
Greig Cephalopolysyndactyly Contiguous Gene Syndrome: Case Report and Literature Review


 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient
2.2. Neuroimaging
2.3. Classical and Molecular Cytogenetic Investigations
3. Results
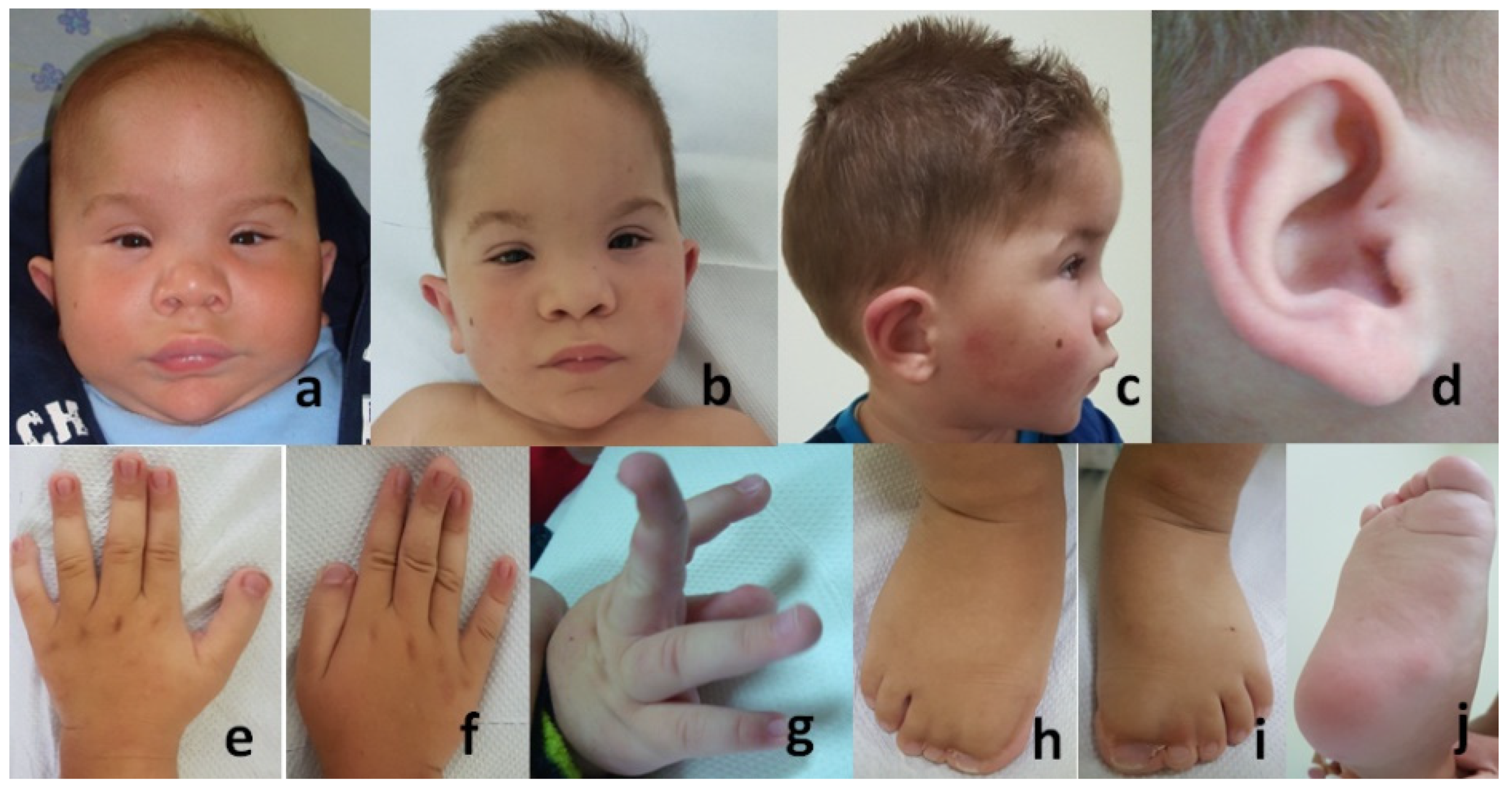
3.1. Morphological Evaluation of the Patient
3.2. Neuroimaging
3.3. Conventional and Molecular Cytogenetic Investigations
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Biesecker, L.G.; Johnston, J.J. Greig Cephalopolysyndactyly Syndrome; Adam, M.P., Ardinger, H.H., Pagon, R.A., Eds.; University of Washington: Seattle, WA, USA, 1993; pp. 1993–2020. [Google Scholar]
- Biesecker, L.G. The Greig cephalopolysyndactyly syndrome. Orphanet. J. Rare Dis. 2008, 3, 10. [Google Scholar] [CrossRef] [Green Version]
- Niida Yo Ozaki, M.; Takase, E.; Yokoyama, T.; Yamada, S. A Girl with Greig Cephalopolysyndactyly Contiguous Gene Deletion Syndrome: The Importance and Usefulness of DNA Microarray Analysis. Genetics 2015, S7, 4. [Google Scholar] [CrossRef]
- Schulz, S.; Volleth, M.; Muschke, P.; Wieland, I.; Wieacker, P. Greig cephalopolysyndactyly (GCPS) contiguous gene syndrome in a boy with a 14 Mb deletion in region 7p13-14 caused by a paternal balanced insertion (5; 7). Appl. Clin. Genet. 2008, 1, 19–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKusick, V.A. Mendelian Inheritance in Man. Available online: http://www.ncbi.nlm.nih.gov/omim (accessed on 25 May 2021).
