WGCNA Analysis of Salt-Responsive Core Transcriptome Identifies Novel Hub Genes in Rice

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and Growth Conditions
2.2. RNA Isolation and qRT-PCR Gene Expression Analysis
2.3. Data Acquisition and RNA-Seq Analysis
2.4. Gene Ontology and KEGG Analysis of the Core DEGs
2.5. Weighted Gene Co-Expression Network Analysis (WGCNA)
3. Results
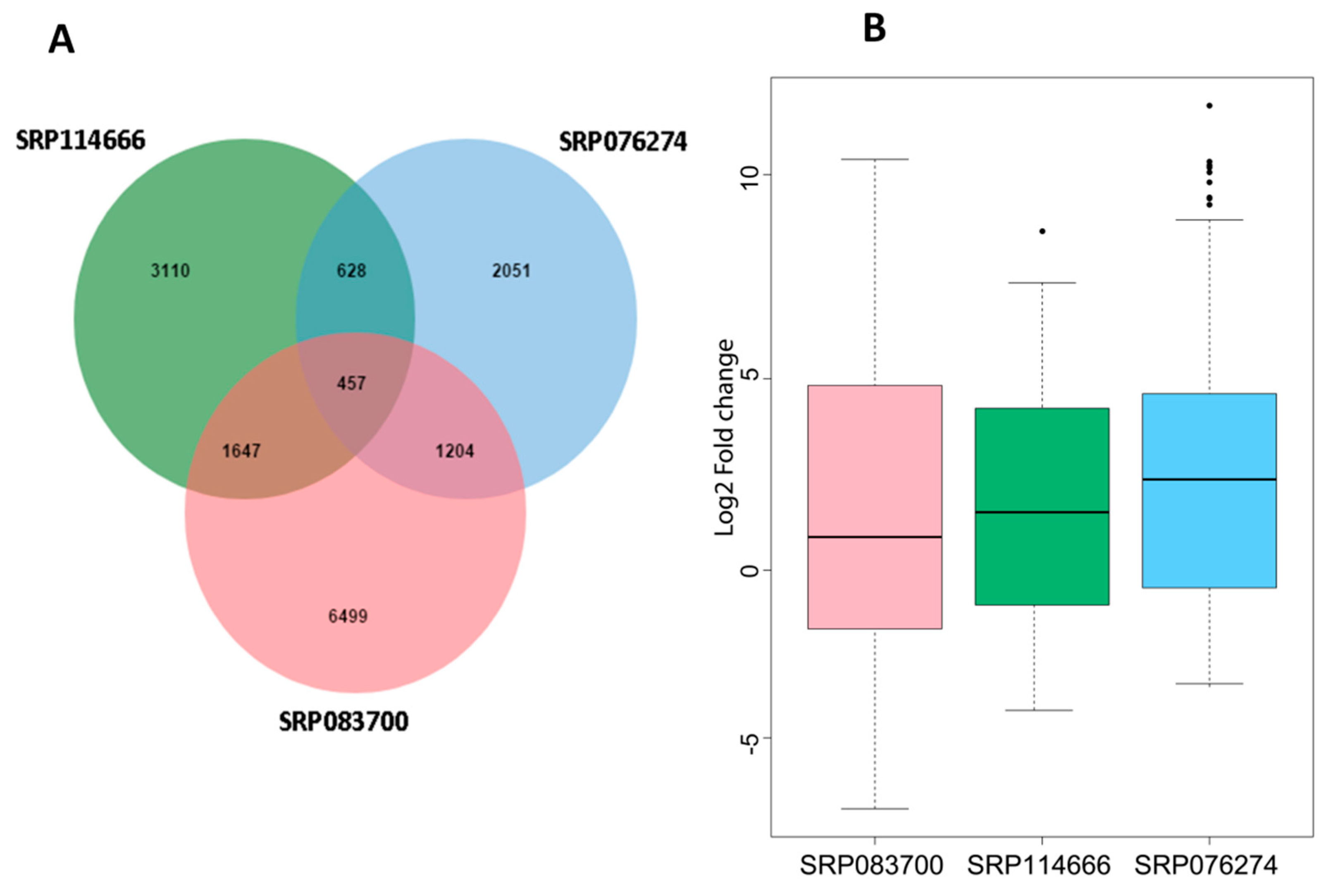
3.1. Identification of the Salt-Responsive Core DEGs in Rice
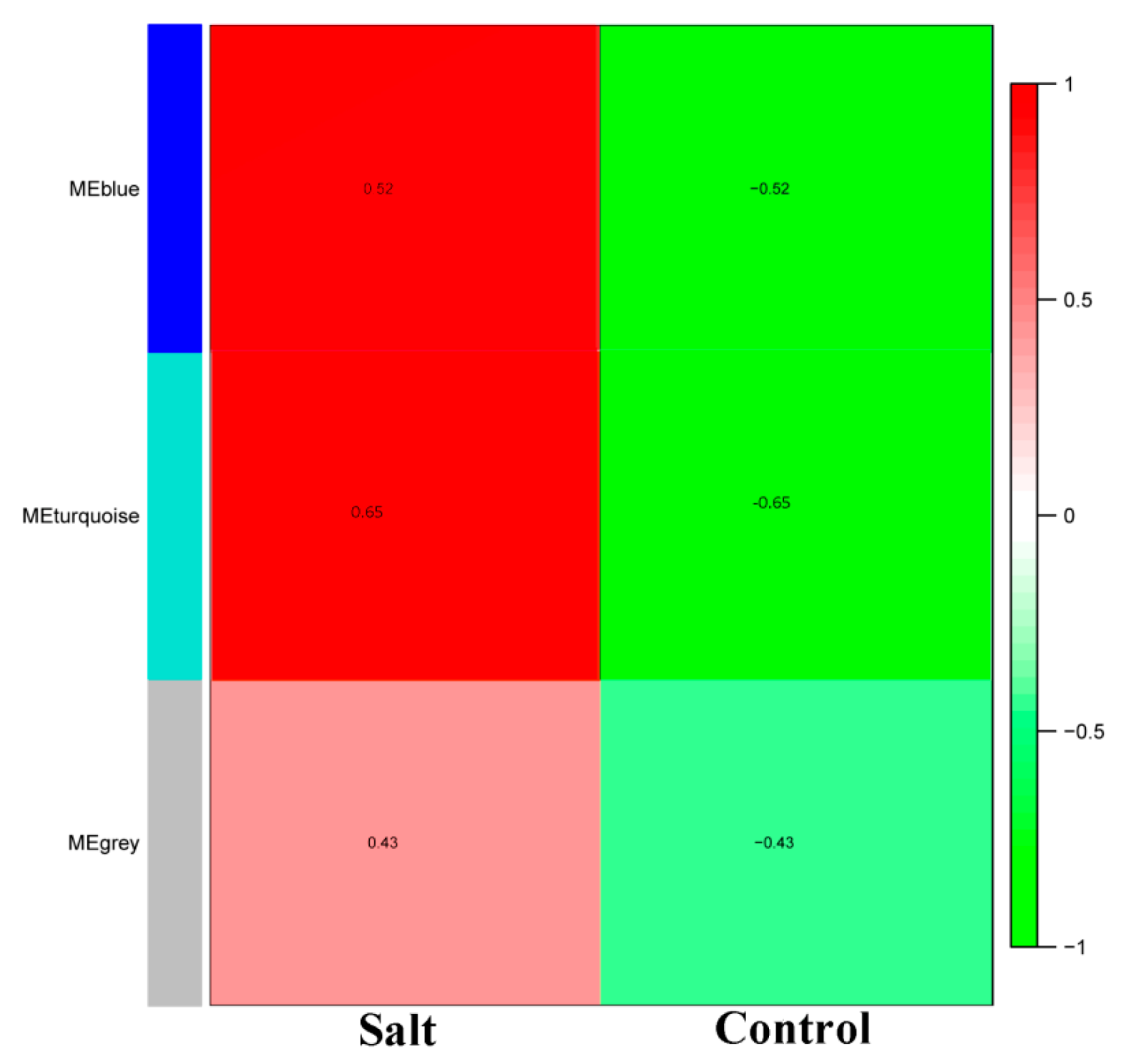
3.2. WGCNA of the Salt-Responsive Core Genome
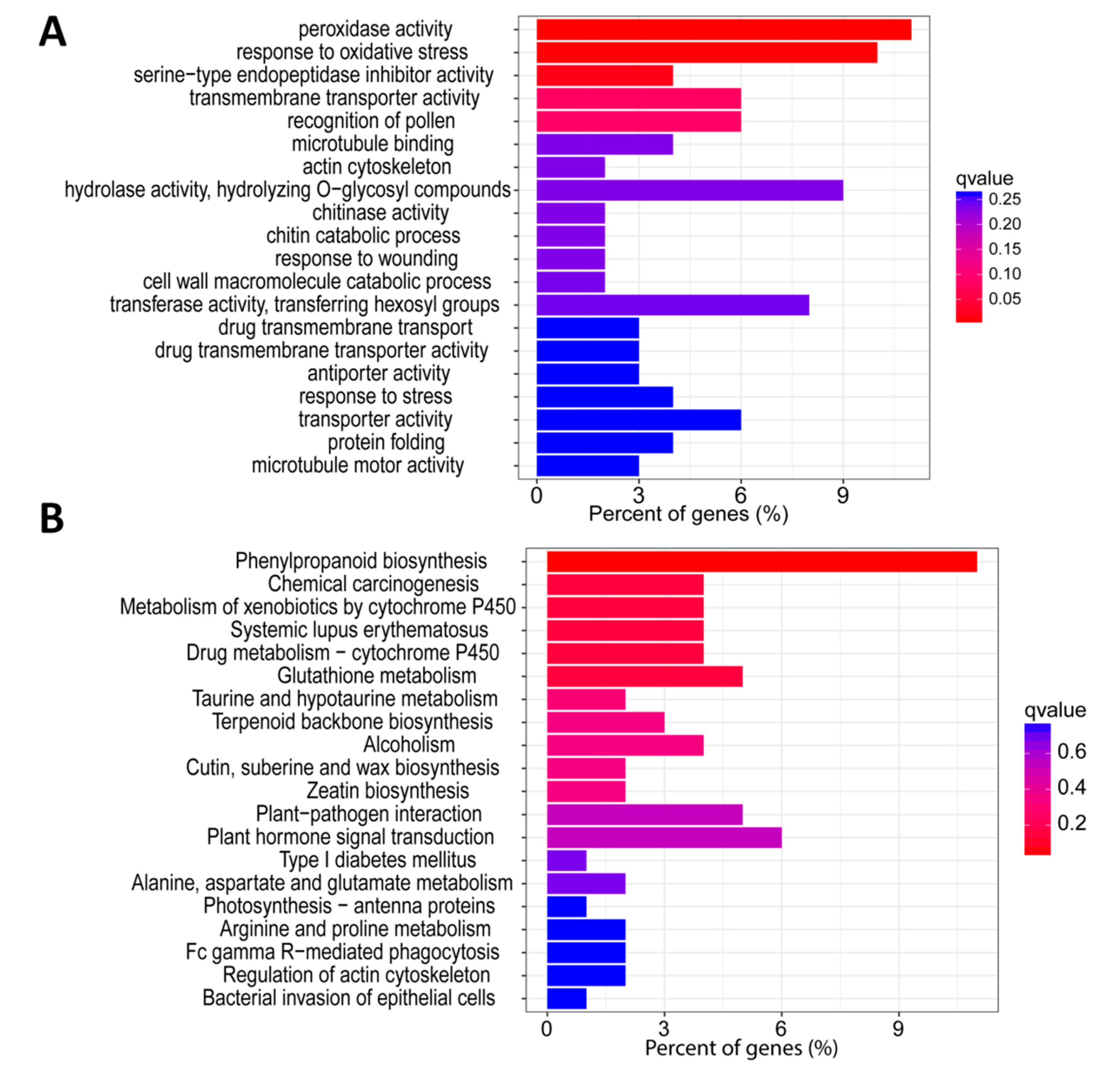
3.3. GO and KEGG Enrichment Analysis of the Detected Co-Expressed Modules
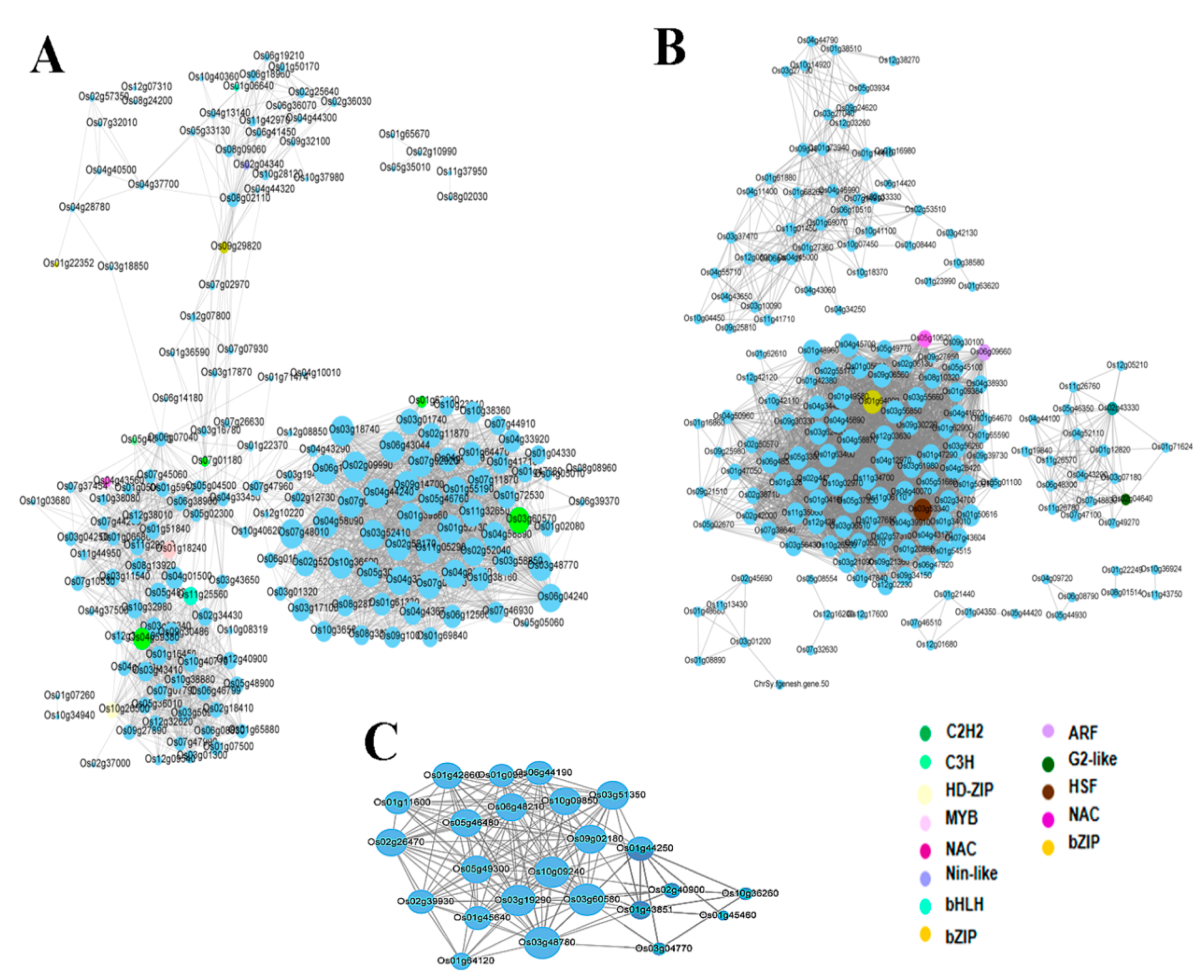
3.4. Networks Displaying Relationships among Genes within Co-Expressed Modules
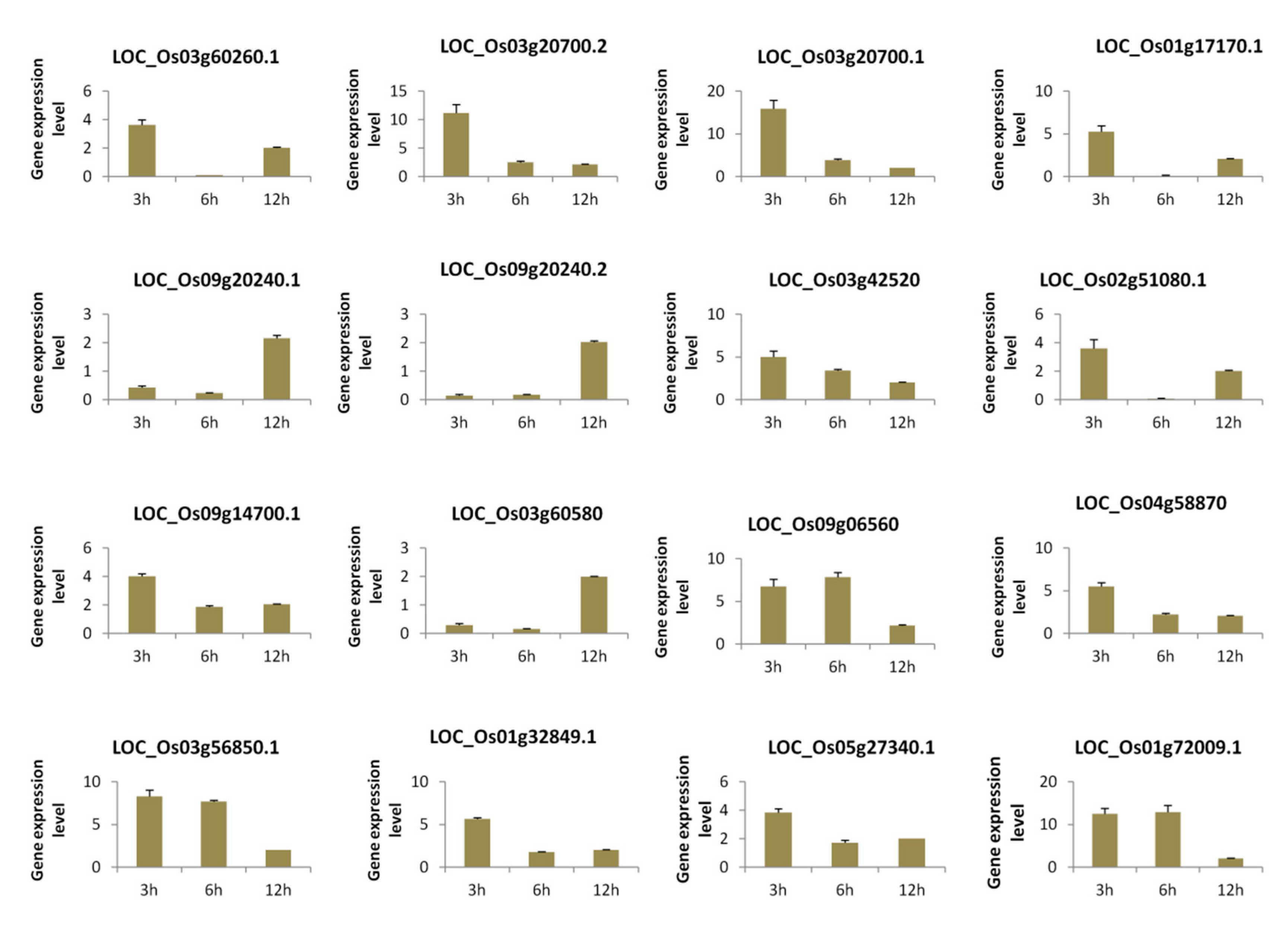
3.5. qRT-PCR Validation of Selected Genes from Each Module under Temporal Salt Stress
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Majeed, A.; Muhammad, Z. Salinity: A Major Agricultural Problem—Causes, Impacts on Crop Productivity and Management Strategies. In Plant Abiotic Stress Tolerance; Hasanuzzaman, M., Hakeem, K., Nahar, K., Alharby, H., Eds.; Springer: Berlin, Germany, 2019; pp. 83–99. ISBN 978-3-030-06117-3. [Google Scholar]
- Munns, R.; Tester, M. Mechanisms of Salinity Tolerance. Annu. Rev. Plant Biol. 2008, 59, 651–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, C.; Wang, Y.; Gu, D.; Nan, J.; Chen, S.; Li, H. Overexpression of S-adenosyl-L-methionine synthetase 2 from sugar beet M14 increased Arabidopsis tolerance to salt and oxidative stress. Int. J. Mol. Sci. 2017, 20, 847. [Google Scholar] [CrossRef] [PubMed]
- Lata, C.; Prasad, M. Role of DREBs in regulation of abiotic stress responses in plants. J. Exp. Bot. 2011, 62, 4731–4748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.S.; Pandey, M.K.; Hemalatha, S. Comparative Studies on the Role of Organic Biostimulant in Resistant and Susceptible Cultivars of Rice Grown under Saline Stress—Organic Biostimulant Alleviate Saline Stress in Tolerant and Susceptible cultivars of Rice. J. Crop Sci. Biotechnol. 2018, 21, 459–467. [Google Scholar] [CrossRef]
- Kumari, S.; Panjabinee Sabharwal, V.; Kushwaha, H.R.; Sopory, S.K.; Singla-Pareek, S.L.; Pareek, A. Transcriptome map for seedling stage specific salinity stress response indicates a specific set of genes as candidate for saline tolerance in Oryza sativa L. Funct. Integr. Genomics 2009, 9. [Google Scholar] [CrossRef] [PubMed]
- Carriger, S.; Vallee, D. More crop per drop. Rice Today 2007, 6, 10–13. [Google Scholar]
- Khan, M.S.; Akther, T.; MubarakAli, D.; Hemalatha, S. An investigation on the role of salicylic acid alleviates the saline stress in rice crop (Oryza sativa (L)). Biocatal. Agric. Biotechnol. 2019, 18, 101027. [Google Scholar] [CrossRef]
