Targeting the RNA-Binding Protein QKI in Myeloid Cells Ameliorates Macrophage-Induced Renal Interstitial Fibrosis

 , , ,
, , ,  ,
,
Abstract
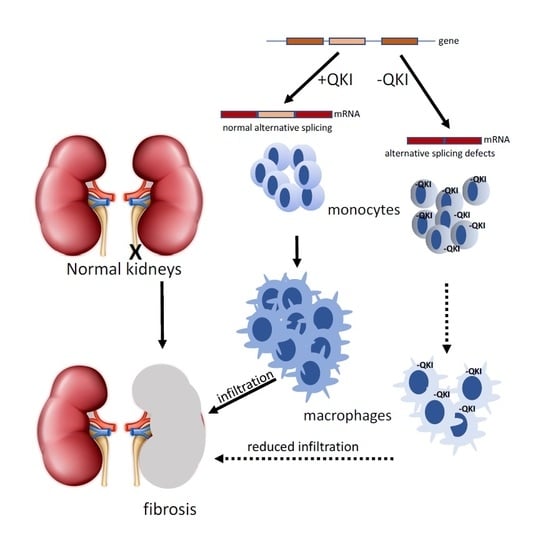
:
1. Introduction
2. Results
2.1. Abundant Expression of the QKI RNA-Binding Protein in the Kidney
2.2. Kidney Injury Induces an Influx of QKI-Expressing Macrophages
2.3. QKI Viable Mice (qkv) Show Decreased Interstitial Fibrosis upon UUO
2.4. QKIFL/FL;LysM-Cre Mice Show Decreased Interstitial Fibrosis upon UUO Kidney Damage
2.5. QKI Mediates Alternative Splicing in Mouse Macrophages
3. Discussion
4. Material and Methods
4.1. Cell Culture
4.2. Antibodies
4.3. Western Blot Analysis
4.4. RNA Isolation, cDNA Synthesis and qRT-PCR Analysis
4.5. Conditional Mouse Design and Genotyping
4.6. Immunohistochemistry
4.7. Statistical Analysis
4.8. Mouse Studies
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bielli, P.; Busà, R.; Paronetto, M.P.; Sette, C. The RNA-binding protein sam68 is a multifunctional player in human cancer. Endocr. Relat Cancer 2011, 18, R91–R102. [Google Scholar] [CrossRef] [Green Version]
- Lukong, K.E.; Chang, K.W.; Khandjian, E.W.; Richard, S. Rna-binding proteins in human genetic disease. Trends Genet. 2008, 24, 416–425. [Google Scholar] [CrossRef]
- Darbelli, L.; Vogel, G.; Almazan, G.; Richard, S. Quaking regulates neurofascin 155 expression for myelin and axoglial junction maintenance. J. Neurosci. 2016, 36, 4106–4120. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Zhou, L.; Tonissen, K.; Tee, R.; Artzt, K. The quaking i-5 (qki-5) has a novel nuclear localization signal and shuttles between the nucleus and the cytoplasm. J. Biol. Chem. 1999, 274, 29202–29210. [Google Scholar] [CrossRef] [Green Version]
- Galarneau, A.; Richard, S. Target RNA motif and target mRNAs of the quaking star protein. Nat. Struct. Mol. Biol. 2005, 12, 691–698. [Google Scholar] [CrossRef]
- van der Veer, E.P.; de Bruin, R.G.; Kraaijeveld, A.O.; de Vries, M.R.; Bot, I.; Pera, T.; Segers, F.M.; Trompet, S.; van Gils, J.M.; Roeten, M.K.; et al. Quaking, an RNA-binding protein, is a critical regulator of vascular smooth muscle cell phenotype. Circ. Res. 2013, 113, 1065–1075. [Google Scholar] [CrossRef] [Green Version]
- Hall, M.P.; Nagel, R.J.; Fagg, W.S.; Shiue, L.; Cline, M.S.; Perriman, R.J.; Donohue, J.P.; Ares, M. Quaking and ptb control overlapping splicing regulatory networks during muscle cell differentiation. RNA 2013, 19, 627–638. [Google Scholar] [CrossRef] [Green Version]
- de Bruin, R.G.; van der Veer, E.P.; Prins, J.; Lee, D.H.; Dane, M.J.; Zhang, H.; Roeten, M.K.; Bijkerk, R.; de Boer, H.C.; Rabelink, T.J. The RNA-binding protein quaking maintains endothelial barrier function and affects ve-cadherin and β-catenin protein expression. Sci. Rep. 2016, 6, 21643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Takakura, N.; Oike, Y.; Imanaka, T.; Araki, K.; Suda, T.; Kaname, T.; Kondo, T.; Abe, K.; Yamamura, K. Defective smooth muscle development in qki-deficient mice. Dev. Growth Differ. 2003, 45, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Ebersole, T.A.; Chen, Q.; Justice, M.J.; Artzt, K. The quaking gene unites signal transduction and RNA-binding in the developing nervous system. Nat. Genet. 1996, 12, 260–265. [Google Scholar] [CrossRef] [PubMed]
