Network Analyses of Integrated Differentially Expressed Genes in Papillary Thyroid Carcinoma to Identify Characteristic Genes



Abstract
:1. Introduction
2. Methods
2.1. Datasets
2.2. Identification of Differentially Expressed Genes
2.3. Construction of Interaction Network of Differentially Expressed Genes
2.4. Analyses of Differentially Expressed Genes Interaction Network
2.4.1. Traditional Topological Properties
2.4.2. Normalized Centrality Measure
2.4.3. Detection of High-Density Subgraphs
2.5. Gene Ontology and Pathway Enrichment Analysis
3. Results and Discussions
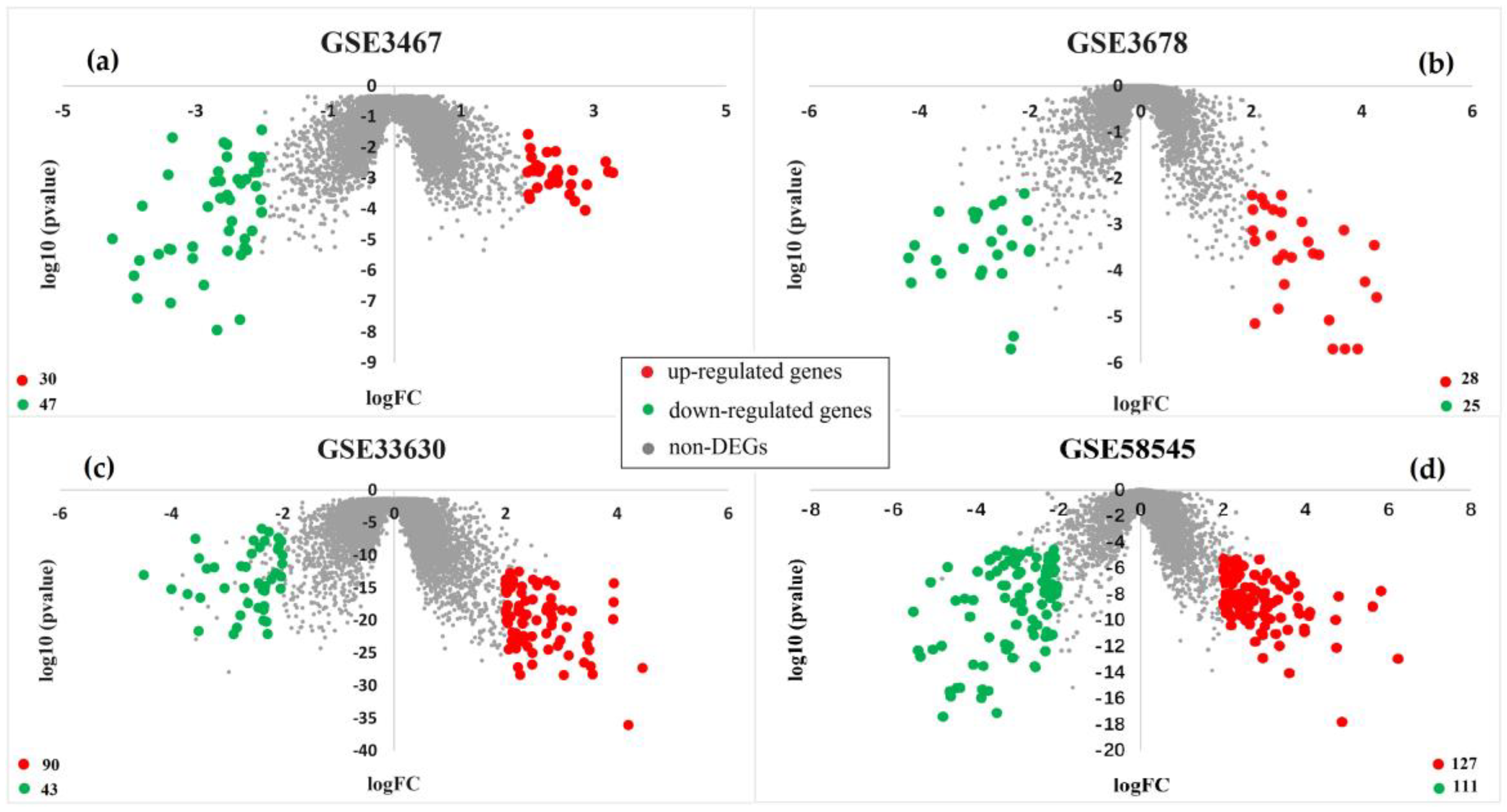
3.1. Identification of Differentially Expressed Genes
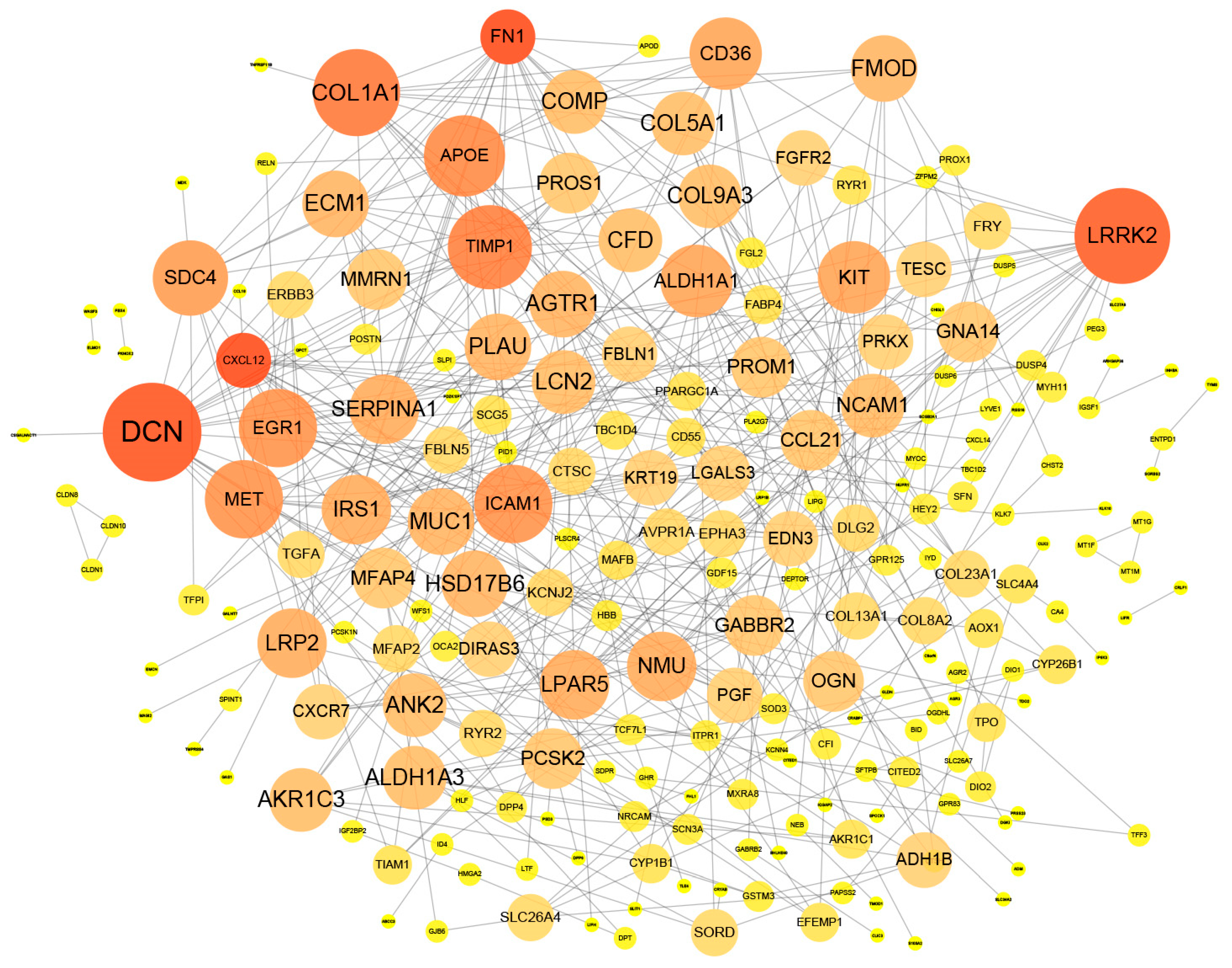
3.2. Analyses of Differentially Expressed Genes Interaction Network
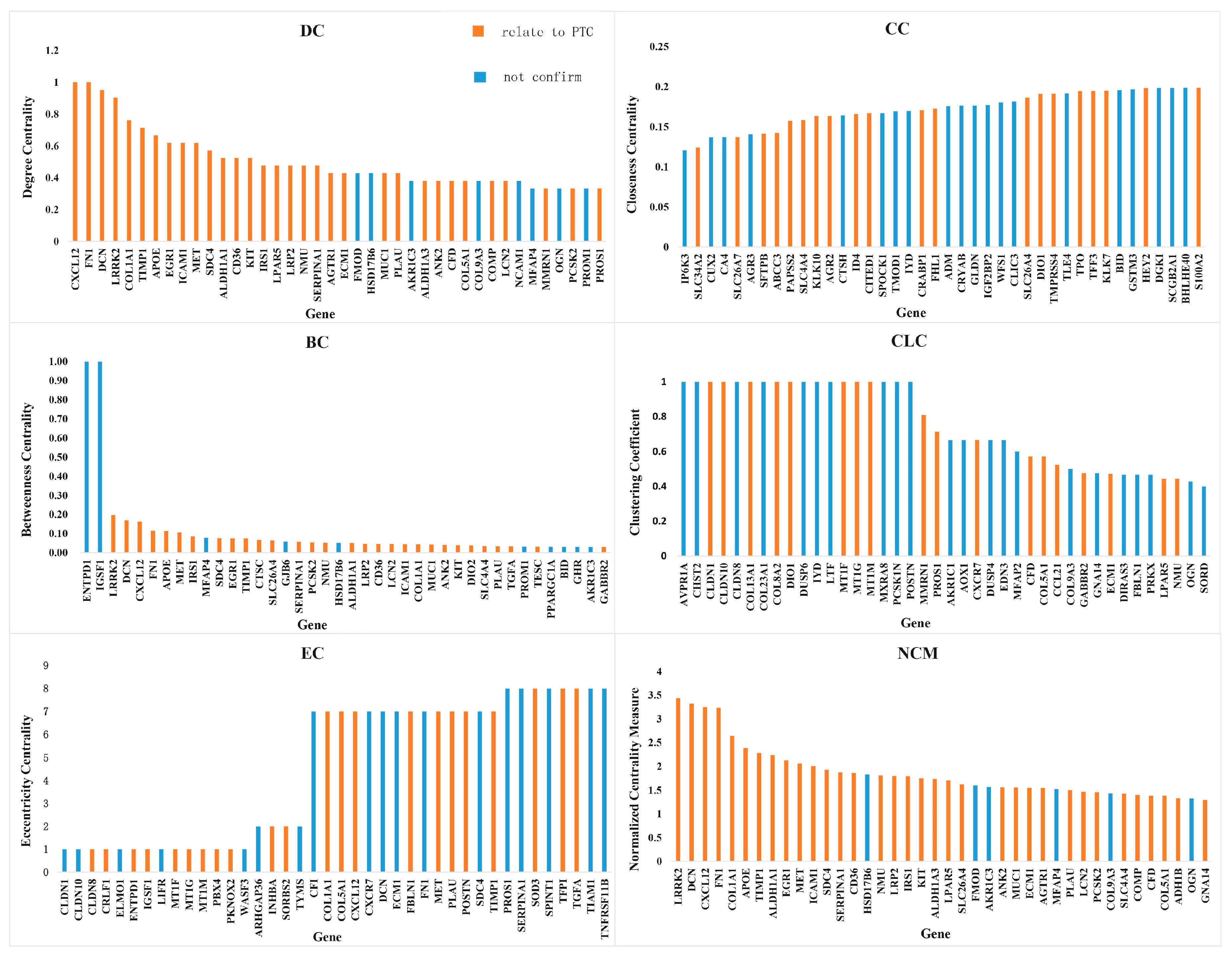
3.3. Analyses of Characteristic Genes
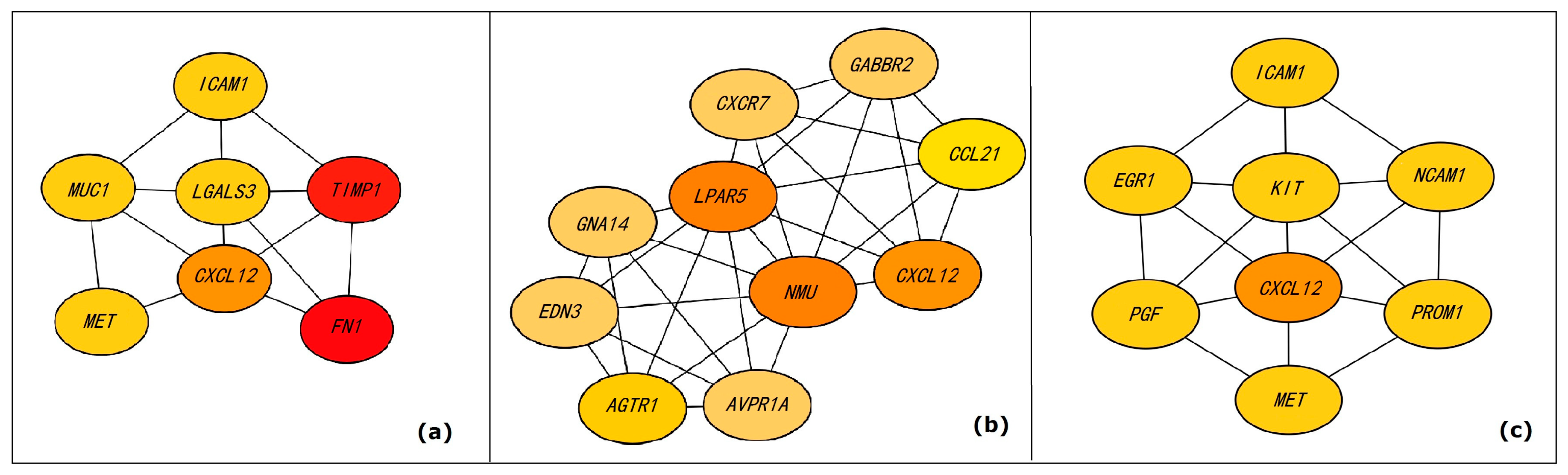
3.4. Analyses of High-Density Subgraphs
3.5. GO Term and KEGG Pathway Enrichment Analysis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Fan, M.; Li, X.; Jiang, W.; Huang, Y.; Li, J.; Wang, Z. A long non-coding RNA, PTCSC3, as a tumor suppressor and a target of miRNAs in thyroid cancer cells. Exp. Ther. Med. 2013, 5, 1143–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, W.Y.; Lee, J.B.; Jung, S.P.; Kim, H.Y.; Woo, S.U.; Son, G.S.; Bae, J.W. Gene expression profiles of papillary thyroid microcarcinoma. Int. Surg. 2017, 102, 39–46. [Google Scholar] [CrossRef]
- El Naggar, A.K. Gene expression profiling of differentiated thyroid neoplasms: Diagnostic and clinical implications. Clin. Cancer Res. 2004, 10, 6586–6597. [Google Scholar]
- Barbara, J.; Malgorzata, W.; Krzysztof, F.; Krzysztof, S.; Michal, J.; Malgorzata, O.-W.; Jan, W.; Agnieszka, C.; Ewa, C.; Dariusz, L. Gene expression profile of papillary thyroid cancer: Sources of variability and diagnostic implications. Cancer Res. 2005, 65, 1587–1597. [Google Scholar]
- He, Z.; Sun, M.; Ke, Y.; Lin, R.; Xiao, Y.; Zhou, S.; Zhao, H.; Wang, Y.; Zhou, F.; Zhou, Y. Identifying biomarkers of papillary renal cell carcinoma associated with pathological stage by weighted gene co-expression network analysis. Oncotarget 2017, 8, 27904–27914. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, J.; Deng, H.W. Identifying gene interaction enrichment for gene expression data. PLoS ONE 2009, 4, e8064. [Google Scholar] [CrossRef]