- Kawahara, Y.; de la Bastide, M.; Hamilton, J.P.; Kanamori, H.; Mccombie, W.R.; Ouyang, S.; Schwartz, D.C.; Tanaka, T.; Wu, J.; Zhou, S.; et al. Improvement of the Oryza sativa nipponbare reference genome using next generation sequence and optical map data. Rice 2013, 6, 4. [Google Scholar] [CrossRef]
- Reddy, I.N.B.L.; Kim, B.-K.; Yoon, I.-S.; Kim, K.-H.; Kwon, T.-R. Salt Tolerance in Rice: Focus on Mechanisms and Approaches. Rice 2017, 24, 123–144. [Google Scholar] [CrossRef]
- Tuteja, N. Abscisic Acid and Abiotic Stress Signaling. Plant Signal. Behav. 2007, 2, 135–138. [Google Scholar] [CrossRef] [Green Version]
- van Dam, S.; Craig, T.; de Magalhães, J.P. GeneFriends: A human RNA-seq-based gene and transcript co-expression database. Nucleic Acids Res. 2015, 43, D1124–D1132. [Google Scholar] [CrossRef] [PubMed]
- Shankar, R.; Bhattacharjee, A.; Jain, M. Transcriptome analysis in different rice cultivars provides novel insights into desiccation and salinity stress responses. Sci. Rep. 2016, 6, 23719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Zhu, J.; Zhang, Y.; Fan, F.; Li, W.; Wang, F.; Zhong, W.; Wang, C.; Yang, J. Comparative transcriptome analysis reveals molecular response to salinity stress of salt-tolerant and sensitive genotypes of indica rice at seedling stage. Sci. Rep. 2018, 8, 2085. [Google Scholar] [CrossRef]
- Chandran, A.K.N.; Kim, J.-W.; Yoo, Y.-H.; Park, H.L.; Kim, Y.-J.; Cho, M.-H.; Jung, K.-H. Transcriptome analysis of rice-seedling roots under soil–salt stress using RNA-Seq method. Plant Biotechnol. Rep. 2019, 1–12. [Google Scholar] [CrossRef]
- Yuan, J.; Li, J.; Yang, Y.; Tan, C.; Zhu, Y.; Hu, L.; Qi, Y.; Lu, Z.J. Stress-responsive regulation of long non-coding RNA polyadenylation in Oryza sativa. Plant J. 2018, 93, 814–827. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Cheng, Y.; Han, S.; Van Handel, B.; Dong, L.; Li, X.; Xie, X. Whole genome sequencing and comparative transcriptome analysis of a novel seawater adapted, salt-resistant rice cultivar-sea rice 86. BMC Genomics 2017, 18, 655. [Google Scholar] [CrossRef]
- Childs, K.L.; Davidson, R.M.; Buell, C.R. Gene coexpression network analysis as a source of functional annotation for rice genes. PLoS ONE 2011, 6, e22196. [Google Scholar] [CrossRef]
- Liang, Y.H.; Cai, B.; Chen, F.; Wang, G.; Wang, M.; Zhong, Y.; Cheng, Z.M.M. Construction and validation of a gene co-expression network in grapevine (Vitis vinifera. L.). Hortic. Res. 2014, 1, 14040. [Google Scholar] [CrossRef] [Green Version]
- Tahmasebi, A.; Ashrafi-Dehkordi, E.; Shahriari, A.G.; Mazloomi, S.M.; Ebrahimie, E. Integrative meta-analysis of transcriptomic responses to abiotic stress in cotton. Prog. Biophys. Mol. Biol. 2019, 146, 112–122. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Sadhukhan, A.; Tazib, T.; Nakano, Y.; Kusunoki, K.; Kamara, M.; Chaffai, R.; Iuchi, S.; Sahoo, L.; Kobayashi, M.; et al. Joint genetic and network analyses identify loci associated with root growth under NaCl stress in Arabidopsis thaliana. Plant Cell Environ. 2016, 39, 918–934. [Google Scholar] [CrossRef]
