
Valeriana rigida Ruiz & Pav. Root Extract: A New Source of Caffeoylquinic Acids with Antioxidant and Aldose Reductase Inhibitory Activities

 , , ,
, , ,  and
and
Abstract
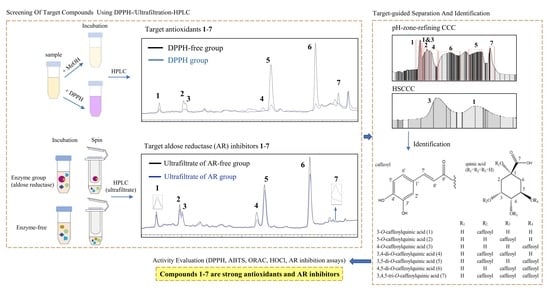
:
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. V. rigida Material and Preparation of Plant Extract
2.3. HPLC Analysis
2.4. Antioxidant Assay
2.4.1. DPPH Radical Scavenging Assay
2.4.2. ABTS Radical Scavenging Assay
2.4.3. Oxygen Radical Absorbance Capacity Assay
2.4.4. Hypochlorous Acid Scavenging Assay
2.5. AR Inhibition Assay
2.6. Screening of Antioxidants from the Extract Using Offline DPPH-HPLC
2.7. Screening of AR Inhibitors from the Extract Using Ultrafiltration-HPLC
2.8. Separation of Target Compounds by pH-Zone-Refining CCC
2.8.1. Screening of pH-Zone-Refining CCC Solvent System
2.8.2. Preparation of pH-Zone-Refining CCC Solvent System and Sample Solution
2.8.3. pH-Zone-Refining CCC Separation
2.9. Separation of Target Compounds 1 and 3 by Conventional HSCCC
2.9.1. Preparation of HSCCC Solvent System and Sample Solution
2.9.2. HSCCC Separation
2.10. Structure Identification
2.11. Quantification of the Major Compounds 5, 6, and 8
2.12. Statistical Analysis
3. Results
3.1. Antioxidant and AR Inhibitory Activity of the 70% MeOH Root Extract of V. rigida
3.2. Screening of Antioxidants and AR Inhibitors from the Extract Using Offline DPPH- and Ultrafiltration-HPLC
3.3. Selection of Solvent System and Separation of Target Compounds by pH-Zone-Refining CCC and Conventional HSCCC
3.4. Identification of the Separated Compounds
3.5. Antioxidant and AR Inhibitory Activity of the Target Compounds 1–7
3.6. Quantification of the Major Compounds 5, 6, and 8
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Al-Waili, N.; Al-Waili, H.; Al-Waili, T.; Salom, K. Natural antioxidants in the treatment and prevention of diabetic nephropathy; a potential approach that warrants clinical trials. Redox Rep. 2017, 22, 99–118. [Google Scholar] [CrossRef]
- Veeresham, C.; Rama Rao, A.; Asres, K. Aldose reductase inhibitors of plant origin. Phytother. Res. 2014, 28, 317–333. [Google Scholar] [CrossRef] [PubMed]
- Maccari, R.; Ottanà, R. Targeting aldose reductase for the treatment of diabetes complications and inflammatory diseases: New insights and future directions. J. Med. Chem. 2015, 58, 2047–2067. [Google Scholar] [CrossRef]
- Rodríguez, M.L.; Pérez, S.; Mena-Mollá, S.; Desco, M.C.; Ortega, Á.L. Oxidative stress and microvascular alterations in diabetic retinopathy: Future Therapies. Oxidative Med. Cell. Longev. 2019, 2019, 4940825. [Google Scholar] [CrossRef] [Green Version]
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [Green Version]
- Harding, J.L.; Pavkov, M.E.; Magliano, D.J.; Shaw, J.E.; Gregg, E.W. Global trends in diabetes complications: A review of current evidence. Diabetologia 2019, 62, 3–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Wu, Z.; Zuo, G.; Lim, S.S.; Yan, H. Defatted Seeds of Oenothera biennis as a Potential Functional Food Ingredient for Diabetes. Foods 2021, 10, 538. [Google Scholar] [CrossRef]
