Identification of miRNAs Associated with Graft Union Development in Pecan [Carya illinoinensis (Wangenh.) K. Koch]

1
Co-Innovation Center for the Sustainable Forestry in Southern China, Nanjing Forestry University, Nanjing 210037, China
2
Institute of Botany, Jiangsu Province and Chinese Academy of Sciences, Nanjing 210014, China
3
Green Universe Pecan Science and Technology Co., Ltd., 38 Muxuyuan Street, Nanjing 210007, China
*
Author to whom correspondence should be addressed.
Forests 2018, 9(8), 472; https://doi.org/10.3390/f9080472
Submission received: 23 May 2018
/
Revised: 14 July 2018
/
Accepted: 31 July 2018
/
Published: 3 August 2018
(This article belongs to the Special Issue Genetic and Morphological Variation in Tropical and Temperate Plant Species)
Abstract
:Pecan [Carya illinoinensis (Wangenh.) K. Koch] is a high-value fruit tree with a long juvenile period. The fruiting process of pecan seedlings can be largely accelerated through grafting. As non-coding small RNAs, plant miRNAs participate in various biological processes through negative regulation of gene expression. To reveal the roles of miRNAs in the graft union development of pecan, four small RNA libraries were constructed from the graft union at days 0, 8, 15, and 30 after grafting. A total of 47 conserved miRNAs belonging to 31 families and 39 novel miRNAs were identified. For identified miRNAs, 584 target genes were bioinformatically predicted, and 266 of them were annotated; 29 miRNAs (including 16 conserved and 13 novel miRNAs) were differentially expressed during the graft process. The expression profiles of 12 miRNA were further validated by quantitative reverse transcription PCR (qRT-PCR). In addition, qRT-PCR revealed that the expression levels of 3 target genes were negatively correlated with their corresponding miRNAs. We found that miRS26 might be involved in callus formation; miR156, miR160, miR164, miR166, and miRS10 might be associated with vascular bundle formation. These results indicate that the miRNA-mediated gene regulations play important roles in the graft union development of pecan.
1. Introduction
Grafting, as an asexual propagation technology, has been applied extensively in fruit trees to aid the adaptation of scion cultivars to potentially disadvantageous soil and climatic conditions, avoid the juvenile period, increase productivity, and improve quality [1]. Successful grafting is a complicated process that involves the initial adhesion of rootstock and scion, callus formation, and vascular connection at the graft union [2]. It has been reported that phytohormones (especially auxin) and antioxidant enzymes are important players during graft union development [3,4,5,6]. At the molecular level, a successful graft is controlled by numerous genes in plants, especially for the genes involved in hormone signaling. cDNA amplified fragment length polymorphism (AFLP) analysis of graft union in hickory [Carya tomentosa (Lam.) Nutt.] indicated that some genes related to signal transduction, metabolism, auxin transportation, wound response, cell cycle, and cell wall synthesis were responsive to grafting [7]. In Arabidopsis, genes involved in hormone signaling, wounding, and cellular debris clearing were induced during graft union development [8]. In grapevine, graft union formation activated the differential expression of genes participated in secondary metabolism, cell wall modification, and signaling [9]. Transcriptomic analysis of graft union in Litchi chinensis Sonn. revealed that 9 unigenes annotated in auxin signaling had higher expression levels in the compatible grafts compared with the incompatible ones [10].
miRNAs, a category of non-coding RNAs with approximate 22 nucleotides (nt), are critical regulatory molecules of gene expression; they induce either post-transcriptional degradation or translational inhibition of their target mRNAs [11]. In plants, miRNAs bind to their target mRNA sequences with perfect or near-perfect complementarity, and negatively regulate gene expression mainly via targeted cleavage [12]. The binding sites of plant miRNAs are almost exclusively located within the open reading frames of their target genes [13]. In recent years, with the development of second-generation sequencing technology, miRNAs could be identified from non-model plants [14,15]. Numerous studies have suggested that miRNAs play regulatory roles in plant resistance to biotic and abiotic stresses, such as cold [16], heat [17], and virus infection [18]. In addition, miRNAs have been confirmed to participate in various development processes [19,20,21]. In grafted plants, miRNA has been reported to be involved in the regulation of scion and rootstock interaction. In watermelon cultivation, grafting is commonly used to increase resistance to environmental stresses. With high-throughput sequencing, Liu et al. [22] found that miRNAs would differentially express in grafted watermelon to regulate plant adaptation to stresses. Li et al. [23] identified grafted-responsive miRNAs in cucumber/pumpkin, pumpkin/cucumber heterografts, and found that miRNAs were involved in regulating physiological process of hetrografts. Khaldun et al. [24] investigated the expression profiles of miRNAs within a distant grafting of tomato/goji, and the result showed that when compared with tomato autografts, tomato/goji heterografts had 43 and 163 differently expressed miRNAs in shoot and fruit, respectively. Although mounting evidence indicates the involvement of miRNAs in scion-rootstock interactions, there was only one published report concerning the functions of miRNAs which participate in the graft process, which was presented in hickory [25].
Pecan [Carya illinoinensis (Wangenh.) K. Koch] is an economic nut tree which belongs to the family Juglandaceae and genus Carya. It has been widely planted in China in recent years. As a woody plant, the juvenile phase of pecan seedlings is very long, lasting about 10 years. To accelerate the fruit bearing process, grafting is widely used in the cultivation of pecan, by which, trees can begin to produce fruit in 5–7 years. In industrial pecan cultivation, grafting success rate is very low; 75% grafting success is considered to be good [26]. Nowadays, in China, using the graft technique of patch budding can sometimes achieve a grafting success of 90% for some cultivars of pecan, such as ‘Pawnee’, ‘Stuart’, and ‘Shaoxing’. However, a low grafting success rate still exists in some cultivars, such as ‘Mahan’ and ‘Jinhua’. To improve the graft survival rate of industrial pecan, a better understanding of the mechanism associated with the graft union development is needed. In our previous studies, morphological, proteomic, and transcriptomic analyses have been conducted in the graft process of pecan [27,28]. In this work, we investigated miRNA expression during the graft process of pecan using RNA-sequencing technology. Four small RNA libraries from the graft union collected at different time points (days 0, 8, 15, and 30 after grafting) were constructed, and the differentially expressed miRNAs were analyzed. Subsequently, the potential roles of these miRNAs and their target genes were discussed.
2. Materials and Methods
2.1. Plant Materials
Pecan homograft was performed through patch grafting in August at the experimental farm at Nanjing Forestry University. The pecan cultivar ‘Pawnee’ was used as scion, and one-year-old seedlings propagated from pecan seeds were used as rootstock. Based on our morphological observation of graft union development, samples from the graft unions (approximately 5 mm in length, the budding segment that includes the tissues of scion, and the developing xylem of rootstock) were collected at day 0 (ungrafted materials, and were used as control), day 8 (the stage of initial callus proliferation), day 15 (the stage of massive callus proliferation along with cambium establishment), and day 30 (the stage of vascular bundles formation). For each sample, three different graft unions were pooled and frozen in liquid nitrogen immediately, and then stored at −80 °C until required for use.
2.2. RNA Extraction and Deep Sequencing of Small RNA
Total RNA was isolated from graft unions at four time points using the trizol reagent (Invitrogen, Carlsbad, CA, USA), following the manufacturer’s instructions, and then digested with RNA-free DNase I (Takara, Kyoto, Japan) to degrade genomic DNA. Sequencing libraries were constructed by NEBNext® UltraTM small RNA Sample Library Prep Kit for Illumina® (NEB, Boston, MA, USA) according to the protocol. Briefly, approximately 1.5 μg RNA was ligated to 5′ and 3′ adapter by T4 RNA ligase for each of the samples. Next, reverse transcription synthetic first chain and PCR amplification was conducted. The resulting PCR products were subjected to polyacrylamide gel electrophoresis, and the 140–160 bp fragments were screened for sequencing. The sequencing raw data was deposited in the NCBI Sequence Read Archive (SRA) with the accession number SRP131300.
2.3. Sequence Analysis and Target Prediction of Pecan miRNA
Following sequencing, raw reads of the four libraries were processed through in-house Perl scripts. In this step, clean reads were obtained by removing low-quality reads and trimming adapter sequences. Reads smaller than 18 nt or longer than 30 nt were also abandoned. By using Bowtie software, clean reads with 18–30 nt in length were subsequently blasted against the Rfam (http://www.sanger.ac.uk/software/Rfam) and Repbase databases (http://www.girinst.org/) to filter rRNA, tRNA, snRNA, snoRNA, other ncRNA and repeats. The remaining sequences were aligned with the miRBase 21.0 database (http://www.mirbase.org/index.shtml) to identify putative conserved miRNAs, allowing no more than two mismatches. The remaining non-annotated reads were mapped to the pecan graft union development’s transcriptome data (accession number SRP118757 and GGRT00000000 in NCBI database) to predict potential novel miRNAs by miRDeep2. The criteria for novel miRNA identification was as follows: (1) miRNA precursors could form hairpin-like structures; (2) miRNA should have a corresponding miRNA * in sequencing data, and the two could form a duplex with 2 nt 3′ overhangs; (3) in miRNA *-deficient cases, candidate miRNAs should derive from multiple and independent libraries [29]. The secondary structures of novel miRNAs were predicted by Randfold software. Putative targets of miRNA were predicted by TargetFinder, and then annotated based on the databases of Nr (NCBI non-redundant protein sequences), Protein family (Pfam) and GO (Gene Ontology). The expression value of putative target genes were obtained from the supplementary materials of our previously published paper (https://www.mdpi.com/2073-4425/9/2/71/s1) [27].