- Rizzolo, K.; Kumar, A.; Kakihara, Y.; Phanse, S.; Minic, Z.; Snider, J.; Stagljar, I.; Zilles, S.; Babu, M.; Houry, W.A. Systems analysis of the genetic interaction network of yeast molecular chaperones. Mol. Omics 2018, 14, 82–94. [Google Scholar] [CrossRef]
- Wei, P.J.; Zhang, D.; Xia, J.; Zheng, C.H. LNDriver: Identifying driver genes by integrating mutation and expression data based on gene-gene interaction network. BMC Bioinform. 2016, 17, 467. [Google Scholar] [CrossRef]
- He, H.; Jazdzewski, K.; Li, W.; Liyanarachchi, S.; Nagy, R.; Volinia, S.; Calin, G.A.; Liu, C.G.; Franssila, K.; Suster, S. The role of microRNA genes in papillary thyroid carcinoma. Proc. Natl. Acad. Sci. USA 2005, 102, 19075–19080. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Qi, B.; Zhou, Q.; Lu, C.; Huang, Q.; Xian, L.; Chen, M. Key genes and pathways in thyroid cancer based on gene set enrichment analysis. Oncol. Rep. 2013, 30, 1391–1397. [Google Scholar] [CrossRef]
- Tomás, G.; Tarabichi, M.; Gacquer, D.; Hébrant, A.; Dom, G.; Dumont, J.E.; Keutgen, X.; Rd, F.T.; Maenhaut, C.; Detours, V. A general method to derive robust organ-specific gene expression-based differentiation indices: Application to thyroid cancer diagnostic. Oncogene 2012, 31, 4490–4498. [Google Scholar] [CrossRef] [PubMed]
- Rusinek, D.; Swierniak, M.; Chmielik, E.; Kowal, M.; Kowalska, M.; Cyplinska, R.; Czarniecka, A.K.; Piglowski, W.; Korfanty, J.; Chekan, M. BRAFV600E-associated gene expression profile: Early changes in the transcriptome, based on a transgenic mouse model of papillary thyroid carcinoma. PLoS ONE 2015, 10, e0143688. [Google Scholar] [CrossRef] [PubMed]
- Smyth, G.K.; Michaud, J.; Scott, H.S. Use of within-Aarray Replicate Spots for Assessing Differential Expression in Microarray Experiments; Oxford University Press: Oxford, UK, 2005. [Google Scholar]
- Chen, W.; Liu, Q.; Lv, Y.; Xu, D.; Chen, W.; Yu, J. Special role of JUN in papillary thyroid carcinoma based on bioinformatics analysis. World J. Surg. Oncol. 2017, 15, 119. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P. The string database in 2017: Quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. CytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8 (Suppl. 4), S11. [Google Scholar] [CrossRef]
- Tadaka, S.; Kinoshita, K. NCMine: Core-peripheral based functional module detection using near-clique mining. Bioinformatics 2016, 32, 3454–3460. [Google Scholar] [CrossRef]