- Testa, R.; Bonfigli, A.R.; Prattichizzo, F.; La Sala, L.; de Nigris, V.; Ceriello, A. The “metabolic memory” theory and the early treatment of hyperglycemia in prevention of diabetic complications. Nutrients 2017, 9, 437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgos-Morón, E.; Abad-Jiménez, Z.; Martinez de Maranon, A.; Iannantuoni, F.; Escribano-López, I.; López-Domènech, S.; Salom, C.; Jover, A.; Mora, V.; Roldan, I. Relationship between oxidative stress, ER stress, and inflammation in type 2 diabetes: The battle continues. J. Clin. Med. 2019, 8, 1385. [Google Scholar] [CrossRef] [Green Version]
- Araki, E.; Nishikawa, T. Oxidative stress: A cause and therapeutic target of diabetic complications. J. Diabetes Investig. 2010, 1, 90–96. [Google Scholar] [CrossRef] [Green Version]
- Soufi, F.G.; Mohammad-Nejad, D.; Ahmadieh, H. Resveratrol improves diabetic retinopathy possibly through oxidative stress—Nuclear factor κb—Apoptosis pathway. Pharmacol. Rep. 2012, 64, 1505–1514. [Google Scholar] [CrossRef]
- Varma, S.; Mizuno, A.; Kinoshita, J. Diabetic cataracts and flavonoids. Science 1977, 195, 205–206. [Google Scholar] [CrossRef]
- Kumar, B.; Gupta, S.K.; Nag, T.C.; Srivastava, S.; Saxena, R.; Jha, K.A.; Srinivasan, B.P. Retinal neuroprotective effects of quercetin in streptozotocin-induced diabetic rats. Exp. Eye Res. 2014, 125, 193–202. [Google Scholar] [CrossRef]
- Lu, Q.; Hao, M.; Wu, W.; Zhang, N.; Isaac, A.T.; Yin, J.; Zhu, X.; Du, L.; Yin, X. Antidiabetic cataract effects of GbE, rutin and quercetin are mediated by the inhibition of oxidative stress and polyol pathway. Acta Biochim. Pol. 2018, 65, 35–41. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.S.; Kim, J.; Lee, Y.M.; Sohn, E.; Jo, K.; Kim, J.S. Inhibitory effects of chlorogenic acid on aldose reductase activity in vitro and cataractogenesis in galactose-fed rats. Arch. Pharm. Res. 2011, 34, 847–852. [Google Scholar]
- Bao, L.; Li, J.; Zha, D.; Zhang, L.; Gao, P.; Yao, T.; Wu, X. Chlorogenic acid prevents diabetic nephropathy by inhibiting oxidative stress and inflammation through modulation of the Nrf2/HO-1 and NF-ĸB pathways. Int. Immunopharmacol. 2018, 54, 245–253. [Google Scholar] [CrossRef]
- Plants of the World Online. Valeriana Rigida Ruiz & Pav. Available online: http://www.plantsoftheworldonline.org/taxon/urn:lsid:ipni.org:names:286650-2 (accessed on 14 April 2021).
- Castillo-Vera, H.; Cochachin, E.; Albán, J. Plantas comercializadas por herbolarios en el mercado del distrito de Cajabamba (Cajamarca, Perú). Boletín Latinoam. Caribe Plantas Med. Aromáticas 2017, 16, 303–318. [Google Scholar]
- Weberling, F.; Bittrich, V. Valerianaceae. In Flowering Plants. Eudicots; Springer: New York, NY, USA, 2016; pp. 385–401. [Google Scholar]
- Bussmann, R.W.; Sharon, D.; Garcia, M. From chamomile to aspirin? Medicinal plant use among clients at Laboratorios Beal in Trujillo, Peru. Ethnob. Res. Appl. 2009, 7, 399–407. [Google Scholar] [CrossRef] [Green Version]
- Bussmann, R.W.; Sharon, D. Medicinal plants of the Andes and the Amazon-The magic and medicinal flora of Northern Peru. Ethnob. Res. Appl. 2018, 15, 1–295. [Google Scholar] [CrossRef] [Green Version]
- Bussmann, R.W.; Glenn, A.; Meyer, K.; Kuhlman, A.; Townesmith, A. Herbal mixtures in traditional medicine in Northern Peru. J. Ethnob. Ethnomed. 2010, 6, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Marles, R.J.; Farnsworth, N.R. Antidiabetic plants and their active constituents. Phytomedicine 1995, 2, 137–189. [Google Scholar] [CrossRef]