2.4. Analysis of Differentially Expressed miRNAs
To calculate the expression levels of miRNAs in four libraries, miRNA counts were first normalized as transcripts per million (TPM) using the following formula: TPM = mapped read count/total reads × 106. Fold changes of miRNA in three comparisons (day 8/day 0, day 15/day 0, and day 30/day 0) were analyzed by IDEG6, and the miRNA were considered to be differentially expressed with the corrected p value (q value) < 0.05 and absolute log2 fold change >1.
2.5. Quantitative Real-Time PCR (qRT-PCR)
To validate the expression profiles of miRNAs, graft unions were collected at days 0, 8, 15, and 30 after grafting, with three biological repetitions. miRNAs were isolated by the Universal Plant microRNA Kit (BioTeke, Beijing, China). The subsequent reverse transcription and real-time PCR were carried out using BioTeke miRNA First Strand cDNA synthesis kit (BioTeke, Beijing, China) and BioTeke miRNA qPCR Detection Kit (BioTeke, Beijing, China), respectively. For target gene detection, total RNAs were extracted from the same samples, as mentioned above. First-strand cDNA synthesis and the following real-time qPCR were conducted by Prime-Script™ II First Strand cDNA synthesis kit (Takara, Dalian, China) and SYBR Premix Ex Taq™ II kit (Takara, Dalian, China), respectively. Primers were designed based on the sequence of corresponding miRNAs and mRNAs, and were available in Table S1. 5.8S rRNA was chosen as an internal reference for miRNA normalization, while the Actin was used as an endogenous reference for mRNA analysis. All qPCR was run in three technical replicates. The relative expression levels of miRNAs and mRNA were calculated using the comparative 2−ΔΔCt method.
To explore tissue-specific expression, miRNAs and total RNAs were extracted from different organs, including wound-induced calluses, xylem, phloem, and leaves. The qPCR primes are listed in Table S1.
3. Results
3.1. Analysis of Small RNA Sequencing
To identify miRNAs associated with graft union development in pecan, four small RNA libraries were constructed from the graft unions harvested at days 0, 8, 15, and 30 after grafting. Deep sequencing produced 20,691,228, 21,849,708, 22,850,876, and 34,439,863 raw reads for the four libraries, respectively (Table 1). After removing low-quality reads, 17,060,180 (day 0), 19,032,782 (day 8), 19,780,849 (day 15), and 28,632,161 (day 30) clean reads were obtained. Among the clean reads, 6,579,996 (day 0), 6,865,905 (day 8), 6,935,788 (day 15), and 7,815,688 (day 30) reads were unique, and 1,506,852 (day 0), 2,537,261 (day 8), 1,759,479 (day 15), and 2,290,283 (day 30) reads could map to the reference unigene (accession number GGRT00000000 in NCBI database). By aligning to Rfam and Repbase database, clean reads were classified into rRNA, snRNA, snoRNA, tRNA, and repeat-associated sRNA for almost all the libraries, except for day 0, which had no snRNA. The remaining unannotated reads were used for conserved miRNA identification and novel miRNAs predication.
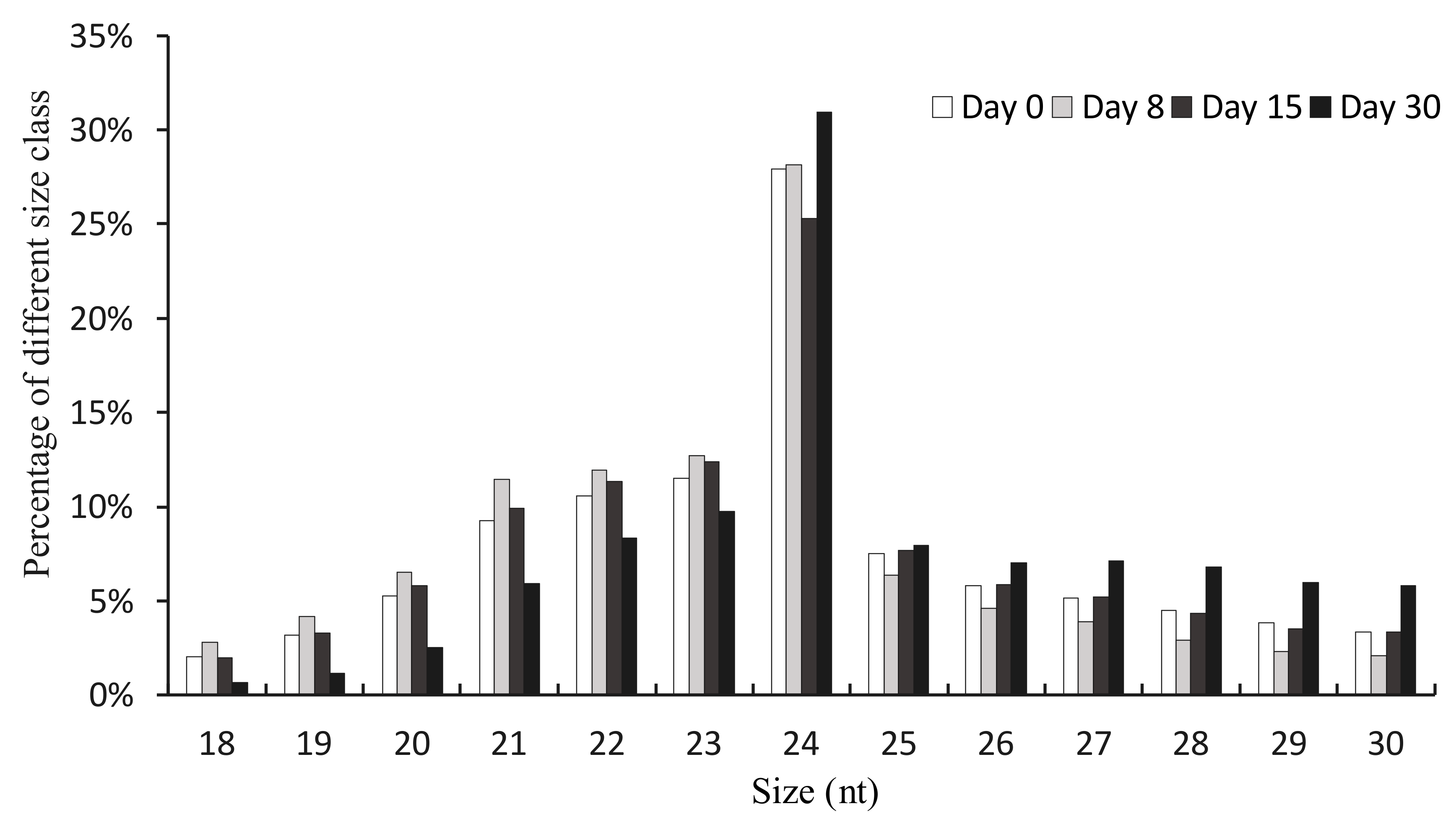
The length distribution of unique clean reads ranging from 18 nt to 30 nt was summarized (Figure 1). We found that the most abundant class was the 24 nt sRNAs, which was consistent with previous studies in hickory [25,30,31]. The second most numerous was 23 nt sRNAs, and the majority of the sRNAs were generally distributed between 21 and 24 nt.
3.2. Identification of Conserved miRNAs in Pecan
To obtain conserved miRNAs in pecan, all unannotated reads in Rfam and Repbase were pooled and used to do a blast against miRbase, allowing two mismatches. Based on miRbase results and hairpin prediction, a total of 47 conserved miRNAs with their corresponding star strands were identified from the four libraries (Table S2). These 47 conserved miRNAs were classified into 31 miRNA families, among them, the miR482 family possessed the maximum members (four), followed by miR166 and miR396, while the remaining families have only one or two members. The 47 miRNAs showed great difference in expression levels, of these, miR159a–b, miR166a–c, and miR319a–b had relatively high expression level, in contrast, members such as miR4998, miR5998, miR6135, miR7504, and miR7717 presented low expression levels.
3.3. Identification of Novel miRNAs in Pecan
To identify novel miRNAs, all the remaining unannotated sRNAs were blasted against our transcriptome data. In total, 39 novel miRNAs corresponding to 39 distinct precursor sequences were obtained from the four libraries (Table S3), and all the precursors of these candidate miRNAs were found to have typical stem-loop structures (Figure S1). Star sequences were detected for all the novel miRNAs, an important evidence of being bona fide miRNAs [29]. The most common base for the first nucleotide of novel miRNAs was Uracil (U), a common pattern observed in other studies [25,32]. The length of these mature miRNAs ranged from 18 nt to 25 nt, and the most common was 24 nt. The range of the minimal free energy (MFE) for these novel miRNA precursors was from −96.9 to −31.8 kcal/mol, with −69.0 kcal/mol on average. The expression level of most novel miRNAs were generally low (TPM < 100), while some miRNAs such as miRS19 and miRS33 presented high level with dynamic TPM > 1000.