- de Bruin, R.G.; Rabelink, T.J.; van Zonneveld, A.J.; van der Veer, E.P. Emerging roles for RNA-binding proteins as effectors and regulators of cardiovascular disease. Eur. Heart J. 2017, 38, 1380–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Bruin, R.G.; Shiue, L.; Prins, J.; de Boer, H.C.; Singh, A.; Fagg, W.S.; van Gils, J.M.; Duijs, J.M.; Katzman, S.; Kraaijeveld, A.O.; et al. Quaking promotes monocyte differentiation into pro-atherogenic macrophages by controlling pre-mRNA splicing and gene expression. Nat. Commun. 2016, 7, 10846. [Google Scholar] [CrossRef]
- Backx, L.; Fryns, J.P.; Marcelis, C.; Devriendt, K.; Vermeesch, J.; Van Esch, H. Haploinsufficiency of the gene quaking (qki) is associated with the 6q terminal deletion syndrome. Am. J. Hum. Genet. 2010, 152A, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Zong, F.-Y.; Fu, X.; Wei, W.-J.; Luo, Y.-G.; Heiner, M.; Cao, L.-J.; Fang, Z.; Fang, R.; Lu, D.; Ji, H. The RNA-binding protein qki suppresses cancer-associated aberrant splicing. PLoS Genet. 2014, 10, e1004289. [Google Scholar] [CrossRef]
- Darbelli, L.; Richard, S. Emerging functions of the quaking RNA-binding proteins and link to human diseases. Wiley Interdiscip. Rev. RNA 2016, 7, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Bandopadhayay, P.; Ramkissoon, L.A.; Jain, P.; Bergthold, G.; Wala, J.; Zeid, R.; Schumacher, S.E.; Urbanski, L.; O’Rourke, R.; Gibson, W.J. Myb-qki rearrangements in angiocentric glioma drive tumorigenicity through a tripartite mechanism. Nat. Genet. 2016, 48, 273–282. [Google Scholar] [CrossRef]
- Lin, S.L.; Castaño, A.P.; Nowlin, B.T.; Lupher, M.L.; Duffield, J.S. Bone marrow ly6chigh monocytes are selectively recruited to injured kidney and differentiate into functionally distinct populations. J. Immunol. 2009, 183, 6733–6743. [Google Scholar] [CrossRef] [Green Version]
- Anders, H.-J.; Ryu, M. Renal microenvironments and macrophage phenotypes determine progression or resolution of renal inflammation and fibrosis. Kidney Int. 2011, 80, 915–925. [Google Scholar] [CrossRef] [Green Version]
- Wada, T.; Sakai, N.; Sakai, Y.; Matsushima, K.; Kaneko, S.; Furuichi, K. Involvement of bone-marrow-derived cells in kidney fibrosis. Clin. Exp. Nephrol. 2011, 15, 8–13. [Google Scholar] [CrossRef] [Green Version]
- Rogers, N.M.; Ferenbach, D.A.; Isenberg, J.S.; Thomson, A.W.; Hughes, J. Dendritic cells and macrophages in the kidney: A spectrum of good and evil. Nat. Rev. Nephrol. 2014, 10, 625–643. [Google Scholar] [CrossRef]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. Inflammatory processes in renal fibrosis. Nat. Rev. Nephrol. 2014, 10, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Neilson, E.G. Mechanisms of tubulointerstitial fibrosis. J. Am. Soc. Nephrol. 2010, 21, 1819–1834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levey, A.S.; Coresh, J. Chronic kidney disease. Lancet 2012, 379, 165–180. [Google Scholar] [CrossRef]