- Sarikurkcu, C.; Jeszka-Skowron, M.; Ozer, M.S. Valeriana dioscoridis aerial parts’ extracts-A new source of phytochemicals with antioxidant and enzyme inhibitory activities. Ind. Crops Prod. 2020, 148, 112273. [Google Scholar] [CrossRef]
- Devi, V.S.; Rao, M.G. Valeriana wallichii–a rich aroma root plant. World J. Pharm. Pharm. Sci. 2014, 3, 1516–1525. [Google Scholar]
- Fraisse, D.; Bred, A.; Felgines, C.; Senejoux, F. Screening and Characterization of Antiglycoxidant Anthocyanins from Vaccinium myrtillus Fruit Using DPPH and Methylglyoxal Pre-Column HPLC Assays. Antioxidants 2020, 9, 512. [Google Scholar] [CrossRef] [PubMed]
- Guillen Quispe, Y.N.; Hwang, S.H.; Wang, Z.; Zuo, G.; Lim, S.S. Screening in vitro targets related to diabetes in herbal extracts from Peru: Identification of active compounds in Hypericum laricifolium Juss. by offline high-performance liquid chromatography. Int. J. Mol. Sci. 2017, 18, 2512. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zuo, G.; Hwang, S.H.; Kwon, S.H.; Kang, Y.H.; Lee, J.Y.; Lim, S.S. Affinity measurement of ligands in Perilla frutescens extract towards α-glucosidase using affinity-based ultrafiltration-high-performance liquid chromatography. J. Chromatogr. B 2019, 1125, 121725. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Kwon, S.H.; Hwang, S.H.; Kang, Y.H.; Lee, J.Y.; Lim, S.S. Competitive binding experiments can reduce the false positive results of affinity-based ultrafiltration-HPLC: A case study for identification of potent xanthine oxidase inhibitors from Perilla frutescens extract. J. Chromatogr. B 2017, 1048, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Hwang, S.H.; Zuo, G.; Kim, S.B.; Lim, S.S. An in vitro affinity-based method for studying herb–drug interactions for direct identification of cytochrome P450 1A2, 3A4, and 2C9 specific ligands from herbal extracts using ultrafiltration-high performance liquid chromatography. RSC Adv. 2018, 8, 8944–8949. [Google Scholar] [CrossRef] [Green Version]
- Ito, Y. Golden rules and pitfalls in selecting optimum conditions for high-speed counter-current chromatography. J. Chromatogr. A 2005, 1065, 145–168. [Google Scholar] [CrossRef]
- Gong, Y.; Huang, X.Y.; Pei, D.; Duan, W.D.; Zhang, X.; Sun, X.; Di, D.L. The applicability of high-speed counter current chromatography to the separation of natural antioxidants. J. Chromatogr. A 2020, 1623, 461150. [Google Scholar] [CrossRef]
- Zhang, Y.; Shi, S.; Wang, Y.; Huang, K. Target-guided isolation and purification of antioxidants from Selaginella sinensis by offline coupling of DPPH-HPLC and HSCCC experiments. J. Chromatogr. B 2011, 879, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Huang, Q.; Zhou, B.; Gong, Z.; Liu, Z.; Shi, S. Preparative isolation and purification of seven main antioxidants from Eucommia ulmoides Oliv. (Du-zhong) leaves using HSCCC guided by DPPH-HPLC experiment. Food Chem. 2013, 139, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Zuo, G.; Wang, Z.; Quispe, Y.N.G.; Hwang, S.H.; Kim, H.Y.; Kang, B.G.; Lim, S.S. Target guided isolation of potential tyrosinase inhibitors from Otholobium pubescens (Poir.) JW Grimes by ultrafiltration, high-speed countercurrent chromatography and preparative HPLC. Ind. Crops Prod. 2019, 134, 195–205. [Google Scholar] [CrossRef]
