNA. N. Engl. J. Med. 2002, 347, 576–580. [Google Scholar] [CrossRef] [PubMed]
- Basse, C.W. Mitochondrial inheritance in fungi. Curr. Opin. Microbiol. 2010, 13, 712–719. [Google Scholar] [CrossRef]
- Wolf, K.; Lang, B.; Burger, G.; Kaudewitz, F. Extrachromosomal-Inheritance in Schizosaccharomyces-Pombe. Mol. Gen. Genet. 1976, 144, 75–81. [Google Scholar] [CrossRef]
- Leducq, J.B.; Nielly-Thibault, L.; Charron, G.; Eberlein, C.; Verta, J.P.; Samani, P.; Sylvester, K.; Hittinger, C.T.; Bell, G.; Landry, C.R. Speciation driven by hybridization and chromosomal plasticity in a wild yeast. Nat. Microbiol. 2016, 1, 15003. [Google Scholar] [CrossRef]
- Leducq, J.B.; Henault, M.; Charron, G.; Nielly-Thibault, L.; Terrat, Y.; Fiumera, H.L.; Shapiro, B.J.; Landry, C.R. Mitochondrial Recombination and Introgression during Speciation by Hybridization. Mol. Biol. Evol. 2017, 34, 1947–1959. [Google Scholar] [CrossRef]
- Yan, Z.; Hull, C.M.; Sun, S.; Heitman, J.; Xu, J.P. The mating type-specific homeodomain genes SXI1 α and SXI2a coordinately control uniparental mitochondrial inheritance in Cryptococcus neoformans. Curr. Genet. 2007, 51, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.X.; Wilson, A.; Xu, J.P. Mitochondrial DNA inheritance in the human fungal pathogen Cryptococcus gattii. Fungal Genet. Biol. 2015, 75, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.R.; Huang, C.Y.; Chen, Q.; Wu, X.L.; Qu, J.B.; Zhang, J.X. Genetic Variability and Population Structure of the Mushroom Pleurotus eryngii var. tuoliensis. PLoS ONE 2013, 8, e83253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Ying, Z.; Pun, N. Mitochondrial recombination in natural populations of the button mushroom Agaricus bisporus. Fungal Genet. Biol. 2012, 55, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.F.; Sha, T.; Zhang, Y.R.; Cao, Y.; Mi, F.; Liu, C.L.; Yang, D.; Tang, X.Z.; He, X.X.; Dong, J.Y.; et al. Frequent heteroplasmy and recombination in the mitochondrial genomes of the basidiomycete mushroom Thelephora ganbajun. Sci. Rep. 2017, 7, 1626. [Google Scholar] [CrossRef]
- Ricardo, P.C.; Francoso, E.; Arias, M.C. Mitochondrial DNA intra-individual variation in a bumblebee species: A challenge for evolutionary studies and molecular identification. Mitochondrion 2020, 53, 243–254. [Google Scholar] [CrossRef]
- White, D.J.; Wolff, J.N.; Pierson, M.; Gemmell, N.J. Revealing the hidden complexities of mtDNA inheritance. Mol. Ecol. 2008, 17, 4925–4942. [Google Scholar] [CrossRef]
- Wallace, D.C.; Chalkia, D. Mitochondrial DNA Genetics and the Heteroplasmy Conundrum in Evolution and Disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a021220. [Google Scholar] [CrossRef]
- Rodriguez-Pena, E.; Verisimo, P.; Fernandez, L.; Gonzalez-Tizon, A.; Barcena, C.; Martinez-Lage, A. High incidence of heteroplasmy in the mtDNA of a natural population of the spider crab Maja brachydactyla. PLoS ONE 2020, 15, e0230243. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Zhang, Y.; Yu, Z.F.; Mi, F.; Liu, C.L.; Tang, X.Z.; Long, Y.X.; He, X.X.; Wang, P.F.; Xu, J.P. Structure, Gene Flow, and Recombination among Geographic Populations of a Russula virescens Ally from Southwestern China. PLoS ONE 2013, 8, e73174. [Google Scholar] [CrossRef] [Green Version]
- Fu, Z.Q.; Liu, G.; Wang, L.X. Assessment of potential human health risk of trace element in wild edible mushroom species collected from Yunnan Province, China. Environ. Sci. Pollut. Res. 2020, 27, 29218–29227. [Google Scholar] [CrossRef] [PubMed]