3.4. Prediction and Functional Annotation of Target Genes of miRNAs
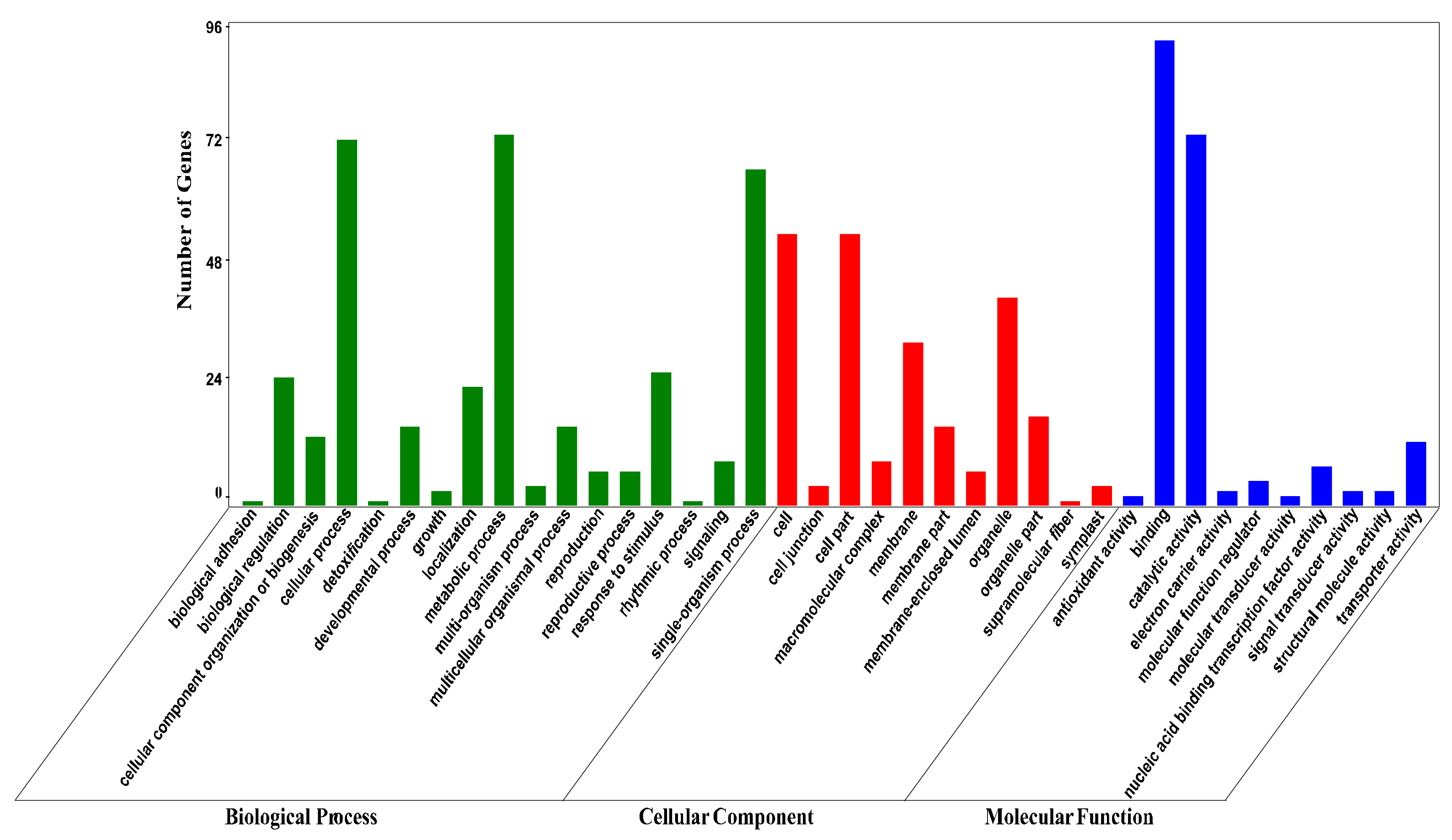
We searched for putative targets by blasting the miRNAs against our transcriptome sequences with perfect or near-perfect complementarity. As a result, a total of 584 targets were predicted for the 86 miRNAs (with an average of 6.8 targets per miRNA), and 266 of them were annotated (Table S4). For functional classification, these targets were subjected to GO (Gene Ontology) analysis. As shown in Figure 2, targets of miRNAs fell into 17 biological processes, with the three most abundant being metabolic process, cellular process, and single-organism process. Targets in the cellular component category were classified into 11 terms, with the three most frequent being cell, cell part, and organelle. With respect to molecular function, targets were assigned to 10 terms, with the two most frequent being binding and catalytic activity.
3.5. Differential Expressed miRNAs during the Graft Process of Pecan
To obtain insight into the possible roles of miRNAs in the graft union development of pecan, differential expressions were analyzed by comparing days 8, 15, 30 to day 0, with the criteria of absolute log2 fold change >1 and q value < 0.05. In total, 29 miRNAs with 16 conserved and 13 novel were considered to be differentially expressed in the three comparisons (Table 2). Of these, 10 miRNAs were differentially expressed in the comparison of day 8/day 0, with 7 down-regulated and 3 up-regulated. Fourteen differential expressed miRNAs were identified in day 15/day 0 comparison, with 4 down-regulated and 10 up-regulated. In the comparison of day 30/day 0, 23 differential expressed miRNAs were found, with 19 down-regulated and 4 up-regulated. There were 10 miRNAs whose expression changed significantly in two comparisons, and 4 miRNAs changed obviously in three comparisons. We compared the differential expressed value between miRNAs and their targets using our transcriptome data, and found that miRNAs were generally negatively correlated with their corresponding targets (Figure 3).
3.6. Differential Expressed miRNAs during the Graft Process of Pecan
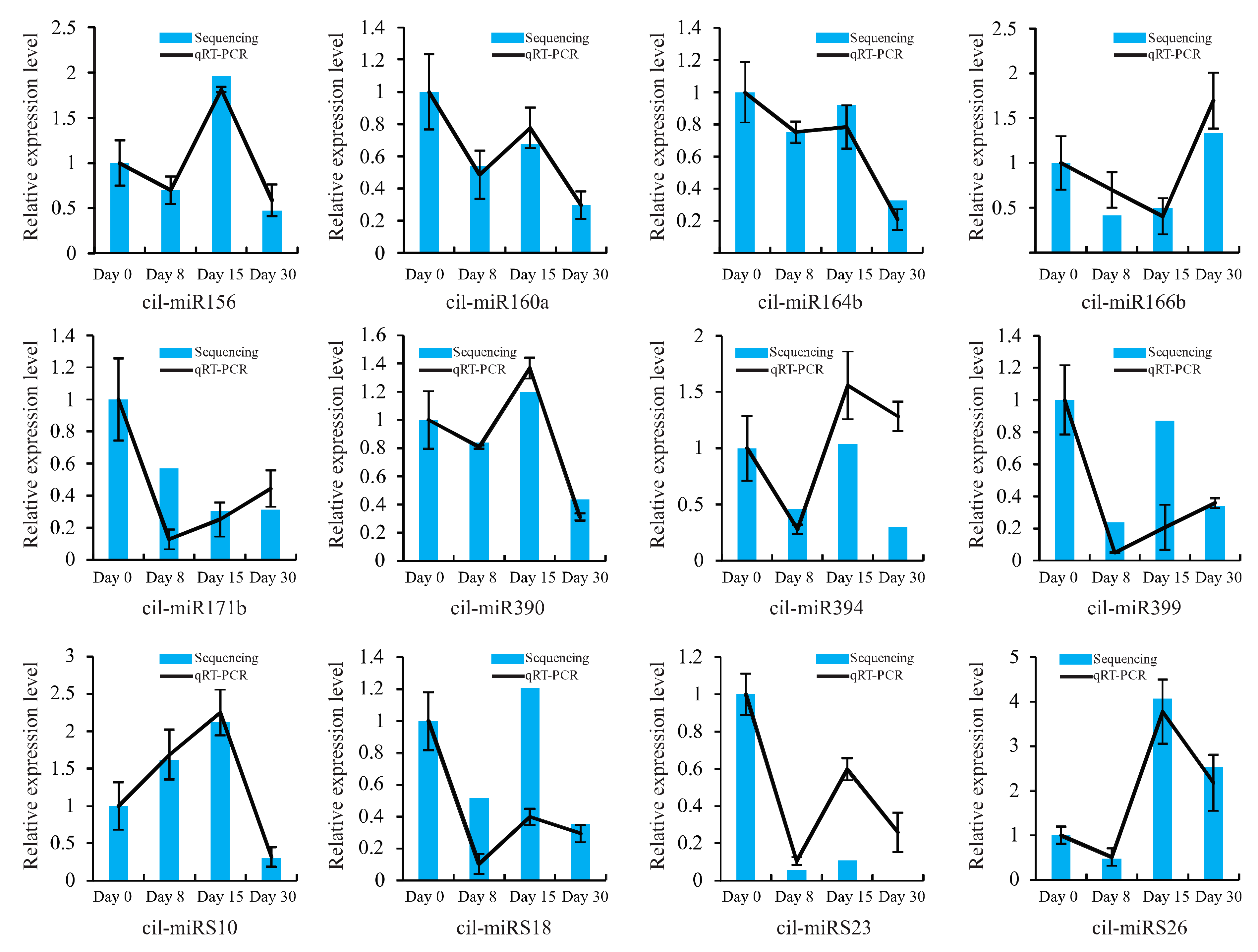
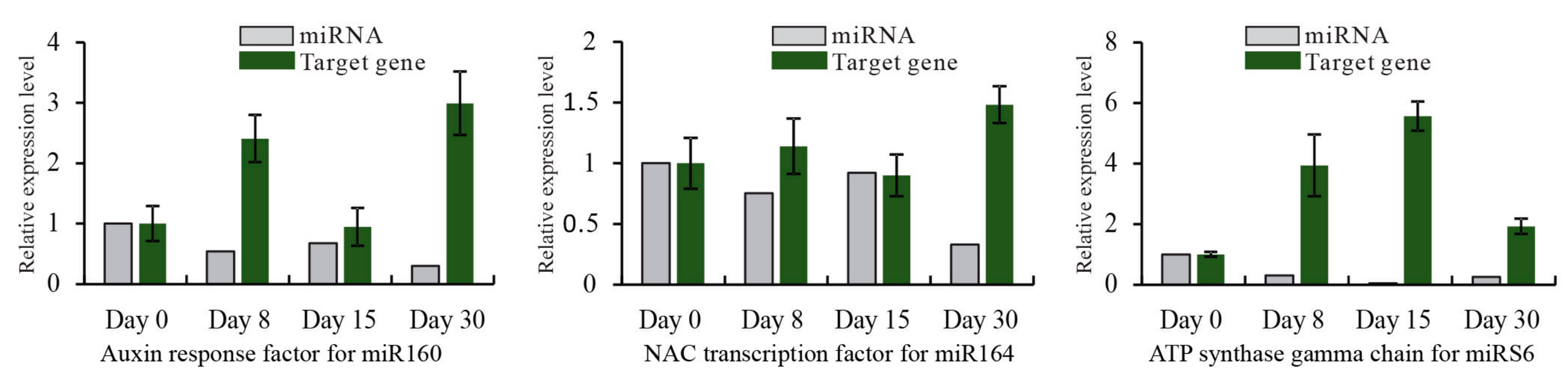
To validate the dynamic expression of miRNAs at different time points after grafting obtained by sequencing, 12 miRNAs, including 8 conserved and 4 novel, were chosen for qRT-PCR analysis (Figure 4). Results showed that most of the expression profiles of studied miRNAs based on qRT-PCR were similar to those detected by high-throughput sequencing, except miR394. The expression of miR394 at day 30 was down-regulated based on high-throughput sequencing, while it was up-regulated detected by qRT-PCR. Also, for specific time points after grafting, the relative expression level of miRNAs detected by these two methods did not match exactly. For instance, sequencing data indicated that the ratio of miRS23 in day 15/day 0 was 0.11, but it was 0.60 with the corresponding qRT-PCR date. This inconsistency might result from the difference in data normalization protocols of the sequencing data and qRT-PCR. The sequencing was normalized to the global abundance of mapped reads sequenced by illumina, while qRT-PCR was normalized to the level of 5.8S rRNA. A correlation analysis of the fold change of miRNA expression between sequencing and qRT-PCR showed a significant similarity with R2 = 0.84 (Figure S2), confirming the reliability of results obtained by sequencing. Additionally, to further validate the dynamic correlation between miRNAs and their targets, the expression of potential targets were also subjected to qRT-PCR assay. Results showed that all the three targets had an inverse expression profile with their corresponding miRNAs (Figure 5).
3.7. Expression Patterns of miRNAs and Their Targets in Different Tissues of Pecan
To understand the main roles of miRNAs and their targets, we analyzed the tissue-specific expression profiles of miRNAs and mRNAs in different organs of pecan. Generally, miRNAs and their targets were negatively correlated, and were preferentially expressed in specific tissues (Figure 6). miR156 showed lower expression level in xylem and phloem, while its target had higher expression values in those tissues. miR160, miR164, miR166, and miRS10 exhibited low expression levels in xylem, while their corresponding targets, expect NAC, were highly expressed in xylem. The target of miRS26 displayed the highest expression in callus.
4. Discussion
Although grafting has been extensively used in horticulture, our knowledge regarding the molecular mechanism of successful graft remains insufficient. Plant miRNAs are non-coding RNAs that play important roles in various biological processes at post-transcriptional level. In this study, we used high throughput sequencing to explore the conserved and novel miRNAs in pecan, and then analyzed the differentially expressed miRNAs to better understand the function of miRNAs in a successful grafting.