- Johnston, J.J.; Olivos-Glander, I.; Turner, J.; Aleck, K.; Bird, L.M.; Mehta, L.; Schimke, R.N.; Heilstedt, H.; Spence, J.E.; Blancato, J.; et al. Clinical and molecular delineation of the Greig cephalopolysyndactyly contiguous gene deletion syndrome and its distinction from acrocallosal syndrome. Am. J. Med. Genet. A 2003, 123A, 236–242. [Google Scholar] [CrossRef]
- Johnston, J.J.; Walker, R.L.; Davis, S.; Facio, F.; Turner, J.T.; Bick, D.P.; Daentl, D.L.; Ellison, J.W.; Meltzer, P.S.; Biesecker, L.G. Zoom-in comparative genomic hybridisation arrays for the characterisation of variable breakpoint contiguous gene syndromes. J. Med. Genet. 2007, 44, e59. [Google Scholar] [CrossRef]
- Johnston, J.J.; Olivos-Glander, I.; Killoran, C.; Elson, E.; Turner, J.T.; Peters, K.F.; Abbott, M.H.; Aughton, D.J.; Aylsworth, A.S.; Bamshad, M.J.; et al. Molecular and Clinical Analyses of Greig Cephalopolysyndactyly and Pallister-Hall Syndromes: Robust Phenotype Prediction from the Type and Position of GLI3 Mutations. Am. J. Hum. Genet. 2005, 76, 609–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettigrew, A.L.; Greenberg, F.; Caskey, C.T.; Ledbetter, D.H. Greig syndrome associated with an interstitial deletion of 7p: Confirmation of the localization of Greig syndrome to 7p13. Hum. Genet. 1991, 87, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Siracusano, M.; Riccioni, A.; Baratta, A.; Baldi, M.; Curatolo, P.; Mazzone, L. Autistic symptoms in Greig cephalopolysyndactyly syndrome: A family case report. J. Med. Case Rep. 2019, 13, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurst, J.A.; Jenkins, D.; Vasudevan, P.C.; Kirchhoff, M.; Skovby, F.; Rieubland, C.; Gallati, S.; Rittinger, O.; Kroisel, P.M.; Johnson, D.; et al. Metopic and sagittal synostosis in Greig cephalopolysyndactyly syndrome: Five cases with intragenic mutations or complete deletions of GLI3. Eur. J. Hum. Genet. 2011, 19, 757–762. [Google Scholar] [CrossRef]
- Kroisel, P.M.; Petek, E.; Wagner, K. Phenotype of five patients with Greig syndrome and microdeletion of 7p13. Am. J. Med. Genet. 2001, 102, 243–249. [Google Scholar] [CrossRef]
- Démurger, F.; Ichkou, A.; Mougou-Zerelli, S.; Le Merrer, M.; Goudefroye, G.; Delezoide, A.-L.; Quélin, C.; Manouvrier, S.; Baujat, G.; Fradin, M.; et al. New insights into genotype–phenotype correlation for GLI3 mutations. Eur. J. Hum. Genet. 2014, 23, 92–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendoza-Londono, R.; Kashork, C.D.; Shaffer, L.G.; Krance, R.; Plon, S.E. Acute lymphoblastic leukemia in a patient with Greig cephalopolysyndactyly and interstitial deletion of chromosome 7 del(7)(p11.2 p14) involving the GLI3 and ZNFN1A1 genes. Genes Chromosomes Cancer 2005, 42, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Chotai, K.A.; Brueton, L.A.; van Herwerden, L.; Garrett, C.; Hinkel, G.K.; Schinzel, A.; Mueller, R.F.; Speleman, F.; Winter, R.M. Six cases of 7p deletion: Clinical, cytogenetic, and molecular studies. Am. J. Med. Genet. 1994, 51, 270–276. [Google Scholar] [CrossRef]