- Dennis, G.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for annotation, visualization, and integrated discovery. Genome Biol. 2003, 4, P3. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Mao, T.; Sato, Y.; Morishima, K. Kegg: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Qiu, J.; Zhang, W.; Zang, C.; Liu, X.; Liu, F.; Ge, R.; Sun, Y.; Xia, Q. Identification of key genes and miRNAs markers of papillary thyroid cancer. Biol. Res. 2018, 51, 45. [Google Scholar] [CrossRef]
- Qiu, J.; Zhang, W.; Xia, Q.; Liu, F.; Li, L.; Zhao, S.; Gao, X.; Zang, C.; Ge, R.; Sun, Y. RNA sequencing identifies crucial genes in papillary thyroid carcinoma (PTC) progression. Exp. Mol. Pathol. 2016, 100, 151–159. [Google Scholar] [CrossRef]
- Qu, T.; Li, Y.P.; Li, X.H.; Chen, Y. Identification of potential biomarkers and drugs for papillary thyroid cancer based on gene expression profile analysis. Mol. Med. Rep. 2016, 14. [Google Scholar] [CrossRef]
- Rosignolo, F.; Sponziello, M.; Durante, C.; Puppin, C.; Mio, C.; Baldan, F.; Di, L.C.; Russo, D.; Filetti, S.; Damante, G. Expression of PAX8 target genes in papillary thyroid carcinoma. PLoS ONE 2016, 11, e0156658. [Google Scholar] [CrossRef] [PubMed]
- Schulten, H.J.; Al-Mansouri, Z.; Baghallab, I.; Bagatian, N.; Subhi, O.; Karim, S.; Al-Aradati, H.; Al-Mutawa, A.; Johary, A.; Meccawy, A.A. Comparison of microarray expression profiles between follicular variant of papillary thyroid carcinomas and follicular adenomas of the thyroid. BMC Genom. 2015, 16, S7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyun Sook, K.; Do Hyung, K.; Yeon, K.J.; Nam Ho, J.; In Kyu, L.; Gu, B.J.; Eui Dal, J. Microarray analysis of papillary thyroid cancers in Korean. Korean J. Intern. Med. 2010, 25, 399–407. [Google Scholar]
- Liang, W.; Sun, F. Identification of key genes of papillary thyroid cancer using integrated bioinformatics analysis. J. Endocrinol. Investig. 2018, 41, 1237–1245. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Liu, J.; Li, C.; Peng, X.; Li, H.; Li, Z. Identification and validation of potential target genes in papillary thyroid cancer. Eur. J. Pharmacol. 2019, 843, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Weber, F.; Shen, L.; Aldred, M.A.; Morrison, C.D.; Frilling, A.; Saji, M.; Schuppert, F.; Broelsch, C.E.; Ringel, M.D.; Eng, C. Genetic classification of benign and malignant thyroid follicular neoplasia based on a three-gene combination. J. Clin. Endocrinol. Metab. 2005, 90, 2512–2521. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Jin, W.X.; Jin, Y.X.; Zheng, Z.C.; Zhou, X.F.; Wang, Q.X.; Ye, D.R.; Sun, Y.H.; Zhang, X.H.; Wang, O.C. Clinical effect of MUC1 and its relevance to BRAF V600E mutation in papillary thyroid carcinoma: A case–control study. Cancer Manag. Res. 2018, 10, 1351–1358. [Google Scholar] [CrossRef]