- Lou, Q.; Chen, L.; Mei, H.; Xu, K.; Wei, H.; Feng, F.; Li, T.; Pang, X.; Shi, C.; Luo, L.; et al. Root Transcriptomic Analysis Revealing the Importance of Energy Metabolism to the Development of Deep Roots in Rice (Oryza sativa L.). Front. Plant Sci. 2017, 8, 1314. [Google Scholar] [CrossRef]
- Amrine, K.C.H.; Blanco-Ulate, B.; Cantu, D. Discovery of Core Biotic Stress Responsive Genes in Arabidopsis by Weighted Gene Co-Expression Network Analysis. PLoS ONE 2015, 10, e0118731. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. TAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Caldana, C.; Scheible, W.R.; Mueller-Roeber, B.; Ruzicic, S. A quantitative RT-PCR platform for high-throughput expression profiling of 2500 rice transcription factors. Plant Methods 2007, 7. [Google Scholar] [CrossRef]
- Anders, S.; McCarthy, D.J.; Chen, Y.; Okoniewski, M.; Smyth, G.K.; Huber, W.; Robinson, M.D. Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat. Protoc. 2013, 8, 1765. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef]
- Smoot, M.; Ono, K.; Ideker, T.; Maere, S. PiNGO: A cytoscape plugin to find candidate genes in biological networks. Bioinformatics 2011, 27, 1030–1031. [Google Scholar] [CrossRef]
- Jeong, H.; Mason, S.P.; Barabasi, A.L.; Oltvai, Z.N. Lethality and centrality in protein networks. Nature 2001, 411, 41–42. [Google Scholar] [CrossRef] [Green Version]
- Yokotani, N.; Ichikawa, T.; Kondou, Y.; Matsui, M.; Hirochika, H.; Iwabuchi, M.; Oda, K. Expression of rice heat stress transcription factor OsHsfA2e enhances tolerance to environmental stresses in transgenic Arabidopsis. Planta 2008, 227, 957–967. [Google Scholar] [CrossRef]
- Dossa, K.; Diouf, D.; Cissé, N. Genome-Wide Investigation of Hsf Genes in Sesame Reveals Their Segmental Duplication Expansion and Their Active Role in Drought Stress Response. Front. Plant Sci. 2016, 7, 1522. [Google Scholar] [CrossRef] [Green Version]
- AbdElgawad, H.; Zinta, G.; Hegab, M.M.; Pandey, R.; Asard, H.; Abuelsoud, W. High Salinity Induces Different Oxidative Stress and Antioxidant Responses in Maize Seedlings Organs. Front. Plant Sci. 2016, 7, 276. [Google Scholar] [CrossRef] [Green Version]
- Walia, H.; Wilson, C.; Wahid, A.; Condamine, P.; Cui, X.; Close, T.J. Expression analysis of barley (Hordeum vulgare L.) during salinity stress. Funct. Integr. Genomics 2006, 6, 143–156. [Google Scholar] [CrossRef]
- Darko, E.; Gierczik, K.; Hudák, O.; Forgó, P.; Pál, M.; Türkösi, E.; Kovács, V.; Dulai, S.; Majláth, I.; Molnár, I.; et al. Differing metabolic responses to salt stress in wheat-barley addition lines containing different 7H chromosomal fragments. PLoS ONE 2017, 12, e0174170. [Google Scholar] [CrossRef]