miRNAs are reported to be widely distributed throughout almost all eukaryotes, and some miRNAs are deeply conserved in plant kingdom [33]. In our work, 47 conserved miRNAs belonging to 31 miRNA families were identified. Of those, miRNAs including miR156, miR159, miR160, miR164, miR166, miR167, miR171, miR172, miR390, miR393, miR394, miR396, miR399, and miR403 were confirmed to be well-conserved in both monocot and dicot model plants [33]. We obtained a total of 39 novel miRNAs in the graft process of pecan. Those newly identified miRNAs might be pecan-specific. We detected that the novel miRNAs generally exhibited a lower expression level than the conserved ones, which was consistent with previous literature [34,35].
A total of 16 conserved and 13 novel miRNAs were differentially expressed during the graft processes. Since successful grafting is a developmental processes involving callus formation and vascular bundle formation, the differential expression of a cascade of miRNAs concerning those processes might suggest their involvement in the graft process as well. Previously, it has been reported that miR159, miR169, miR171, and miR172 were identified as being responsive to embryogenic callus formation of Larix leptolepis Gordon [36]. miR396 expressed at high level would attenuate cell proliferation in the developing leaves of Arabidopsis thaliana [37]. miR166 has been reported to be involved in xylem development of Acacia mangium (Willd.) [38]. In Arabidopsis, miR166 was found to be involved in vascular development through negatively regulating the expression of ATHB15, a class III homeodomain-leucine zipper (HD-ZIP III) gene [39]. miRNAs including miR156, miR159, miR160, miR172, miR390, and miR482 have been confirmed to participate in the graft process of hickory [25]. Consistent with these previous research, miR156, miR160, miR166, miR171, miR390, miR396, and miR482 showed significantly differential expression for our research, suggesting they might function for graft union development.
The putative target of the differentially expressed miR156, squamosa promoter-binding protein-like (SPL), encodes a plant-specific transcription factor that functions in multiple biological processes, including plant architecture, leaf development, juvenile-to-adult transition, flower and fruit development, as well as gibberellin (GA) signaling [40]. Among its divergent functions, SPL responses to GA signaling by affecting the genes involved in GAs biosynthesis. Studies have verified that GAs are important regulators in xylem differentiation [41]. In our study, miR156 was significantly down-regulated at day 30, which might induce the up-regulation of SPL during the stage of vasculature formation. It was presumed that the miR156-SPL interaction might involve in vascular bundle formation through regulating GA signaling indirectly.
A putative target of miR160 is auxin response factor (ARF). In the graft process, auxin has been confirmed to be critical in regulating callus formation and vascular development [8,42]. Auxin signaling is transduced via ARFs to regulate the expression of genes containing auxin response elements (AuxREs) in their promoter areas [43]. In Arabidopsis, ARF6 and ARF8 mutants reduced cell proliferation in response to cutting [42], and ARF5 mutants showed abnormality in vascular development [44]. In the present study, we hypothesized that the down-regulated miR160a-b at day 30 may induce the accumulation of ARF, resulting in vascular connection.
A putative gene targeted by miR164 was the NAC transcription factor, which was in accordance with Arabidopsis [45], Medicago truncatula Gaertn [46], and Triticum aestivum L. [34]. NAC transcription factors are the master regulators in controlling secondary cell wall formation [47], and overexpression NAC1 in Arabidopsis was shown to produce thicker stems than the untransformed control plants [48]. Previous studies have reported that secondary cell-wall formation was indispensable for vascular system development [49]; thus, the down-regulated miR164b at day 30 in this work might stimulate NAC1 expression to regulate vascular development.
miR166b belongs to the miR166 family, and targets the homeobox-leucine zipper (HD-ZIP) gene. Members of HD-ZIP gene family have been reported to function in various stress conditions, such as drought, salinity, and wounding [50,51]. The class III HD-ZIP gene family plays important roles in vascular bundle development. It was reported to be highly expressed in cambium tissue [52,53]. In Arabidopsis, the class III HD-ZIP proteins were also showed to control cambium activity through inducing axial cell elongation and xylem differentiation [54]. Overexpressing a populus class III HD-ZIP gene would lead to ectopic formation of vascular cambium within cortical parenchyma in poplar [55]. For a successful grafting, the formation of vascular bundles results from the promotion of vascular cambium activity. In our study, miR166 was down-regulated at day 15, suggesting class III HD-ZIP might be up-regulated at the stage of new cambium establishment. It is speculated that the increased class III HD-ZIP may stimulate the cambium activity for the subsequent xylem formation. Interestingly, we found that miR166 was significantly down-regulated at day 8 as well, indicating that the initial xylem differentiation might happen before new cambium establishment, as demonstrated by the reports of Pina [2].