- Wang, C.; Sun, W.; Wang, X.; Jin, Y.; Zhao, S.; Luo, M.; Tong, S. Large-scale separation of baicalin and wogonoside from Scutellaria baicalensis Georgi by the combination of pH-zone-refining and conventional counter-current chromatography. J. Chromatogr. A 2019, 1601, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y. pH-zone-refining counter-current chromatography: Origin, mechanism, procedure and applications. J. Chromatogr. A 2013, 1271, 71–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, G.L.; Kim, H.Y.; Guillen Quispe, Y.N.; Wang, Z.Q.; Hwang, S.H.; Shin, K.O.; Lim, S.S. Efficient Separation of Phytochemicals from Muehlenbeckia volcanica (Benth.) Endl. by Polarity-Stepwise Elution Counter-Current Chromatography and Their Antioxidant, Antiglycation, and Aldose Reductase Inhibition Potentials. Molecules 2021, 26, 224. [Google Scholar] [CrossRef]
- Yang, Y.; Yin, C.; Huo, F.; Chao, J.; Zhang, Y.; Jin, S. Simple 1, 8-diaminonaphthalene-based fluorescence chemosensor for hypochlorites and its practical application. Sen. Actuators B Chem. 2014, 199, 226–231. [Google Scholar] [CrossRef]
- Ramirez, M.A.; Borja, N.L. Epalrestat: An aldose reductase inhibitor for the treatment of diabetic neuropathy. Pharmacotherapy 2008, 28, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhu, L.Y.; Shen, H.; Zhang, H.M.; Jia, X.B.; Yan, R.; Li, S.L.; Xu, H.X. A critical view on spike recovery for accuracy evaluation of analytical method for medicinal herbs. J. Pharm. Biomed. Anal. 2012, 62, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Marston, A.; Hostettmann, K. Developments in the application of counter-current chromatography to plant analysis. J. Chromatogr. A 2006, 1112, 181–194. [Google Scholar] [CrossRef]
- Wan, C.; Li, S.; Liu, L.; Chen, C.; Fan, S. Caffeoylquinic acids from the aerial parts of Chrysanthemum coronarium L. Plants 2017, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Abdel Motaal, A.; Ezzat, S.M.; Tadros, M.G.; El-Askary, H.I. In vivo anti-inflammatory activity of caffeoylquinic acid derivatives from Solidago virgaurea in rats. Pharm. Biol. 2016, 54, 2864–2870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nawal, H.; Atta, E. Cytotoxic and Antioxidant Activity of Marrubium Vulgare and Its Flavonoid Constituents. In Proceedings of the 2nd International Conference on Chemical, Environmental and Biological Sciences, Dubai, United Arab Emirates, 17–18 March 2013; pp. 40–42. [Google Scholar]
- Al-Attraqchi, O.H.; Deb, P.K.; Al-Attraqchi, N.H.A. Review of the Phytochemistry and Pharmacological Properties of Valeriana officinalis. Curr. Tradit. Med. 2020, 6, 260–277. [Google Scholar] [CrossRef]
- Jugran, A.K.; Rawat, S.; Bhatt, I.D.; Rawal, R.S. Valeriana jatamansi: An herbaceous plant with multiple medicinal uses. Phytother. Res. 2019, 33, 482–503. [Google Scholar] [CrossRef]
- Dhiman, B.; Sharma, P.; Pal, P.K. Biology, chemical diversity, agronomy, conservation and industrial importance of Valeriana jatamansi: A natural sedative. J. Appl. Res. Med. Arom. Plants 2020, 16, 100243. [Google Scholar] [CrossRef]
- Wang, P.C.; Ran, X.H.; Luo, H.R.; Ma, Q.Y.; Liu, Y.Q.; Zhou, J.; Zhao, Y.X. Phenolic compounds from the roots of Valeriana officinalis var. latifolia. J. Braz. Chem. Soc. 2013, 24, 1544–1548. [Google Scholar]
- Sundaresan, N.; Ilango, K. Review on Valeriana Species-Valeriana wallichii and Valeriana jatamansi. J. Pharm. Sci. Res. 2018, 10, 2697–2701. [Google Scholar]