- Khan, M.S.; Akther, T.; Hemalatha, S. Impact of Panchagavya on Oryza sativa L. Grown Under Saline Stress. J. Plant Growth Regul. 2017, 36, 702–713. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Zeng, A.; Chen, P.; Korth, K.L.; Ping, J.; Thomas, J.; Wu, C.; Srivastava, S.; Pereira, A.; Hancock, F.; Brye, K.; et al. RNA sequencing analysis of salt tolerance in soybean (Glycine max). Genomics 2018, 111, 629–635. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, P.; Cui, F.; Zhang, F.; Luo, X.; Xie, J. Transcriptome Analysis of Salt Stress Responsiveness in the Seedlings of Dongxiang Wild Rice (Oryza rufipogon Griff.). PLoS ONE 2016, 11, e0146242. [Google Scholar] [CrossRef]
- Luo, Y.; Reid, R.; Freese, D.; Li, C.; Watkins, J.; Shi, H.; Zhang, H.; Loraine, A.; Song, B.-H. Salt tolerance response revealed by RNA-Seq in a diploid halophytic wild relative of sweet potato. Sci. Rep. 2017, 7, 9624. [Google Scholar] [CrossRef]
- Bahieldin, A.; Atef, A.; Sabir, J.S.M.; Gadalla, N.O.; Edris, S.; Alzohairy, A.M.; Radhwan, N.A.; Baeshen, M.N.; Ramadan, A.M.; Eissa, H.F.; et al. RNA-Seq analysis of the wild barley (H. spontaneum) leaf transcriptome under salt stress. C. R. Biol. 2015, 338, 285–297. [Google Scholar] [CrossRef]
- Zhang, J.; Zhao, W.; Fu, R.; Fu, C.; Wang, L.; Liu, H.; Li, S.; Deng, Q.; Wang, S.; Zhu, J.; et al. Comparison of gene co-networks reveals the molecular mechanisms of the rice (Oryza sativa L.) response to Rhizoctonia solani AG1 IA infection. Funct. Integr. Genomics 2018, 8, 545–557. [Google Scholar] [CrossRef]
- Choudhury, F.K.; Rivero, R.M.; Blumwald, E.; Mittler, R. Reactive oxygen species, abiotic stress and stress combination. Plant J. 2017, 90, 856–867. [Google Scholar] [CrossRef] [PubMed]
- Sewelam, N.; Kazan, K.; Schenk, P.M. Global Plant Stress Signaling: Reactive Oxygen Species at the Cross-Road. Front. Plant Sci. 2016, 7, 187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kibria, M.G.; Hossain, M.; Murata, Y.; Hoque, M.A. Antioxidant Defense Mechanisms of Salinity Tolerance in Rice Genotypes. Rice Sci. 2017, 24, 155–162. [Google Scholar] [CrossRef]
- Hazman, M.; Hause, B.; Eiche, E.; Nick, P.; Riemann, M. Increased tolerance to salt stress in OPDA-deficient rice ALLENE OXIDE CYCLASE mutants is linked to an increased ROS-scavenging activity. J. Exp. Bot. 2015, 66, 3339–3352. [Google Scholar] [CrossRef] [PubMed]
- Moazzzam Jazi, M.; Seyedi, S.M.; Ebrahimie, E.; Ebrahimi, M.; De Moro, G.; Botanga, C. A genome-wide transcriptome map of pistachio (Pistacia vera L.) provides novel insights into salinity-related genes and marker discovery. BMC Genomics 2017, 18, 627. [Google Scholar] [CrossRef]
- Nadernejad, N.; Ahmadimoghadam, A.; Hosseinifard, J.; Pourseyedi, S. Phenylalanin Ammonia-Lyase Activity, Total Phenolic and Flavonoid Content in Flowers, Leaves, Hulls and Kernels of Three Pistachio (Pistacia vera L.) Cultivars. Environ. Sci. 2012, 12, 807–814. [Google Scholar]