A putative target of miRS10, cinnamoyl-CoA reductase (CCR), is a gene dedicated to monolignol biosynthesis. Down-regulation of CCR in poplar exhibited up to 50% reduced lignin level in outer xylem [56]. Since lignin is essential for vascular development, the down-regulated miRS10 at day 30 might induce the up-regulation of CCR, and then lead to the lignification of vasculature during the graft process of pecan.
A predicted target of miRS26 was D-type cyclin (CYCD). CYCD is a critical regulator that promotes the progression of cell cycle by binding to cyclin-dependent kinases A, which plays vital role in cell proliferation [57]. It was found to be induced by auxin and cytokinin [58]. Arabidopsis hypocotyl explants of overexpressing CYCD4 showed faster induction of callus than the control explants on a media with lower auxin concentration [59]. In this study, miRS26 was down-regulated at day 8, a stage during initial callus formation, and then up-regulated at the following time-points, while CYCD was up-regulated throughout the grafting process, indicating that a negative correlation between miRS26 and CYCD at days 15 and 30 did not exist. However, the expressions of miRS26 and CYCD in different tissues indicated that they were negatively correlated. Considering the negative interaction exiting between miRS26 and CYCD during the stage of initial callus formation, and CYCD displaying highest expression abundance in callus tissue, we deduced that miRS26 might play a vital role in stimulating callus proliferation during graft union development.
In our study, the tissue-specific expression profiles of miRNAs and their targets might indirectly suggest their specific roles for the graft union development. The low expression of miRNAs in xylem tissues, such as miR156, miR160, miR164, miR166, and miRS10 might be indicative of their specific roles during vascular development. miRS26 showed low expression in callus tissues, suggest its possible involvement in callus formation for a successful graft.
5. Conclusions
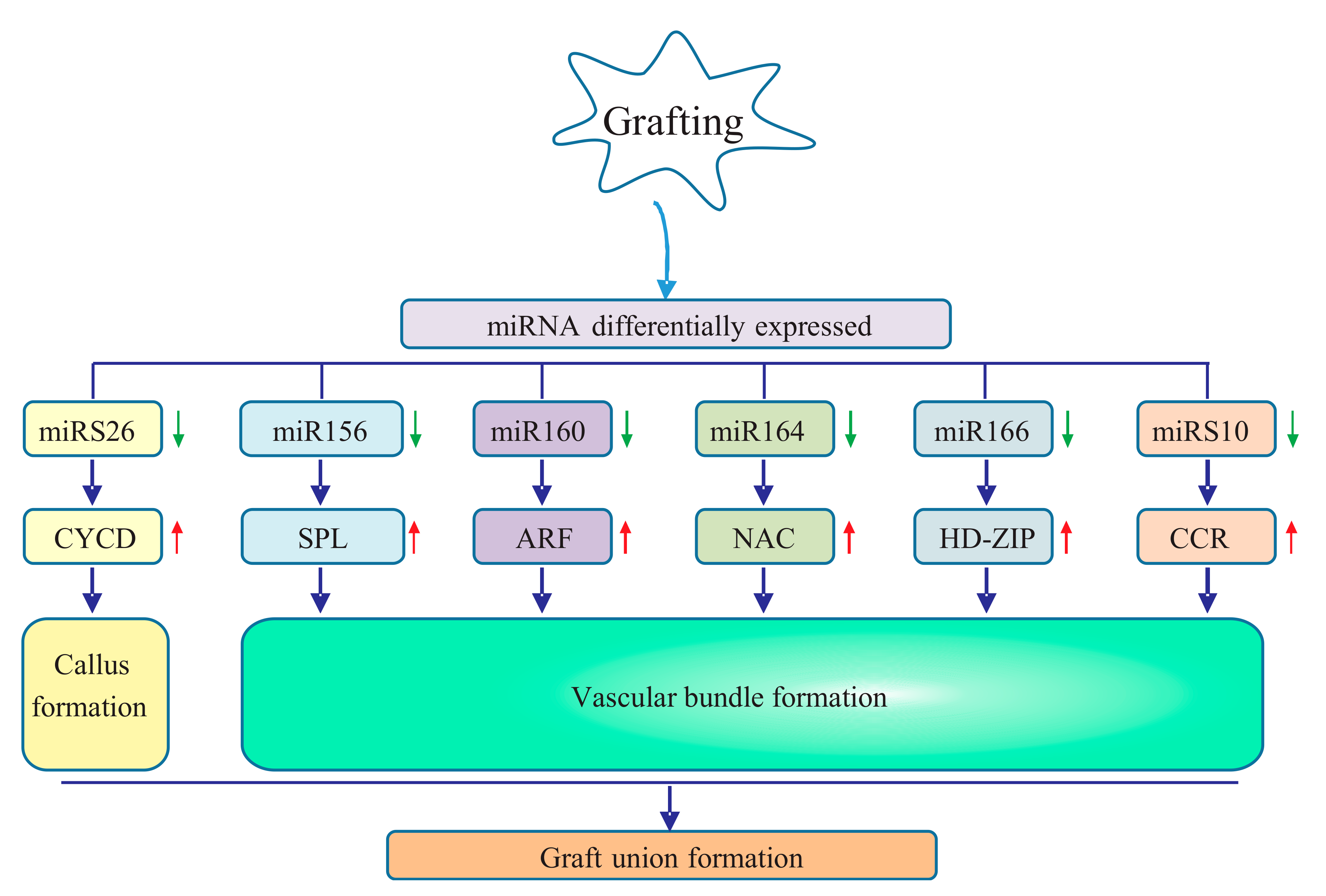
Our study constructed four sRNA libraries from the graft unions of pecan collected at days 0, 8, 15, and 30 after grafting. We identified a total of 47 conserved miRNAs belonging to 31 families and 39 novel miRNAs. Among them, 29 miRNAs with 16 conserved and 13 novel were differentially expressed in the graft process, suggesting their critical roles in successful grafting. Particularly, for the graft union development of pecan, miRS26 might play an important role in callus formation; miR166, miR156, miR160, miR164, and miRS10 might contribute to vascular bundle formation (Figure 7).
Supplementary Materials
The following are available online at https://www.mdpi.com/1999-4907/9/8/472/s1, Figure S1: The hairpin structures of novel miRNAs. The mature miRNAs are in red, and the miRNA *s are in blue, Figure S2: Correlation analysis between sequencing and qRT-PCR. Scatter plots show fold-change measured by sequencing and qRT-PCR, Table S1: Primers used in this study, Table S2: Conserved miRNAs identified in pecan, Table S3: Novel miRNAs identified in pecan, Table S4: Target genes of identified miRNAs in pecan.
Author Contributions
F.P. conceived and designed the study. Z.M. performed the data analysis and wrote the manuscript. G.F. and W.S. carried out qRT-PCR. Z.L. was involved in sample collection. All authors read and approved the final manuscript.
Funding
The authors appreciate the financial support from the SanXin project of Jiangsu province (LYSX(2016)44), the state bureau of forestry 948 project (2015-4-16) and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Conflicts of Interest
The authors declare that they have no competing interests.