- González-Trujano, M.E.; Contreras-Murillo, G.; López-Najera, C.A.; Hidalgo-Flores, F.J.; Navarrete-Castro, A.; Sánchez, C.G.; Magdaleno-Madrigal, V.M. Anticonvulsant activity of Valeriana edulis roots and valepotriates on the pentylenetetrazole-induced seizures in rats. J. Ethnopharmacol. 2020, 265, 113299. [Google Scholar] [CrossRef]
- Chen, H.W.; Wei, B.-J.; He, X.H.; Liu, Y.; Wang, J. Chemical Components and Cardiovascular Activities of Valeriana spp. Evid. Based Complement. Altern. Med. 2015, 2015, 947619. [Google Scholar] [CrossRef] [Green Version]
- Nishiya, K.; Kimura, T.; Takeya, K.; Itokawa, H. Sesquiterpenoids and iridoid glycosides from Valeriana fauriei. Phytochemistry 1992, 31, 3511–3514. [Google Scholar] [CrossRef]
- Indy Tamayose, C.; Dos Santos, E.A.; Roque, N.; Costa-Lotufo, L.V.; Pena Ferreira, M.J. Caffeoylquinic acids: Separation method, antiradical properties and cytotoxicity. Chem. Biodivers. 2019, 16, e1900093. [Google Scholar] [CrossRef]
- Li, X.; Li, K.; Xie, H.; Xie, Y.; Li, Y.; Zhao, X.; Jiang, X.; Chen, D. Antioxidant and cytoprotective effects of the Di-O-Caffeoylquinic acid family: The mechanism, structure–activity relationship, and conformational effect. Molecules 2018, 23, 222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakajima, Y.; Shimazawa, M.; Mishima, S.; Hara, H. Water extract of propolis and its main constituents, caffeoylquinic acid derivatives, exert neuroprotective effects via antioxidant actions. Life Sci. 2007, 80, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, N.P.; Salva, T.d.J.G.; Bragagnolo, N. Influence of coffee genotype on bioactive compounds and the in vitro capacity to scavenge reactive oxygen and nitrogen species. J. Agric. Food Chem. 2015, 63, 4815–4826. [Google Scholar] [CrossRef] [PubMed]
- Podrez, E.A.; Abu-Soud, H.M.; Hazen, S.L. Myeloperoxidase-generated oxidants and atherosclerosis. Free Radic. Biol. Med. 2000, 28, 1717–1725. [Google Scholar] [CrossRef]
- Green, P.S.; Mendez, A.J.; Jacob, J.S.; Crowley, J.R.; Growdon, W.; Hyman, B.T.; Heinecke, J.W. Neuronal expression of myeloperoxidase is increased in Alzheimer’s disease. J. Neurochem. 2004, 90, 724–733. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, K.; Das, S.; Aich, K.; Goswami, S.; Chowdhury, S.; Bhattacharyya, M. A novel sensor to estimate the prevalence of hypochlorous (HOCl) toxicity in individuals with type 2 diabetes and dyslipidemia. Clin. Chim. Acta 2016, 458, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Kurata, R.; Yahara, S.; Yamakawa, O.; Yoshimoto, M. Simple high-yield purification of 3, 4, 5-tri-O-caffeoylquinic acid from sweetpotato (Ipomoea batatas L.) leaf and its inhibitory effects on aldose reductase. Food Sci. Technol. Res. 2011, 17, 87–92. [Google Scholar] [CrossRef] [Green Version]
- Panahi, Y.; Khalili, N.; Sahebi, E.; Namazi, S.; Karimian, M.S.; Majeed, M.; Sahebkar, A. Antioxidant effects of curcuminoids in patients with type 2 diabetes mellitus: A randomized controlled trial. Inflammopharmacology 2017, 25, 25–31. [Google Scholar] [CrossRef]