- Gao, S.; Ouyang, C.; Wang, S.; Xu, Y.; Tang, L.; Chen, F. Effects of salt stress on growth, antioxidant enzyme and phenylalanine ammonia-lyase activities in Jatropha curcas L. seedlings. Plant Soil Environ. 2008, 54, 374–381. [Google Scholar] [CrossRef] [Green Version]
- Dehghan, S.; Sadeghi, M.; Pöppel, A.; Fischer, R.; Lakes-Harlan, R.; Kavousi, H.R.; Vilcinskas, A.; Rahnamaeian, M. Differential inductions of phenylalanine ammonia-lyase and chalcone synthase during wounding, salicylic acid treatment, and salinity stress in safflower, Carthamus tinctorius. Biosci. Rep. 2014, 34, e00114. [Google Scholar] [CrossRef]
- Gholizadeh, A.; Kohnehrouz, B.B. Activation of phenylalanine ammonia lyase as a key component of the antioxidative system of salt-challenged maize leaves. Braz. J. Plant Physiol. 2010, 22, 217–223. [Google Scholar] [CrossRef] [Green Version]
- Gill, S.S.; Tuteja, N. Cadmium stress tolerance in crop plants: Probing the role of sulfur. Plant Signal. Behav. 2011, 6, 215–222. [Google Scholar] [CrossRef]
- Rabhi, M.; Barhoumi, Z.; Ksouri, R.; Abdelly, C.; Gharsalli, M. Interactive effects of salinity and iron deficiency in Medicago ciliaris. C. R. Biol. 2007, 6, 215–222. [Google Scholar]
- Mostafa, H.; Mohammad, G.; Maryam, H. Effect of sulfur and iron fertilizers on yield, yield components and nutrient uptake in sesame (Sesamum indicum L.) under water stress. Afr. J. Biotechnol. 2016, 10, 8816–8822. [Google Scholar] [CrossRef]
- Okçu, G.; Kaya, M.D.; Atak, M. Effects of salt and drought stresses on germination and seedling growth of pea (Pisum sativum L.). Turk. J. Agric. For. 2005, 29, 237–242. [Google Scholar]
- Thalmann, M.; Santelia, D. Starch as a determinant of plant fitness under abiotic stress. New Phytol. 2017, 214, 943–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onaga, G.; Wydra, K. Advances in Plant Tolerance to Abiotic Stresses. In Plant Genomics; Abdurakhmonov, I.Y., Ed.; IntechOpen: London, UK, 2016; pp. 1–13. [Google Scholar] [CrossRef]
- Krasensky, J.; Jonak, C. Drought, salt, and temperature stress-induced metabolic rearrangements and regulatory networks. J. Exp. Bot. 2012, 63, 1593–1608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rooks, D.S.; Huang, J.; Bierbaum, B.E.; Bolus, S.A.; Rubano, J.; Connolly, C.E.; Alpert, S.; Iversen, M.D.; Katz, J.N. Effect of preoperative exercise on measures of functional status in men and women undergoing total hip and knee arthroplasty. Arthritis Rheum. 2006, 55, 700–708. [Google Scholar] [CrossRef] [PubMed]
- Yuenyong, W.; Chinpongpanich, A.; Comai, L.; Chadchawan, S.; Buaboocha, T. Downstream components of the calmodulin signaling pathway in the rice salt stress response revealed by transcriptome profiling and target identification. BMC Plant Biol. 2018, 18, 335. [Google Scholar] [CrossRef]
- Yang, S.-Q.; Li, W.-Q.; Miao, H.; Gan, P.-F.; Qiao, L.; Chang, Y.-L.; Shi, C.-H.; Chen, K.-M. REL2, A Gene Encoding An Unknown Function Protein which Contains DUF630 and DUF632 Domains Controls Leaf Rolling in Rice. Rice 2016, 9, 37. [Google Scholar] [CrossRef]