- Wei, X.; Zhang, Y.; Yu, S.; Li, S.; Jiang, W.; Zhu, Y.; Xu, Y.; Yang, C.; Tian, G.; Mi, J. PDLIM5 identified by label-free quantitative proteomics as a potential novel biomarker of papillary thyroid carcinoma. Biochem. Biophys. Res. Commun. 2018, 499, 338–344. [Google Scholar] [CrossRef]
- Katsuhiro, T.; Hiroshi, S.; Junichi, K.; Tsunehisa, N.; Sumiko, O.; Yutaka, Y.; Shigeru, Y. Inhibition of infiltration and angiogenesis by thrombospondin-1 in papillary thyroid carcinoma. Clin. Cancer Res. 2002, 8, 1125–1131. [Google Scholar]
- Arnaldi, L.A.; Borra RCMaciel, R.M.; Cerutti, J.M. Gene expression profiles reveal that DCN, DIO1, and DIO2 are underexpressed in benign and malignant thyroid tumors. Thyroid 2005, 15, 210–221. [Google Scholar] [CrossRef]
- Lisi, S.; Botta, R.; Rotondo Dottore, G.; Leo, M.; Latrofa, F.; Vitti, P.; Marinò, M. Intracellular retention of thyroglobulin in the absence of the low-density lipoprotein receptor-associated protein (RAP) is likely due to premature binding to megalin in the biosynthetic pathway. J. Endocrinol. Investig. 2016, 39, 1039–1044. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Yan, F.; Xu, J.; Bao, Y.; Zhu, J.; Wang, X.; Wu, J.; Li, Y.; Pu, W.; Liu, Y.; et al. Identification and validation of the methylation biomarkers of non-small cell lung cancer (NSCLC). Clin. Epigenet. 2015, 7, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, J.; Qian, X.; Song, B.; An, X.; Cai, T.; Zuo, Z.; Ding, D.; Lu, Y.; Li, H. Integrated bioinformatics analysis reveals that the expression of cathepsin S is associated with lymph node metastasis and poor prognosis in papillary thyroid cancer. Oncol. Rep. 2018, 40, 111–122. [Google Scholar] [PubMed]
- Tang, J.; Kong, D.; Cui, Q.; Wang, K.; Zhang, D.; Yuan, Q.; Liao, X.; Gong, Y.; Wu, G. Bioinformatic analysis and identification of potential prognostic microRNAs and mRNAs in thyroid cancer. PeerJ 2018, 6, e4674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kai-Chun, C.; Chun-Tzu, H.; Kai-Yuan, C.; Kuo-Jen, C.; Wen-Chuan, W.; Jau-Ling, S.; Yu-Jen, W.; Cheng-Hsien, C. Proteomic surveillance of putative new autoantigens in thyroid orbitopathy. Br. J. Ophthalmol. 2015, 99, 1571–1576. [Google Scholar]
- Lim, A.S.P.; Gaiteri, C.; Yu, L.; Sohail, S.; Swardfager, W.; Tasaki, S.; Schneider, J.A.; Paquet, C.; Stuss, D.T.; Masellis, M.; et al. Seasonal plasticity of cognition and related biological measures in adults with and without Alzheimer disease: Analysis of multiple cohorts. PLoS Med. 2018, 15, e1002647. [Google Scholar] [CrossRef] [PubMed]