- Debeer, P.; Devriendt, K.; De Smet, L.; Deravel, T.; Gonzalez-Meneses, A.; Grzeschik, K.-H.; Fryns, J.-P. The spectrum of hand and foot malformations in patients with Greig cephalopolysyndactyly. J. Child. Orthop. 2007, 1, 143–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, P.G.; Hersh, J.H.; Yen, F.F.; Barch, M.J.; Kleinert, H.E.; Kunz, J.; Kalff-Suske, M. Greig cephalopolysyndactyly syndrome: Altered phenotype of a microdeletion syndrome due to the presence of a cytogenetic abnormality. Clin. Genet. 1997, 52, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Schwarzbraun, T.; Windpassinger, C.; Ofner, L.; Vincent, J.B.; Cheung, J.; Scherer, S.W.; Wagner, K.; Kroisel, P.M.; Petek, E. Genomic analysis of five chromosome 7p deletion patients with Greig cephalopolysyndactyly syndrome (GCPS). Eur. J. Med. Genet. 2006, 49, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Zneimer, S.M.; Cotter, P.D.; Stewart, S.D. Telomere-telomere (end to end) fusion of chromosomes 7 and 22 with an interstitial deletion of chromosome 7p11.2→p15.1: Phenotypic consequences and possible mechanisms. Clin Genet. 2000, 58, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Fenton, T.R.; Kim, J.H. A systematic review and meta-analysis to revise the Fenton Growth chart for preterm infants. BMC Pediatr. 2013, 13, 59. [Google Scholar] [CrossRef] [Green Version]
- Joubert, K.; Gy, G.; Darvay, S.; Ágfalvi, R. Results of the Hungarian Longitudinal Child Growth Study: From Birth to the Age of 18 Years (I). January 2010. Available online: https://edit.elte.hu/xmlui/bitstream/handle/10831/10549/Kutjel83_honlapra.pdf?sequence=1 (accessed on 17 October 2021).
- Allanson, J.E.; Cunniff, C.; Hoyme, H.E.; McGaughran, J.; Muenke, M.; Neri, G. Elements of morphology: Standard terminology for the head and face. Am. J. Med. Genet. A 2009, 149A, 6–28. [Google Scholar] [CrossRef] [Green Version]
- Biesecker, L.G.; Aase, J.M.; Clericuzio, C.; Gurrieri, F.; Temple, I.K.; Toriello, H. Elements of morphology: Standard terminology for the hands and feet. Am. J. Med. Genet. A 2009, 149A, 93–127. [Google Scholar] [CrossRef] [Green Version]
- Carey, J.C.; Cohen MMJr Curry, C.J.; Devriendt, K.; Holmes, L.B.; Verloes, A. Elements of morphology: Standard terminology for the lips, mouth, and oral region. Am. J. Med. Genet. A 2009, 149A, 77–92. [Google Scholar] [CrossRef]
- Hennekam, R.C.; Biesecker, L.G.; Allanson, J.E.; Hall, J.G.; Opitz, J.M.; Temple, I.K.; Carey, J.C.; Elements of Morphology Consortium. Elements of morphology: General terms for congenital anomalies. Am. J. Med. Genet. A 2013, 161A, 2726–2733. [Google Scholar] [CrossRef]
- Hamilton, M.J.; Suri, M. CDK13-related disorder. Adv. Genet. 2019, 3, 163–182. [Google Scholar] [CrossRef]
- Bostwick, B. CDK13-Related Disorder; Adam, M.P., Ardinger, H.H., Pagon, R.A., Eds.; University of Washington: Seattle, WA, USA, 1993; Available online: https://www-ncbi-nlm-nih-gov.ezproxy.u-pec.fr/books/NBK536784/ (accessed on 31 January 2019).