- Schmidt, R.; Schippers, J.H.M.; Welker, A.; Mieulet, D.; Guiderdoni, E.; Mueller-Roeber, B. Transcription factor OsHsfC1b regulates salt tolerance and development in Oryza sativa ssp. japonica. AoB Plants 2012, 2012, pls011. [Google Scholar] [CrossRef]
- Nietzsche, M.; Schießl, I.; Börnke, F. The complex becomes more complex: Protein-protein interactions of SnRK1 with DUF581 family proteins provide a framework for cell- and stimulus type-specific SnRK1 signaling in plants. Front. Plant Sci. 2014, 5, 54. [Google Scholar] [CrossRef]
- Trivedi, D.K.; Gill, S.S.; Yadav, S.; Tuteja, N. Genome-wide analysis of glutathione reductase (GR) genes from rice and Arabidopsis. Plant Signal. Behav. 2013, 8, e23021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharjee, A.; Khurana, J.P.; Jain, M. Characterization of Rice Homeobox Genes, OsHOX22 and OsHOX24, and Over-expression of OsHOX24 in Transgenic Arabidopsis Suggest Their Role in Abiotic Stress Response. Front. Plant Sci. 2016, 7, 627. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.Z. OsLEA3, a late embryogenesis abundant protein gene from rice, confers tolerance to water deficit and salt stress to transgenic rice. Russ. J. Plant Physiol. 2008, 55, 530–537. [Google Scholar] [CrossRef]
- Nguyen, N.H.; Nguyen, C.T.T.; Jung, C.; Cheong, J.-J. AtMYB44 suppresses transcription of the late embryogenesis abundant protein gene AtLEA4-5. Biochem. Biophys. Res. Commun. 2019, 511, 931–934. [Google Scholar] [CrossRef] [PubMed]
- Zafar, S.A.; Hussain, M.; Raza, M.; Muhu-Din Ahmed, H.G.; Rana, I.A.; Sadia, B.; Atif, R.M. Genome wide analysis of heat shock transcription factor (HSF) family in chickpea and its comparison with Arabidopsis. Plant Omics 2016, 9, 136–141. [Google Scholar] [CrossRef]
- Dossa, K.; Mmadi, M.A.; Zhou, R.; Zhang, T.; Su, R.; Zhang, Y.; Wang, L.; You, J.; Zhang, X. Depicting the Core Transcriptome Modulating Multiple Abiotic Stresses Responses in Sesame (Sesamum indicum L.). Int. J. Mol. Sci. 2019, 20, 3930. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, H.; Araki, T.; Meshi, T.; Iwabuchi, M. Expression of a subset of the Arabidopsis Cys(2)/His(2)-type zinc-finger protein gene family under water stress. Gene 2000, 248, 23–32. [Google Scholar] [CrossRef]
- Kim, J.C.; Lee, S.H.; Cheong, Y.H.; Yoo, C.M.; Lee, S.I.; Chun, H.J.; Yun, D.J.; Hong, J.C.; Lee, S.Y.; Lim, C.O.; et al. A novel cold-inducible zinc finger protein from soybean, SCOF-1, enhances cold tolerance in transgenic plants. Plant J. 2001, 25, 247–259. [Google Scholar] [CrossRef]
- Katiyar, A.; Smita, S.; Lenka, S.K.; Rajwanshi, R.; Chinnusamy, V.; Bansal, K.C. Genome-wide classification and expression analysis of MYB transcription factor families in rice and Arabidopsis. BMC Genomics 2012, 13, 544. [Google Scholar] [CrossRef]