- Stein, L. Copy number and gene expression alterations in radiation-induced papillary thyroid carcinoma from Chernobyl pediatric patients. Thyroid 2010, 20, 475–487. [Google Scholar] [CrossRef]
- Maro, G.D.; Salerno, P.; Unger, K.; Orlandella, F.M.; Monaco, M.; Chiappetta, G.; Thomas, G.; Oczko-Wojciechowska, M.; Masullo, M.; Jarzab, B. Anterior gradient protein 2 promotes survival, migration and invasion of papillary thyroid carcinoma cells. Mol. Cancer 2014, 13, 160. [Google Scholar] [CrossRef] [Green Version]
- Griffith, O.L.; Adrienne, M.; Jones, S.J.M.; Wiseman, S.M. Meta-analysis and meta-review of thyroid cancer gene expression profiling studies identifies important diagnostic biomarkers. J. Clin. Oncol. 2006, 24, 5043–5051. [Google Scholar] [CrossRef]
- Tailor, P.D.; Kodeboyina, S.K.; Bai, S.; Patel, N.; Sharma, S.; Ratnani, A.; Copland, J.A.; She, J.X.; Sharma, A. Diagnostic and prognostic biomarker potential of kallikrein family genes in different cancer types. Oncotarget 2018, 9, 17876–17888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, Y.; Yoshida, H.; Tomoda, C.; Uruno, T.; Miya, A.; Kobayashi, K.; Matsuzuka, F.; Kakudo, K.; Kuma, K.; Miyauchi, A. Expression of S100A2 and S100A6 in thyroid carcinomas. Histopathology 2010, 46, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Nasr, M.R.; Mukhopadhyay, S.; Zhang, S.; Katzenstein, A.A. Immunohistochemical markers in diagnosis of papillary thyroid carcinoma: Immunohistochemical markers in diagnosis of papillary thyroid carcinoma: Utility of HBME1 combined with CK19 immunostaining. Mod. Pathol. 2006, 19, 1631–1637. [Google Scholar] [CrossRef]
- Electron, K.; Miao, P.; Emily, R.; Quan-Yang, D.; Clark, O.H.; Alex, M.M. ECM1 and TMPRSS4 are diagnostic markers of malignant thyroid neoplasms and improve the accuracy of fine needle aspiration biopsy. Ann. Surg. 2005, 242, 353–363. [Google Scholar]
- Abend, M.; Pfeiffer, R.M.; Ruf, C.; Hatch, M.; Bogdanova, T.I.; Tronko, M.D.; Hartmann, J.; Meineke, V.; Mabuchi, K.; Brenner, A.V. Iodine-131 dose-dependent gene expression: Alterations in both normal and tumour thyroid tissues of post-Chernobyl thyroid cancers. Br. J. Cancer 2013, 109, 2286–2294. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, P.; Ponny, S.R.; Xu, H.; Bai, S.; Smallridge, R.; Copland, J.; Sharma, A. Cell cycle M-phase genes are highly upregulated in anaplastic thyroid carcinoma. Thyroid 2017, 27, 236–252. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Lin, J.D.; Chen, J.T.; Chang, C.M.; Weng, H.F.; Hsueh, C.; Chien, H.P.; Yu, J.S. Integrated analysis of fine-needle-aspiration cystic fluid proteome, cancer cell secretome, and public transcriptome datasets for papillary thyroid cancer biomarker discovery. Oncotarget 2018, 9, 12079–12100. [Google Scholar] [CrossRef] [PubMed]
- Smallridge, R.C.; Ana-Maria, C.; Yan, W.; Asmann, Y.W.; Casler, J.D.; Serie, D.J.; Reddi, H.V.; Cradic, K.W.; Michael, R.; Grebe, S.K.; et al. RNA sequencing identifies multiple fusion transcripts, differentially expressed genes, and reduced expression of immune function genes in BRAF (V600E) mutant vs BRAF wild-type papillary thyroid carcinoma. J. Clin. Endocrinol. Metab. 2014, 99, E338–E347. [Google Scholar] [CrossRef]
- Mateus Camargo, B.F.; Fabio Albuquerque, M.; Clóvis Ant?Nio, P.; Silvia Regina, R.; Luiz Paulo, K. High diagnostic accuracy based on CLDN10, HMGA2, and LAMB3 transcripts in papillary thyroid carcinoma. J. Clin. Endocrinol. Metab. 2015, 100, 890–899. [Google Scholar]
- Jiao, F.; Lv, H.; Guan, H.; Ma, X.; Ji, M.; He, N.; Shi, B.; Peng, H. High diagnostic accuracy based on CLDN10, HMGA2, and LAMB3 transcripts in papillary thyroid carcinoma. J. Clin. Endocrinol. Metab. BMC Cancer 2013, 13, 462. [Google Scholar]
- Zhang, H.; Teng, X.; Liu, Z.; Lei, Z.; Zhen, L. Gene expression profile analyze the molecular mechanism of CXCR7 regulating papillary thyroid carcinoma growth and metastasis. J. Exp. Clin. Cancer Res. 2015, 34, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Janssen, S.F.; Gorgels, T.G.; Spek, P.J.V.D.; Jansonius, N.M.; Bergen, A.A. In silico analysis of the molecular machinery underlying aqueous humor production: Potential implications for glaucoma. J. Clin. Bioinform. 2013, 3, 21. [Google Scholar] [CrossRef] [PubMed]
- Dziegiel, P.; Pula, B.; Kobierzycki, C.; Stasiolek, M.; Podhorska-Okolow, M. The Role of Metallothioneins in Carcinogenesis; Springer International Publishing: Berlin, Germany, 2016; Volume 218, pp. 29–63. [Google Scholar]
- Passon, N.; Bregant, E.; Sponziello, M.; Dima, M.; Rosignolo, F.; Durante, C.; Celano, M.; Russo, D.; Filetti, S.; Damante, G. Somatic amplifications and deletions in genome of papillary thyroid carcinomas. Endocrine 2015, 50, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, N.; Akbani, R.; Aksoy, B.A.; Ally, A.; Arachchi, H.; Asa, S.; Auman, J.T.; Balasundaram, M.; Balu, S.; Baylin, S. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014, 159, 676–690. [Google Scholar] [CrossRef] [PubMed]