- Geissmann, F.; Manz, M.G.; Jung, S.; Sieweke, M.H.; Merad, M.; Ley, K. Development of monocytes, macrophages, and dendritic cells. Science 2010, 327, 656–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, C.; Pamer, E.G. Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 2011, 11, 762–774. [Google Scholar] [CrossRef] [Green Version]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef]
- Bijkerk, R.; de Bruin, R.G.; van Solingen, C.; van Gils, J.M.; Duijs, J.M.; van der Veer, E.P.; Rabelink, T.J.; Humphreys, B.D.; van Zonneveld, A.J. Silencing of microRNA-132 reduces renal fibrosis by selectively inhibiting myofibroblast proliferation. Kidney Int. 2016, 89, 1268–1280. [Google Scholar] [CrossRef]
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Goforth, M.H.; Morel, C.R.; Subramanian, V.; Mukundan, L.; Eagle, A.R.; Vats, D.; Brombacher, F.; Ferrante, A.W. Macrophage-specific ppar&ggr; controls alternative activation and improves insulin resistance. Nature 2007, 447, 1116–1120. [Google Scholar]
- Darbelli, L.; Choquet, K.; Richard, S.; Kleinman, C.L. Transcriptome profiling of mouse brains with qki-deficient oligodendrocytes reveals major alternative splicing defects including self-splicing. Sci. Rep. 2017, 7, 7554. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Richard, S. Structure-function analysis of qk1: A lethal point mutation in mouse quaking prevents homodimerization. Mol. Cell Biol. 1998, 18, 4863–4871. [Google Scholar] [CrossRef] [Green Version]
- Kanasty, R.; Dorkin, J.R.; Vegas, A.; Anderson, D. Delivery materials for siRNA therapeutics. Nat. Mater. 2013, 12, 967–977. [Google Scholar] [CrossRef] [PubMed]
- Shu, D.; Shu, Y.; Haque, F.; Abdelmawla, S.; Guo, P. Thermodynamically stable RNA three-way junction for constructing multifunctional nanoparticles for delivery of therapeutics. Nat. Nanotechnol. 2011, 6, 658–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, Y.; Cinier, M.; Shu, D.; Guo, P. Assembly of multifunctional phi29 pRNA nanoparticles for specific delivery of siRNA and other therapeutics to targeted cells. Methods 2011, 54, 204–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundaram, P.; Kurniawan, H.; Byrne, M.E.; Wower, J. Therapeutic RNA aptamers in clinical trials. European J. Pharm. Sci. 2013, 48, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P. Post-transcriptional control of cytokine production. Nat. Immunol. 2008, 9, 353–359. [Google Scholar] [CrossRef]
- Chen, C.-Y.; Gherzi, R.; Ong, S.-E.; Chan, E.L.; Raijmakers, R.; Pruijn, G.J.; Stoecklin, G.; Moroni, C.; Mann, M.; Karin, M. Au binding proteins recruit the exosome to degrade are-containing mRNAs. Cell 2001, 107, 451–464. [Google Scholar] [CrossRef] [Green Version]
- Leppek, K.; Schott, J.; Reitter, S.; Poetz, F.; Hammond, M.C.; Stoecklin, G. Roquin promotes constitutive mRNA decay via a conserved class of stem-loop recognition motifs. Cell 2013, 153, 869–881. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Hu, Y.; Nunez, S.; Foulkes, A.S.; Cieply, B.; Xue, C.; Gerelus, M.; Li, W.; Zhang, H.; Rader, D.J. Transcriptome-wide analysis reveals modulation of human macrophage inflammatory phenotype through alternative splicing. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1434–1447. [Google Scholar] [CrossRef]