- Gomes, M.B.; Negrato, C.A. Alpha-lipoic acid as a pleiotropic compound with potential therapeutic use in diabetes and other chronic diseases. Diabetol. Metab. Synd. 2014, 6, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, B.; Gupta, S.K.; Nag, T.C.; Srivastava, S.; Saxena, R. Green tea prevents hyperglycemia-induced retinal oxidative stress and inflammation in streptozotocin-induced diabetic rats. Ophthalmic Res. 2012, 47, 103–108. [Google Scholar] [CrossRef]
- Ding, M.; Bhupathiraju, S.N.; Chen, M.; van Dam, R.M.; Hu, F.B. Caffeinated and decaffeinated coffee consumption and risk of type 2 diabetes: A systematic review and a dose-response meta-analysis. Diabetes Care 2014, 37, 569–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, M.R.; Kunimasa, K.; Kumazawa, S.; Nakayama, T.; Kaji, K.; Uto, Y.; Hori, H.; Nagasawa, H.; Ohta, T. Correlation between antiangiogenic activity and antioxidant activity of various components from propolis. Mol. Nutr. Food Res. 2009, 53, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zuo, G.L.; Wang, C.Y.; Kim, H.Y.; Lim, S.S.; Tong, S.Q. An Off-Line DPPH-GC-MS Coupling Countercurrent Chromatography Method for Screening, Identification, and Separation of Antioxidant Compounds in Essential Oil. Antioxidants 2020, 9, 702. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Hwang, S.H.; Lim, S.S. Characterization of DHDP, a novel aldose reductase inhibitor isolated from Lysimachia christinae. J. Funct. Food. 2017, 37, 241–248. [Google Scholar] [CrossRef]
- Wang, Z.; Quispe, Y.N.G.; Hwang, S.H.; Zuo, G.; Lim, S.S. Pistafolin B is the major aldose reductase inhibitor of the pods of tara [Caesalpinia spinose (Molina) Kuntze]. Ind. Crops Prod. 2018, 122, 709–715. [Google Scholar] [CrossRef]
- Wang, Z.; Shen, S.; Cui, Z.; Nie, H.; Han, D.; Yan, H. Screening and isolating major aldose reductase inhibitors from the seeds of evening primrose (Oenothera biennis). Molecules 2019, 24, 2709. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Li, L.; Chen, S.; Xu, Y.; Xia, Q.; Guo, Y.; Liu, X.; Tang, Y.; Zhang, T.; Chen, Y. Identification of inhibitors of the antibiotic-resistance target New Delhi metallo-β-lactamase 1 by both nanoelectrospray ionization mass spectrometry and ultrafiltration liquid chromatography/mass spectrometry approaches. Anal. Chem. 2013, 85, 7957–7965. [Google Scholar] [CrossRef]
- Zhang, H.; Xu, C.; Tian, Q.; Zhang, Y.; Zhang, G.; Guan, Y.; Tong, S.; Yan, J. Screening and characterization of aldose reductase inhibitors from Traditional Chinese medicine based on ultrafiltration-liquid chromatography mass spectrometry and in silico molecular docking. J. Ethnopharmacol. 2021, 264, 113282. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, C.; Yu, M.; Zhang, Z.; Qi, Y.; Wang, J.; Wu, G.; Li, S.; Yu, J.; Hu, Y. Application of accelerated solvent extraction coupled with high-performance counter-current chromatography to extraction and online isolation of chemical constituents from Hypericum perforatum L. J. Chromatogr. A 2011, 1218, 2827–2834. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.; Yan, J.; Guan, Y.X. Preparative separation of isomeric caffeoylquinic acids from Flos Lonicerae by pH-zone-refining counter-current chromatography. J. Chromatogr. A 2008, 1212, 48–53. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample a | HOCl Scavenging Activity | AR Inhibitory Activity | ||||
|---|---|---|---|---|---|---|
| Concentration (µg/mL) | Inhibition (%) b | IC50 c (µg/mL) | Concentration (µg/mL) | Inhibition (%) | IC50 (µg/mL) | |