- Shang, X.; Zhong, X.; Tian, X. Metabolomics of papillary thyroid carcinoma tissues: Potential biomarkers for diagnosis and promising targets for therapy. Tumor Biol. 2016, 37, 11163–11175. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GEO | Platform | Normal | Tumor | Reference |
|---|---|---|---|---|
| GSE3467 | GPL570 | 9 | 9 | He et al. [9] |
| GSE3678 | GPL570 | 7 | 7 | He et al. [10] |
| GSE33630 | GPL570 | 49 | 45 | Tomás et al. [11] |
| GSE58545 | GPL96 | 27 | 18 | Rusinek et al. [12] |
| Measures | Count 1 | Genes 2 |
|---|---|---|
| Degree Centrality | 32 | AGTR1 [34] ALDH1A1 [30] COL1A1 [21] COMP [25] CXCL12 [26] DCN [32] ECM1 [24] FN1 [20] ICAM1 [20] IRS1 [26] KIT [24] LCN2 [23] LRP2 [33] LRRK2 [2] MET [21] MUC1 [29] PLAU [20] SDC4 [2] SERPINA1 [21] TIMP1 [20] CD36 [31] EGR1 [38] ALDH1A3 [37] NMU [36] ANK2 [39] APOE [35] CFD [27] COL5A1 [20] LPAR5 [36] PCSK2 [28] PROS1 [2] MMRN1 [27] |
| Closeness Centrality | 20 | AGR2 [40] CITED1 [2] CRABP1 [41] KLK10 [42] KLK7 [25] S100A2 [43] SFTPB [44] SLC26A4 [30] SLC34A2 [25] TFF3 [28] TMPRSS4 [45] TPO [41] DIO1 [32] ID4 [45] PAPSS2 [39] SLC4A4 [25] ABCC3 [46] FHL1 [38] HEY2 [46] SLC26A7 [47] |
| Betweenness Centrality | 30 | ALDH1A1 [30] COL1A1 [21] CTSC [48] CXCL12 [26] DCN [32] FN1 [20] ICAM1 [20] IRS1 [26] KIT [24] LCN2 [23] LRP2 [33] LRRK2 [2] MET [22] MUC1 [29] PLAU [21] SDC4 [2] SERPINA1 [22] SLC26A4 [30] TGFA [41] TIMP1 [19] DIO2 [32] CD36 [31] ANK2 [39] APOE [35] GABBR2 [24] PCSK2 [28] SLC4A4 [25] TESC [39] EGR1 [38] NMU [36] |
| Clustering Coefficient | 18 | CCL21 [49] CLDN1 [20] CLDN10 [50] ECM1 [24] DIO1 [32] MT1G [51] CFD [27] COL13A1 [22] COL5A1 [21] CXCR7 [52] GABBR2 [24] MMRN1 [27] PROS1 [2] COL8A2 [53] LPAR5 [36] MT1F [54] MT1M [54] NMU [36] |
| Eccentricity Centrality | 22 | CLDN1 [20] CLDN10 [50] MT1G [51] COL1A1 [20] CXCL12 [26] DCN [32] ECM1 [24] FN1 [20] MET [22] PLAU [20] SDC4 [2] TIMP1 [20] SERPINA1 [22] TGFA [41] TNFRSF11B [22] ELMO1 [55] IGSF1 [55] MT1F [54] SORBS2 [39,48] COL5A1 [21] CXCR7 [52] PROS1 [2] |
| Normalized Centrality Measure | 34 | ALDH1A1 [30] CD36 [31] COL1A1 [20] COMP [25] CXCL12 [26] DCN [32] ECM1 [24] FN1 [20] ICAM1 [20] IRS1 [26] KIT [24] LCN2 [23] LRP2 [33] LRRK2 [2] MET [22] MUC1 [29] PLAU [21] SDC4 [2] SERPINA1 [22] SLC26A4 [30] TIMP1 [20] AGTR1 [34] ANK2 [39] APOE [35] CFD [27] COL5A1 [21] LPAR5 [36] PCSK2 [28] SLC4A4 [25] ADH1B [37] GNA14 [37] NMU [36] ALDH1A3 [37] EGR1 [38] |