- Jakoby, M.; Weisshaar, B.; Dröge-Laser, W.; Vicente-Carbajosa, J.; Tiedemann, J.; Kroj, T.; Parcy, F. bZIP transcription factors in Arabidopsis. Opin. TRENDS Plant Sci. 2002, 7, 106–111. [Google Scholar] [CrossRef]
- Zou, M.; Guan, Y.; Ren, H.; Zhang, F.; Chen, F. A bZIP transcription factor, OsABI5, is involved in rice fertility and stress tolerance. Plant Mol. Biol. 2008, 66, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Ying, S.; Zhang, D.F.; Fu, J.; Shi, Y.S.; Song, Y.C.; Wang, T.Y.; Li, Y. Cloning and characterization of a maize bZIP transcription factor, ZmbZIP72, confers drought and salt tolerance in transgenic Arabidopsis. Planta 2012, 235, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Mao, B.; Ou, S.; Wang, W.; Liu, L.; Wu, Y.; Chu, C.; Wang, X. OsbZIP71, a bZIP transcription factor, confers salinity and drought tolerance in rice. Plant Mol. Biol. 2014, 84, 19–36. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Bao, X.; Zhi, Y.; Wu, Q.; Guo, Y.; Yin, X.; Zeng, L.; Li, J.; Zhang, J.; He, W.; et al. Overexpression of a MYB Family Gene, OsMYB6, Increases Drought and Salinity Stress Tolerance in Transgenic Rice. Front. Plant Sci. 2019, 10, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nuruzzaman, M.; Sharoni, A.M.; Kikuchi, S. Roles of NAC transcription factors in the regulation of biotic and abiotic stress responses in plants. Front. Microbiol. 2013, 4, 248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, Y.; Zhang, H.; Huang, L.; Li, D.; Song, F. Overexpression of a Stress-Responsive NAC Transcription Factor Gene ONAC022 Improves Drought and Salt Tolerance in Rice. Front. Plant Sci. 2016, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Dai, M.; Yao, J.; Xiao, B.; Li, X.; Zhang, Q.; Xiong, L. Overexpressing a NAM, ATAF, and CUC (NAC) transcription factor enhances drought resistance and salt tolerance in rice. Proc. Natl. Acad. Sci. USA 2006, 103, 12987–12992. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SRA Study | SRA Accession | Tissue | Treatment | Sequencing Platform | Cultivar | References |
|---|---|---|---|---|---|---|
| SRP076274 | SRR3647326 | Leaf | Unstressed-110d old seedling | Illumina HiSeq 2500 | Nipponbare | - |
| SRP076274 | SRR3647327 | Leaf | Salt (200 mM)-110d old seedling | Illumina HiSeq 2500 | Nipponbare | - |
| SRP076274 | SRR3647328 | Leaf | Unstressed-110d old seedling | Illumina HiSeq 2500 | Nipponbare | - |
| SRP076274 | SRR3647329 | Leaf | Salt (200 mM)-110d old seedling | Illumina HiSeq 2500 | Nipponbare | - |
| SRP076274 | SRR3647330 | Leaf | Unstressed-110d old seedling | Illumina HiSeq 2500 | Nipponbare | - |
| SRP076274 | SRR3647331 | Leaf | Salt (200 mM)-110d old seedling | Illumina HiSeq 2500 | Nipponbare | - |
| SRP076274 | SRR3647334 | Leaf | Unstressed-110d old seedling | Illumina HiSeq 2500 | Nipponbare | - |
| SRP076274 | SRR3647335 | Leaf | Salt (200 mM)-110d old seedling | Illumina HiSeq 2500 | Nipponbare | - |