References
- Mudge, K.; Janick, J.; Scofield, S.; Goldschmidt, E.E. A history of grafting. Hortic. Rev. 2009, 437–493. [Google Scholar]
- Pina, A.; Errea, P. A review of new advances in mechanism of graft compatibility–incompatibility. Sci. Hortic. 2005, 106, 1–11. [Google Scholar] [CrossRef]
- Aloni, B.; Karni, L.; Deventurero, G.; Levin, Z.; Cohen, R.; Katzir, N.; Lotan-Pompan, M.; Edelstein, M.; Aktas, H.; Turhan, E. Physiological and biochemical changes at the rootstock-scion interface in graft combinations between cucurbita rootstocks and a melon scion. J. Hortic. Sci. Biotechnol. 2008, 83, 777–783. [Google Scholar] [CrossRef]
- Fernández-García, N.; Carvajal, M.; Olmos, E. Graft union formation in tomato plants: Peroxidase and catalase involvement. Ann. Bot. 2004, 93, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Asahina, M.; Iwai, H.; Kikuchi, A.; Yamaguchi, S.; Kamiya, Y.; Kamada, H.; Satoh, S. Gibberellin produced in the cotyledon is required for cell division during tissue reunion in the cortex of cut cucumber and tomato hypocotyls. Plant Physiol. 2002, 129, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Melnyk, C.W.; Schuster, C.; Leyser, O.; Meyerowitz, E.M. A developmental framework for graft formation and vascular reconnection in arabidopsis thaliana. Curr. Biol. 2015, 25, 1306–1318. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.S.; Chu, H.L.; Jin, S.H.; Huang, Y.J.; Wang, Z.J.; Chen, M.; Huang, J.Q. Cdna-aflp analysis of gene expression in hickory (carya cathayensis) during graft process. Tree Physiol. 2009, 30, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Yan, B.; Sun, J.; Jia, P.; Zhang, Z.; Yan, X.; Chai, J.; Ren, Z.; Zheng, G.; Liu, H. Graft-union development: A delicate process that involves cell–cell communication between scion and stock for local auxin accumulation. J. Exp. Bot. 2012, 63, 4219–4232. [Google Scholar] [CrossRef] [PubMed]
- Cookson, S.J.; Moreno, M.J.C.; Hevin, C.; Mendome, L.Z.N.; Delrot, S.; Trossatmagnin, C.; Ollat, N. Graft union formation in grapevine induces transcriptional changes related to cell wall modification, wounding, hormone signalling, and secondary metabolism. J. Exp. Bot. 2013, 64, 2997–3008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Zhao, J.; Hu, F.; Qin, Y.; Wang, X.; Hu, G. Transcriptome changes between compatible and incompatible graft combination of litchi chinensis by digital gene expression profile. Sci. Rep. 2017, 7, 3954. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. Micrornas: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Mallory, A.C.; Vaucheret, H. Erratum: Functions of micrornas and related small rnas in plants. Nat. Genet. 2006, 38, S31. [Google Scholar] [CrossRef]
- Millar, A.A.; Waterhouse, P.M. Plant and animal micrornas: Similarities and differences. Funct. Integr. Genom. 2005, 5, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Jiang, Y.; Wang, N.; Xia, B.; Jiang, Y.; Li, X.; Zhang, Z.; Li, Y.; Wang, R. Identification and differential regulation of micrornas in response to methyl jasmonate treatment in lycoris aurea by deep sequencing. BMC Genom. 2016, 17, 789. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Wang, W.; Sun, X.; Liang, Z.; Wang, F. Conserved and novel heat stress-responsive micro rnas were identified by deep sequencing in s accharina japonica (l aminariales, p haeophyta). Plant Cell Environ. 2015, 38, 1357–1367. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, T.H.; Gentile, A.; Vilela, R.D.; Costa, G.G.; Dias, L.I.; Endres, L.; Menossi, M. Micrornas associated with drought response in the bioenergy crop sugarcane (Saccharum spp.). PLoS ONE 2012, 7, e46703. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Hu, T.; Amombo, E.; Fu, J. Genome-wide identification of heat stress-responsive small rnas in tall fescue (Festuca arundinacea) by high-throughput sequencing. J. Plant Physiol. 2017, 213, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Jiang, D.; Zhang, C.; Tan, H.; Li, Y.; Lv, S.; Hou, X.; Cui, X. Genome-wide identification of turnip mosaic virus -responsive micrornas in non-heading chinese cabbage by high-throughput sequencing. Gene 2015, 571, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Ma, L.; Geng, Y.; Hao, C.; Chen, X.; Zhang, X. Small rna and degradome sequencing reveal complex roles of mirnas and their targets in developing wheat grains. PLoS ONE 2015, 10, e0139658. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Qiu, Y.; Zhang, X.; Chen, X.; Shen, D.; Wang, H.; Li, X. Genome-wide identification of micrornas associated with taproot development in radish (Raphanus sativus L.). Gene 2015, 569, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Han, S.; Li, W.; Zhou, J.; Li, X.; Qi, L. Mirna regulation in fast- and slow-growing hybrid larix trees. Trees 2012, 26, 1597–1604. [Google Scholar] [CrossRef]
- Liu, N.; Yang, J.; Guo, S.; Xu, Y.; Zhang, M. Genome-wide identification and comparative analysis of conserved and novel micrornas in grafted watermelon by high-throughput sequencing. PLoS ONE 2013, 8, e57359. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, Y.; Bai, L.; Zhang, T.; He, C.; Yan, Y.; Yu, X. Grafting-responsive mirnas in cucumber and pumpkin seedlings identified by high-throughput sequencing at whole genome level. Physiol. Plant. 2014, 151, 406–422. [Google Scholar] [CrossRef] [PubMed]
- Khaldun, A.; Huang, W.; Lv, H.; Liao, S.; Zeng, S.; Wang, Y. Comparative profiling of mirnas and target gene identification in distant-grafting between tomato and lycium (goji berry). Front. Plant Sci. 2016, 7, 1475. [Google Scholar] [CrossRef] [PubMed]
- Sima, X.; Jiang, B.; Fang, J.; He, Y.; Fang, Z.; Saravana Kumar, K.M.; Ren, W.; Qiu, L.; Chen, X.; Zheng, B. Identification by deep sequencing and profiling of conserved and novel hickory micrornas involved in the graft process. Plant Biotechnol. Rep. 2015, 9, 115–124. [Google Scholar] [CrossRef]
- Nesbitt, M.L. Effect of scionwood packing moisture and cut-end scaling on pecan graft success. Horttechnology 2002, 12, 257–260. [Google Scholar]
- Mo, Z.; Feng, G.; Su, W.; Liu, Z.; Peng, F. Transcriptomic analysis provides insights into grafting union development in pecan (Carya illinoinensis). Genes 2018, 9, 71. [Google Scholar] [CrossRef] [PubMed]
- Mo, Z.; He, H.; Su, W.; Peng, F. Analysis of differentially accumulated proteins associated with graft union formation in pecan (Carya illinoensis). Sci. Hortic. 2017, 224, 126–134. [Google Scholar] [CrossRef]
- Meyers, B.C.; Axtell, M.J.; Bartel, B.; Bartel, D.P.; Baulcombe, D.; Bowman, J.L.; Cao, X.; Carrington, J.C.; Chen, X.; Green, P.J. Criteria for annotation of plant micrornas. Plant Cell 2008, 20, 3186–3190. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Huang, J.; Huang, Y.; Li, Z.; Zheng, B. Discovery and profiling of novel and conserved micrornas during flower development in carya cathayensis via deep sequencing. Planta 2012, 236, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Huang, R.; Sun, Z.; Zhang, T.; Huang, J. Identification and profiling of conserved and novel micrornas involved in oil and oleic acid production during embryogenesis in carya cathayensis sarg. Funct. Integr. Genom. 2017, 17, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, Y.; Ren, Y.; Xu, J.; Zhang, Z.; Wang, Y. Genome-wide identification of cold-responsive and new micrornas in populus tomentosa by high-throughput sequencing. Biochem. Biophys. Res. Commun. 2012, 417, 892–896. [Google Scholar] [CrossRef] [PubMed]
- Jonesrhoades, M.W.; Bartel, D.P.; Bartel, B. Micrornas and their regulatory roles in plants. Annu. Rev. Plant Biol. 2006, 57, 19–53. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Xin, Z.; Wang, Z.; Yang, Q.; Guo, S.; Guo, X.; Cao, L.; Lin, T. Identification and comparative analysis of differentially expressed mirnas in leaves of two wheat (Triticum aestivum L.) genotypes during dehydration stress. BMC Plant Biol. 2015, 15, 21. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, R.; Vaucheret, H.; Trejo, J.; Bartel, D. A diverse and evolutionarily fluid set of micrornas in arabidopsis thaliana. Genes Dev. 2006, 20, 3407–3425. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.G.; Zhou, J.; Han, S.Y.; Yang, W.H.; Li, W.F.; Wei, H.L.; Li, X.M.; Qi, L.W. Four abiotic stress-induced mirna families differentially regulated in the embryogenic and non-embryogenic callus tissues of Larix leptolepis. Biochem. Biophys. Res. Commun. 2010, 398, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, R.E.; Mecchia, M.A.; Debernardi, J.M.; Schommer, C.; Weigel, D.; Palatnik, J.F. Control of cell proliferation in arabidopsis thaliana by microrna mir396. Development 2010, 137, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.S.; Wickneswari, R. Characterization of micrornas expressed during secondary wall biosynthesis in Acacia mangium. PLoS ONE 2012, 7, e49662. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Jung, J.H.; Reyes, J.L.; Kim, Y.S.; Kim, S.Y.; Chung, K.S.; Kim, J.A.; Lee, M.; Lee, Y.; Narry Kim, V. Microrna-directed cleavage of athb15 mrna regulates vascular development in arabidopsis inflorescence stems. Plant J. 2005, 42, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.B.; Zhang, Z.L.; Liu, D.M.; Zhang, K.; Li, A.L.; Mao, L. Squamosa promoter-binding protein-like transcription factors: Star players for plant growth and development. J. Integr. Plant Biol. 2010, 52, 946–951. [Google Scholar] [CrossRef] [PubMed]
- Milhinhos, A.; Miguel, C.M. Hormone interactions in xylem development: A matter of signals. Plant Cell Rep. 2013, 32, 867–883. [Google Scholar] [CrossRef] [PubMed]