| DEGs | ID | Term | Count 1 | Genes 2 |
|---|---|---|---|---|
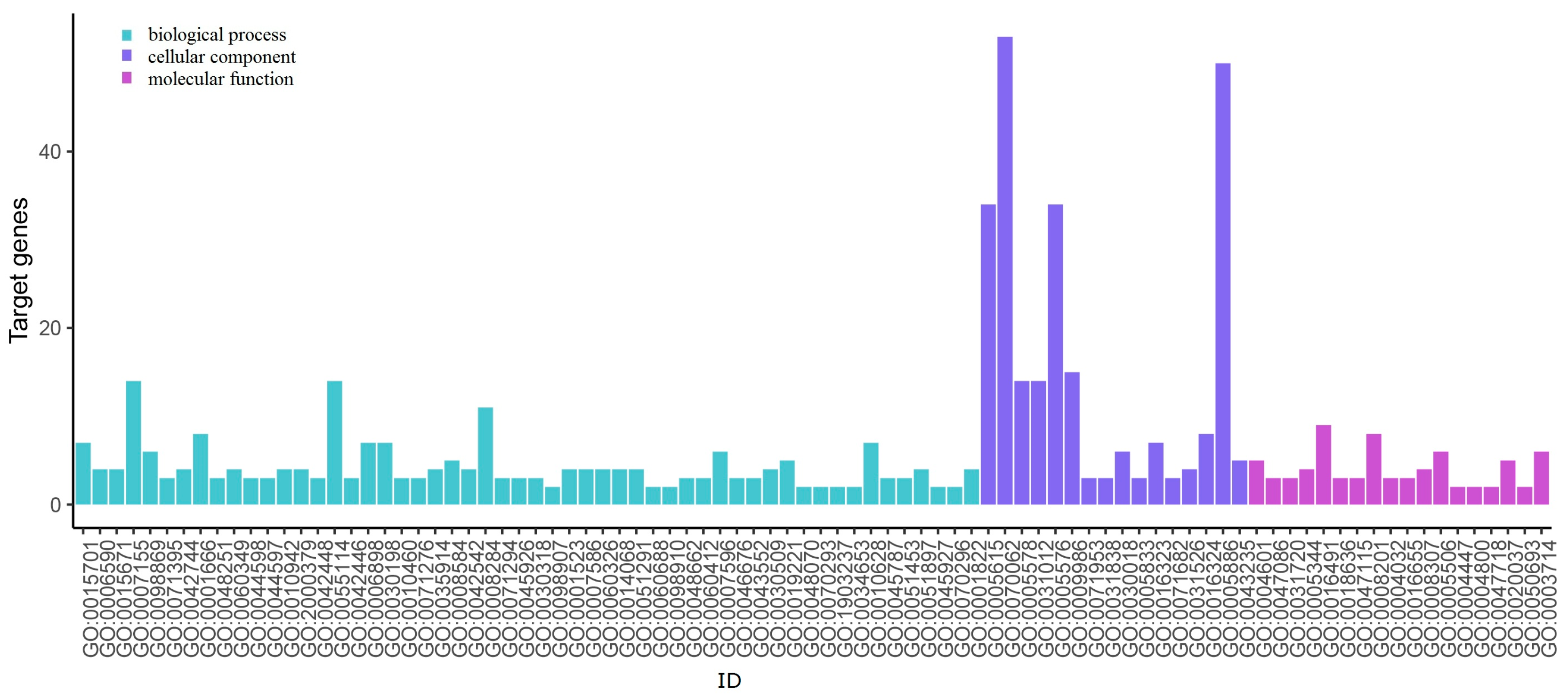
| Up-regulated | GO:0070062 | extracellular exosome | 53 | TIMP1 FN1 APOE COMP SERPINA1 NMU ECM1 COL5A1 LCN2 COL1A1 PLAU MET |
| GO:0005886 | plasma membrane | 50 | GNA14 AGTR1 SLC4A4 IRS1 CD36 KIT LRP2 SLC26A4 ANK2 | |
| GO:0005615 | extracellular space | 34 | FMOD KIT DCN CXCL12 OGN CFD CD36 | |
| GO:0005576 | extracellular region | 34 | FMOD DCN CXCL12 OGN COL9A3 CFD MFAP4 | |
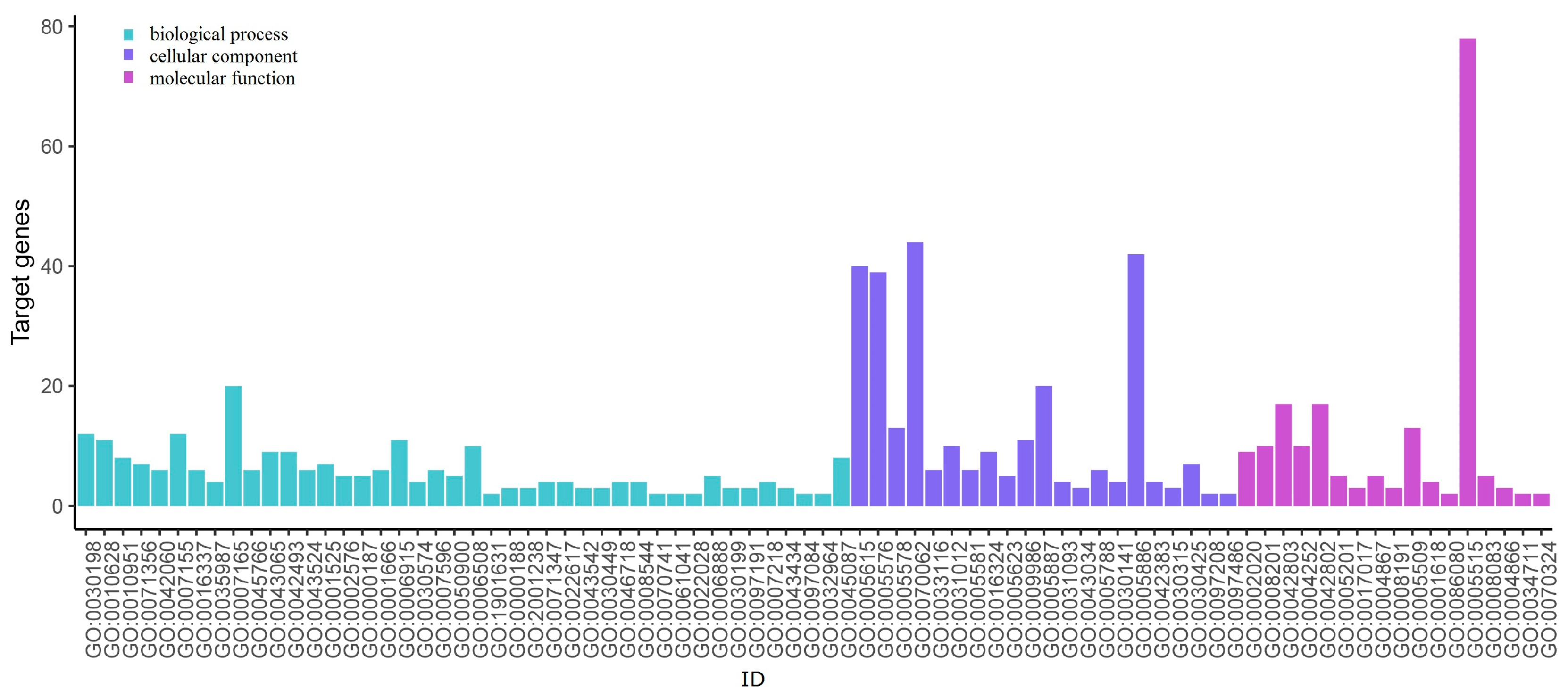
| Down-regulated | GO:0005515 | protein binding | 78 | APOE COL1A1 COMP ECM1 FN1 ICAM1 LRRK2 MUC1 NMU PCSK2 PLAU SDC4 SERPINA1 TIMP1 MET |
| GO:0070062 | extracellular exosome | 44 | SDC4 TIMP1 APOE COMP ALDH1A3 SERPINA1 FN1 MUC1 ICAM1 ECM1 COL5A1 LCN2 LRRK2 PLAU | |
| GO:0005886 | plasma membrane | 42 | SDC4 APOE ICAM1 MET LPAR5 LRRK2 PLAU | |
| GO:0005615 | extracellular space | 40 | TIMP1 PCSK2 APOE COMP SERPINA1 FN1 MUC1 ICAM1 ECM1 LCN2 COL1A1 LRRK2 PLAU | |
| GO:0005576 | extracellular region | 39 | TIMP1 APOE COMP SERPINA1 NMU FN1 MET ECM1 COL5A1 LCN2 COL1A1 PLAU |
| Term | ID | Input Number | p-Value | Input Genes |
|---|---|---|---|---|
| Cytokine-cytokine receptor interaction | hsa04060 | 12 | 0.0002 | KIT MET CXCL12 LIFR CXCL14 ACKR3 TNFRSF11B TNFRSF12A CCL18 CCL21 GHR INHBA |
| PI3K-Akt signaling pathway | hsa04151 | 12 | 0.0017 | MET FN1 RELN KIT FGFR2 LPAR5 COMP COL1A1 PGF IRS1 GHR COL9A3 |
| Metabolic pathways | hsa01100 | 24 | 0.0267 | AKR1C3 TYMS GALNT7 TPO ADH1B LIPG ALDH1A1 GALE OGDHL TUSC30 PLA2G73 SORD CSGALNACT1 PAPSS2 GATM CYP26B1 DGKI ALDH1A3 ALOX15B HGD TDO2 AOX1 HSD17B6 IMPA2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shang, J.; Ding, Q.; Yuan, S.; Liu, J.-X.; Li, F.; Zhang, H. Network Analyses of Integrated Differentially Expressed Genes in Papillary Thyroid Carcinoma to Identify Characteristic Genes. Genes 2019, 10, 45. https://doi.org/10.3390/genes10010045
Shang J, Ding Q, Yuan S, Liu J-X, Li F, Zhang H. Network Analyses of Integrated Differentially Expressed Genes in Papillary Thyroid Carcinoma to Identify Characteristic Genes. Genes. 2019; 10(1):45. https://doi.org/10.3390/genes10010045
Chicago/Turabian StyleShang, Junliang, Qian Ding, Shasha Yuan, Jin-Xing Liu, Feng Li, and Honghai Zhang. 2019. "Network Analyses of Integrated Differentially Expressed Genes in Papillary Thyroid Carcinoma to Identify Characteristic Genes" Genes 10, no. 1: 45. https://doi.org/10.3390/genes10010045