- Hua, R.; Chen, Z.; Fu, W. An Overview of Wild Edible Fungi Resource Conservation and Its Utilization in Yunnan. J. Agric. Sci. 2017, 9, 158–169. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; Sun, S.; Shahid, M.; Xu, J.P. Environment factors can influence mitochondrial inheritance in the fungus Cryptococcus neoformans. Fungal Genet. Biol. 2007, 44, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Bolchacova, E.; Voigt, K.; Crous, P.W.; et al. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hibbett, D.; Abarenkov, K.; Koljalg, U.; Opik, M.; Chai, B.; Cole, J.; Wang, Q.; Crous, P.; Robert, V.; Helgason, T.; et al. Sequence-based classification and identification of Fungi. Mycologia 2016, 108, 1049–1068. [Google Scholar] [CrossRef]
- Lindner, D.L.; Banik, M.T. Intragenomic variation in the ITS rDNA region obscures phylogenetic relationships and inflates estimates of operational taxonomic units in genus Laetiporus. Mycologia 2011, 103, 731–740. [Google Scholar] [CrossRef] [Green Version]
- Jie, C.; Magalie, M.; Xu, J.; Wang, S.; Marie, F.O.; Zhao, R.; Hyde, K.D.; Philippe, C.; Carter, D.A. Genetic Analyses of the Internal Transcribed Spacer Sequences Suggest Introgression and Duplication in the Medicinal Mushroom Agaricus subrufescens. PLoS ONE 2016, 11, e0156250. [Google Scholar] [CrossRef]
- Zhou, J.L.; Xu, J.P.; Jiao, A.G.; Yang, L.; Chen, J.; Callac, P.; Liu, Y.; Wang, S.X. Patterns of PCR Amplification Artifacts of the Fungal Barcode Marker in a Hybrid Mushroom. Front. Microbiol. 2019, 10, 02686. [Google Scholar] [CrossRef]
- Kuhn, G.; Hijri, M.; Sanders, I.R. Evidence for the evolution of multiple genomes in arbuscular mycorrhizal fungi. Nature 2001, 414, 745–748. [Google Scholar] [CrossRef]
- Jie, G.; Rx, A.; Nan, W.A.; Jz, A.; Xs, A.; Hw, B.; Jt, B.; Ac, A. Rapid identification of Amanita citrinoannulata poisoning using colorimetric and real-time fluorescence and loop-mediated isothermal amplification (LAMP) based on the nuclear ITS region. Food Chem. Mol. Sci. 2022, 4, 100082. [Google Scholar] [CrossRef]
- Koch, R.A.; Herr, J.R. Global Distribution and Richness of Armillaria and Related Species Inferred From Public Databases and Amplicon Sequencing Datasets. Front. Microbiol. 2021, 12, 733159. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Gao, J.; Tian, E.J.; Yu, W.J.; Li, H.; Zhang, J.; Xie, R.B.; Chen, A.L. Rapid Russula senecis identification assays using loop-mediated isothermal amplification based on real-time fluorescence and visualization. Appl. Microbiol. Biot. 2022, 106, 1227–1239. [Google Scholar] [CrossRef] [PubMed]
- Nicklas, J.A.; Brooks, E.M.; Hunter, T.C.; Single, R.; Branda, R.F. Development of a quantitative PCR (TaqMan) assay for relative mitochondrial DNA copy number and the common mitochondrial DNA deletion in the rat. Environ. Mol. Mutagen. 2004, 44, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Stephens, M.; Donnelly, P. A comparison of Bayesian methods for haplotype reconstruction from population genotype data. Am. J. Hum. Genet. 2003, 73, 1162–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozas, J.; Ferrer-Mata, A.; Sanchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sanchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.P.; Sha, T.; Li, Y.C.; Zhao, Z.W.; Yang, Z.L. Recombination and genetic differentiation among natural populations of the ectomycorrhizal mushroom Tricholoma matsutake from southwestern China. Mol. Ecol. 2008, 17, 1238–1247. [Google Scholar] [CrossRef]
- Leigh, J.W.; Bryant, D. POPART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Hubisz, M.J.; Falush, D.; Stephens, M.; Pritchard, J.K. Inferring weak population structure with the assistance of sample group information. Mol. Ecol. Resour. 2009, 9, 1322–1332. [Google Scholar] [CrossRef] [Green Version]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar] [CrossRef]