| SRP083700 | SRR4098173 | Root | Unstressed-2 weeks old seedling | Illumina HiSeq 2000 | japonica rice | Yuan et al. [16] |
| SRP083700 | SRR4098191 | Root | Salt (300 mM NaCl for 12 h)-2 weeks old seedling | Illumina HiSeq 2000 | japonica rice | Yuan et al. [16] |
| SRP114666 | SRR5889379 | Root | Unstressed-seedling | Illumina HiSeq 2500 | Sea Rice 86 | Chen et al. [17] |
| SRP114666 | SRR5889380 | Root | Unstressed-seedling | Illumina HiSeq 2500 | Sea Rice 86 | Chen et al. [17] |
| SRP114666 | SRR5889381 | Root | Unstressed-seedling | Illumina HiSeq 2500 | Sea Rice 86 | Chen et al. [17] |
| SRP114666 | SRR5889382 | Root | Salt (sea water for 30 d)-seedling | Illumina HiSeq 2500 | Sea Rice 86 | Chen et al. [17] |
| SRP114666 | SRR5889383 | Root | Salt (sea water for 30 d)-seedling | Illumina HiSeq 2500 | Sea Rice 86 | Chen et al. [17] |
| SRP114666 | SRR5889384 | Root | Salt (sea water for 30 d)-seedling | Illumina HiSeq 2500 | Sea Rice 86 | Chen et al. [17] |
| Modules | Gene_id | Arabidopsis Orthologs | Predicted Functions |
|---|---|---|---|
| Blue | LOC_Os03g15270 | AT5G16080 | Carboxyesterase 17 |
| LOC_Os05g27340 | AT5G01750 | Unknown | |
| LOC_Os01g72009 | AT5G04080 | Unknown | |
| LOC_Os07g48710 | AT2G41010 | Calmodulin (CAM)-binding protein of 25 kDa | |
| LOC_Os02g13800 | AT3G24520 | Heat shock transcription factor C1 | |
| LOC_Os02g43770 | AT3G60320 | Protein of unknown function (DUF630 and DUF632) | |
| Grey | LOC_Os02g51080 | AT1G74470 | Pyridine nucleotide-disulphide oxidoreductase family protein |
| LOC_Os03g20700 | AT5G13630 | Magnesium-chelatase subunit chlH, chloroplast, putative/Mg-protoporphyrin IX chelatase, | |
| LOC_Os01g17170 | AT3G56940 | Dicarboxylate diiron protein CRD1 | |
| LOC_Os09g20240 | AT1G78020 | Unknown DUF581 | |
| Turquoise | LOC_Os03g42520 | AT1G07985.1 | Expressed protein |
| LOC_Os03g60260 | AT1G07985.1 | Aromatic and neutral transporter 1 | |
| LOC_Os03g17790 | AT2G38905.1 | Low temperature and salt responsive protein family | |
| LOC_Os04g48270 | AT3G61060.1 | Phloem protein PP2-A13 | |
| LOC_Os08g23870 | AT5G06760.1 | Late Embryogenesis Abundant 4-5, LEA4-5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, M.; Xie, H.; Wei, X.; Dossa, K.; Yu, Y.; Hui, S.; Tang, G.; Zeng, X.; Yu, Y.; Hu, P.; et al. WGCNA Analysis of Salt-Responsive Core Transcriptome Identifies Novel Hub Genes in Rice. Genes 2019, 10, 719. https://doi.org/10.3390/genes10090719
Zhu M, Xie H, Wei X, Dossa K, Yu Y, Hui S, Tang G, Zeng X, Yu Y, Hu P, et al. WGCNA Analysis of Salt-Responsive Core Transcriptome Identifies Novel Hub Genes in Rice. Genes. 2019; 10(9):719. https://doi.org/10.3390/genes10090719
Chicago/Turabian StyleZhu, Mingdong, Hongjun Xie, Xiangjin Wei, Komivi Dossa, Yaying Yu, Suozhen Hui, Guohua Tang, Xiaoshan Zeng, Yinghong Yu, Peisong Hu, and et al. 2019. "WGCNA Analysis of Salt-Responsive Core Transcriptome Identifies Novel Hub Genes in Rice" Genes 10, no. 9: 719. https://doi.org/10.3390/genes10090719