- Sifrim, A.; Hitz, M.-P.; Wilsdon, A.; Breckpot, J.; Al Turki, S.H.; Thienpont, B.; McRae, J.; Fitzgerald, T.W.; Singh, T.; Swaminathan, G.J.; et al. Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nat. Genet. 2016, 48, 1060–1065. [Google Scholar] [CrossRef]
- Greifenberg, A.K.; Hönig, D.; Pilarova, K.; Düster, R.; Bartholomeeusen, K.; Bösken, C.A.; Anand, K.; Blazek, D.; Geyer, M. Structural and Functional Analysis of the Cdk13/Cyclin K Complex. Cell Rep. 2016, 14, 320–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamilton, M.J.; Caswell, R.; Canham, N.; Cole, T.; Firth, H.V.; Foulds, N.; Heimdal, K.; Hobson, E.; Houge, G.; Joss, S.; et al. Heterozygous mutations affecting the protein kinase domain of CDK13 cause a syndromic form of developmental delay and intellectual disability. J. Med Genet. 2017, 55, 28–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Küry, S.; Van Woerden, G.M.; Besnard, T.; Onori, M.P.; Latypova, X.; Towne, M.C.; Cho, M.T.; Prescott, T.E.; Ploeg, M.A.; Sanders, S.; et al. De Novo Mutations in Protein Kinase Genes CAMK2A and CAMK2B Cause Intellectual Disability. Am. J. Hum. Genet. 2017, 101, 768–788. [Google Scholar] [CrossRef] [PubMed]
- Rizzi, S.; Spagnoli, C.; Salerno, G.G.; Frattini, D.; Caraffi, S.G.; Trimarchi, G.; Moratti, C.; Pascarella, R.; Garavelli, L.; Fusco, C. Severe intellectual disability, absence of language, epilepsy, microcephaly and progressive cerebellar atrophy related to the recurrent de novo variant p (P139L) of the CAMK2B gene: A case report and brief review. Am. J. Med. Genet. Part A 2020, 182, 2675–2679. [Google Scholar] [CrossRef]
- Heiman, P.; Drewes, S.; Ghaloul-Gonzalez, L. A familial case of CAMK2B mutation with variable expressivity. SAGE Open Med. Case Rep. 2021, 9. [Google Scholar] [CrossRef]
- Hiatt, S.M.; Neu, M.B.; Ramaker, R.C.; Hardigan, A.A.; Prokop, J.W.; Hancarova, M.; Prchalova, D.; Havlovicova, M.; Prchal, J.; Stranecky, V.; et al. De novo mutations in the GTP/GDP-binding region of RALA, a RAS-like small GTPase, cause intellectual disability and developmental delay. PLoS Genet. 2018, 14, e1007671. [Google Scholar] [CrossRef] [Green Version]
- Sava, C.N.; Ritli, L.; Balmoş, A.B.; Iuhas, A.R.; Marian, P.; Motorca, M.A.; Lele, L.A.; Straciuc, O.; Zaha, D.C.; Jurcă, M.C.; et al. Unusual extramedullary relapses in a case of common B-cell acute lymphoblastic leukemia. Case Rep. Rev. Lit. 2019, 60, 249–254. [Google Scholar]


{kind=link}
| Results | Size of the Deleted Segment (Mb) | OMIM Genes | |
|---|---|---|---|
| Karyotype | 46,XY, del (7)(p13-p15) | ||
| aCGH | arr[hg38]7p14.2-p11.2 (35830920-54201451)x1 | 18.04 | ANLN, NME8, POU6F2, MPLKIP, GLI3, BLVRA, GCK, NPC1L1, OGDH, CCM2, ADCY1, IKZF1, DDC, PGAM2, CDK13, AEBP1, CAMK2B, SFRP4, PKD1L1, VPS41, SUGCT, RALA |
| LOH | - | 18.37 | ANLN, NME8, POU6F2, MPLKIP, GLI3, BLVRA, PGAM2, GCK, NPC1L1, OGDH, CCM2, ADCY1, IKZF1, DDC, CDK13, AEBP1, CAMK2B, SFRP4, PKD1L1, VPS41, SUGCT, RALA, SEPTIN7 |
| References | Chromosomal Localisation | Size of the Deleted Segment (Mb) | Array Coordinates |
|---|---|---|---|
| Present case | 7p14.2-p11.2 | 18.37 | arr[hg38](35830920_54201451)del |
| Niida Y. et al [3] (2015) | 7p14.1-p12.3 | 6.2 | arr[hg19](41076615_47282889)del |
| Demurger F. et al. [13] (2015) | 7p13-p15 | 7 | arr[hg19](38521704_45810267)del |