| Extract | 25 | 67.00 ± 2.65 | 16.52 | 0.625 | 61.45 ± 2.34 b | 0.478 |
| 12.5 | 42.74 ± 1.58 | 0.313 | 36.25 ± 0.63 | |||
| 6.25 | 28.38 ± 2.25 | 0.156 | 26.47 ± 3.02 | |||
| Trolox | 25 | 97.07 ± 5.45 | 8.52 | - | - | - |
| 12.5 | 66.53 ± 2.54 | - | - | |||
| 6.25 | 40.76 ± 0.39 | - | - | |||
| Quercetin | - d | - | - | 7.556 | 67.05 ± 3.22 | 4.536 |
| - | - | 3.778 | 45.98 ± 4.49 | |||
| - | - | 1.889 | 34.78 ± 0.51 | |||
| Quercitrin | - | - | - | 0.112 | 74.57 ± 1.11 | 0.046 |
| - | - | 0.056 | 62.33 ± 0.44 | |||
| - | - | 0.028 | 32.57 ± 2.70 | |||
| Epalrestat | - | - | - | 0.040 | 91.81 ± 1.84 | 0.016 |
| - | - | 0.020 | 79.28 ± 1.35 | |||
| - | - | 0.010 | 21.66 ± 1.25 | |||
| Solvent System (v/v) | Addition of Acid or Base | K(upper/lower) Value | ||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | ||
| EtOAc/H2O, 1:1 | 208 mM formic acid | 0.49 | 0.95 | 1.07 | 3.49 | 7.71 | 12.24 | 23.71 |
| 30 mM ammonia | 0.02 | 0.01 | 0.02 | 0.02 | 0.01 | 0.01 | 0.02 | |
| EtOAc/n-BuOH/H2O, 4:1:5 | 208 mM formic acid | 1.36 | 2.43 | 3.05 | 4.86 | 14.23 | 22.17 | 27.56 |
| 30 mM ammonia | 0.00 | 0.01 | 0.01 | 0.02 | 0.01 | 0.01 | 0.03 | |
| EtOAc/n-BuOH/H2O, 3:2:5 | 208 mM formic acid | 2.03 | 5.48 | 4.76 | 16.14 | 35.91 | 41.13 | 26.44 |
| 30 mM ammonia | 0.05 | 0.03 | 0.05 | 0.03 | 0.01 | 0.05 | 0.22 | |
| EtOAc/n-BuOH/H2O, 2:3:5 | 208 mM formic acid | 2.49 | 6.86 | 5.22 | 20.24 | 38.56 | 45.95 | 29.51 |
| 30 mM ammonia | 0.01 | 0.01 | 0.01 | 0.04 | 0.12 | 0.13 | 0.40 | |
| HSCCC System (v/v) | K(upper/lower) Value | |
|---|---|---|
| 1 | 3 | |
| n-BuOH/H2O, 1:1 | 0.04 | 0.13 |
| n-BuOH/H2O, 1:1 + 8.7 mM acetic acid | 0.73 | 1.57 |
| Sample | TE (µmol Trolox Equivalents Per µmol Compound) | ||
|---|---|---|---|
| DPPH | ABTS | ORAC | |
| 3-CQA (1) | 0.26 ± 0.00 e | 0.28 ± 0.02 f | 1.47 ± 0.10 c |
| 4-CQA (3) | 0.83 ± 0.05 c | 0.84 ± 0.02 d | 3.54 ± 0.11 a |
| 5-CQA (2) | 0.49 ± 0.01 d | 0.58 ± 0.03 e | 2.21 ± 0.04 ab |
| 3,4-diCQA (4) | 1.09 ± 0.09 b | 1.06 ± 0.06 c | 3.26 ± 0.10 a |
| 3,5-diCQA (5) | 0.86 ± 0.08 c | 0.89 ± 0.05 d | 2.13 ± 0.07 b |
| 4,5-diCQA (6) | 1.23 ± 0.07 a | 1.22 ± 0.03 b | 3.70 ± 0.34 a |
| 3,4,5-triCQA (7) | 1.28 ± 0.04 a | 1.31 ± 0.05 a | 1.95 ± 0.15 b |
| Sample a | HOCl Scavenging Activity | AR inhibitory Activity | ||||
|---|---|---|---|---|---|---|
| Concentration (µM) | Inhibition (%) b | IC50 c (µM) | Concentration (µM) | Inhibition (%) b | IC50 (µM) | |
| 3-CQA (1) | 100 | 70.79 ± 2.40 | 68.73 | 6.25 | 60.06 ± 1.22 | 7.80 |
| 50 | 38.98 ± 1.33 | 3.125 | 45.84 ± 1.75 | |||
| 25 | 18.77 ± 0.92 | 1.5625 | 29.35 ± 3.40 | |||
| 4-CQA (3) | 100 | 84.70 ± 2.82 | 58.13 | 6.25 | 54.23 ± 2.18 | 4.83 |
| 50 | 47.18 ± 1.01 | 3.125 | 42.88 ± 3.09 | |||
| 25 | 18.64 ± 1.72 | 1.5625 | 30.95 ± 2.76 | |||
| 5-CQA (2) | 100 | 91.90 ± 0.55 | 58.80 | 1.563 | 63.81 ± 2.55 | 0.91 |
| 50 | 41.27 ± 1.08 | 0.781 | 44.21 ± 1.17 | |||
| 25 | 15.40 ± 1.47 | 0.391 | 30.80 ± 2.18 | |||
| 3,4-diCQA (4) | 12.5 | 59.15 ± 1.19 | 5.78 | 0.391 | 68.67 ± 0.68 | 0.22 |
| 6.25 | 50.28 ± 0.19 | 0.195 | 50.40 ± 3.34 | |||
| 3.125 | 46.68 ± 2.87 | 0.098 | 18.86 ± 2.43 | |||
| 3,5-diCQA (5) | 12.5 | 62.86 ± 1.23 | 2.93 | 0.391 | 75.60 ± 1.33 | 0.15 |
| 6.25 | 56.13 ± 3.01 | 0.195 | 62.33 ± 1.33 | |||
| 3.125 | 49.56 ± 1.23 | 0.098 | 35.37 ± 1.33 | |||