- Mi, F.; Zhang, Y.; Yang, D.; Tang, X.Z.; Wang, P.F.; He, X.X.; Zhang, Y.R.; Dong, J.Y.; Cao, Y.; Liu, C.L.; et al. Evidence for Inbreeding and Genetic Differentiation among Geographic Populations of the Saprophytic Mushroom Trogia venenata from Southwestern China. PLoS ONE 2016, 11, e0149507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, D.Y.; Xu, J.P.; Dong, J.Y.; Li, H.X.; Wang, D.; Gu, J.; Zhang, K.Q.; Zhang, Y. Historical Differentiation and Recent Hybridization in Natural Populations of the Nematode-Trapping Fungus Arthrobotrys oligospora in China. Microorganisms 2021, 9, 1919. [Google Scholar] [CrossRef] [PubMed]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. Clumpak: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, N.A. DISTRUCT: A program for the graphical display of population structure. Mol. Ecol. Notes 2004, 4, 137–138. [Google Scholar] [CrossRef]
- Caspermeyer, J. MEGA Evolutionary Software Re-Engineered to Handle Today’s Big Data Demands. Mol. Biol. Evol. 2016, 33, 1887. [Google Scholar] [CrossRef] [Green Version]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis. Nuclc. Acids. Symp. 1999, 41, 95–98. [Google Scholar] [CrossRef]
- Yang, X.Q.; Ma, S.Y.; Peng, Z.X.; Wang, Z.Q.; Yu, Z. Diversity of Plectosphaerella within aquatic plants from southwest China, with P. endophytica and P. sichuanensis spp. nov. MycoKeys 2021, 80, 57–75. [Google Scholar] [CrossRef]
- Cabon, M.; Li, G.J.; Saba, M.; Kolarik, M.; Jancovicova, S.; Khalid, A.N.; Moreau, P.A.; Wen, H.A.; Pfister, D.H.; Adarncik, S. Phylogenetic study documents different speciation mechanisms within the Russula globispora lineage in boreal and arctic environments of the Northern Hemisphere. IMA Fungus 2019, 10, 5. [Google Scholar] [CrossRef]
- Jensen, A.M.; O’Neil, N.P.; Iwaniuk, A.N.; Burg, T.M. Landscape effects on the contemporary genetic structure of Ruffed Grouse (Bonasa umbellus) populations. Ecol. Evol. 2019, 9, 5572–5592. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Chai, H.; Chen, M.; Zhang, X. Study on micro intervention and propagation promotion technology for Thelephora ganbajun. Edible Med. Mushrooms 2017, 25, 297–302. [Google Scholar]
- Tikochinski, Y.; Carreras, C.; Tikochinski, G.; Vilaca, S.T. Population-specific signatures of intra-individual mitochondrial DNA heteroplasmy and their potential evolutionary advantages (vol 81, 514, 2020). Sci. Rep. 2020, 10, 211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.H.; Ke, H.M.; Lin, C.I.; Lee, T.J.; Chung, C.L.; Tsai, I.J. Evidence of Extensive Intraspecific Noncoding Reshuffling in a 169-kb Mitochondrial Genome of a Basidiomycetous Fungus. Genome Biol. Evol. 2019, 11, 2774–2788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doublet, V.; Souty-Grosset, C.; Bouchon, D.; Cordaux, R.; Marcade, I. A Thirty Million Year-Old Inherited Heteroplasmy. PLoS ONE 2008, 3, e0002938. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, H.T.; Lehtinen, S.K.; Spelbrink, J.N. No sex please, we’re mitochondria: A hypothesis on the somatic unit of inheritance of mammalian mtDNA. Bioessays 2000, 22, 564–572. [Google Scholar] [CrossRef]
- Polovina, E.S.; Parakatselaki, M.E.; Ladoukakis, E.D. Paternal leakage of mitochondrial DNA and maternal inheritance of heteroplasmy in Drosophila hybrids. Sci. Rep. 2020, 10, 2599. [Google Scholar] [CrossRef]
- Nguyen, T.H.M.; Sondhi, S.; Ziesel, A.; Paliwal, S.; Fiumera, H.L. Mitochondrial-nuclear coadaptation revealed through mtDNA replacements in Saccharomyces cerevisiae. BMC Evol. Biol. 2020, 20, 128. [Google Scholar] [CrossRef]
- Lee, H.Y.; Chou, J.Y.; Cheong, L.; Chang, N.H.; Yang, S.Y.; Leu, J.Y. Incompatibility of nuclear and mitochondrial genomes causes hybrid sterility between two yeast species. Cell 2008, 135, 1065–1073. [Google Scholar] [CrossRef] [Green Version]