- Liu, H.; Lorenzini, P.A.; Zhang, F.; Xu, S.; Wong, M.S.M.; Zheng, J.; Roca, X. Alternative splicing analysis in human monocytes and macrophages reveals mbnl1 as major regulator. Nucleic Acids Res. 2018, 46, 6069–6086. [Google Scholar] [CrossRef]
- Matsuoka, Y.; Li, X.; Bennett, V. Adducin: Structure, function and regulation. Cell Mol. Life Sci. 2000, 57, 884–895. [Google Scholar] [CrossRef]
- Winograd-Katz, S.E.; Fassler, R.; Geiger, B.; Legate, K.R. The integrin adhesome: From genes and proteins to human disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 273–288. [Google Scholar] [CrossRef]
- Imhof, B.A.; Aurrand-Lions, M. Adhesion mechanisms regulating the migration of monocytes. Nat. Rev. Immunol. 2004, 4, 432–444. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Xia, Y.; Jiang, R.; Chen, D.; Xu, J.; Deng, L.; Huang, X.; Wang, X.; Sun, B. Ptpro plays a dual role in hepatic ischemia reperfusion injury through feedback activation of nf-kappab. J. Hepatol. 2014, 60, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Gordon, S. The m1 and m2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waddell, S.J.; Popper, S.J.; Rubins, K.H.; Griffiths, M.J.; Brown, P.O.; Levin, M.; Relman, D.A. Dissecting interferon-induced transcriptional programs in human peripheral blood cells. PLoS ONE 2010, 5, e9753. [Google Scholar] [CrossRef] [PubMed]
- Bossi, D.; Carlomagno, F.; Pallavicini, I.; Pruneri, G.; Trubia, M.; Raviele, P.R.; Marinelli, A.; Anaganti, S.; Cox, M.C.; Viale, G.; et al. Functional characterization of a novel fgfr1op-ret rearrangement in hematopoietic malignancies. Mol. Oncol. 2014, 8, 221–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, H.; Wu, Q.; Cowell, J.K.; Ren, M. Fgfr1op2-fgfr1 induced myeloid leukemia and t-cell lymphoma in a mouse model. Haematologica 2016, 101, e91–e94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballerini, P.; Struski, S.; Cresson, C.; Prade, N.; Toujani, S.; Deswarte, C.; Dobbelstein, S.; Petit, A.; Lapillonne, H.; Gautier, E.F.; et al. Ret fusion genes are associated with chronic myelomonocytic leukemia and enhance monocytic differentiation. Leukemia 2012, 26, 2384–2389. [Google Scholar] [CrossRef] [Green Version]
- Giannandrea, M.; Parks, W.C. Diverse functions of matrix metalloproteinases during fibrosis. Dis. Model Mech. 2014, 7, 193–203. [Google Scholar] [CrossRef] [Green Version]
- Duffield, J.S.; Forbes, S.J.; Constandinou, C.M.; Clay, S.; Partolina, M.; Vuthoori, S.; Wu, S.; Lang, R.; Iredale, J.P. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J. Clin. Investig. 2005, 115, 56–65. [Google Scholar] [CrossRef] [Green Version]