| 4,5-diCQA (6) | 12.5 | 65.80 ± 4.06 | 4.61 | 0.391 | 65.87 ± 2.21 | 0.23 |
| 6.25 | 51.23 ± 2.14 | 0.195 | 44.36 ± 4.45 | |||
| 3.125 | 48.80 ± 5.62 | 0.098 | 23.73 ± 2.70 | |||
| 3,4,5-triCQA (7) | 25 | 84.32 ± 3.39 | 11.68 | 0.781 | 69.85 ± 0.77 | 0.40 |
| 12.5 | 52.93 ± 1.02 | 0.391 | 49.73 ± 0.94 | |||
| 6.25 | 35.39 ± 1.17 | 0.195 | 27.56 ± 2.43 | |||
| Acacetin (8) | - d | - | - | 100 | 36.30 ± 3.78 | - |
| - | - | 50 | 24.00 ± 0.63 | |||
| - | - | 25 | 12.30 ± 4.22 | |||
| Trolox | 100 | 97.07 ± 5.45 | 34.03 | - | - | - |
| 50 | 66.53 ± 2.54 | - | - | |||
| 25 | 40.76 ± 0.39 | - | - | |||
| Quercetin | - | - | - | 25 | 67.05 ± 3.22 | 15.01 |
| - | - | 12.5 | 45.98 ± 4.49 | |||
| - | - | 6.25 | 34.78 ± 0.51 | |||
| Quercitrin | - | - | - | 0.25 | 74.57 ± 1.11 | 0.10 |
| - | - | 0.125 | 62.33 ± 0.44 | |||
| - | - | 0.063 | 32.57 ± 2.70 | |||
| Epalrestat | - | - | - | 0.125 | 91.81 ± 1.84 | 0.05 |
| - | - | 0.063 | 79.28 ± 1.35 | |||
| - | - | 0.031 | 21.66 ± 1.25 | |||
| Parameter | Compound | |||
|---|---|---|---|---|
| 3,5-diCQA (5) | 4,5-diCQA (6) | Acacetin (8) | ||
| Calibration curve (n = 3), r2 | y = 0.184x − 0.0692, r2 = 0.9996 | y = 0.1297x − 0.4051, r2 = 0.9991 | y = 0.4122x + 0.9041, r2 = 0.9987 | |
| Linear range (µg/mL) | 12.50–400.00 | 12.50–400.00 | 6.25–400.00 | |
| LOD (µg/mL, S/N = 3) | 0.05 | 0.06 | 0.20 | |
| LOQ (µg/mL, S/N = 10) | 0.17 | 0.20 | 0.65 | |
| Precision on the day in relative standard deviation, % (n = 6) | 50.00 µg/mL | 8.70 | 6.64 | 6.45 |
| 200.00 µg/mL | 7.60 | 5.80 | 6.24 | |
| Precision between days in relative standard deviation, % (n = 3) | 50.00 µg/mL | 3.30 | 4.73 | 4.62 |
| 200.00 µg/mL | 7.40 | 4.10 | 3.57 | |
| Analyte | C0 (µg/mL) | Vo (mL) | C1 (µg/mL) | V1 (mL) | C2 (µg/mL) | V2 (mL) | Recovery (%) | Content (mg/g) |
|---|---|---|---|---|---|---|---|---|
| 3,5-diCQA (5) | 100 | 0.2 | 66.47 ± 3.95 | 0.2 | 82.89 ± 3.80 | 0.4 | 99.32 ± 7.60 | 66.47 ± 3.95 |
| 300 | 0.2 | 66.47 ± 3.95 | 0.2 | 184.62 ± 6.54 | 0.4 | 100.93 ± 4.36 | ||
| 4,5-diCQA (6) | 100 | 0.2 | 112.95 ± 2.63 | 0.2 | 107.08 ± 3.44 | 0.4 | 101.21 ± 6.88 | 112.95 ± 2.63 |
| 300 | 0.2 | 112.95 ± 2.63 | 0.2 | 206.16 ± 5.85 | 0.4 | 99.79 ± 3.90 | ||
| Acacetin (8) | 100 | 0.2 | 23.26 ± 0.85 | 0.2 | 60.69 ± 2.83 | 0.4 | 98.11 ± 5.66 | 23.26 ± 0.85 |
| 300 | 0.2 | 23.26 ± 0.85 | 0.2 | 155.63 ± 6.20 | 0.4 | 96.00 ± 4.13 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zuo, G.; Kim, H.-Y.; Guillen Quispe, Y.N.; Wang, Z.; Kim, K.-H.; Gonzales Arce, P.H.; Lim, S.-S. Valeriana rigida Ruiz & Pav. Root Extract: A New Source of Caffeoylquinic Acids with Antioxidant and Aldose Reductase Inhibitory Activities. Foods 2021, 10, 1079. https://doi.org/10.3390/foods10051079
Zuo G, Kim H-Y, Guillen Quispe YN, Wang Z, Kim K-H, Gonzales Arce PH, Lim S-S. Valeriana rigida Ruiz & Pav. Root Extract: A New Source of Caffeoylquinic Acids with Antioxidant and Aldose Reductase Inhibitory Activities. Foods. 2021; 10(5):1079. https://doi.org/10.3390/foods10051079
Chicago/Turabian StyleZuo, Guanglei, Hyun-Yong Kim, Yanymee N. Guillen Quispe, Zhiqiang Wang, Kang-Hyuk Kim, Paul H. Gonzales Arce, and Soon-Sung Lim. 2021. "Valeriana rigida Ruiz & Pav. Root Extract: A New Source of Caffeoylquinic Acids with Antioxidant and Aldose Reductase Inhibitory Activities" Foods 10, no. 5: 1079. https://doi.org/10.3390/foods10051079