| 9 | arr[hg19](35674000_37280000) _(46116000_46598000)del | ||
| Jane A Hurst et al [11] (2011) | 7p13-p14.1 | 6.0 | arr[hg18](39013006_39213707)del |
| 6.8 | arr[hg18](39130081_45492392)del | ||
| 7p12.3-p14.1 | 8.3 | arr[hg18](40845981_40855164) _(49136714_49160830)del | |
| Solveig Schulz et a [4] (2008) | 7p13-7p14 | 14 | NA |
| Debeer Philippe et al. [16] (2007) | 7p14.3 | NA | NA |
| 7p14.3 | |||
| Jennifer J Johnston et al. [6,7] (2003, 2007) | 7p14.1 | 1.8 | NA |
| 7p14.1-7p13 | 3.2 | ||
| 7p14.1 | 4.1 | ||
| 7p14.1-7p13 | 5.2 | ||
| 7p14.1-7p13 | 5.9 | ||
| 7p14.2-7p14.1 | 6.3 | ||
| 7p14.1-7p12.3 | 8.4 | ||
| 7p14.2-7p13 | 9.8 | ||
| 7p14.2-7p13 | 10.3 | ||
| Kroisel PM et al [12] Schwarbraun T. et al. [18] (2001, resp. 2005) | 7p13 | 4.5-15 | NA |
| 7p12.3-p13 | |||
| 7p12.3-p13 | |||
| 7p12.3-p14.2 | |||
| 7p11.2-p13 | |||
| Williams PG et al. [17] (1997) | 7p13-p15.1 | NA | NA |
| Zneimer SM et al. [19] (2000) | 45,XY,der(22;7) (p13;p22.3)del (7)-(p11.2-p15.1) | NA | NA |
| References | Patients’s Age | Size of the Deleted Segment (Mb) | Phenotype Associated OMIM Genes | Neurodevelopment |
|---|---|---|---|---|
| Present case * | 3 years followed to age 6 | 18.37 | ANLN, NME8, POU6F2, MPLKIP, GLI3, BLVRA, GCK, NPC1L1, OGDH, CCM2, ADCY1, IKZF1, DDC, PGAM2, CDK13, AEBP1, CAMK2B, SFRP4, PKD1L1, VPS41, SUGCT, RALA | Severe intellectual disability, speech and developmental delay |
| Niida Y. et al. * [3] (2015) | 2 years | 6.2 | GLI3, GCK, CCM2, AEBP1, CAMK2B, PGAM2, BLVRA, ADCY1, NPC1L1, OGDH | Developmental delay |
| Demurger F. et al.* [13] (2015) | NA | 7 | GLI3, AEBP1, MPLKIP, GCK, CCM2, CDK13, CAMK2B, VPS41, BLVRA, PGAM2, ADCY1, SUGCT, POU6F2, NPC1L1, RALA, OGDH | Developmental delay |
| NA | 9 | GLI3, GCK, CDK13, MPLKIP AEBP1, CCM2, CAMK2B, SFRP4, VPS41, PGAM2, BLVRA, NME8, SUGCT, ADCY1, NPC1L1, POU6F2, ANLN, RALA, OGDH | Developmental delay | |
| Jane A Hurst et al.* [11] (2011) | 2 years | 6.0 | GLI3, GCK, CCM2, CDK13, MPLKIP, AEBP1, CAMK2B, BLVRA, PGAM2, SUGCT, RALA, POU6F2, NPC1L1, OGDH | Developmental delay |
| 5 years | 6.8 | GLI3, GCK, CCM2, CDK13, MPLKIP, AEBP1, CAMK2B, BLVRA, PGAM2, SUGCT, RALA, NPC1L1, POU6F2, OGDH | Severe intellectual disability, speech and developmental delay | |
| 15 years | 8.3 | GLI3, GCK, CCM2, AEBP1, CAMK2B, PKD1L1, BLVRA, PGAM2, SUGCT, ADCY1, NPC1L1, OGDH | Severe intellectual disability, speech and delayed development |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kozma, K.; Bembea, M.; Jurca, C.M.; Ioana, M.; Streață, I.; Şoşoi, S.Ş.; Pirvu, A.; Petchesi, C.D.; Szilágyi, A.; Sava, C.N.; et al. Greig Cephalopolysyndactyly Contiguous Gene Syndrome: Case Report and Literature Review. Genes 2021, 12, 1674. https://doi.org/10.3390/genes12111674
Kozma K, Bembea M, Jurca CM, Ioana M, Streață I, Şoşoi SŞ, Pirvu A, Petchesi CD, Szilágyi A, Sava CN, et al. Greig Cephalopolysyndactyly Contiguous Gene Syndrome: Case Report and Literature Review. Genes. 2021; 12(11):1674. https://doi.org/10.3390/genes12111674
Chicago/Turabian StyleKozma, Kinga, Marius Bembea, Claudia M. Jurca, Mihai Ioana, Ioana Streață, Simona Ş. Şoşoi, Andrei Pirvu, Codruța D. Petchesi, Ariana Szilágyi, Cristian N. Sava, and et al. 2021. "Greig Cephalopolysyndactyly Contiguous Gene Syndrome: Case Report and Literature Review" Genes 12, no. 11: 1674. https://doi.org/10.3390/genes12111674