- De Chiara, M.; Friedrich, A.; Barre, B.; Breitenbach, M.; Schacherer, J.; Liti, G. Discordant evolution of mitochondrial and nuclear yeast genomes at population level. BMC Biol. 2020, 18, 49. [Google Scholar] [CrossRef]
- Dunbar, D.R.; Moonie, P.A.; Jacobs, H.T.; Holt, I.J. Different Cellular Backgrounds Confer a Marked Advantage to Either Mutant or Wild-Type Mitochondrial Genomes. Proc. Natl. Acad. Sci. USA 1995, 92, 6562–6566. [Google Scholar] [CrossRef] [Green Version]
- Parakatselaki, M.E.; Ladoukakis, E.D. mtDNA Heteroplasmy: Origin, Detection, Significance, and Evolutionary Consequences. Life 2021, 11, 633. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | Geographic Population | Long. (E) | Lat. (N) | Sample Size | Number of Haplotypes (No. of Private Haplotypes) | ITS Haplotype Diversity |
|---|---|---|---|---|---|---|
| Baoshan | Baoshan (BS) | 99.09 | 25.06 | 35 | 7 (2) | 0.509 |
| Changning (CN) | 99.36 | 24.49 | 6 | 4 | 0.727 | |
| Chuxiong | Chuxiong (CX) | 101.31 | 25.02 | 5 | 1 | 0 |
| Lufeng (LF) | 102.04 | 25.09 | 20 | 7 (1) | 0.553 | |
| Nanhua (NH) | 101.16 | 25.11 | 22 | 5 | 0.633 | |
| Dali | Dali (DL) | 100.16 | 25.36 | 9 | 6 (2) | 0.745 |
| Midu (MD) | 100.29 | 25.2 | 5 | 2 | 0.356 | |
| Songgui (SG) | 100.12 | 26.21 | 8 | 7 (2) | 0.892 | |
| Xiangyun (XY) | 100.33 | 25.29 | 13 | 11 (2) | 0.837 | |
| Wenxian (WX) | 100.24 | 25.58 | 8 | 4 | 0.667 | |
| Fengyi (FY) | 100.19 | 26.41 | 5 | 2 | 0.533 | |
| Honghe | Gejiu (GJ) | 103.09 | 23.21 | 18 | 11 (1) | 0.846 |
| Kaiyuan (KY) | 103.16 | 23.42 | 48 | 22 (10) | 0.824 | |
| Jianshui (JS) | 102.79 | 23.64 | 9 | 5 (3) | 0.758 | |
| Luxi (LX) | 103.76 | 24.52 | 12 | 3 | 0.301 | |
| Mile (ML) | 103.41 | 24.41 | 7 | 6 (1) | 0.780 | |
| Kunming | Songming(SM) | 103.02 | 25.2 | 9 | 3 | 0.392 |
| Jinning (JN) | 102.35 | 24.4 | 13 | 5 | 0.646 | |
| Luquan (LQ) | 102.28 | 25.33 | 16 | 8 (3) | 0.736 | |
| Shilin (SL) | 103.2 | 24.45 | 36 | 11 (3) | 0.788 | |
| Xundian (XD) | 103.15 | 25.33 | 9 | 3 (1) | 0.523 | |
| Yiliang (YL) | 103.08 | 24.55 | 31 | 15 (6) | 0.723 | |
| Lincang | Lincang (LC) | 99.397 | 23.52 | 15 | 6 | 0.697 |
| Qujing | Luliang (LL) | 104.64 | 25.04 | 12 | 5 (1) | 0.728 |
| Shizong (SZ) | 103.59 | 24.49 | 43 | 11 (4) | 0.607 | |
| Weishan | Ershan (ES) | 102.24 | 24.1 | 14 | 3 | 0.423 |
| Yuxi | Yimen (YM) | 102.09 | 24.4 | 31 | 12 (2) | 0.697 |
| Xuanwei | Longtan (LT) | 104.01 | 26.41 | 12 | 6 (4) | 0.725 |
| Reshui (RS) | 103.77 | 26.07 | 10 | 5 (2) | 0.732 | |
| Wude (WD) | 103.72 | 26.26 | 8 | 5 (1) | 0.733 | |
| Total | 30 | 489 | 70 (51) | 0.781 |
| α | β | α/β | |
|---|---|---|---|
| ITS sequence difference | −0.101 (p = 0.02) | 0.084 (p = 0.02) | −0.111 (p = 0.01) |
| Geographical distance | −0.067 (p = 0.06) | 0.03 (p = 0.11) | −0.082 (p = 0.02) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Xu, J.; Wang, S.; Wang, P.; Rao, W.; Hou, B.; Zhang, Y. Genetic Differentiation and Widespread Mitochondrial Heteroplasmy among Geographic Populations of the Gourmet Mushroom Thelephora ganbajun from Yunnan, China. Genes 2022, 13, 854. https://doi.org/10.3390/genes13050854
Li H, Xu J, Wang S, Wang P, Rao W, Hou B, Zhang Y. Genetic Differentiation and Widespread Mitochondrial Heteroplasmy among Geographic Populations of the Gourmet Mushroom Thelephora ganbajun from Yunnan, China. Genes. 2022; 13(5):854. https://doi.org/10.3390/genes13050854
Chicago/Turabian StyleLi, Haixia, Jianping Xu, Shaojuan Wang, Pengfei Wang, Wanqin Rao, Bin Hou, and Ying Zhang. 2022. "Genetic Differentiation and Widespread Mitochondrial Heteroplasmy among Geographic Populations of the Gourmet Mushroom Thelephora ganbajun from Yunnan, China" Genes 13, no. 5: 854. https://doi.org/10.3390/genes13050854