- Atabai, K.; Jame, S.; Azhar, N.; Kuo, A.; Lam, M.; McKleroy, W.; DeHart, G.; Rahman, S.; Xia, D.D.; Melton, A.C. Mfge8 diminishes the severity of tissue fibrosis in mice by binding and targeting collagen for uptake by macrophages. J. Clin. Investig. 2009, 119, 3713–3722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucattelli, M.; Cavarra, E.; De Santi, M.; Tetley, T.; Martorana, P.; Lungarella, G. Collagen phagocytosis by lung alveolar macrophages in animal models of emphysema. Eur. Respir. J. 2003, 22, 728–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madsen, D.H.; Leonard, D.; Masedunskas, A.; Moyer, A.; Jürgensen, H.J.; Peters, D.E.; Amornphimoltham, P.; Selvaraj, A.; Yamada, S.S.; Brenner, D.A. M2-like macrophages are responsible for collagen degradation through a mannose receptor–mediated pathway. J. Cell Biol. 2013, 202, 951–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Kwon, J.; Popov, Y.; Gajdos, G.B.; Ordog, T.; Brekken, R.A.; Mukhopadhyay, D.; Schuppan, D.; Bi, Y.; Simonetto, D.; et al. Vascular endothelial growth factor promotes fibrosis resolution and repair in mice. Gastroenterology 2014, 146, 1339–1350 e1331. [Google Scholar] [CrossRef] [Green Version]
- Fallowfield, J.A.; Mizuno, M.; Kendall, T.J.; Constandinou, C.M.; Benyon, R.C.; Duffield, J.S.; Iredale, J.P. Scar-associated macrophages are a major source of hepatic matrix metalloproteinase-13 and facilitate the resolution of murine hepatic fibrosis. J. Immunol. 2007, 178, 5288–5295. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, P.; Pellicoro, A.; Vernon, M.A.; Boulter, L.; Aucott, R.L.; Ali, A.; Hartland, S.N.; Snowdon, V.K.; Cappon, A.; Gordon-Walker, T.T. Differential ly-6c expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc. Natl. Acad. Sci. USA 2012, 109, E3186–E3195. [Google Scholar] [CrossRef] [Green Version]
- Aldigier, J.C.; Kanjanbuch, T.; Ma, L.-J.; Brown, N.J.; Fogo, A.B. Regression of existing glomerulosclerosis by inhibition of aldosterone. J. Am. Soc. Nephrol. 2005, 16, 3306–3314. [Google Scholar] [CrossRef] [Green Version]
- Fioretto, P.; Steffes, M.W.; Sutherland, D.E.; Goetz, F.C.; Mauer, M. Reversal of lesions of diabetic nephropathy after pancreas transplantation. N. Engl. J. Med. 1998, 339, 69–75. [Google Scholar] [CrossRef]
- Pichaiwong, W.; Hudkins, K.L.; Wietecha, T.; Nguyen, T.Q.; Tachaudomdach, C.; Li, W.; Askari, B.; Kobayashi, T.; O’Brien, K.D.; Pippin, J.W.; et al. Reversibility of structural and functional damage in a model of advanced diabetic nephropathy. J. Am. Soc. Nephrol. 2013, 24, 1088–1102. [Google Scholar] [CrossRef] [Green Version]
- Sugimoto, H.; LeBleu, V.S.; Bosukonda, D.; Keck, P.; Taduri, G.; Bechtel, W.; Okada, H.; Carlson, W.; Bey, P.; Rusckowski, M. Activin-like kinase 3 is important for kidney regeneration and reversal of fibrosis. Nat. Med. 2012, 18, 396–404. [Google Scholar] [CrossRef] [Green Version]
- Bijkerk, R.; van Solingen, C.; de Boer, H.C.; van der Pol, P.; Khairoun, M.; de Bruin, R.G.; van Oeveren-Rietdijk, A.M.; Lievers, E.; Schlagwein, N.; van Gijlswijk, D.J. Hematopoietic microRNA-126 protects against renal ischemia/reperfusion injury by promoting vascular integrity. J. Am. Soc. Nephrol. 2014, 25, 1710–1722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Manufacturer | Cat. Nr. | Application |
|---|---|---|---|
| Rabbit α-QKI-5 | Millipore | AB9904 | WB + IF |