- Pitaksaringkarn, W.; Ishiguro, S.; Asahina, M.; Satoh, S. Arf6 and arf8 contribute to tissue reunion in incised arabidopsis inflorescence stems. Plant Biotechnol. 2014, 31, 49–53. [Google Scholar] [CrossRef]
- Li, S.; Xie, Z.; Hu, C.; Zhang, J. A review of auxin response factors (arfs) in plants. Front. Plant Sci. 2016, 7, 47. [Google Scholar] [CrossRef] [PubMed]
- Hardtke, C.S.; Berleth, T. The arabidopsis gene monopteros encodes a transcription factor mediating embryo axis formation and vascular development. EMBO J. 1998, 17, 1405–1411. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.S.; Xie, Q.; Fei, J.F.; Chua, N.H. Microrna directs mrna cleavage of the transcription factor nac1 to downregulate auxin signals for arabidopsis lateral root development. Plant Cell 2005, 17, 1376–1386. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Chen, L.; Wang, T.; Tian, Q.; Zhang, W. Identification of drought-responsive micrornas in medicago truncatula by genome-wide high-throughput sequencing. BMC Genom. 2011, 12, 367. [Google Scholar]
- Zhong, R.; Lee, C.; Zhou, J.; McCarthy, R.L.; Ye, Z.-H. A battery of transcription factors involved in the regulation of secondary cell wall biosynthesis in arabidopsis. Plant Cell 2008, 20, 2763–2782. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Frugis, G.; Colgan, D.; Chua, N. Arabidopsis nac1 transduces auxin signal downstream of tir1 to promote lateral root development. Genes Dev. 2000, 14, 3024–3036. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Zhong, R. Molecular control of wood formation in trees. J. Exp. Bot. 2015, 66, 4119–4131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manavella, P.A.; Dezar, C.A.; Bonaventure, G.; Baldwin, I.T.; Chan, R.L. Hahb4, a sunflower hd-zip protein, integrates signals from the jasmonic acid and ethylene pathways during wounding and biotic stress responses. Plant J. 2008, 56, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ma, Q.; Jin, X.; Peng, X.; Liu, J.; Deng, L.; Yan, H.; Sheng, L.; Jiang, H.; Cheng, B. A novel maize homeodomain–leucine zipper (hd-zip) i gene, zmhdz10, positively regulates drought and salt tolerance in both rice and arabidopsis. Plant Cell Physiol. 2014, 55, 1142–1156. [Google Scholar] [CrossRef] [PubMed]
- Baima, S.; Possenti, M.; Matteucci, A.E.; Altamura, M.M.; Ruberti, I. The arabidopsis athb-8 hd-zip protein acts as a differentiation-promotingtranscription factor of the vascular meristems. Plant Physiol. 2001, 126, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Schrader, J.; Sandberg, G. A high-resolution transcript profile across the wood-forming meristem of poplar identifies potential regulators of cambial stem cell identity. Plant Cell 2004, 16, 2278–2292. [Google Scholar] [CrossRef] [PubMed]
- Ilegems, M.; Douet, V.; Meylanbettex, M.; Uyttewaal, M.; Brand, L.; Bowman, J.L.; Stieger, P.A. Interplay of auxin, kanadi and class iii hd-zip transcription factors in vascular tissue formation. Development 2010, 137, 975–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robischon, M.; Du, J.; Miura, E.; Groover, A. The populus class iii hd zip, poprevoluta, influences cambium initiation and patterning of woody stems. Plant Physiol. 2011, 155, 1214–1225. [Google Scholar] [CrossRef] [PubMed]
- Leple, J.; Dauwe, R.; Morreel, K.; Storme, V.; Lapierre, C.; Pollet, B.; Naumann, A.; Kang, K.; Kim, H.; Ruel, K. Downregulation of cinnamoyl-coenzyme a reductase in poplar: Multiple-level phenotyping reveals effects on cell wall polymer metabolism and structure. Plant Cell 2007, 19, 3669–3691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeuchi, M.; Sugimoto, K.; Iwase, A. Plant callus: Mechanisms of induction and repression. Plant Cell 2013, 25, 3159–3173. [Google Scholar] [CrossRef] [PubMed]
- Fehér, A.; Magyar, Z. Coordination of cell division and differentiation in plants in comparison to animals. Acta Biol. Szeged. 2015, 59, 275–289. [Google Scholar]
- Kono, A.; Ohno, R.; Umeda-Hara, C.; Uchimiya, H.; Umeda, M. A distinct type of cyclin d, cycd4;2, involved in the activation of cell division in arabidopsis. Plant Cell Rep. 2006, 25, 540–545. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Size distribution of sRNAs from the libraries of days 0, 8, 15, and 30 in pecan. For each library, sRNAs were based on the total unique clean reads.
Figure 1.
Size distribution of sRNAs from the libraries of days 0, 8, 15, and 30 in pecan. For each library, sRNAs were based on the total unique clean reads.

Figure 2.
GO annotation of targets of identified miRNAs. Targets were functionally categorized by biological process, cellular component and molecular function according to the ontological definitions of the GO terms.
Figure 2.
GO annotation of targets of identified miRNAs. Targets were functionally categorized by biological process, cellular component and molecular function according to the ontological definitions of the GO terms.

Figure 3.
Expression profile of some miRNAs and their targets in the graft process. Columns in the heatmap reprent different comparisons (experiment/control: day 8/day 0, day 15/day 0, and day 30/day 0). Comparisons were made to calculate expression changes (fold change). Rows in the heatmap symbolize miRNAs or target genes. The data in the heapmap are the value of log2 (fold change). Red and green indicate up-regulation and down-regulation respectively.
Figure 3.
Expression profile of some miRNAs and their targets in the graft process. Columns in the heatmap reprent different comparisons (experiment/control: day 8/day 0, day 15/day 0, and day 30/day 0). Comparisons were made to calculate expression changes (fold change). Rows in the heatmap symbolize miRNAs or target genes. The data in the heapmap are the value of log2 (fold change). Red and green indicate up-regulation and down-regulation respectively.

Figure 4.
qRT-PCR validation of miRNAs in the graft process of pecan. The histograms and lines indicate miRNA expression results obtained by sequencing and qRT-PCR, respectively. The x-axis represents samples collected at different time points after grafting, while the y-axis represents the relative expression level of miRNAs. The expression levels of miRNAs are normalized to the level of 5.8S rRNA. The normalized miRNA levels at day 0 are arbitrarily set to 1. Data from qRT-PCR are means of three replicates and bars represent SE (standard error).
Figure 4.
qRT-PCR validation of miRNAs in the graft process of pecan. The histograms and lines indicate miRNA expression results obtained by sequencing and qRT-PCR, respectively. The x-axis represents samples collected at different time points after grafting, while the y-axis represents the relative expression level of miRNAs. The expression levels of miRNAs are normalized to the level of 5.8S rRNA. The normalized miRNA levels at day 0 are arbitrarily set to 1. Data from qRT-PCR are means of three replicates and bars represent SE (standard error).

Figure 5.
The expression of miRNAs and their targets. The relative expression levels of miRNAs and their corresponding target genes are shown in grey and green histograms, respectively. The x-axis represents samples collected at different time points after grafting, while the y-axis represents the relative expression level of miRNAs and their target genes. The expression level of miRNAs and target genes are normalized to the level of 5.8S rRNA and Actin gene. For each miRNA and target gene, the expression levels at day 0 are arbitrarily set to 1. Data from qRT-PCR are means of three replicates and bars represent SE.
Figure 5.
The expression of miRNAs and their targets. The relative expression levels of miRNAs and their corresponding target genes are shown in grey and green histograms, respectively. The x-axis represents samples collected at different time points after grafting, while the y-axis represents the relative expression level of miRNAs and their target genes. The expression level of miRNAs and target genes are normalized to the level of 5.8S rRNA and Actin gene. For each miRNA and target gene, the expression levels at day 0 are arbitrarily set to 1. Data from qRT-PCR are means of three replicates and bars represent SE.

Figure 6.
Expression of miRNAs and their targets in different tissues of pecan. The relative expression levels of miRNAs and their corresponding target genes are shown in blue and green histograms, respectively. The x-axis represents different tissues collected from pecan, while the y-axis represents the relative expression level of miRNAs and their target genes. The expression level of miRNAs and target gene are normalized to the level of 5.8S rRNA and Actin gene. For each miRNA and target gene, the expression levels at day 0 are arbitrarily set to 1. Data are means of three replicates and bars represent SE. SPL, squamosa promoter-binding protein-like; ARF, auxin response factor; NAC, NAC transcription factor; HD-ZIP, homeobox-leucine zipper; CCR, cinnamoyl-CoA reductase; CYCD, D-type cyclin.
Figure 6.
Expression of miRNAs and their targets in different tissues of pecan. The relative expression levels of miRNAs and their corresponding target genes are shown in blue and green histograms, respectively. The x-axis represents different tissues collected from pecan, while the y-axis represents the relative expression level of miRNAs and their target genes. The expression level of miRNAs and target gene are normalized to the level of 5.8S rRNA and Actin gene. For each miRNA and target gene, the expression levels at day 0 are arbitrarily set to 1. Data are means of three replicates and bars represent SE. SPL, squamosa promoter-binding protein-like; ARF, auxin response factor; NAC, NAC transcription factor; HD-ZIP, homeobox-leucine zipper; CCR, cinnamoyl-CoA reductase; CYCD, D-type cyclin.

Figure 7.