| Rabbit α-QKI-6 | Millipore | AB9906 | IF |
| Rabbit α-QKI-7 | Millipore | AB9908 | IF |
| Rat α-Meca-32 | BD Pharmingen | 550563 | IF |
| Rat α-F4/80 | Abcam | ab6640 | IF |
| Mouse α-Pan-QKI | Neuromab | 75–168 | WB |
| Mouse α-QKI-5 | Neuromab | 73–232 | WB |
| Mouse α-QKI-6 | Neuromab | 73–190 | WB |
| Mouse α-QKI-7 | Neuromab | 73–200 | WB |
| Rabbit α-CD206 | Abcam | ab64693 | WB |
| Mouse α-αSMA | Abcam | ab7817 | WB |
| Rabbit α-HistoneH3 | Abcam | ab1791 | WB |
| mRNA Expression and Splicing Primers | 5′–3′ |
|---|---|
| FW-QKI5 | GTGTATTAGGTGCGGTGGCT |
| REV-QKI5 | ATAGGTTAGTTGCCGGTGGC |
| FW-QKI6 | ACCTAGTGGTGTATTAGGTATGGCT |
| REV-QKI6 | CCGGAGGCTGCTGAGACTA |
| FW-QKI7 | ACATTGGCACCAGCTACATCA |
| REV-QKI7 | CAGCAAGTCAATGGGCTGAAAT |
| FW-EMR1 | CCTGGACGAATCCTGTGAAG |
| REV-EMR1 | GGTGGGACCACAGAGAGTTG |
| FW-CD115 | AGAGTGATGTGTGGTCCTAC |
| REV-CD115 | GTTAGCATAGTCCTGGTCTC |
| FW-COL1A1 | TGACTGGAAGAGCGGAGAGT |
| REV-COL1A1 | GTTCGGGCTGATGTACCAGT |
| FW-aSMA | CTGACAGAGGCACCACTGAA |
| REV-aSMA | CATCTCCAGAGTCCAGCACA |
| FW-exon2-QKI | GGTGGGACCCATTGTTCAGT |
| REV-exon5-QKI | AGGCTGTCTTCACCTTCAGC |
| FW-GAPDH | ACTCCCACTCTTCCACCTTC |
| REV-GAPDH | CACCACCCTGTTGCTGTAG |
| FW-ACTB | AGGTCATCACTATTGGCAACGA |
| REV-ACTB | CCAAGAAGGAAGGCTGGAAAA |
| FW-ADD3-splicing | CCACCTCCTGGAAGGAGAAC |
| REV-ADD3-splicing | CATGGAGGTGAAGCTCTTGGA |
| FW-PTPRO-splicing | ATGTGGAGCTGGCACGTTTG |
| REV-PTPRO-splicing | ACGGGGTTTGTTAGTTTCCTCT |
| FW-FGFR1OP2-splicing | CATGGCCAGCAAGAAAGATGAC |
| REV-FGFR1OP2-splicing | TTTGGTCAACATGTGCTTGC |
| FW-REPS1-splicing | AGCCAGGTGAGGTAGGTTACT |
| REV-REPS1-splicing | CTGCATGTGGATTTTGCTTGGA |
| Genotyping Primers | 5′–3′ |
|---|---|
| FW-QKI-flox | ACAGAGGCTTTTCCTGACCA |
| REV-QKI-flox | TTCAGAACCCCCACATTACC |
| FW-QKI-recombination | CCTGGAATGGTGCTTTCCTA |
| REV-QKI-recombination | TTCAGAACCCCCACATTACC |
| FW-quaking viable genotyping | TCTAAAGAGCATTTTCGAAGT |
| REV-quaking viable genotyping | TTGCTAACTGAATATTACT |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Bruin, R.G.; Vogel, G.; Prins, J.; Duijs, J.M.J.G.; Bijkerk, R.; van der Zande, H.J.P.; van Gils, J.M.; de Boer, H.C.; Rabelink, T.J.; van Zonneveld, A.J.; et al. Targeting the RNA-Binding Protein QKI in Myeloid Cells Ameliorates Macrophage-Induced Renal Interstitial Fibrosis. Epigenomes 2020, 4, 2. https://doi.org/10.3390/epigenomes4010002
de Bruin RG, Vogel G, Prins J, Duijs JMJG, Bijkerk R, van der Zande HJP, van Gils JM, de Boer HC, Rabelink TJ, van Zonneveld AJ, et al. Targeting the RNA-Binding Protein QKI in Myeloid Cells Ameliorates Macrophage-Induced Renal Interstitial Fibrosis. Epigenomes. 2020; 4(1):2. https://doi.org/10.3390/epigenomes4010002
Chicago/Turabian Stylede Bruin, Ruben G., Gillian Vogel, Jurrien Prins, Jacques M. J. G. Duijs, Roel Bijkerk, Hendrik J. P. van der Zande, Janine M. van Gils, Hetty C. de Boer, Ton J. Rabelink, Anton Jan van Zonneveld, and et al. 2020. "Targeting the RNA-Binding Protein QKI in Myeloid Cells Ameliorates Macrophage-Induced Renal Interstitial Fibrosis" Epigenomes 4, no. 1: 2. https://doi.org/10.3390/epigenomes4010002