Putative regulatory mechanism involving differentially expressed miRNAs and their targets in graft union formation of pecan. The upper arrow indicates upregulation, and the down arrow represents downregulation. CYCD, D-type cyclin; SPL, squamosa promoter-binding protein-like; ARF, auxin response factor; NAC, NAC transcription factor; CCR, cinnamoyl-CoA reductase; HD-ZIP, homeobox-leucine zipper.
Figure 7.
Putative regulatory mechanism involving differentially expressed miRNAs and their targets in graft union formation of pecan. The upper arrow indicates upregulation, and the down arrow represents downregulation. CYCD, D-type cyclin; SPL, squamosa promoter-binding protein-like; ARF, auxin response factor; NAC, NAC transcription factor; CCR, cinnamoyl-CoA reductase; HD-ZIP, homeobox-leucine zipper.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Analysis of small RNAs from libraries of days 0, 8, 15, and 30 in pecan.
| Libraries | Day 0 | Day 8 | Day 15 | Day 30 |
|---|---|---|---|---|
| Raw reads | 20,691,228 | 21,849,708 | 22,850,876 | 34,439,863 |
| Clean reads | 17,060,180 | 19,032,782 | 19,780,849 | 28,632,161 |
| Unique reads | 6,579,996 | 6,865,905 | 6,935,788 | 7,815,688 |
| rRNA | 7,103,064 | 6,569,083 | 8,616,736 | 12,273,558 |
| snRNA | 0 | 1 | 1 | 54 |
| snoRNA | 4481 | 32,307 | 6889 | 6924 |
| tRNA | 278,816 | 217,321 | 280,911 | 279,464 |
| Repbase | 19,799 | 31,233 | 19,735 | 24,141 |
| Unannotated reads | 9,654,020 | 12,182,837 | 10,856,577 | 16,048,020 |
| Mapped reads | 1,506,852 | 2,537,261 | 1,759,479 | 2,290,283 |
Note: Raw reads, reads generated from squencing platform; Clean reads, reads after quality control; Unique reads, clean reads after clustering; rRNA, ribosomal ribonucleic acid; snRNA, small nuclear ribonucleic acid; tRNA, transfer RNA; Repbase, repeat sequence; Unannotated reads, reads can not align to Rfam and Repbase databases; Mapped reads, the unannotated reads that can map to reference unigenes.
Table 2.
Differentially expressed miRNAs during the graft process of pecan.
| miRNA | Mature_Sequence (5′→3′) | TPM | Log2 Fold Change (Treatment vs. Control) | Putative Target | |||||
|---|---|---|---|---|---|---|---|---|---|
| Day 0 | Day 8 | Day 15 | Day 30 | Day 8/Day 0 | Day 15/Day 0 | Day 30/Day 0 | |||
| cil-miR156 | uugacagaagauagagagcac | 1053.64 | 738.88 | 2066.88 | 496.77 | −0.51 | 0.97 | −1.08 | SPL |
| cil-miR160a | ugccuggcucccuguaugcc | 174.90 | 94.66 | 118.25 | 51.99 | −0.89 | −0.56 | −1.75 | ARF |
| cil-miR160b | ugccuggcucccugaaugcc | 191.96 | 103.34 | 185.09 | 66.43 | −0.89 | −0.05 | −1.53 | ARF |
| cil-miR164a | caugugcucuagcucuccagc | 25.59 | 21.04 | 87.41 | 20.22 | −0.28 | 1.77 | −0.34 | NAC |
| cil-miR164b | uggagaagcagggcacgugca | 2337.62 | 1761.75 | 2154.29 | 768.26 | −0.41 | −0.12 | −1.61 | NAC |
| cil-miR166b | ucggaccaggcuucauucccc | 79,283.01 | 33,244.46 | 39,632.89 | 105,566.42 | −1.25 | −1.00 | 0.41 | Class III HD-ZIP |
| cil-miR171b | agguauugauguggcucaauu | 473.50 | 270.84 | 143.96 | 147.30 | −0.81 | −1.72 | −1.68 | |
| cil-miR390 | aagcucaggagggauagcgcc | 1612.45 | 1354.18 | 1933.20 | 704.72 | −0.25 | 0.26 | −1.19 | |
| cil-miR394 | uuggcauucuguccaccucc | 695.32 | 320.80 | 719.81 | 207.95 | −1.12 | 0.05 | −1.74 | F-box only protein 6-like |
| cil-miR396b | guucaauaaagcugugggaug | 38,707.31 | 95,055.53 | 110,470.14 | 30,583.10 | 1.30 | 1.51 | −0.34 | Serine carboxypeptidase-like |
| cil-miR399 | agggcuucucuccuuuggcagg | 76.78 | 18.41 | 66.84 | 25.99 | −2.06 | −0.20 | −1.56 | |
| cil-miR482a | ggaaugggcuguuugggauga | 30,943.67 | 36,341.98 | 39,209.03 | 6830.58 | 0.23 | 0.34 | −2.18 | |
| cil-miR482b | aaugggaagauaggaaagaac | 4146.30 | 3168.52 | 3578.48 | 1614.50 | −0.39 | −0.21 | −1.36 | |
| cil-miR482c | uggacaugggugaauugguaag | 16,060.51 | 11,832.64 | 35,229.52 | 6726.61 | −0.44 | 1.13 | −1.26 | |
| cil-miR818 | cacgacgucgguuuauuuaacagg | 25.59 | 12.76 | 41.13 | 92.42 | −1.00 | 0.68 | 1.85 | |
| cil-miR860a | cauaucuuugacuauguacugau | 29.86 | 103.34 | 174.81 | 118.42 | 1.79 | 2.55 | 1.99 | |
| cil-miRS2 | uuuuguaagaucucuguguag | 452.17 | 707.33 | 1007.73 | 872.23 | 0.65 | 1.16 | 0.95 | Syntaxin-131 |
| cil-miRS6 | caaggaaaauaggcuuuugug | 221.82 | 68.37 | 10.28 | 57.76 | −1.70 | −4.43 | −1.94 | ATP synthase gamma chain |
| cil-miRS7 | caucaguuugugggauuacuuu | 46.92 | 44.70 | 174.81 | 69.32 | −0.07 | 1.90 | 0.56 | |
| cil-miRS8 | aguguggucgggaacccggaacu | 81.05 | 52.59 | 359.90 | 31.77 | −0.62 | 2.15 | −1.35 | |
| cil-miRS9a | ugacagaagagagagagcac | 34.13 | 23.67 | 25.71 | 0.00 | −0.53 | −0.41 | −6.64 | SPL |
| cil-miRS9b | ugacagaagagagagagcac | 34.13 | 23.67 | 25.71 | 0.00 | −0.53 | −0.41 | −6.64 | SPL |
| cil-miRS10 | ugacagaagagagugagcac | 1847.06 | 2987.08 | 3928.10 | 557.42 | 0.69 | 1.09 | −1.73 | Cinnamoyl-CoA reductase 1 |
| cil-miRS18 | caaaaugacuagucaaugau | 81.05 | 42.07 | 97.69 | 28.88 | −0.95 | 0.27 | −1.49 | Esterase-like isoform X1 |
| cil-miRS23 | uguuacuaguuuggcuuugauacu | 46.92 | 2.63 | 5.14 | 0.00 | −4.16 | −3.19 | −6.64 | |
| cil-miRS26 | uuuucguugcuauaaauuggu | 34.13 | 16.38 | 138.82 | 86.65 | −1.06 | 2.02 | 1.34 | cyclin-D1-1 isoform X1 |
| cil-miRS29 | gguggcuggauugaaucc | 230.35 | 199.84 | 534.72 | 5.78 | −0.20 | 1.21 | −5.32 | WD-40 repeat family protein |
| cil-miRS33 | caaaagugauuguauguaacauu | 3911.68 | 3512.98 | 3583.62 | 10,232.88 | −0.16 | −0.13 | 1.39 | Cell division control protein |
| cil-miRS38 | uggcugccaugcaucgucuagc | 4850.14 | 13,539.17 | 7979.60 | 8863.87 | 1.48 | 0.72 | 0.87 | |
Note: SPL, squamosa promoter-binding protein-like; NAC, NAC transcription factor; HD-ZIP, homeobox-leucine zipper; ATP, adenine triphosphate; CoA, coenzyme A; WD, tryptophan-aspartic acid.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mo, Z.; Feng, G.; Su, W.; Liu, Z.; Peng, F. Identification of miRNAs Associated with Graft Union Development in Pecan [Carya illinoinensis (Wangenh.) K. Koch]. Forests 2018, 9, 472. https://doi.org/10.3390/f9080472
AMA Style
Mo Z, Feng G, Su W, Liu Z, Peng F. Identification of miRNAs Associated with Graft Union Development in Pecan [Carya illinoinensis (Wangenh.) K. Koch]. Forests. 2018; 9(8):472. https://doi.org/10.3390/f9080472
Chicago/Turabian StyleMo, Zhenghai, Gang Feng, Wenchuan Su, Zhuangzhuang Liu, and Fangren Peng. 2018. "Identification of miRNAs Associated with Graft Union Development in Pecan [Carya illinoinensis (Wangenh.) K. Koch]" Forests 9, no. 8: 472. https://doi.org/10.3390/f9080472
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.